

Abstract
The COVID-19 pandemic has triggered the first widespread vaccination campaign against a coronavirus. Many vaccinated subjects are previously naive to SARS-CoV-2; however, almost all have previously encountered other coronaviruses (CoVs), and the role of this immunity in shaping the vaccine response remains uncharacterized. Here, we use longitudinal samples and highly multiplexed serology to identify mRNA-1273 vaccine-induced antibody responses against a range of CoV Spike epitopes, in both phylogenetically conserved and non-conserved regions. Whereas reactivity to SARS-CoV-2 epitopes shows a delayed but progressive increase following vaccination, we observe distinct kinetics for the endemic CoV homologs at conserved sites in Spike S2: these become detectable sooner and decay at later time points. Using homolog-specific antibody depletion and alanine-substitution experiments, we show that these distinct trajectories reflect an evolving cross-reactive response that can distinguish rare, polymorphic residues within these epitopes. Our results reveal mechanisms for the formation of antibodies with broad reactivity against CoVs.
Keywords: multiplexed serology, SARS-CoV-2, vaccination
Graphical abstract
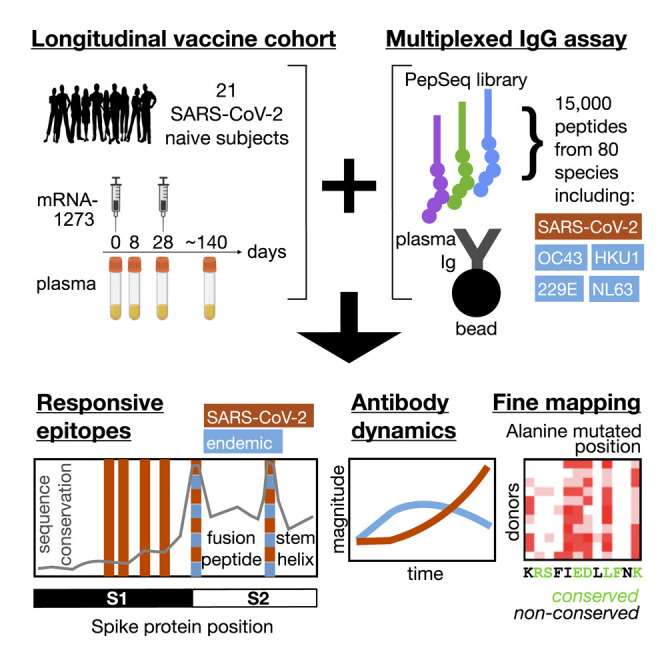
Elko et al. use a multiplexed assay to track epitope-specific antibody responses to COVID-19 vaccination. They identify conserved sites where antibodies cross-recognize endemic coronaviruses, but with divergent kinetics compared with SARS-CoV-2. The evolving recognition of polymorphic residues within otherwise conserved sites may influence the formation of broadly neutralizing antibodies.
Introduction
Prior to the COVID-19 pandemic, widespread human immunity to viruses of the Coronaviridae family was limited to four viruses: the alphacoronaviruses HCoV-229E and HCoV-NL63 and the betacoronaviruses HCoV-OC43 and HCoV-HKU1. Now, global immunization to a fifth coronavirus—SARS-CoV-2—is underway, through widespread natural infection and, increasingly, through vaccination with formulations designed to immunize against the Spike protein, sometimes alongside other viral proteins. To date, the most effective and widely studied vaccines contain a modified mRNA encoding a stabilized full-length SARS-CoV-2 Spike protein (such as mRNA-1273; Baden et al., 2021), which generates robust T and B cell responses, including high titers of neutralizing antibodies that correlate with protection against disease (Bergwerk et al., 2021). It remains incompletely understood how pre-pandemic immunity to coronaviruses shapes this newly acquired immunity against SARS-CoV-2.
One of the hallmarks of the polyclonal adaptive immune response is the generation of memory against a variety of epitopes of an infecting virus, sometimes including reactivity to conserved regions that can be recruited and matured in subsequent responses triggered by infections with heterologous viruses (White, 2021; Wong et al., 2020). Cross-reactive antibodies and memory B cells of this type support accelerated responses but can have divergent functional consequences, ranging from life-threatening antibody-dependent enhancement (exemplified by dengue virus; Halstead and O’Rourke, 1977) to “imprinting” that may constrain the specificity of subsequent responses (exemplified by influenza virus; Davenport et al., 1953; Gostic et al., 2016; Halstead and O’Rourke, 1977) and the maturation of antibodies with broad neutralizing capacity (exemplified by HIV; Liao et al., 2013).
The S1 subunit of the SARS-CoV-2 Spike protein diverges considerably in sequence from the corresponding regions of the four endemic seasonal coronaviruses, allowing the pandemic virus to evade most neutralizing antibodies raised by pre-pandemic coronavirus (CoV) exposures (Poston et al., 2021). In contrast, the S2 subunit, containing the fusion apparatus, is more conserved and contains regions of high sequence identity to both circulating betacoronaviruses and members of the alphacoronavirus genus. Consistent with this conservation, antibody responses that cross-recognize Spike proteins from pandemic and endemic CoVs have been reported (Aguilar-Bretones et al., 2021; Anderson et al., 2021), and in some cases mapped to defined epitopes within S2 (Ladner et al., 2021; Pinto et al., 2021; Shrock et al., 2020; Song et al., 2021). These cross-reactive epitopes include the fusion peptide (FP) region, whose sequence is conserved across SARS-CoV-2 and the four endemic HCoVs, and the stem-helix (SH) region, conserved across many betacoronaviruses and involved in the conformational rearrangement that mediates membrane fusion. At the SH epitope, clones with the ability to broadly neutralize across the betacoronavirus genus have been isolated (Pinto et al., 2021); however, antibodies like these appear to comprise only a small fraction of the overall neutralizing response to SARS-CoV-2 Spike (Piccoli et al., 2020). A contribution to SARS-CoV-2 neutralization by antibodies binding the FP epitope has also been reported (Poh et al., 2020).
Here, we study the interaction between immunity to endemic CoVs and SARS-CoV-2, using longitudinal epitope-resolved profiling of plasma immunoglobulin G (IgG) antibodies prior to and following vaccination with the mRNA-1273 vaccine. The highly-multiplexed assay platform PepSeq allows large programmable libraries of DNA-barcoded peptide constructs to be synthesized and assayed in massively parallel reactions (Kozlov et al., 2012; Ladner et al., 2021). We use this system to generate a human virome-wide 15,000-plex library of 30-mer peptides representing the four endemic HCoVs and SARS-CoV/SARS-CoV-2 (both members of the SARS-related human CoV species [SARSr-CoV]), map cross-reactive and non-cross-reactive Spike epitopes targeted by COVID-19 vaccination, and study the evolution of the corresponding antibody specificities over time.
Results
Epitope-resolved antibody responses to CoV spike proteins induced by vaccination
A cohort of 21 subjects with no prior SARS-CoV-2 infection history and undetectable anti-SARS-CoV-2 antibodies was recruited prior to receiving both doses of the mRNA-1273 vaccine. Blood was drawn from each subject at baseline and subsequently at post-vaccination days ∼8 (range 7–10), ∼28 (range 24–42, preceding the second dose), and ∼140 (range 133–166) (Figure 1A and Table S1). From a negative baseline, all subjects showed increases in total Spike-binding and Spike:receptor-blocking antibodies over time, beginning as early as day 8 in some cases and uniformly progressing to strong positivity by the final time point (Figure S1). To resolve the kinetics of IgG responses against conserved and non-conserved Spike epitopes, we analyzed plasma samples using the PepSeq platform (Ladner et al., 2021), in which plasma was incubated with a library containing thousands of DNA-barcoded peptides, immunoprecipitated onto protein G beads, and then analyzed through bulk amplification and high-throughput sequencing of the DNA tags to identify the IgG-binding peptides.
Figure 1.

