

SUMMARY
External and intrinsic factors regulate the transcriptional profile of T helper 17 (TH17) cells, thereby affecting their pathogenic potential and revealing their context-dependent plasticity. The stimulator of interferon genes (STING), a component of the intracellular DNA-sensing pathway, triggers immune responses but remains largely unexplored in T cells. Here, we describe an intrinsic role of STING in limiting the TH17 cell pathogenic program. We demonstrate that non-pathogenic TH17 cells express higher levels of STING than those activated under pathogenic conditions. Activation of STING induces interleukin-10 (IL-10) production in TH17 cells, decreasing IL-17A and IL-23R expression in a type I interferon (IFN)-independent manner. Mechanistically, STING-induced IL-10 production partially requires aryl hydrocarbon receptor (AhR) signaling, while the decrease of IL-17A expression occurs due to a reduction of Rorγt transcriptional activity. Our findings reveal a regulatory function of STING in the TH17 cell activation program, proposing it as a valuable target to limit TH17-cell-mediated inflammation.
Graphical Abstract
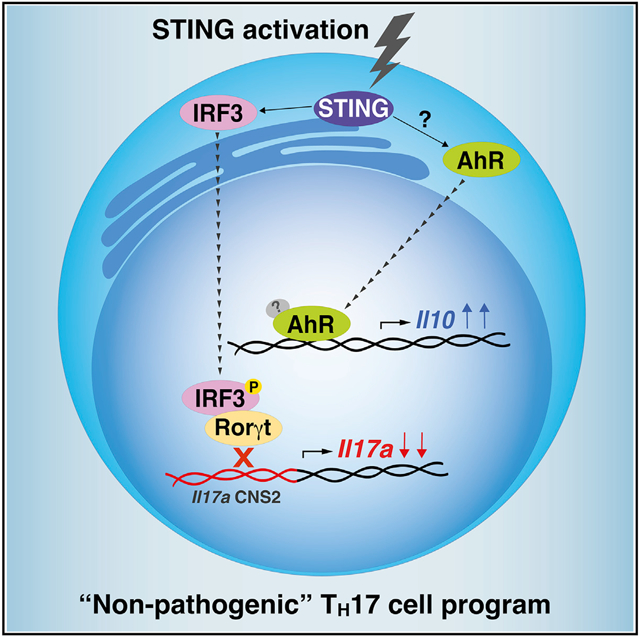
In brief
TH17 cells display a spectrum of pathogenic states depending on environmental and intrinsic cues. Damasceno et al. demonstrate that STING activation induces a non-pathogenic TH17 profile. Mechanistically, STING impairs Rorγt-mediated Il17a transcription, thereby reducing IL-17A production. Besides that, STING activation promotes IL-10 expression through AhR signaling pathway.
INTRODUCTION
T helper (TH) cells are prone to undergo transcriptional, epigenetic, and post-translational modifications induced by environmental and intrinsic factors that affect their phenotype and function (Chang et al., 2014). T helper type 17 (TH17) cells are implicated in the host immune response against pathogens, but they also mediate autoimmune inflammation, giving rise to the dual nature of “non-pathogenic” and “pathogenic” TH17 cell populations and highlighting its complex context-dependent plasticity (Stockinger and Omenetti, 2017). For instance, TH17 cells can transdifferentiate into interleukin-10 (IL-10)-producing TH17 cells after reaching the inflammatory site (Gagliani et al., 2015). Therefore, understanding the molecular mechanisms involved in TH17 cell plasticity might expand the perspectives for treating TH17-mediated inflammatory diseases.
Recognition of microbial nucleic acids is a mechanism of the innate immune system to respond against pathogens (Tan et al., 2018). The adaptor protein stimulator of interferon genes (STING) links nucleic acid cytosolic sensing with immune cell effector functions (Motwani et al., 2019). STING is activated by microbial-derived cyclic dinucleotides or generated by sensors like cGAS following recognition of cytosolic double-stranded DNA (Burdette et al., 2011; Sun et al., 2013), but it can also directly bind to DNA (Abe et al., 2013). Once activated, STING induces the expression of type I interferon (IFN) via the TBK1-IRF3 signaling pathway, contributing to antiviral response (Ishikawa and Barber, 2008; Zhong et al., 2008). Although most studies reveal the importance of STING for antimicrobial immune responses, there is some evidence that it negatively regulates inflammation. For instance, colitis-bearing STING-deficient mice develop severe disease associated with higher levels of IL-17 in the colonic tissue and decreased IL-10 expression in lymph nodes (Canesso et al., 2018). Also, STING activation is involved in the production of IL-10 needed for intestinal immune homeostasis (Ahn et al., 2017). However, while the expression of STING in T cells is documented (Cerboni et al., 2017; Gulen et al., 2017; Larkin et al., 2017), its intrinsic role in TH17 cells remains unclear.
Here, we unveil the role of STING as an intrinsic checkpoint inhibitor that restrains the TH17 cell pathogenic program.
RESULTS AND DISCUSSION
STING activation switches TH17 effector cytokine profile
To determine the role of STING in TH17 cells, we initially analyzed its expression profile in T cells. We found that both naive and T cell receptor (TCR)-activated CD4 T cells (TH0) express STING mRNA levels (encoded by the Tmem173 gene), which were higher in TH17 cells generated in vitro (Figure 1A). Protein levels of STING also increased over the time of TH17 cell differentiation (Figure 1B). To determine the role of STING in TH17 cells, we differentiated TH17 cells with IL-6 and trans-forming growth factor beta (TGF-β) in the presence of DMXAA, a murine STING agonist (Prantner et al., 2012). We also used cyclic dinucleotides c-di-AMP and c-di-GMP, which are naturally signaling molecules in bacteria that activate STING (Burdette et al., 2011). Upon activation, STING recruits TBK1 that phosphorylates both STING and IRF3, leading to type I IFN production (Liu et al., 2015; Tanaka and Chen, 2012; Zhang et al., 2019). Accordingly, we observed that all these STING signaling cascade components were activated in TH17 cells treated with DMXAA (Figure 1C). Unexpectedly, the addition of DMXAA, c-di-AMP, or c-di-GMP to the TH17 cell cultures reduced the production of IL-17A while increasing the frequency of the IL-10-expressing TH17 cell population compared with the control group (Figure 1D). Since IL-10-producing TH17 cells have been described as restraining inflammation rather than promoting disease (McGeachy et al., 2007), we hypothesized that STING activation might redirect TH17 cells to a less pathogenic phenotype.
Figure 1. STING regulates effector cytokine profile TH17 cells.

(A) Tmem173 mRNA expression in naive or TCR-activated CD4 T (TH0) and TH17 cells at 48 h of culture. Fold change relative to naive cells (n = 3).
(B) Kinetics of STING protein expression in TH17 cell during differentiation determined by immunoblot. β-actin was used as the loading control.