Identification of conserved and non-conserved antibody epitopes recognized following COVID-19 vaccination
(A) A cohort of 21 subjects with no prior SARS-CoV-2 infection history and undetectable SARS-CoV-2 antibodies prior to vaccination received two doses of the mRNA-1273 vaccine and gave blood samples at 0, ∼8, ∼28, and ∼140 days relative to the first dose.
(B) Plasma from each time point was analyzed for IgG reactivity by PepSeq with a 15,000-peptide human virome library (“HV2”). Z scores for the 421 HV2 peptides designed from members of the Coronaviridae family were analyzed across subjects and time points to identify peptides showing significant time-differential signal. Each dot represents an individual peptide: the y axis shows the FDR-adjusted ANOVA p value (−log10Padj) across all time points, and the x axis shows the maximum log2 fold change (log2maxFC), which was calculated by dividing the maximum Z score at days 8, 28, and 140 by the Z score at day 0. The key in the lower right shows the number of peptides for each CoV species (note: HCoV-OC43 is a member of the Beta-CoV-1 species and SARS-CoV-2 is a member of the SARSr-CoV species). Dashed lines indicate thresholds at Padj = 0.05 and maxFC = 1.45.
(C) Mapping of differentially recognized peptides (passing both Padj and maxFC thresholds shown in [B]) to a multiple sequence alignment of Spike proteins from SARS-CoV-2, HCoV-OC43, HCoV-HKU1, HCoV-229E, and HCoV-NL63. Plots show structural features or cleavage sites (dotted green lines) of SARS-CoV-2 Spike (top) and sliding-9-mer window amino acid sequence identity to SARS-CoV-2 Spike for the endemic Spike proteins (bottom). The differentially recognized peptides map to eight epitopes in SARS-CoV-2 Spike (vertical orange boxes), two epitopes in HCoV-OC43 (blue), one epitope in HCoV-HKU1 (pink), one epitope in HCoV-229E (green), and one epitope in HCoV-NL63 (gold). Epitopes detected in the endemic CoV proteins all occur at two regions of high sequence conservation with SARS-CoV-2. The following features of Spike are highlighted: NTD, N-terminal domain; RBD, receptor binding domain; SD1, subdomain 1; SD2, subdomain 2; FP, fusion peptide; HR1, heptad repeat 1; SH, stem helix; HR2, heptad repeat 2; TM, transmembrane region.
For these studies, we designed and synthesized a PepSeq library (“HV2”) containing 15,000 30-mer peptides representing 80 common human-infecting viral species. These sequences were each selected based on some a priori evidence of reactivity, including (1) an empirical PepSeq screen conducted on a larger library of peptides, (2) published epitopes reported in the Immune Epitope Database, and (3) homology to known reactive epitopes in related species (see STAR Methods for details). Within the HV2 library, 421 peptides were designed from five CoV species (SARSr-CoV [which includes SARS-CoV and SARS-CoV-2], betacoronavirus 1 [which includes HCoV-OC43], HCoV-229E, HCoV-NL63, and HCoV-HKU1), of which 174 were derived from Spike sequences. An additional 60 sequences were included, in which two 30-mer peptides containing SARS-CoV-2 Spike epitopes were subjected to saturation alanine scanning (discussed further below). Because of the high levels of sequence identity between SARS-CoV and SARS-CoV-2, we did not attempt to differentiate between responses against these two members of the same virus species (SARSr-CoV). Moreover, no MERS-CoV peptides were included in this library as MERS-CoV infections are rare in humans and generally geographically restricted.
To identify peptides recognized by vaccine-responsive antibodies, we used ANOVA to compare the responses to each of the CoV peptides over time in each subject. Vaccine-responsive peptides were defined as those with false discovery rate (FDR)-corrected p < 0.05 and average fold induction across the cohort (maximum post-vaccine value relative to day 0) of >1.45, selected as a threshold met by none of the 247 non-Spike-derived peptides (Figure 1B). At this threshold, 23 of the 41 SARS-CoV-2 Spike peptides were strongly responsive (Figure 1B), with average fold inductions across the cohort ranging up to ∼64. In addition, 15 Spike peptides from other CoVs (nine from HCoV-OC43, one from HCoV-HKU1, two from HCoV-229E, and three from HCoV-NL63) were significantly vaccine responsive, but more weakly so, with fold inductions ranging up to ∼8.
Mapping of vaccine-responsive peptides to a multiple sequence alignment of the five Spike proteins revealed eight epitope regions (Figure 1C, vertical orange boxes): four in the S1 subunit and four in the S2 subunit (see Figure S2 for rendering on a 3D model), several of which have been previously described as targets of anti-SARS-CoV-2 reactivity (Ladner et al., 2021; Shrock et al., 2020). In our vaccine cohort, peptides derived from SARS-CoV-2 were reactive at all eight of these epitopes. In contrast, reactivity to the endemic HCoVs was restricted to two regions in the S2 subunit: the FP and the SH regions (Figure 1C, blue, pink, green, gold shading). In each case, the pattern of reactivity detected by PepSeq reflects epitope sequence conservation with SARS-CoV-2 (Figure 1C, black traces). Specifically, peptides from the FP regions of all five human-infecting CoVs were recognized by the vaccine response. The FP region is highly conserved across both the alpha- and the betacoronavirus genera. In contrast, the vaccine response recognized only SARS-CoV-2- and HCoV-OC43-derived peptides from the SH region, consistent with patterns of amino acid sequence conservation. For selected epitopes, we confirmed the fidelity of the DNA-barcoded ribosomally synthesized PepSeq probes using unbarcoded chemically synthesized peptides of the same sequences, using both peptide ELISA (Figure S3) and peptide-specific antibody depletion assays (Figure S4).
Differential kinetics of the vaccine response across epitope classes
Next, we studied the kinetics of the vaccine-induced responses across the identified epitopes. Vaccine-responsive peptides were organized into the eight epitopes described above (six non-conserved and two conserved; sequences for each are listed in Table S3) and the maximum signal for each was calculated at each time point in each subject. Time-resolved reactivity patterns partitioned into two groups: a “late-progressive” pattern for SARS-CoV-2 epitopes and an “early peak” pattern for endemic epitopes (Figures 2A and 2B). Specifically, reactivity to each of the eight SARS-CoV-2 epitopes was non-significantly increased at day 8, intermediately and variably increased at day 28, and universally and maximally increased at day 140 (Figure 2B). In contrast, reactivity to endemic homologs at the FP and SH epitopes was elevated as early as day 8, was maximal at day 28, and then declined from day 28 to day 140 (Figure 2B).
Figure 2.

Divergent kinetics of the responses to SARS-CoV-2 compared with endemic Spike antigens following vaccination
(A and B) PepSeq Z scores for the six non-cross-reactive epitopes (A) and two cross-reactive epitopes (B) across the vaccinated cohort at time points of approximately 0, 8, 28, and 140 days post-vaccination. Shown is the maximum Z score from the collection of peptides overlapping each vaccine-responsive epitope (Figure 1). The title of each plot indicates the focal epitope, named according to the features shown in Figure 1C. Each point represents the mean of two technical replicates.
(C) IgG reactivity against full-length SARS-CoV-2 and HCoV-OC43 Spike proteins in vaccinated subjects across time, detected using a fluorescent bead assay. Limit of detection for the assay is indicated by the horizontal black dotted line. Across all panels, orange boxes indicate the response to the respective SARS-CoV-2 peptide/protein, whereas gray boxes indicate reactivity to the respective endemic peptide/protein, and blue boxes indicate the response to the respective HCoV-OC43 peptide/protein. †The “endemic” category includes peptides from HCoV-OC43, HCoV-HKU1, HCoV-229E, and HCoV-NL63 for the FP epitope in (B). The limits of the boxes correspond to the first and third quartiles, the black lines inside each box correspond to the median, and the whiskers extend to points that lie within 1.5 interquartile ranges of the first and third quartiles. Days 0, 8, and 140, n = 21; day 28, n = 18. Comparisons between the indicated time point and day 0, t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Comparisons between SARS-CoV-2 and endemic epitopes within individual time points, t test: ###p < 0.001.
To determine how these epitope-level kinetics compared with the anti-Spike IgG antibody response more generally, we analyzed the same samples using multiplexed assays in which plasma IgG reactivity to full-length SARS-CoV-2 and HCoV-OC43 Spike proteins was quantified using target-conjugated fluorescent magnetic beads (MagPix, Luminex Corp.). Similar to the results of the epitope-resolved assay, we observed significant vaccine-induced antibody responses to both targets, but with different kinetics. Whereas the response to SARS-CoV-2 Spike consistently increased from day 0 to day 140, the response to the endemic betacoronavirus HCoV-OC43 was elevated at day 28 but returned to baseline by day 140 (Figure 2C). Therefore, we conclude that the response kinetics for individual peptide epitopes detected by PepSeq reflect those of the broader composite responses against the pandemic and endemic Spike proteins.
Quantification of antibody cross-reactivity at the FP and SH epitopes over time
The induction of a response against conserved regions of endemic CoV Spike proteins following vaccination with SARS-CoV-2 Spike suggests the existence of IgG antibodies capable of cross-recognizing both endemic and SARS-CoV-2 homologs at each of the FP and SH epitopes. To formally test this hypothesis, we performed cross-depletion experiments in which plasma was pre-treated with beads bearing pooled FP and SH peptides from either SARS-CoV-2 or HCoV-OC43 (hereafter the “depleting species”) to deplete virus-specific subsets of antibodies, prior to being assayed by PepSeq. In this scheme, reactivity to peptides from the depleting species was expected to decrease (relative to non-depleted samples), and any reduction in reactivity to peptides from the non-depleting species would imply the existence of antibodies capable of cross-recognizing both species (Figure 3A).
Figure 3.

Cross-reactive antibodies dominate the responses to FP and SH across time, with the emergence of late monoreactivity to SARS-CoV-2 FP
(A) To quantify the extent of cross-reactivity between SARS-CoV-2 and HCoV-OC43 at the two conserved Spike epitopes, vaccinee plasma samples were treated with beads bearing biotinylated FP + SH peptide pools of either the SARS-CoV-2 (orange) or the HCoV-OC43 (blue) sequences to deplete binding antibodies. The resulting samples were assayed by PepSeq, wherein a reduction in signal corresponding to the non-depleting species would indicate the presence of cross-reactive IgG antibodies.
(B) Change in PepSeq signal for the SARS-CoV-2 and/or HCoV-OC43 homologs of the SD1 (SARS-CoV-2 only, top), FP (middle), or SH (bottom) epitopes at the indicated post-vaccination time point, after depletion by FP + SH peptide pools of either the SARS-CoV-2 (orange) or the HcoV-OC43 (blue) sequences. The limits of the boxes correspond to the first and third quartiles, the black lines inside each box correspond to the median, and the whiskers extend to points that lie within 1.5 interquartile ranges of the first and third quartiles. Plots include only subject:time point combinations with detectable reactivity for either SARS-CoV-2 or HCoV-OC43 at the focal epitope (Z > 10). Each point represents the mean of two technical replicates. Number of samples included in each boxplot range from 1 to 10 as indicated; t test: ∗∗p < 0.01, ∗∗∗p < 0.001.
While we observed no change in signal for epitopes not included in the depleting pool (Figure 3B, top row), substantial decreases were evident in the reactivity to both the SARS-CoV-2 and the HCoV-OC43 homologs following depletion with FP and SH peptides from either species, revealing substantial cross-reactivity (Figure 3B, middle and bottom rows). Indeed, for the SH epitope, no difference was detectable at any time point in the extent of signal reduction for the depleting species compared with the non-depleting species, indicating that cross-reactive antibodies dominate the response. Similarly, signal reduction was unchanged between the depleting and the non-depleting species for the FP epitope, except at the final time point, where significantly less of the response to the SARS-CoV-2 epitope was depleted by the HCoV-OC43 peptide (Figure 3B, middle). We conclude that the responses against these two conserved epitopes are dominated across time by antibodies that cross-recognize both species, with the exception of the late response to FP, in which SARS-CoV-2 monoreactivity becomes detectable.
Evolving specificity at cross-reactive epitopes
To examine the temporal trajectories of endemic versus SARS-CoV-2 IgG specificity in individual subjects, we derived a “specificity” index for each of the cross-reactive epitopes, which represents the ratio of reactivity to the SARS-CoV-2 versus endemic peptides. To avoid noise, we restricted this analysis to cases in which there was some overall detectable reactivity at the region of interest by excluding those donor-time point combinations in which no peptide (from any species) at the focal epitope exceeded a Z-score threshold (STAR Methods). Applying this criterion, we detected FP reactivity in 3, 8, 10, and 15 donors, and SH reactivity in 4, 4, 8, and 15 donors, respectively, at days 0, 8, 28, and 140 (Figure 4A). Although this index varied widely across donors at each time point, there was a significant overall increase over time for both the FP and the SH epitopes. More specifically, when trajectories were resolved to the level of individual donors, we observed that, although responses began at a range of specificity levels, they progressively increased toward SARS-CoV-2 from day ∼8 onward.
Figure 4.

Temporal evolution of antibody specificity at the FP and SH epitopes in vaccinated and convalescent subjects
(A) Log-transformed ratio of PepSeq Z scores for the SARS-CoV-2 and endemic HCoV homologs (“specificity index”) of IgG reactivity against the FP and SH epitopes, plotted against days post-vaccination. Shown are data points in which reactivity against the focal epitope exceeds the threshold of detection (Z score ≥ 10) (in either the SARS-CoV-2 or the HCoV homologs), and lines connect data points from the same subjects.
(B) Specificity index of the antibody responses at the FP and SH epitopes in unvaccinated COVID-19 convalescent subjects dichotomized by days post-infection and analyzed using the ratio described in (A). Each point represents the mean of two technical replicates. Number of samples included in each boxplot range from 10 to 16 as indicated; t test: ∗p < 0.05, ∗∗∗p < 0.001.
To determine whether a similar temporal shift in the specificity of the antibody response at these two cross-reactive epitopes also occurs in the setting of SARS-CoV-2 infection, we studied a cohort of 46 COVID-19 convalescent subjects from whom plasma was collected at a range of time points following infection (range 17–142 days, one sample per subject) (Figures 4B and Table S2). These samples were assayed by PepSeq using the HV2 library and analyzed for FP and SH specificity using the same metric as described for the vaccinated cohort. Donor-level longitudinal analysis was unavailable, as the convalescent cohort lacked serial collections; however, dichotomization of subjects according to the number of days between diagnosis and plasma collection revealed significant increases in the specificity index in the later cohort for both the FP and the SH epitopes (p = 0.0006 and 0.044, respectively).
Fine mapping of spike residues recognized by evolving, cross-reactive responses
To study the mechanisms by which antibodies may cross-recognize—yet also evolve to distinguish—the SARS-CoV-2 and endemic homologs of the FP and SH epitopes, we used alanine-substitution scanning to map sequence dependencies of the vaccine-induced IgG responses at amino acid resolution (Figure 5 ). For each position in both epitopes, we calculated a “residue dependency index,” representing the log2 ratio of signal from the wild-type peptide versus a mutant peptide with alanine substituted at the focal site. Analysis across Spike positions 810–839 (containing FP) and 1,136–1,165 (containing SH) in day 140 post-vaccination samples revealed core regions of dependency for each epitope (positions 814–825 for FP and 1,148–1,156 for SH) that were largely consistent across donors (Figure 5A). For each epitope, the core closely matched the region of maximum sequence identity across CoV species, consistent with the observed cross-reactivity. However, for both epitopes we also observed some dependence on positions that were polymorphic between viruses: positions K814 and I818 in the FP epitope were involved in binding for most donors, and in >90% of subjects the anti-SH response was heavily dependent on the Y1155 residue. Two polymorphic sites outside of the core binding region (D830 and I834) also contributed to antibody binding in most donors at the FP epitope. Overall, the contribution of polymorphic sites, within otherwise conserved regions, to antibody binding provides a mechanism by which the response may distinguish closely related homologs at each of these cross-reactive epitope regions.