(C) Immunoblot analysis of STING downstream signaling components in control (medium) and DMXAA-treated (10 μM) TH17 cells collected at 72 h of culture. β-actin or GAPDH was used as the loading control.
(D) Flow-cytometric analysis of IL-10 and IL-17A expression in TH17 cells differentiated with increasing concentrations of DMXAA, c-di-AMP, or c-di-GMP for 72 h (n = 3–5).
Data are representative of at least two independent experiments and are shown as mean ± SEM. *p < 0.05 determined by one-way ANOVA followed by Tukey’s post hoc test compared with naive (A) or control (D) groups.
STING expression and activity are inversely associated with TH17 cell pathogenicity
Accumulating studies demonstrate that TH17 cells are a heterogeneous subset with a spectrum of functional states ranging from non-pathogenic (or conventional) to a pathogenic phenotype (Stockinger and Omenetti, 2017). IL-6 and TGF-β are critical inducers of conventional TH17 cells (cTH17), which fail to promote tissue inflammation (Bettelli et al., 2006; McGeachy et al., 2007). On the other hand, although IL-23 does not stimulate TH17 differentiation per se, it is crucial for inducing a pro-inflammatory pathogenic program in TH17 cells (Langrish et al., 2005; McGeachy et al., 2009). The generation of TH17 cells with a pathogenic profile (pTH17) in vitro is obtained by activation with the cytokines IL-1β, IL-6, and IL-23 in a TGF-β-independent manner (Ghoreschi et al., 2010). We used both approaches to verify differential expression and activity of the STING pathway between cTH17 and pTH17 cells. We found that the Tmem173 mRNA was intensely expressed after 48 h of culture in cTH17 cells, declining at 72 h but still high compared with at 24 h. Contrariwise, pTH17 only showed a slight and transient rise in Tmem173 expression (Figure 2A). Accordingly, STING protein expression was more pronounced in cTH17 than in pTH17 cells (Figure 2B). Of note, IL-1β, IL-6, or IL-23 alone was insufficient to increase STING expression to that level observed in cTH17 cells (Figure S1A), suggesting that the combination of IL-6 and TGF-β is the optimal signal required for STING expression in TH17 cells.
Figure 2. STING restrains the TH17 cell pathogenic program.

(A) Kinetic of Tmem173 mRNA expression in cTH17 and pTH17 cells (n = 3). Fold change relative to cTH17 at 24 h.
(B) STING protein expression in cTH17 and pTH17 cells at 72 h of culture. β-actin was used as the loading control.
(C) Flow-cytometric analysis of IL-10 and IL-17A expression in WT and STING-deficient cTH17 and pTH17 cells differentiated with DMXAA (10 mM) for 72 h (n = 3).
(D) Il10, Il23r, and Il17a mRNA expression in TH17 cells cultured as in (C) at 48 h of culture. Fold change relative to cTH17 WT control (medium).
(E) Flow-cytometric analysis of IL-10 and IL-17A expression in cTH17 cell cultured with C-176 (1 μM) overnight followed by the addition of DMXAA for 72 h (n = 3).
(F) Ifnb1, Il17a, Il23r, Il10, and Cd5l mRNA expression in cTH17 cells cultured as in (E) (n = 4). Fold change relative to control.
Data are representative of at least two independent experiments and are shown as mean ± SEM. *p < 0.05 determined by one-way ANOVA (E and F) or two-way ANOVA (A, C, and D) followed by Tukey’s post hoc test.
See also Figure S1.
We next evaluated the role of STING in both TH17 cell profiles. As shown above, STING activation increased IL-10 production concomitantly with diminished expression of IL-17A (Figure 2C). These data were corroborated by the increase of Il10 and reduction of Il17a and Il23r mRNA levels (Figure 2D), the latter being a key marker of TH17 cell pathogenic phenotype (McGeachy et al., 2009). These effects were abolished in STING-deficient T cells (Figures 2C and 2D). Similarly, treatment with C-176, a potent and selective STING inhibitor (Haag et al., 2018), blocked the effect of agonist-induced STING activation on the TH17 cell cytokine profile (Figure 2E). Also, C-176 blocked DMXAA effects on the mRNA expression of Ifnb1, Il17a, Il23r, Il10, and Cd5l (Figure 2F), the latter being a regulator of lipid metabolism linked with non-pathogenic TH17 cells (Wang et al., 2015).
Additionally, we used cells from STINGGt mice that harbor a mutated inactive STING protein (Sauer et al., 2011) and found that STINGGt TH17 cells phenocopied the cytokine profile found in STING-deficient TH17 cells (Figure S1B). Interestingly, STING-deficient TH17 cells showed even higher levels of IL-17A than wild-type (WT) cells, suggesting that STING is being activated during T cell activation in vitro. We speculated that nucleic acids released by T cells that die over the activation process or mitochondrial DNA released after cellular stress (Imanishi et al., 2014; West et al., 2015) might be potential endogenous ligands for STING activation in our setting, which merits further investigation.
STING-mediated IL-10 expression in TH17 cells partially depends on aryl hydrocarbon receptor (AhR) signaling
STING-mediated IL-10 expression is essential for controlling colitis (Ahn et al., 2017). However, how STING induces IL-10 expression was still unaddressed. Once STING is activated, it triggers the production of IFNβ (Ishikawa and Barber, 2008). Moreover, it is known that IFNβ can upregulate IL-10 expression and reduce the inflammatory response elicited by TH17 cells (Ramgolam et al., 2009; Zhang et al., 2011). We then asked whether STING restrains TH17 pathogenicity by enhancing IFN-β-IFNAR signaling. Indeed, STING activation increased Ifnb1 mRNA expression in cTH17 cells (Figure 3A). However, DMXAA still promoted IL-17A decreased levels and enhanced IL-10 production in IFNAR-deficient T cells, indicating that STING regulates TH17 cell pathogenicity through an IFNβ-IFNAR-independent mechanism (Figure 3B).
Figure 3. STING-mediated IL-10 expression in TH17 cells is partially dependent on AhR.

(A) Ifnb1 mRNA expression in DMXAA-treated WT or STING-deficient cTH17 cells at 48 h of culture (n = 3). Fold change relative to WT control.
(B) Flow cytometric analysis of IL-10 and IL-17A expression in WT or IFNAR-deficient cTH17 cells cultured with DMXAA for 72 h (n = 3).
(C) Maf, Ahr, Cyp1a1, and Ahrr mRNA expression in DMXAA-treated WT or STING-deficient cTH17 cells at 48 h of culture (n = 3). Fold change relative to WT control.
(D and E) STING and AhR protein expression in TH subsets at 72 h of culture (left). AhR protein expression in WT and STING-deficient cTH17 and pTH17 cells (right). β-actin was used as the loading control.
(F) AhR protein expression in DMXAA-treated cTH17 cells at different time points. β-actin was used as the loading control.
(G) Flow-cytometric analysis of IL-10 and IL-17A expression in WT and STING-deficient cTH17 cells cultured with DMXAA and/or CH223191 (30 μM) for 72 h (n = 3).