Figure 5.

Responses to FP and SH epitopes depend on both conserved and polymorphic residues
(A) PepSeq analysis of vaccinated subjects (rows) at day 140 for sets of HV2 peptides (columns) in which each residue of SARS-CoV-2 Spike at positions 810–839 (containing FP region, left) or positions 1,136–1,165 (containing SH region, right) was individually substituted to alanine. Color scale indicates the log2(ratio) of PepSeq Z scores in each subject for the wild-type peptide, divided by the respective alanine-substituted peptide (“residue dependency index”). Boxes enclose the inferred core binding regions, and the sequence letters colored in green below indicate positions that are conserved between all species shown. Stars indicate polymorphic positions that are involved in binding. Shown are subjects for whom the response to the respective wild-type peptide exceeds a Z score threshold of 10: respectively, 11 and 18 subjects for FP and SH.
(B) Analysis of the alanine-substituted FP (top) and SH (bottom) peptide sets described in (A), now comparing the responses at day 28 versus day 140 for subjects (each in a different color) whose reactivity to the wild-type peptides at both time points exceeded a Z score threshold of 10: respectively, 5 and 9 subjects for FP and SH. The y axis shows the log2 change (from the day 28 to the day 140 time point) in residue dependency (as defined in [A]), at the position shown on the x axis. Dotted horizontal lines demarcate the 99th percentile range of variation observed in a set of 1,830 control data points from the same nine subjects at 11 non-CoV epitopes to which stable (non-vaccine-induced) responses were detected and that were also each represented as sets of 30 alanine mutants and analyzed in the same way.
(C) Example scatterplots showing peptide-level reactivity at day 28 versus day 140 in two subjects with evidence for temporal changes in residue-level specificity within the SH epitope. Alanine-mutated SH peptides are highlighted in red/purple (corresponding to their respective donor’s color in [B]), the wild-type SH peptide in black, and non-SH peptides in gray. Peptides with significant temporal deviation relative to wild type (as calculated in [B]) are shown in cyan (increasing dependency on the substituted position) or orange (decreasing dependency on the substituted position) and annotated with the coordinate of the substituted residue. Dashed lines show the fit for mutant peptides without significant temporal deviation.
To study the evolution of fine specificity at the FP and SH epitopes, we compared residue dependencies between day 28 and day 140 for a subset of five and nine subjects with robustly detectable reactivity (Z ≥ 10) at both days (Figures 5B and 5C). Using a set of stably recognized non-CoV viral epitopes that were also each represented in the form of 30 alanine mutants in the HV2 library, we set a threshold that bounded 99% of changes in residue dependency between day 28 and day 140 observed under conditions presumed to be non-maturing for those epitopes (horizontal dotted lines in Figure 5B; see STAR Methods). In total, 29 and 28 events (of a total 150 and 270 subject/position combinations) were observed above this threshold for FP and SH, respectively (p < 1 × 10−9), of which 25 (86%) and 22 (79%) occurred in the inferred core regions. In some cases, residues that were essential for binding at day 28 became largely dispensable at day 140 (e.g., purple dots below the upper dotted line at SH residues S1147 and K1149 in Figure 5B), and conversely, we observed examples where positions with small contributions to binding at day 28 became substantially more important at day 140 (e.g., blue dots above the upper dotted line at SH residues D1153, Y1155, and F1156 in Figure 5B). For both epitopes, the latter category included polymorphic positions—K814 (three donors), F817 (four donors), and D830 (two donors) in FP and Y1155 (three donors) in SH–indicating their increased involvement as the response evolves. We conclude that evolution of the vaccine response to cross-reactive epitopes includes temporal changes in the fine specificity of the response, including at positions that are polymorphic between CoV species.
Discussion
Using longitudinal samples and highly multiplexed serology, we analyzed the circulating IgG antibody response following COVID-19 vaccination and its evolution over time at epitope-level resolution. We showed that vaccination elicits robust antibody responses against a range of linear epitopes from SARS-CoV-2 Spike, as well as against several Spike epitopes from other HCoVs, and that these responses partition into two classes. The first class is directed against non- or minimally conserved regions of the Spike protein and involves responses that are undetectable until day 28 post-vaccination, but then dramatically increase after the second vaccine dose. We did not observe reactivity against homologous HCoV peptides for epitopes in this class. The second class of antibodies is directed against two sites in the S2 subunit—FP and SH—that are highly conserved with Spike proteins from endemic HCoVs. Whereas reactivity to the SARS-CoV-2 sequences at these two epitopes follows the same kinetics as the first class (i.e., undetectable until day 28 and then increasing through day 140), binding to the corresponding endemic homolog epitopes is observed as soon as day 8 post-vaccination and then decreases from day 28 to day 140 (Figure 2). This epitope-resolved analysis is restricted to the subset of IgG antibodies binding linear epitopes; however, our observation of similar patterns with full-length SARS-CoV-2 and HCoV-OC43 Spike proteins (Figure 2C) suggests that the responses directed to other epitopes may follow similar kinetics.
A parsimonious explanation for these disparate kinetics among responses recognizing closely conserved homologs is that they reflect the recruitment and affinity maturation of cross-reactive memory B cell clones formed during prior encounters with the endemic HCoVs. Consistent with this model, we have previously demonstrated the existence of antibodies reactive against both the FP and the SH epitopes in SARS-CoV-2 naive donors (Ladner et al., 2021). Moreover, at both epitopes, our current results provide several lines of direct evidence for IgG antibodies capable of cross-recognizing both SARS-CoV-2 and endemic peptide sequences. Most fundamental is our observation that responses against the endemic versions of both epitopes—as well as against the full-length HCoV-OC43 Spike protein—are significantly induced following vaccination (Figure 1), indicating that antibodies stimulated by the SARS-CoV-2 immunogen also bind the endemic homologs, consistent with prior studies (Amanat et al., 2021). More specifically, in cross-depletion experiments focused on both the FP and the SH epitopes (Figure 3), we observe that beads coated with peptides corresponding to either species generally deplete equivalent fractions of the IgG reactive to the SARS-CoV-2 and endemic homologs. We observed this pattern at all post-vaccine time points for SH and at the two earliest time points for FP, indicating that the responses at these sites are generally dominated by cross-reactive antibodies. The exception to this rule is the response to SARS-CoV-2 FP at day 140, a substantial component of which is depletable by the SARS-CoV-2 sequence but not the endemic homolog. This could result from the emergence of a late monospecific reactivity at this epitope, perhaps via the acquisition of somatic mutations that reduce the endemic-specific binding of cross-reactive clones and/or the emergence of new clones specific for SARS-CoV-2 alone.
Consistent with the observed cross-reactivity, fine mapping at the FP and SH regions of Spike (Figure 5) confirms that the core IgG recognition sites reside in regions of high amino acid sequence identity, across much of the betacoronavirus genus in the case of SH and spanning the alpha- and betacoronavirus genera in the case of FP. Within each core recognition site, the SARS-CoV-2 epitopes differ by two or three amino acids from their endemic homologs, indicating residues whose differential participation in antibody binding can allow the response to distinguish between species. In support of this mechanism, our alanine-scanning results reveal several examples in which the response shows selectively increased dependence on non-conserved residues as it evolves from day 28 to day 140. The ability to focus the evolving specificity of these responses on a small minority of non-conserved residues is consistent with the sustained germinal center responses observed after infection (Dugan et al., 2021; Sokal et al., 2021) and vaccination (Turner et al., 2021) and highlights the remarkable discriminating power of such responses, which can serve as an important safeguard against the production of autoantibodies in cases of molecular mimicry between self and non-self (Burnett et al., 2018). Future studies tracking the sequences of individual cross-reactive antibody clones as they evolve, including in combination with structural analysis, will enable more detailed resolution of the molecular basis of the changing specificity between Spike S2 homologs.
The functional consequences of the cross-reactive responses to Spike S2 remain to be characterized; however, previous studies in convalescent patients (Poh et al., 2020; Pinto et al., 2021; Sauer et al., 2021) and immunized animals (Ravichandran et al., 2021; Wang et al., 2021) have shown that antibodies against both the FP and the SH epitopes have neutralizing activity against SARS-CoV-2. Exposure and incorporation of the FP region of Spike into the host cell membrane are essential steps in virus entry into cells; the corresponding site in the HA2 subunit of influenza A hemagglutinin is also conserved and has been shown to be targeted by antibodies with broad neutralizing activity (Corti et al., 2011), suggesting that such viral structures may be common targets for broadly reactive antibodies. FP is also known to be the target of an immunodominant CD4 T cell response to SARS-CoV-2 that cross-reacts with endemic CoVs (Low et al., 2021; Loyal et al., 2021).
Pinto and colleagues describe monoclonal antibodies recognizing the precise SH epitope we identify here, and their studies demonstrated that those antibodies are capable of neutralizing multiple members of the betacoronavirus genus, albeit with modest potency (Pinto et al., 2021; Sauer et al., 2021). Interestingly, however, they observed a prevalence of antibodies directed to this epitope (∼20% of vaccinees, ∼20% of convalescent subjects) that is substantially lower than the rate described here (>90% of vaccinees) and in previous work (∼50%–80% of convalescent subjects (Kaslow et al., 2014; Ladner et al., 2021; Shrock et al., 2020)). Together with evidence that antibodies against non-receptor binding domain targets generally account for <10% of the overall neutralizing activity in convalescent donors (Piccoli et al., 2020), these observations suggest that antibodies with measurable neutralizing function against multiple human-infecting CoV species may arise commonly following SARS-CoV-2 infection or vaccination, but play a limited role in overall neutralization. In addition, neutralization capacities may vary between clones binding to the same epitope. Therefore, more work is needed to fully understand the protective potential of responses against the FP and SH regions.
Our work suggests that, in addition to protection against SARS-CoV-2, COVID-19 vaccination may induce a boost in immunity against seasonal endemic CoVs, mediated by cross-reactive epitopes such as the two studied here. While future epidemiological studies will be required to determine the extent to which this response may protect against endemic HCoV infection, our results suggest that any such protection may be of limited duration, at least to the extent that it is antibody mediated. Nevertheless, it may be possible to exploit the cross-reactivity characterized here through rational immunization or therapeutic strategies designed to elicit escape-resistant immunity against diverse viruses within the Coronaviridae family.
Limitations of the study
Although we robustly detect and characterize a number of vaccine-responsive epitopes, our assay is based on unmodified linear 30-mer peptides and so is insensitive to antibodies binding conformational epitopes or requiring post-translational modifications. In addition, we have studied only bulk responses in polyclonal sera, rather than individual B cell clones. Therefore, although our depletion analysis confirms the presence of cross-reactive antibodies, we cannot quantify the degree to which clonal affinity maturation versus changes in the composition of a polyclonal repertoire contribute to the evolution of specificity that we observe.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human Plasma | Healthy participants were recruited from a local research institution and the surrounding community prior to vaccination. | WIRB#1299650 |
| Human Plasma | Unvaccinated COVID-19 convalescent individuals were recruited from participating clinical sites. | IRB#20204 |
| Chemicals, peptides, and recombinant proteins | ||
| 4 NTPs (ATP, CTP, GTP, UTP) | Lucigen | Cat# RN02825 |
| Protease Inhibitor Cocktail | VWR | Cat# G6521 |
| DYNAL MyOne Dynabeads Streptavidin T1 | Thermo Fisher Scientific | Cat# 65604D |
| AcTEV Protease | Thermo Fisher Scientific | Cat# 12575015 |
| RNase Inhibitor, Murine | New England Biolabs | Cat# M0314L |
| ProtoScript II Reverse Transcriptase | New England Biolabs | Cat# M0368X |
| RNase H | New England Biolabs | Cat# M02971 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491L |
| T4 RNA Ligase 2 | New England Biolabs | Cat# M0239L |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| Critical commercial assays | ||
| Ampliscribe T7-Flash Transcription Kit | Lucigen | Cat# ASF3507 |
| PURExpress In Vitro Protein Synthesis Kit | New England Biolabs | Cat# E6800L |
| SCoV-2 Detect™ IgG | InBios | Cat# COVE-G |
| SARS-CoV-2 Surrogate Virus Neutralization Test | GenScript | Cat# L00847-A |
| Deposited data | ||
| PepSeq read count data | Open Science Framework | https://osf.io/67r3d/ |
| Oligonucleotides | ||
| Custom 15,000 oligo library CCTATACTTCCAAGGCGCAxxxxxxxxxxxx xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxxxGGTGACTCTCTGTCTTGGC |
Agilent Technologies | G7225A |
| DNA amplification primer (Forward): GCG AATTAATACGACTCACTATAGGGCTTAA GTATAAGGAGGAAAAAATATGGGAGAA AACCTATACTTCCAAGGCGCA |
Integrated DNA Technologies | N/A |
| DNA amplification primer (Reverse): GCT CCTGCTGCATTTCCGTTCAGCAGACGC AGCAGCCAAGACAGAGAGTCACC |
Integrated DNA Technologies | N/A |
| Biotinylated oligo for bead purification: TT TTTCATATTTTTTCCTCCTTATACTTAAGCCC |
Integrated DNA Technologies | N/A |
| Software and algorithms | ||
| Peptide design algorithm | Zenodo | https://doi.org/10.5281/zenodo.6613749 |
| PepSIRF (version 1.3.2) | Zenodo | https://doi.org/10.5281/zenodo.6609194 |
| Custom Python scripts (PepSIRF/extensions) | Zenodo | https://doi.org/10.5281/zenodo.6613749 |
| R version 4.0.2 | R project | https://www.r-project.org/ |
| PyMol 2.3.2 | Schrodinger LLC | https://pymol.org |
| Other | ||
| NextSeq 500/500 Mid Output Kit v2.5 (150 Cycle) | Illumina | Cat# 20024904 |
| NextSeq 500/500 Mid Output Kit v2.5 (75 Cycle) | Illumina | Cat# 20024906 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Altin (jaltin@tgen.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study subjects
Under an IRB-approved study (WIRB#1299650), 21 healthy participants were recruited from a local research institution and the surrounding community. Subjects donated blood prior to their first dose (baseline) of the Moderna COVID-19 vaccine (mRNA-1273), approximately 8 days following the first dose (“day 8”), just prior to the second dose (“day 28”), and then approximately 140 days from baseline (“day 140”). The characteristics and exact collection timepoints for each donor are listed in Table S1.
Under a separate IRB-approved study (IRB#20204, NCT04497779), unvaccinated COVID-19 convalescent individuals were recruited from participating clinical sites. Participants all experienced mild COVID-19 symptoms and donated blood within 7–142 days of their initial PCR-based diagnosis. Age, gender, diagnosis, and sample collection dates are listed in Table S2.