(H) Il17a, Il23r, Il10, Il22, and Cyp1a1 mRNA expression in cTH17 cells cultured as in (G) (n = 4). Fold change relative to WT control. Data are displayed in a heatmap.
(I) Ifnb1, Cyp1a1, Il22, Il17a, Il23r, and Il10 mRNA expression in WT and AhR-deficient cTH17 cells cultured with DMXAA for 72 h (n = 4). Fold change relative to WT control.
Data are representative of at least two independent experiments and are shown as mean ± SEM. *p < 0.05 determined by two-way ANOVA (A–C, G, and I) followed by Tukey’s post hoc test.
B lymphocyte-induced maturation protein 1 (Blimp-1), encoded by the Prdm1 gene, is a transcriptional repressor that has been shown to promote IL-10 expression in TH1 cells and TH17 cells (Heinemann et al., 2014; Neumann et al., 2014). To investigate whether STING-mediated IL-10 production in TH17 cells requires Blimp-1, we cultured WT or Blimp-1-deficient naive CD4 T cells (CD4-Cre Prdm1fl/fl) under TH17-skewing conditions with DMXAA. The absence of Blimp-1 did not affect the STING-driven IL-10 expression in TH17 cells (Figure S2), ruling out a role of Blimp-1 in this process.
AhR is a ligand-activated transcriptional factor, activated by xenobiotic compounds and endogenous ligands, involved in TH17 cell generation (Quintana et al., 2008; Veldhoen et al., 2008). Besides, AhR can also cooperate with cMaf to induce IL-10 production in TR1 cells (Apetoh et al., 2010). We found that DMXAA did not affect Maf and Ahr gene expression in cTH17 cells, but it increased Cyp1a1 and Ahrr mRNA expression, which are transcriptional targets of AhR (Figure 3C), suggesting that STING activation triggers AhR transcriptional activity. Interestingly, STING and AhR expressions were higher in cTH17 cells than in other CD4 T cell populations (Figure 3D). Furthermore, STING-deficient cTH17 and pTH17 cells showed reduced levels of AhR expression (Figure 3E). By analyzing a temporal expression of AhR protein levels after STING activation, we did not observe substantial differences until 72 h, when it was slightly decreased (Figure 3F), probably by degradation due to its increased activity (Davarinos and Pollenz, 1999). We, therefore, hypothesized that AhR might cooperate with STING signaling to induce a non-pathogenic program.
To test this hypothesis, we cultured cTH17 in the presence of a STING agonist with or without CH223191, an AhR antagonist (Kim et al., 2006). Notably, AhR inhibition partially reduced the ability of DMXAA to increase IL-10 cytokine production (Figure 3G). Accordingly, AhR inhibition reduced STING-induced rise in the Il10 mRNA levels and abrogation of Il22 and Cyp1a1, both readouts of AhR signaling (Figure 3H). As previously reported (Veldhoen et al., 2009), AhR inhibition reduced TH17 cell differentiation, and this effect was even augmented in the presence of DMXAA, as shown by the reduction of Il17a and Il23r mRNA expression (Figures 3G and 3H). We confirmed these findings using STING-activated AhR-deficient cTH17 cells, in which mRNA levels for Cyp1a1, Il22, Il17a, Il23r, and Il10 phenocopied the pharmacological blockade of AhR (Figure 3I). Of note, AhR deficiency did not affect the DMXAA-induced Ifnb1 mRNA expression (Figure 3I). On the other hand, in TH1 cells, which barely express AhR (Quintana et al., 2008; Veldhoen et al., 2008), DMXAA did not affect IL-10 expression, although it slightly reduced the frequency of IFNγ-expressing TH1 cells (Figure S3). Collectively, these data indicate that IL-10 production induced by activation of STING in TH17 cells is partially dependent on AhR.
STING activation reduces Rorγt-mediated Il17a transcription
Since exogenous IL-10 inhibits TH17 cell inflammatory functions (Huber et al., 2011; Zhang et al., 2011), we hypothesized that IL-10 produced by TH17 cells upon STING activation would affect the IL-17A expression in an autocrine/paracrine manner. However, the reduction of IL-17A expression by DMXAA was maintained in IL-10-lacking TH17 cells (Figure 4A). Moreover, activation of STING still induced downregulation of Il17a and Il23r mRNA in IL-10-deficient cells (Figure 4B). Thus, STING regulates pathogenicity of TH17 cells independent of an IL-10-induced suppressive environment.
Figure 4. STING activation limits IL-17A expression through regulation of Rorγt transcriptional activity.

(A) Flow-cytometric analysis of WT and IL-10-deficient cTH17 cells cultured with DMXAA for 72 h (n = 4).
(B) Il10, Cd5l, Il17a, Il23r, and Ifnb1 mRNA expression in cTH17 cultured as in (A) (n = 3). Fold change relative to WT control. Data are displayed in a heatmap.
(C and D) Rorγt protein expression in cTH17 cells cultured with DMXAA for 72 h as determined by flow cytometry (left; n = 4) and immunoblot (right). β-actin was used as the loading control.
(E) Ifnb1, Il17a, Il23r, Il10, and Cd5l mRNA expression in TH17 cells after a second round of culture with rmIL-23 and DMXAA for 72 h (n = 4). Fold change relative to control.
(F) Flow-cytometric analysis of IL-17A and Rorγt expression in TH17 cells cultured as in (E) with C-176 for 72 h (n = 5).
(G) Immunoprecipitation of Rorγt and IRF3. Control and DMXAA-treated cTH17 cell lysates were subjected to IP with anti-Rorγt or immunoglobulin G (IgG) control, and immunoblot was performed as indicated. GAPDH was used as the input loading control.
(H) Immunoblot analysis of phospho-IRF3, IRF3, and Rorγt in cytoplasmic and nuclear fractions from control and DMXAA-treated cTH17 cells. GAPDH and nucleophosmin (NPM) were used as the cytoplasmic and nuclear loading control, respectively.
(I) ChIP-qPCR analysis of Rorγt binding to the Il17a CNS2 enhancer region in control and DMXAA-treated cTH17 for 72 h (n = 3). Data are depicted as fold enrichment to isotype IgG control.
(J) Flow-cytometric analysis of IL-17A-TdTom+ cTH17 cells cultured with DMXAA or c-di-AM(PS)2(Rp,Rp) (15 μM) for 72 h (n = 3).
(K) Ifnb1, Il10, and Il23r mRNA expression in sorted DMXAA-treated IL-17A-TdTom+ cTH17 cells at 72 h of culture (n = 4). Fold change relative to control.
Data are representative of at least two independent experiments and are shown as mean ± SEM. *p < 0.05 determined by two-tailed Student’s t test (C, E, and I), one-way ANOVA (F and J), or two-way ANOVA (A and K) followed by Tukey’s post hoc test.
See also Figure S4.