Method details
ELISA assays
Total SARS-CoV-2 Spike-binding or RBD:ACE2 inhibiting antibody levels were quantified in plasma using ELISA kits: SCoV-2 Detect™ IgG (InBios) or SARS-CoV-2 Surrogate Virus Neutralization Test (GenScript), respectively, according to the manufacturer’s instructions. Peptide-specific ELISAs were performed using chemically synthesized N-terminally biotinylated 20-mer peptides (Sigma) with sequences from SARS-CoV-2 epitopes SH ([Btn]_LQPELDSFKEELDKYFKNHT) and SD2 ([Btn]_EVPVAIHADQLTPTWRVYSTGSNVFQTRAG). NeutrAvidin-coated plates (Pierce) were coated with peptide diluted to 5 μg/mL in PBS and incubated overnight at 4°C. Next, plates were blocked with 1X Blocker BSA (Thermo) in PBS for 1 h at room temperature (RT). Plasma samples diluted in Superblock TBST (Pierce) were added to wells and incubated for 1 h at 37°C. Goat anti-human IgG-HRP (Abcam) was applied to allwells and incubated for 30 min at 37°C. Ultra TMB (Thermo) was added and plates were incubated at RT in darkness for 12–15 min; the colorimetric reaction was stopped with sulfuric acid (Thermo). Plates were read on a BioRad xMark spectrophotometer at 450 nm. Plates were washed 6X with 1X TBST20 (Pierce) following each step save after TMB addition. Assay background was subtracted from sample wells prior to analysis.
HV2 library design
The Human Virome version 2 (“HV2”) PepSeq library is comprised of 15,000 unique 30mer peptides and was designed to encompass the most reactive linear epitopes from 80 virus species commonly known to infect humans, including five species of CoVs (SARSr-CoV, HCoV-OC43, HCoV-229E, HCoV-NL63 and HCoV-HKU1). Most of the peptides in HV2 (70%; 10,536/15,000) were selected from our original HV (10,475) or SCV2 (61) libraries (Ladner et al., 2021) based on empirical observations of reactivity in human sera. An additional 3,566 peptides (24% of HV2) were selected to ensure that we included homologous regions from all viruses that belong to the same genus.
To increase coverage of underrepresented viruses in HV2, we included publicly reported linear epitopes present in IEDB (https://www.iedb.org/). Specifically, we downloaded 913 linear B cell epitopes covering 17 virus species: Alphapapillomavirus 7, Alphapapillomavirus 9, Chikungunya virus, Human gammaherpesvirus 8, Human immunodeficiency virus 1, Human mastadenovirus A, Human polyomavirus 1, Human polyomavirus 2, Japanese encephalitis virus, Lymphocytic choriomeningitis mammarenavirus, Mumps rubulavirus, Orthohepevirus A, Primate T-lymphotropic virus 1, Rabies lyssavirus, Rubella virus, West Nile virus, and Zika virus. All IEDB epitopes were ≤48 aa long. For each epitope >30 aa, two peptides were designed, one starting at the N terminus and one ending at the C terminus. For each epitope ≤30 aa, a single peptide was designed, with extra residues obtained, when necessary, from the longer protein sequence to which the epitope was linked. The final HV2 design included 404 peptides designed with this approach.
One of our goals with the design of the HV2 PepSeq library was to ensure the inclusion of homologous peptides from closely related viruses. Specifically, we focused on 12 genera, each of which contained ≥2 virus species in our design: Alphacoronavirus, Alphapapillomavirus, Betacoronavirus, Betapapillomavirus, Betapolyomavirus, Enterovirus, Flavivirus, Mastadenovirus, Roseolovirus, Rotavirus, Rubulavirus, and Simplexvirus. For each genus, we generated protein-level alignments with one representative from each species. Then, for each HV2 peptide selected from HV1, SCV2 or IEDB, we designed peptides covering the homologous region from all congeners. However, these new peptides were only included in the final HV2 design if they were distinct from all existing design peptides (<70% of 5mers shared). The final HV2 design included 3,566 peptides designed with this approach.
To finely map antibody binding sites within a subset of peptides, we also include 30 derivative peptides, one with an alanine (or glycine, if alanine was the wild-type amino acid) in place of the wild-type residue at each residue (“alanine scans”). Specifically, we included these alanine scans for 16 unique peptides, including peptides covering the SARS-CoV-2 FP and SH epitopes. The final HV2 design included 480 peptides designed with this approach.
Finally, we included 14 negative control peptides derived from an assortment of eukaryotic proteins of exotic species (e.g., coelacanth, coral).
PepSeq library synthesis and assay
The HV2 library was encoded as a library of 15,000 DNA oligonucleotides and used to synthesize a corresponding ‘PepSeq’ library of DNA-barcoded peptides for multiplexed analysis of plasma antibodies, as previously described (Ladner et al., 2021). Briefly, the oligonucleotide library was PCR-amplified and then used to generate mRNA in an in vitro transcription reaction. The product was ligated to a hairpin oligonucleotide adaptor bearing a puromycin molecule tethered by a PEG spacer and used as a template in an in vitro translation reaction. Finally, a reverse transcription reaction, primed by the adaptor hairpin, was used to generate cDNA, and the original mRNA was removed using RNAse. To perform serological assays, the resulting DNA-barcoded peptide library was added to diluted plasma and incubated overnight. The binding reaction was applied to pre-washed protein G-bearing beads, washed, eluted, and indexed using barcoded DNA oligos. Following PCR cleanup, products were pooled, quantified, and sequenced using an Illumina NextSeq instrument.
PepSeq data analysis
PepSeq sequencing data were processed and analyzed as previously described (Ladner et al., 2021) using PepSIRF v1.4.0 (Fink et al., 2020), as well as custom scripts (https://github.com/LadnerLab/PepSIRF/tree/master/extensions). First, the reads were demultiplexed and assigned to peptides using the PepSIRF demux module, allowing for one mismatch in each index sequence and two mismatches in the variable DNA tag region. The PepSIRF norm module was then used to normalize counts to reads per million (RPM). RPM normalized reads from seven buffer-only control samples were subsequently used to create bins for Z-score calculation using the PepSIRF bin module. To normalize for different starting peptide abundances within each bin, reads were further normalized by subtracting the average RPM from the buffer only controls (--diff option in norm module). Z-scores were calculated using the PepSIRF zscore module using the 75% highest density interval within each bin. Finally, the PepSIRF enrich module was used to calculate peptides that were enriched in each sample using a minimum Z-score threshold of 10.
For epitope level analysis (Figure 2), each sample was run in duplicate and epitope reactivity was calculated using the peptide with the maximum Z-score in either of the two replicates. If multiple peptides contained the target epitope sequence, the peptide with the highest signal was selected.
Reduction in reactivity following peptide depletion was calculated by comparing depleted and non-depleted Z-scores across all HV2 peptides. For each sample, the change in Z-score was calculated by taking the difference between the depleted Z-score and the non-depleted Z-score for each peptide of interest. Only samples with average peptide Z-scores above 10 prior to depletion were analyzed. If multiple peptides contained the epitope of interest, the peptide with the largest difference was used.
To assess the importance of each residue within 30mer peptides containing the FP and SH epitopes, we compared the Z-scores for peptides containing alanine substitutions at each peptide residue to the Z-score of the wild-type peptide. We refer to this metric as the “residue dependency index” and it was calculated as follows: log2(ZWT + 3) - log2(Zala + 3), where ZWT and Zala are the Z-scores for the wild-type and alanine substituted peptides, respectively. To look at changes over time in antibody binding profiles, we compared estimates of this residue dependency index between days 28 and 140 post vaccination. To evaluate the significance of changes in the residue dependency index within the SARS-CoV-2 peptides, we also calculated the same metric for residues contained within non-CoV peptides for which alanine mutants were also included in HV2 (i.e., negative controls). In total, we considered 11 negative control peptides, each designed from a different common human-infecting virus. Subject/peptide combinations were included as negative controls in the analysis if the wild-type peptide had a Z-score ≥ 10 at both days 28 and 140 post-vaccination (i.e., strong reactivity at both timepoints) and if the Z-score fold change between these timepoints was less than 1.5 (i.e., no substantial change in reactivity between timepoints).
Species-specific antibody depletion
In selected cases, species-specific subsets of antibodies were depleted from plasma samples using bead-bound peptides prior to the PepSeq assay. Pairs of chemically synthesized N-terminally biotinylated 20-mer FP + SH peptides (Sigma) with sequences from SARS-CoV-2 (PSKRSFIEDLLFNKVTLADA + LQPELDSFKEELDKYFKNHT) or HCoV-OC43 (ASSRSAIEDLLFDKVKLSDV + SIPNLPDFKEELDQWFKNQT) were pooled at an equimolar ratio (100 μg/mL), added to 180 μL of pre-washed Streptavidin beads (Thermo) and incubated at room temperature for 15 min on a rotator. Peptide-coated beads were then washed 3 times and re-suspended in superblock (Thermo). Next, 60 μL of serum/plasma was added to 60 μL of peptide-coated beads and incubated for 15 min on a rotator. Serum was removed from the beads and re-applied to a fresh peptide-bearing bead aliquot; this process was repeated for a total of 3 depletion cycles prior to use in the PepSeq assay.
Full-length spike protein reactivity (MagPix)
The SARS-CoV-2 spike protein (Isolate WA1) was kindly provided by InBios International, Inc. (Seattle Washington). The HCOV-OC43 spike (S1+S2 ECD His Tag) protein was purchased from Sino Biological. Nucleocapsid proteins (used as controls) were synthesized in-house. Purified His-tagged proteins from SARS-CoV-2 (spike and nucleocapsid), HCoV-NL63 (nucleocapsid), and HCov-OC43 (spike) viruses along with immunoglobulin G (ThermoFisher Scientific, Waltham, MA) and R-Phycoerythrin (AnaSpec, Fremont, CA) were covalently conjugated to different fluorescently labeled MagPlex® microspheres using the two-step carbodiimide coupling chemistry at pH 5–6 using Sulfo-NHS (N-hydroxysulfosuccinimide) and EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) (Luminex xMAP® Cookbook, 4th Ed.). The purified His-tagged proteins were conjugated at a ratio of 5 μg protein per one million beads. The immunoglobulins and R-Phycoerythrin conjugated at a ratio of 0.5 μg protein per one million beads. Protein-conjugated microspheres were stored in PBS-TBN (0.05% Tween, 1.0% BSA, 0.1% Sodium Azide) at 4°C and protected from light. His-tagged protein bead conjugation was confirmed using monoclonal anti-6-histidine antibody (Abcam, Boston, MA) conjugated to biotin and Streptavidin, R-Phycoerythrin Conjugate (SAPE, Life technologies). The antibody conjugation was confirmed using goat anti-human IgG biotin (Abcam, Boston, MA) and SAPE.
Protein-conjugated beads were diluted in 1X Blocker BSA solution (Fisher Scientific, Hampton, NH) and mixed at a ratio of 1,000 beads per bead region per assay. Beads were washed two times with wash buffer (11.9 mM Phosphate, pH 7.4, 137 mM NaCl, 2.7 mM KCl, and 0.05% Tween 20) and the wash buffer was removed after beads were bound to a plate magnet. Serum was diluted 500 or 2,500-fold in 1X Block BSA solution and 100 μL diluted serum was added to the beads. Serum and beads were incubated for 1 h with shaking at RT. The beads were washed 3 times with wash buffer and 100 μL of 2 mg/mL Goat anti-human IgG (Abcam, Boston, MA) biotin-conjugated secondary antibody diluted in 1X Block BSA was added to the beads. The secondary antibody and beads were incubated for 1 h with shaking at RT and washed 3 times with wash buffer after incubation. Beads were then incubated with 4 mg/mL SAPE (Life Technologies) diluted in 1X Blocker BSA for 0.5 h with shaking at RT and washed 3 times with wash buffer after incubation. Beads were suspended in 1X Blocker BSA and the signal intensities of different fluorescently labeled MagPlex® microspheres were read on a MAGPIX® system (Luminex, Austin, TX); median fluorescence intensity (MFI) units per bead region was calculated using xPONENT® software (Luminex). MFI intensities were corrected based on sample dilution to ensure linearity of the assay.
Quantification and statistical analysis
For epitope discovery, PepSeq Z-scores (calculated as described above) from CoV peptides were log2 transformed and compared across the 4 timepoints, matched by subject using repeated-measures ANOVA. p-values were corrected for the False Discovery Rate across all 421 CoV peptides, and a corrected alpha threshold value of 0.05 was used. To evaluate the responses at defined epitope regions, paired T-tests were used to compare log2 transformed Z-scores for focal peptides between each post-vaccine timepoint and baseline, for homologous peptides within timepoints, or to compare the MagPix intensities from full-length SARS-CoV-2 or HCoV-OC43 Spike proteins between each post-vaccine timepoint and baseline. For the cross-sectional convalescent cohort, an unpaired T-test was used to compare the response maturity ratio for the 2 epitopes of interest between donor groups dichotomized by time since COVID diagnosis.
Acknowledgments
We are grateful to all vaccinated and convalescent blood donors, to Carmel Plude for phlebotomy, to Danielle Metz and Bethine Moore for clinical research coordination, and to Haley Nunnallay for technical assistance in generating the MagPix data. This work was supported by the NIAID (U24AI152172, U24AI152172-01S1, U24AI152172-IOF, PI: J.A.A.), the California Institute for Regenerative Medicine (CLIN2COVID19-11775, PI: J.A.Z), the State of Arizona Technology and Research Initiative Fund (TRIF, administered by the Arizona Board of Regents, through Northern Arizona University), the Flinn Foundation, and The Cowden Endowment for Microbiology. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California.
Author contributions
Conceptualization, E.A.E., P.S.K., J.T.L., and J.A.A.; data curation, E.A.E., H.L.M., E.J.K., J.T.L., and J.A.A.; formal analysis, E.A.E., J.T.L., and J.A.A.; methodology, E.A.E., E.J.K., S.T.C., A.P., E.W.S., J.T.L., and J.A.A.; investigation, E.A.E., G.A.N., S.T.C., N.G.S., C.E.H., J.L.E., A.S.B., S.N.H., F.R., and K.R.C.; visualization, E.A.E., J.T.L., and J.A.A.; software, E.A.E., E.J.K., J.T.L., and J.A.A.; validation, E.A.E., J.Y., and J.W.S.; project administration, E.A.E., H.L.M., P.S.K., E.W.S., and J.A.A.; writing – original draft, E.A.E. and J.A.A.; writing – review & editing, E.A.E., H.L.M., S.T.C., V.L.V., P.S.K., K.R.C., J.Y., E.W.S., J.A.Z., J.T.L., and J.A.A.; funding acquisition, P.S.K., J.T.L., and J.A.A.; resources, V.L.V., A.A.C., J.Y., E.W.S., D.A.B., G.C.Y., S.R.M., and J.A.Z.; supervision, H.L.M., P.S.K., E.W.S., J.T.L., and J.A.A.
Declaration of interests
The authors declare that they have no competing interests.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: July 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111022.