TH17 cell differentiation and function require the activity of Rorγt, a transcriptional factor responsible for the expression of lineage signature genes such as Il17a and Il23r (Ivanov et al., 2006; Yang et al., 2008). We found that despite the STING activation causing a reduction in IL-17A production, it did not affect the expression of Rorγt (Figures 4C and 4D). Furthermore, when we exposed fully differentiated cTH17 cells (100% of Rorγt+ T cells) to IL-23 in the second round of culture, DMXAA still decreased Il17a and Il23r mRNA levels while augmenting the expression of Il10 and Cd5l mRNAs (Figure 4E). Of interest, the treatment of cell cultures with STING inhibitor (C-176) along with the agonist in the second round of culture was sufficient to revert IL-17A levels (Figure 4F). Together, these data suggest that STING activation might reduce Rorγt transcriptional activity rather than its expression during TH17 cell differentiation.
The canonical STING signaling cascade culminates in IRF3 phosphorylation and its subsequent translocation into the nucleus, triggering IFNβ production (Tanaka and Chen, 2012). STING activation induced the phosphorylation of IRF3 in cTH17 cells (Figure 1C), but IFNAR signaling was dispensable for STING-induced IL-17A reduction (Figure 3B). Of note, it was reported that IRF3 restrains IL-17A expression in CD8 T cells through direct interaction with Rorγt, hampering its recruitment to the conserved noncoding sequence 2 (CNS2) enhancer region in the Il17a locus (De Lendonck et al., 2013). In fact, the binding of Rorγt on the CNS2 enhancer region of the Il17a locus plays an indispensable role in initiating Il17a transcription (Wang et al., 2012). Hence, we postulated that STING-driven activation of IRF3 could be a mechanism by which STING activation reduces the Il17a expression in TH17 cells. Accordingly, we observed that IRF3 forms a complex with Rorγt in cTH17 cells by immunoprecipitation analysis, but STING activation did not change the extent of this physical interaction (Figure 4G). Upon activation, IRF3 undergoes dimerization and phosphorylation, which is essential to form complexes with coactivators CBP/p300 in the nucleus and enable the transcription of its target genes (Chen et al., 2008). We found that DMXAA increased the expression of phosphorylated IRF3 almost exclusively in the nucleus of cTH17 cells, which coincides with the high levels of Rorγt in the same compartment (Figure 4H). Importantly, the phosphorylation of IRF3 was abrogated in STING-deficient cells (Figure S4).
To confirm whether STING activation affects Rorγt transcriptional function, we performed the chromatin immunoprecipitation (ChIP) assay and analyzed the recruitment of Rorγt to the Il17a CNS2 enhancer region. Remarkably, DMXAA reduced the ability of Rorγt to bind to the CNS2 region (Figure 4I). As supported by a previous study (De Lendonck et al., 2013), it likely occurs via STING downstream activation of IRF3. Of note, we noticed basal levels of inactive IRF3 in the nuclear compartment of cTH17 cells (Figures 4H and S4). This is consistent with studies showing that, before its activation, IRF3 is primarily found in the cytoplasm in a monomeric autoinhibitory state, and it can shuttle between cytoplasm and nucleus due to its constitutive active nuclear localization and nuclear-export signals. Nevertheless, only the phosphorylated dimeric form of IRF3 can induce transcriptional activity (Kumar et al., 2000; Zhu et al., 2015). Thus, it is likely that only the phosphorylated nuclear IRF3 is required to inhibit Rorγt-driven Il17a transcription after STING activation.
Finally, by crossing Il17a-Cre mice (Hirota et al., 2011) with TdTomato conditional reporter mice (Madisen et al., 2010), we generated a system in which TdTomato fluorescence emission only occurs whether the cell activates the Il17a transcription program (TdTom+), reflecting transcriptional activity on Il17a locus. We observed that STING agonists reduced the frequency of IL-17A-TdTom+ T cells compared with control, indicating that STING downstream signaling reduces Il17a transcription (Figure 4J). We also sorted IL-17A-TdTom+ cells from TH17 cultures and found that STING activation reduced Il23r mRNA levels while increasing the expression of Il10 (Figure 4K).
STING signaling in undifferentiated T cells can cause antiproliferative and pro-apoptotic effects (Cerboni et al., 2017; Gulen et al., 2017; Larkin et al., 2017). In this context, apart from the TCR signal, the cytokine milieu has crucial implications over the transcriptional program that governs T cell survival and functions. For instance, IL-6 has been described to rescue T cells from death (Ayroldi et al., 1998). Thus, the apoptotic effect of STING signaling might be counterbalanced by pro-surviving signals of cytokines in T cells undergoing differentiation, especially in a disease setting. In this realm, we revealed here an intrinsic role of STING in restraining the TH17 cell pathogenic program. We demonstrated that STING activation induced the production of IL-10 in TH17 cells, decreasing IL-17A expression in a type I IFN-independent manner. This effect occurred partially through AhR-dependent signaling for the production of IL-10 along with IRF3-mediated reduction of Rorγt transcriptional activity for IL-17A. However, we cannot exclude the involvement of additional mechanisms. For instance, STING can also activate the nuclear factor κB (NF-κB) signaling pathway (Abe and Barber, 2014), which is considered an important transcriptional factor for IL-10 production in macrophages (Saraiva et al., 2005). Whether STING activation could regulate IL-10 expression via NF-κB activation remains to be determined.
STING has gained attention due to the complex and diverse biological functions it can exert upon activation by cytosolic double-stranded DNA (dsDNA) or cyclic dinucleotides (Li et al., 2017). Besides, recent reports show that some transmembrane carriers can enable an intercellular shuttle of cyclic dinucleotides into immune cells (Concepcion et al., 2022; Luteijn et al., 2019; Ritchie et al., 2019). Thus, it is reasonable to consider that cyclic dinucleotides released from host cells or even self-DNA from damaged tissue might activate STING in TH17 cells, reprograming their responses to a non-pathogenic profile as a mechanism to counteract chronic and autoimmune inflammation. In support of that, STING deficiency results in increased TH17 cell tissue infiltration in a model of chronic pancreatitis (Zhao et al., 2019).
Limitations of the study
Our data imply that STING activation restrains the TH17 pathogenic state. However, the major limitation of the current study is that we have not explored the TH17-cell-intrinsic regulatory role of STING in a disease setting. Future in vivo experiments using appropriate conditional knockout mice and animal models are needed to confirm the extent and relevance of our findings in an inflammatory process. Another caveat is that we could not clarify how STING activates AhR signaling to induce IL-10 production in TH17 cells, which merits further investigation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jose C. Alves-Filho (jcafilho@usp.br).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 wild-type (WT), IL-10 KO (B6.129P2-Il10tm1Cgn/J), IFNAR KO (B6(Cg)-Ifnar1tm1.2Ees/J), STINGGt (C57BL/6J-Tmem173gt/J), TdTomato reporter (Ai14; B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J), Il17a-Cre (Il17atm1.1(icre)Stck/J), Prdm1-floxed (B6.129-Prdm1tm1Clme/J), CD4-Cre (Tg(Cd4-Cre)1Cwi/BfluJ) mice were obtained from Jackson Laboratories. STING KO mice (Ishikawa and Barber, 2008) were kindly provided by Dr. Sergio Costa Oliveira (Federal University of Minas Gerais, Brazil), and AhR KO mice (Fernandez-Salguero et al., 1995) were kindly provided by Dr. Bernhard Ryffel (CNRS Orleans, France). All mice were maintained in specific-pathogen-free conditions at the Ribeirao Preto Medical School under controlled temperature (22–25°C), 12-h light-dark cycle, and provided with water and food ad libitum. Eight- to ten-week-old age- and sex-matched male and female mice were used for the experiments. All experiments were performed with permission from and in accordance with the Ethics Committee on Animal Use guidelines of Ribeirao Preto Medical School, University of Sao Paulo (protocol number 095/2019).
METHOD DETAILS
In vitro T cell differentiation
CD4+ T cells were enriched from mice lymph nodes and spleen with anti-CD4 microbeads by using an AutoMACS magnetic cell sorter (Miltenyi Biotec) according to the manufacturer’s protocol. Thereafter, naive CD4+CD62LhighCD44lowT cells were purified using a FACSAria III cell sorter (BD Biosciences). The sort-purified naive CD4 T cells were TCR-activated with plate-bound anti-CD3ε (4 μg/mL) and anti-CD28 (2 μg/mL) (BD Biosciences) in complete IMDM (supplemented with 10% FBS, L-glutamine, penicillin-streptomycin and β–mercaptoethanol). Skewing conditions were as follows: 1 ng/mL rhTGF-β1 (eBioscience) plus 25 ng/mL rmIL-6 (R&D Systems) for cTH17; 25 ng/mL rmIL-6, 20 ng/mL rm-IL-1β and 30 ng/mL rmIL-23 (all from R&D Systems) for pTH17; 1 ng/mL rhTGF-β1 for iTreg; 20 ng/mL rmIL-12 and 20 ng/mL rmIL-2 (both from R&D Systems) for TH1. When indicated, CH223191 (30 μM; Tocris), DMXAA (3–30 μM; Invivogen), C-176 (1 μM; Sigma-Aldrich), c-di-AM(PS)2 (Rp,Rp) (15 μM; Invivogen), c-di-AMP or c-di-GMP (both 30–100 μM; Invivogen) were used.
RNA extraction and quantitative real-time PCR
Total RNA from CD4 T cells was obtained using the RNeasy Isolation Kit according to the manufacturer’s instructions (Qiagen). Total RNA was quantified, followed by conversion to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time PCR (qPCR) was performed with SYBR Green PCR Master Mix (Applied Biosystems) using a StepOnePlus Real-Time PCR machine (Applied Biosystems). Gene expression was determined relative to Gapdh, and fold change was calculated using the 2−ΔΔT threshold cycle method. Heat maps were generated using the Morpheus web interface (https://software.broadinstitute.org/morpheus). Gene expression correlates with color intensity and data (replicates mean) were row-normalized. A list of primer pairs used for RT-qPCR is presented in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NA/LE Hamster Anti-Mouse CD3ε (clone 145-2C11) | BD Biosciences | Cat# 553057; RRID:AB_394590 |
| NA/LE Hamster Anti-Mouse CD28 (clone 37.51) | BD Biosciences | Cat# 553294; RRID:AB_394763 |
| Rat Anti-Mouse CD4-FITC (clone RM4-5) | BD Biosciences | Cat# 553047; RRID:AB_394583 |
| Rat Anti-Mouse CD4-PerCP-Cy5.5 (clone RM4-5) | BD Biosciences | Cat# 550954; RRID:AB_393977 |
| Rat Anti-Mouse CD4-APC (clone RM4-5) | BD Biosciences | Cat# 553051; RRID:AB_398528 |
| Rat Anti-Mouse CD44-APC (clone IM7) | BD Biosciences | Cat# 559250; RRID:AB_398661 |
| Rat Anti-Mouse CD62L-PE (clone MEL-14) | BD Biosciences | Cat# 553151; RRID:AB_394666 |
| Rat Anti-Mouse IL17A-BV421 (clone TC11-18H10) | BD Biosciences | Cat# 563354; RRID:AB_2687547 |
| Rat Anti-Mouse IL-17A-PE (clone TC11-18H10) | BD Biosciences | Cat# 559502; RRID:AB_397256 |
| Rat Anti-Mouse IL-17A-APC (clone TC11-18H10) | BD Biosciences | Cat# 560184; RRID:AB_1645204 |
| Rat Anti-Mouse IL-10-APC (clone JES5-16E3) | BD Biosciences | Cat# 554468; RRID:AB_398558 |
| Rat Anti-Mouse IL-10-BV421 (clone JES5-16E3) | BD Biosciences | Cat# 563276; RRID:AB_2738111 |
| Mouse Anti-Mouse Rorγt-AF647 (clone Q31-378) | BD Biosciences | Cat# 562682; RRID:AB_2687546 |
| Mouse Anti-Mouse Rorγt-PerCP-Cy5.5 (clone Q31-378) | BD Biosciences | Cat# 562683; RRID:AB_2737720 |
| Rat Anti-Mouse IL-10-PE (clone JES5-16E3) | eBioscience | Cat# 12-7101-82; RRID:AB_466176 |
| Rat Anti-Mouse IFNγ-FITC (clone XMG1.2) | eBioscience | Cat# 11-7311-82; RRID:AB_465412 |
| Rabbit Anti-Phospho-IRF3 (S396) (clone D6O1M) | Cell Signaling | Cat# 29047; RRID:AB_2773013 |
| Rabbit Anti-IRF3 (clone D83B9) | Cell Signaling | Cat# 4302; RRID:AB_1904036 |
| Rabbit Anti-Phospho-TBK1 (S172) (clone D52C2) | Cell Signaling | Cat# 5483; RRID:AB_10693472 |