Supplemental information
Data and code availability
The raw peptide count data and Supplemental Tables from this study have been deposited in the Open Science Framework (https://osf.io/67r3d/), https://doi.org/10.17605/OSF.IO/67R3D. All custom code is available via GitHub (https://github.com/LadnerLab). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aguilar-Bretones M., Westerhuis B.M., Raadsen M.P., de Bruin E., Chandler F.D., Okba N.M.A., Haagmans B.L., Langerak T., Endeman H., van den Akker J.P.C., et al. Seasonal coronavirus–specific B cells with limited SARS-CoV-2 cross-reactivity dominate the IgG response in severe COVID-19. J. Clin. Invest. 2021;131:e150613. doi: 10.1172/jci150613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F., Thapa M., Lei T., Ahmed S.M.S., Adelsberg D.C., Carreño J.M., Strohmeier S., Schmitz A.J., Zafar S., Zhou J.Q., et al. SARS-CoV-2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell. 2021;184:3936–3948.e10. doi: 10.1016/j.cell.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E.M., Goodwin E.C., Verma A., Arevalo C.P., Bolton M.J., Weirick M.E., Gouma S., McAllister C.M., Christensen S.R., Weaver J., et al. Seasonal human coronavirus antibodies are boosted upon SARS-CoV-2 infection but not associated with protection. Cell. 2021;184:1858–1864.e10. doi: 10.1016/j.cell.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwerk M., Gonen T., Lustig Y., Amit S., Lipsitch M., Cohen C., Mandelboim M., Levin E.G., Rubin C., Indenbaum V., et al. Covid-19 breakthrough infections in vaccinated health care workers. N. Engl. J. Med. 2021;385:1474–1484. doi: 10.1056/NEJMoa2109072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett D.L., Langley D.B., Schofield P., Hermes J.R., Chan T.D., Jackson J., Bourne K., Reed J.H., Patterson K., Porebski B.T., et al. Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science. 2018;360:223–226. doi: 10.1126/science.aao3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D., Voss J., Gamblin S.J., Codoni G., Macagno A., Jarrossay D., Vachieri S.G., Pinna D., Minola A., Vanzetta F., et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- Davenport F.M., Hennessy A.V., Francis T., Jr. Epidemiologic and immunologic significance of age distribution of antibody to antigenic variants of influenza virus. J. Exp. Med. 1953;98:641–656. doi: 10.1084/jem.98.6.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan H.L., Stamper C.T., Li L., Changrob S., Asby N.W., Halfmann P.J., Zheng N.-Y., Huang M., Shaw D.G., Cobb M.S., et al. Profiling B cell immunodominance after SARS-CoV-2 infection reveals antibody evolution to non-neutralizing viral targets. Immunity. 2021;54:1290–1303.e7. doi: 10.1016/j.immuni.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink Z.W., Martinez V., Altin J., Ladner J.T. PepSIRF: A flexible and comprehensive tool for the analysis of data from highly-multiplexed DNA-barcoded peptide assays. arXiv. 2020 https://arxiv.org/abs/2007.05050 Preprint at. [Google Scholar]
- Gostic K.M., Ambrose M., Worobey M., Lloyd-Smith J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science. 2016;354:722–726. doi: 10.1126/science.aag1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead S.B., O’Rourke E.J. Antibody-enhanced dengue virus infection in primate leukocytes. Nature. 1977;265:739–741. doi: 10.1038/265739a0. [DOI] [PubMed] [Google Scholar]
- Kaslow R.A., Stanberry L.R., Le Duc J.W. Springer; 2014. Viral Infections of Humans: Epidemiology and Control. [Google Scholar]
- Kozlov I.A., Thomsen E.R., Munchel S.E., Villegas P., Capek P., Gower A.J., Pond S.J.K., Chudin E., Chee M.S. A highly scalable peptide-based assay system for proteomics. PLoS One. 2012;7:e37441. doi: 10.1371/journal.pone.0037441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner J.T., Henson S.N., Boyle A.S., Engelbrektson A.L., Fink Z.W., Rahee F., D’ambrozio J., Schaecher K.E., Stone M., Dong W., et al. Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses. Cell Rep. Med. 2021;2:100189. doi: 10.1016/j.xcrm.2020.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H.-X., Lynch R., Zhou T., Gao F., Alam S.M., Boyd S.D., Fire A.Z., Roskin K.M., Schramm C.A., Zhang Z., et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.S., Vaqueirinho D., Mele F., Foglierini M., Jerak J., Perotti M., Jarrossay D., Jovic S., Perez L., Cacciatore R., et al. Clonal analysis of immunodominance and cross-reactivity of the CD4 T cell response to SARS-CoV-2. Science. 2021;372:1336–1341. doi: 10.1126/science.abg8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyal L., Braun J., Henze L., Kruse B., Dingeldey M., Reimer U., Kern F., Schwarz T., Mangold M., Unger C., et al. Cross-reactive CD4 T cells enhance SARS-CoV-2 immune responses upon infection and vaccination. Science. 2021;374:eabh1823. doi: 10.1126/science.abh1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli L., Park Y.-J., Tortorici M.A., Czudnochowski N., Walls A.C., Beltramello M., Silacci-Fregni C., Pinto D., Rosen L.E., Bowen J.E., et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183:1024–1042.e21. doi: 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Sauer M.M., Czudnochowski N., Low J.S., Tortorici M.A., Housley M.P., Noack J., Walls A.C., Bowen J.E., Guarino B., et al. Broad betacoronavirus neutralization by a stem helix-specific human antibody. Science. 2021;373:1109–1116. doi: 10.1126/science.abj3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y.-P., Chee R.S.-L., Fong S.-W., Yeo N.K.-W., Lee W.-H., Torres-Ruesta A., et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020;11:2806. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston D., Weisblum Y., Wise H., Templeton K., Jenks S., Hatziioannou T., Bieniasz P. Absence of severe acute respiratory syndrome coronavirus 2 neutralizing activity in prepandemic sera from individuals with recent seasonal coronavirus infection. Clin. Infect. Dis. 2021;73:e1208–e1211. doi: 10.1093/cid/ciaa1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran S., Tang J., Grubbs G., Lee Y., Pourhashemi S., Hussaini L., Lapp S.A., Jerris R.C., Singh V., Chahroudi A., et al. SARS-CoV-2 immune repertoire in MIS-C and pediatric COVID-19. Nat. Immunol. 2021;22:1452–1464. doi: 10.1038/s41590-021-01051-8. [DOI] [PubMed] [Google Scholar]
- Sauer M.M., Tortorici M.A., Park Y.-J., Walls A.C., Homad L., Acton O.J., Bowen J.E., Wang C., Xiong X., de van der Schueren W., et al. Structural basis for broad coronavirus neutralization. Nat. Struct. Mol. Biol. 2021;28:478–486. doi: 10.1038/s41594-021-00596-4. [DOI] [PubMed] [Google Scholar]
- Shrock E., Fujimura E., Kula T., Timms R.T., Lee I.-H., Leng Y., Robinson M.L., Sie B.M., Li M.Z., Chen Y., et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science. 2020;370:eabd4250. doi: 10.1126/science.abd4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal A., Chappert P., Barba-Spaeth G., Roeser A., Fourati S., Azzaoui I., Vandenberghe A., Fernandez I., Meola A., Bouvier-Alias M., et al. Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell. 2021;184:1201–1213.e14. doi: 10.1016/j.cell.2021.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G., He W.-T., Callaghan S., Anzanello F., Huang D., Ricketts J., Torres J.L., Beutler N., Peng L., Vargas S., et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat. Commun. 2021;12:2938. doi: 10.1038/s41467-021-23074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J.S., O’Halloran J.A., Kalaidina E., Kim W., Schmitz A.J., Zhou J.Q., Lei T., Thapa M., Chen R.E., Case J.B., et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature. 2021;596:109–113. doi: 10.1038/s41586-021-03738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., van Haperen R., Gutiérrez-Álvarez J., Li W., Okba N.M.A., Albulescu I., Widjaja I., van Dieren B., Fernandez-Delgado R., Sola I., et al. A conserved immunogenic and vulnerable site on the coronavirus spike protein delineated by cross-reactive monoclonal antibodies. Nat. Commun. 2021;12:1715. doi: 10.1038/s41467-021-21968-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White H.N. B-cell memory responses to variant viral antigens. Viruses. 2021;13 doi: 10.3390/v13040565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R., Belk J.A., Govero J., Uhrlaub J.L., Reinartz D., Zhao H., Errico J.M., D’Souza L., Ripperger T.J., Nikolich-Zugich J., et al. Affinity-restricted memory B cells dominate Recall responses to heterologous flaviviruses. Immunity. 2020;53:1078–1094.e7. doi: 10.1016/j.immuni.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw peptide count data and Supplemental Tables from this study have been deposited in the Open Science Framework (https://osf.io/67r3d/), https://doi.org/10.17605/OSF.IO/67R3D. All custom code is available via GitHub (https://github.com/LadnerLab). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.