| Rabbit Anti-TBK1 (clone D1B4) | Cell Signaling | Cat# 3504; RRID:AB_2255663 |
| Rabbit Anti-Phospho-STING (S365) (clone D8F4W) | Cell Signaling | Cat# 72971; RRID:AB_2799831 |
| Rabbit Anti-STING (clone D2P2F) | Cell Signaling | Cat# 13647; RRID:AB_2732796 |
| Rabbit Anti-AhR (polyclonal) | Enzo Life Sciences | Cat# BML-SA210; RRID:AB_10540536 |
| Rabbit Anti-Rorγt (clone EPR20006) | Abcam | Cat# ab207082; RRID:AB_2889310 |
| Anti-Rorγt (clone AFKJS-9) | Invitrogen | Cat# 14-6988-82; RRID:AB_1834475 |
| Mouse Anti-GAPDH (clone D4C6R) | Cell Signaling | Cat# 97166; RRID:AB_2756824 |
| Mouse Anti-βactin (clone 8H10D10) | Cell Signaling | Cat# 3700; RRID:AB_2242334 |
| Rabbit Anti-NPM (polyclonal) | Cell Signaling | Cat# 3542; RRID:AB_2155178 |
| Normal Rabbit IgG | Cell Signaling | Cat# 2729; RRID:AB_1031062 |
| Goat Anti-rabbit IgG HRP | Sigma-Aldrich | Cat# A0545; RRID:AB_257896 |
| Rabbit Anti-Mouse IgG HRP | Sigma-Aldrich | Cat# A9044; RRID:AB_258431 |
| Biological samples | ||
| HyClone™ Fetal Bovine Serum | GE Healthcare | Cat# SV30160.03 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS (Phosphate buffered saline) 1X | Corning | Cat# 21-040 |
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Cat# 130-117-043 |
| Laemmli sample buffer | Bio-Rad | Cat# 161-0737 |
| 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels | Bio-Rad | Cat# 4561094 |
| Trans-Blot Turbo Mini Nitrocellulose Transfer Packs | Bio-Rad | Cat #1704158 |
| β-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| IMDM (Iscove’s Modification of DMEM) | Corning | Cat# 15-016 |
| L-Glutamine | Corning | Cat# 25-005 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat# P4333 |
| Foxp3/Transcription Factor Staining Buffer Set | Invitrogen | Cat# 00-5523-00 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | Cat# A9418 |
| β-Mercaptoethanol for cell culture | GIBCO | Cat# 21985023 |
| Fixable Viability Dye eFluor™ 780 | Invitrogen | Cat# 65-0865-14 |
| Power SYBR Green Master Mix | Applied Biosystems | Cat# 4368708 |
| iQ™ SYBR® Green Supermix | Bio-Rad | Cat# 1708886 |
| PMA | Sigma-Aldrich | Cat# P1585 |
| Ionomycin | Sigma-Aldrich | Cat# I0634 |
| BD GolgiStop™ (containing Monensin) | BD Biosciences | Cat# 554724 |
| Recombinant mIL-6 | R&D Systems | Cat# 406-ML |
| Recombinant mIL-1β | R&D Systems | Cat# 401-ML |
| Recombinant mIL-23 | R&D Systems | Cat# 1887-ML |
| Recombinat hTGF-β1 | eBioscience | Cat# 14-8348-62 |
| Recombinant mIL-12 | R&D Systems | Cat# 419-ML |
| Recombinant mIL-2 | R&D Systems | Cat# 402-ML |
| Protease/Phosphatase Inhibitor Cocktail (100X) | Cell Signaling | Cat# 5872 |
| ECL Prime | GE Healthcare | Cat# RPN2236 |
| DMXAA | Invivogen | Cat# tlrl-dmx |
| c-di-AMP | Invivogen | Cat# tlrl-nacda |
| c-di-GMP | Invivogen | Cat# tlrl-nacdg |
| 2′3′-c-di-AM(PS)2 (Rp,Rp) | Invivogen | Cat# tlrl-nacda2r |
| C-176 | Sigma-Aldrich | Cat# SML2559 |
| CH223191 | Tocris | Cat# 3858 |
| Critical commercial assays | ||
| RNA Isolation RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| Bicinchoninic Acid (BCA) kit for Protein Determination | Sigma-Aldrich | Cat# BCA1 |
| Pierce™ Co-Immunoprecipitation Kit | Thermo Scientific | Cat# 26149 |
| NE-PER Nuclear and Cytoplasmic Extraction Kit | Thermo Scientific | Cat# 78835 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368813 |
| MAGnify™ Chromatin Immunoprecipitation System | Applied Biosystems | Cat# 492024 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | N/A |
| Mouse: IL-10 KO (B6.129P2-Il10tm1Cgn/J) | The Jackson Laboratory | Stock No: 002251 |
| Mouse: IFNAR KO (B6(Cg)-Ifnar1tm1.2Ees/J) | The Jackson Laboratory | Stock No: 028288 |
| Mouse: STINGGt (C57BL/6J-Tmem173gt/J) | The Jackson Laboratory | Stock No: 017537 |
| Mouse: Il17a-Cre (Il17atm1.1(icre)Stck/J) | The Jackson Laboratory | Stock No: 016879 |
| Mouse: Ai14 (B6.Cg-Gt(ROSA)26Sortm14 (CAG-tdTomato)Hze/J) | The Jackson Laboratory | Stock No: 007914 |
| Mouse: Prdm1 floxed (B6.129-Prdm1tm1Clme/J) | The Jackson Laboratory | Stock No: 008100 |
| Mouse: CD4-Cre Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | Stock No: 017336 |
| Mouse: STING KO | Gift from Dr. Sergio C. Oliveira | (Ishikawa and Barber, 2008) |
| Mouse: AhR KO | Gift from Dr. Bernhard Ryffel | (Fernandez-Salguero et al., 1995) |
| Oligonucleotides | ||
| Il23r fwd: 5′-GCCAAGAAGACCATTCCCGA-3′ Il23r rev: 5′-TCAGTGCTACAATCTTCTTCAGAGGACA-3′ | Merck | N/A |
| Il17a fwd: 5′-GCTCCAGAAGGCCCTCAG-3′ Il17a rev: 5′-CTTTCCCTCCGCATTGACA-3′ | Merck | N/A |
| Il22 fwd: 5′-CAGCTCCTGTCACATCAGCGGT-3′ Il22 rev: 5′-AGGTCCAGTTCCCCAATCGCCT-3′ | Merck | N/A |
| Ifnb1 fwd: 5′-CAGCTCCAAGAAAGGACGAAC-3′ Ifnb1 rev: 5′-GGCAGTGTAACTCTTCTGCAT-3′ | Merck | N/A |
| Maf fwd: 5′-AGCAGTTGGTGACCATGTCG-3′ Maf rev: 5′-TGGAGATCTCCTGCTTGAGG-3′ | Merck | N/A |
| Ahr fwd:5′-CAAATCAGAGACTGGCAGGA-3′ Ahr rev: 5′-AGAAGACCAAGGCATCTGCT-3′ | Merck | N/A |
| Ahrr fwd: 5′-ACAGGGCAGACATTGTGGTT-3′ Ahrr rev: 5′-CCTGAGGCACAGACATGAAG-3′ | Merck | N/A |
| Cd5l fwd: 5′-GAGGACACATGGATGGAATGT-3′ Cd5l rev: 5′-ACCCTTGTGTAGCACCTCCA-3′ | Merck | N/A |
| Cyp1a1 fwd: 5′-GTTCTTGGAGCTTCCCCGAT-3′ Cyp1a1 rev: 5′-CTGACACGAAGGCTGGAAGT-3′ | Merck | N/A |
| Il10 fwd: 5′-ATAACTGCACCCACTTCCCA-3′ Il10 rev: 5′-GGGCATCACTTCTACCAGGT-3′ | Merck | N/A |
| Gapdh fwd: 5′-CATCTTCTTGTGCAGTGCCA-3′ Gapdh rev: 5′-CGGCCAAATCCGTTCAC-3′ | Merck | N/A |
| Tmem173: IDT assay ID Mm.PT.58.7864131.g | IDT | N/A |
| Gapdh: IDT assay ID Mm.PT.39a.1 | IDT | N/A |
| Il17a CNS2 (ChIP) fwd: 5′-CCGTTTAGACTTGAAACCCAGTC-3′ Il17a CNS2 (ChIP) rev: 5′-GTACCTATGTGTTAGGAGGCGC-3′ | Eurofins Genomics | N/A |
| Software and algorithms | ||
| FACSuite™ software | BD Biosciences | N/A |
| BD FACSDiva™ software | BD Biosciences | N/A |
| FlowJo™ v10 | BD Biosciences | https://www.flowjo.com |
| StepOne Software v2.3 | Applied Biosystems | N/A |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| Image Lab v6.1 | Bio-Rad | https://www.bio-rad.com |
| Prism 8 | GraphPad | https://www.graphpad.com |
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed with the MAGnify™ Chromatin Immunoprecipitation System (Life Technologies). Briefly, naive CD4 T cells were cultured under cTH17-skewing conditions in the presence or absence of 10 μM DMXAA for 72 h. 4.5 × 106 cells were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M glycine. Samples were lysed and chromatin sheared using a Bioruptor Plus (Diagenode) sonication system to obtain 200- to 500-bp fragments (14 cycles, 30″ on, 30″ off, high setting). Chromatin-protein complexes were immunoprecipitated with either 5 μg anti-Rorγt (Invitrogen) or IgG control that were pre-bound to Protein A/G Dynabeads™ (Invitrogen) overnight on a rotator at 4°C. Afterward, cross-linking was reversed, and DNA was eluted and purified according to the supplier’s instructions. Purified DNA was used for qPCR analysis with iQ™SYBR Green real-time PCR kit (Bio-Rad) using primers encompassing the Il17a CNS2 enhancer region, listed in the key resources table. Data were calculated (2−(ΔCT IP − ΔCT control)) and presented as fold enrichment over IgG control.
Flow cytometry
For intracellular cytokine staining, T cells were stimulated for 4 h with phorbol 12-myristate 13-acetate (PMA) (50 ng/mL; Sigma-Aldrich) and ionomycin (500 ng/mL; Sigma-Aldrich) in the presence of a protein-transport inhibitor containing monensin (GolgiStop 1.5 μg/mL; BD Biosciences). For surface staining, cells were incubated with a fixable viability dye (Invitrogen) to exclude dead cells and fluorochrome-labeled monoclonal antibodies for the indicated markers. Thereafter, cells were fixed and permeabilized with the Foxp3 Fixation/Permeabilization Kit (Invitrogen), followed by intracellular staining. Data were acquired on FACSVerse or FACSCanto II machines (BD Biosciences) and analyzed using FlowJo software (BD Biosciences).
Immunoblot and immunoprecipitation analysis
Whole-cell lysates were obtained using RIPA lysis buffer (Sigma-Aldrich) containing protease and phosphatase inhibitor cocktail (Cell Signaling). Protein concentration was determined with the BCA protein assay (Sigma-Aldrich), and then cell lysates were mixed in Laemmli Sample Buffer (Bio-Rad) containing β–mercaptoethanol (95°C, 5 min). For separation by electrophoresis, 10 μg of total protein was loaded onto Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad) and transferred to a nitrocellulose membrane using the Trans-Blot Turbo Transfer Pack (Bio-Rad). Membranes were treated with block buffer (5% non-fat milk in 0.1% TBS-Tween-20) at room temperature for 1 h and then probed overnight at 4°C with primary antibodies against STING (1:1000), IRF3 (1:1000), TBK1 (1:1000), phospho(S365)-STING (1:500), phospho(S396)-IRF3 (1:500), phospho(S172)-TBK1 (1:500) (all from Cell Signaling), AhR (1:3000; Enzo Life Sciences) or Rorγt (1:1000; Abcam). βactin or GAPDH (1:1000; Cell Signaling) served as loading controls. All primary antibodies were detected with appropriate HRP-conjugated secondary antibodies (Sigma-Aldrich; 1:5000). Detection was performed using ECL prime reagent (GE Healthcare) and chemiluminescence signals were recorded on the ChemiDoc XRS+ imaging system (Bio-Rad). Data were analyzed with Image Lab (Bio-Rad). Cytoplasmic and nuclear fractionation was performed by using the NE-PER Extraction kit (Thermo Scientific) according to the manufacturer’s recommendations. GAPDH and NPM (1:1000; Cell Signaling) were used as cytoplasmic and nuclear loading controls, respectively. Immunoprecipitation was performed using the Pierce co-IP kit (Thermo Scientific) following the manufacturer’s protocol. Briefly, anti-Rorγt (Abcam) was immobilized using AminoLink Plus coupling resin. Equal amounts of cell lysates were precleared, followed by incubation with the antibody-coupled resin overnight at 4°C. The immunoprecipitate was eluted and subsequent immunoblot analysis was performed as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 8.0 software. Comparisons for two groups were calculated using unpaired two-tailed Student’s t-tests. Multiple-group comparisons were performed with either one-way ANOVA or two-way ANOVA followed by Tukey’s post hoc test. p value <0.05 was considered significant. Data are depicted as means ± SEM.
Supplementary Material
Highlights.
The expression of STING is inversely associated with TH17 cell pathogenic state
AhR signaling is involved in the STING-driven IL-10 expression in TH17 cells
STING activation impairs Rorγt binding to Il17a CNS2 enhancer region
ACKNOWLEDGMENTS
We thank all members of the Laboratory of Inflammation and Pain at Ribeirao Preto Medical School for technical support. This work was supported by a São Paulo Research Foundation (FAPESP) grant (no.13/08216-2 - Center for Research in Inflammatory Diseases), a National Council for Scientific and Technological Development (CNPq) grant (nos. 430823/2018-5 and 315880/2021-0), and a National Institutes of Health (NIH) grant (no. R01 AI116453). This work was also supported by FAPESP fellowships to L.E.A.D. (no. 18/17542-4), G.C.M.C. (no. 19/15070-0), and G.A.P. (no.20/04170-1).
Footnotes
DECLARATION OF INTERESTS
All authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110838.
REFERENCES
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol 88, 5328–5341. 10.1128/jvi.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, Ahn J, Gutman D, and Barber GN (2013). STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 50, 5–15. 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Son S, Oliveira SC, and Barber GN (2017). STING-dependent signaling underlies IL-10 controlled inflammatory colitis. Cell Rep. 21, 3873–3884. 10.1016/j.celrep.2017.11.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, and Kuchroo VK (2010). The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol 11, 854–861. 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Cannarile L, D’ Adamio F, Grohmann U, Delfino DV, and Riccardi C (1998). Interleukin-6 (IL-6) prevents activation-induced cell death: IL-2-Independent inhibition of fas/fasL expression and cell death. Blood 92, 4212–4219. 10.1182/blood.v92.11.4212.423k42_4212_4219. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canesso MCC, Lemos L, Neves TC, Marim FM, Castro TBR, Veloso É, Queiroz CP, Ahn J, Santiago HC, Martins FS, et al. (2018). The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. 11, 820–834. 10.1038/mi.2017.88. [DOI] [PubMed] [Google Scholar]
- Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, Picard C, Neven B, Fischer A, Amigorena S, et al. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med 214, 1769–1785. 10.1084/jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nat. Immunol 15, 1104–1115. 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE, and Lin K (2008). Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J. Mol. Biol 379, 251–260. 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion AR, Wagner LE, Zhu J, Tao AY, Yang J, Khodadadi-Jamayran A, Wang Y-H, Liu M, Rose RE, Jones DR, et al. (2022). The volume-regulated anion channel LRRC8C suppresses T cell function by regulating cyclic dinucleotide transport and STING–p53 signaling. Nat. Immunol 23, 287–302. 10.1038/s41590-021-01105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davarinos NA, and Pollenz RS (1999). Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem 274, 28708–28715. 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Pineau T, Hilbert D, McPhail T, Lee SST, Lee S, Kimura S, Nebert DW, Nebert D, Rudikoff S, et al. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726. 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, De Zoete MR, Licona-Limón P, Paiva RS, Ching T, et al. (2015). Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225. 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. (2010). Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 467, 967–971. 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat. Commun 8, 427. 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, and Ablasser A (2018). Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- Heinemann C, Heink S, Petermann F, Vasanthakumar A, Rothhammer V, Doorduijn E, Mitsdoerffer M, Sie C, Da Costa OP, Buch T, et al. (2014). IL-27 and IL-12 oppose pro-inflammatory IL-23 in CD4+ T cells by inducing Blimp1. Nat. Commun 5, 3770. 10.1038/ncomms4770. [DOI] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12, 255–263. 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, Gagliani N, Esplugues E, O’Connor W, Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, et al. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565. 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Ishihara C, Badr MESG, Hashimoto-Tane A, Kimura Y, Kawai T, Takeuchi O, Ishii KJ, Taniguchi S, Noda T, et al. (2014). Nucleic acid sensing by T cells initiates Th2 cell differentiation. Nat. Commun 5, 3566. 10.1038/ncomms4566. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Kim S-H, Henry EC, Kim D-K, Kim YH, Shin KJ, Han MS, Lee TG, Kang JK, Gasiewicz TA, Ryu SH, and Suh PG (2006). Novel compound 2-Methyl-2 H -pyrazole-3-carboxylic acid (2-methyl-4- o -tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol. Pharmacol 69, 1871–1878. 10.1124/mol.105.021832. [DOI] [PubMed] [Google Scholar]
- Kumar KP, McBride KM, Weaver BK, Dingwall C, and Reich NC (2000). Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol 20, 4159–4168. 10.1128/mcb.20.11.4159-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, and Cua DJ (2005). IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med 201, 233–240. 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A (2017). Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol 199, 397–402. 10.4049/jimmunol.1601999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lendonck LY, Tonon S, Nguyen M, Vandevenne P, Welsby I, Martinet V, Molle C, Charbonnier LM, Leo O, and Goriely S (2013). Interferon regulatory factor 3 controls interleukin-17 expression in CD8 T lymphocytes. Proc. Natl. Acad. Sci. U S A 110, E3189–E3197. 10.1073/pnas.1219221110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wilson HL, and Kiss-Toth E (2017). Regulating STING in health and disease. J. Inflamm 14, 11–21. 10.1186/s12950-017-0159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 80, 347. 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, McWhirter SM, Katibah GE, Corn JE, Woodward JJ, and Raulet DH (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. 10.1038/s41586-019-1553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, and Cua DJ (2007). TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat. Immunol 8, 1390–1397. 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, and Cua DJ (2009). The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17–producing effector T helper cells in vivo. Nat. Immunol 10, 314–324. 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, and Fitzgerald KA (2019). DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet 20, 657–674. 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- Neumann C, Heinrich F, Neumann K, Junghans V, Mashreghi MF, Ahlers J, Janke M, Rudolph C, Mockel-Tenbrinck N, Kühl AA, et al. (2014). Role of blimp-1 in programing th effector cells into IL-10 producers. J. Exp. Med 211, 1807–1819. 10.1084/jem.20131548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, and Vogel SN (2012). 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem 287, 39776–39788. 10.1074/jbc.m112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, and Weiner HL (2008). Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Ramgolam VS, Sha Y, Jin J, Zhang X, and Markovic-Plese S (2009). IFN-β inhibits Human Th17 cell differentiation. J. Immunol 183, 5418–5427. 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 75, 372–381.e5. 10.1016/j.molcel.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva M, Christensen JR, Tsytsykova AV, Goldfeld AE, Ley SC, Kioussis D, and O′Garra A (2005). Identification of a macrophage-specific chromatin signature in the IL-10 locus. J. Immunol 175, 1041–1046. 10.4049/jimmunol.175.2.1041. [DOI] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, Von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, and Vance RE (2011). The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun 79, 688–694. 10.1128/iai.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, and Omenetti S (2017). The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol 17, 535–544. 10.1038/nri.2017.50. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sun L, Chen J, and Chen ZJ (2018). Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol 72, 447–478. 10.1146/annurev-micro-102215-095605. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20. 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, and Stockinger B (2008). The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109. 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Christensen J, O’Garra A, and Stockinger B (2009). Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med 206, 43–49. 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Zu Horste GM, Pawlak M, et al. (2015). CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163, 1413–1427. 10.1016/j.cell.2015.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, Ojeda SS, Kang HS, Schluns KS, Gui J, Jetten AM, and Dong C (2012). Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity 36, 23–31. 10.1016/j.immuni.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West P, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. 10.1016/j.bpj.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. (2008). T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 28, 29–39. 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai X, and Chen ZJ (2019). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. 10.1038/s41586-019-1000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yuan S, Cheng G, and Guo B (2011). Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One 6, e28432. 10.1371/journal.pone.0028432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Manohar M, Wei Y, Pandol SJ, and Habtezion A (2019). STING signalling protects against chronic pancreatitis by modulating Th17 response. Gut 68, 1827–1837. 10.1136/gutjnl-2018-317098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, and Shu HB (2008). The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor Activation. Immunity 29, 538–550. 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zhu M, Fang T, Li S, Meng K, and Guo D (2015). Bipartite nuclear localization signal controls nuclear import and DNA-binding activity of IFN regulatory factor 3. J. Immunol 195, 289–297. 10.4049/jimmunol.1500232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.