

Abstract
Three classes of G protein-coupled receptors (GPCR) partners, – G proteins, GPCR kinases, and arrestins, – preferentially bind active GPCRs. Our analysis suggests that the structures of GPCRs bound to these interaction partners available today do not reveal a clear conformational basis for signaling bias, which would have enabled the rational design of biased GRCR ligands. In view of this, three possibilities are conceivable: there are no generalizable GPCR conformations conducive to binding a particular type of partner; subtle differences in the orientation of individual residues and/or their interactions not easily detectable in the receptor-transducer structures determine partner preference; or the dynamics of GPCR binding to different types of partners rather than the structures of the final complexes might underline transducer bias.
Keywords: GPCR, G protein, arrestin, GRK, biased signaling
Distinct branches of GPCR-driven signaling.
GPCRs (see Glossary) are membrane signaling proteins activated by light, hormones, neurotransmitters, peptides, proteins, extracellular ions, etc. The human genome encodes >800 different GPCRs, which account for >2% of protein-coding genes [1]. GPCRs are targeted by about a third of therapeutically used drugs. These receptors got their name because their signaling via heterotrimeric G proteins was discovered first. Four subfamilies of G proteins (Gs/olf, Gi/o, Gq/11, and G12/13) link GPCRs to distinct intracellular effectors. A single receptor can couple to more than one type of G protein [2–5]. Arrestins, another class of GPCR-binding partners, were initially discovered as negative regulators of the G protein-dependent signaling via homologous desensitization (reviewed in [6]). The data accumulated over the last ~20 years indicate that GPCR-bound arrestins also regulate several branches of signaling (reviewed in [7, 8]), with potential therapeutic implications [9, 10]. For most GPCRs, the binding of arrestins is contingent upon receptor phosphorylation by GPCR kinases (GRKs) [11]. Although receptor-bound GRKs have not been reported to participate in cell signaling directly, receptor phosphorylation by distinct GRKs apparently plays a critical role in controlling the signaling [12]. Thus, the molecular mechanism of GPCR-GRK interactions is an important determinant of the signaling outcome. The Tyr2195.58Ala β2AR mutant (we use Ballesteros-Weinstein nomenclature [13]) is an instructive example: it is not phosphorylated by GRKs, fails to recruit arrestin-2, internalize, or signal via arrestins, while G protein signaling remains intact [12]. Thus, receptor phosphorylation by GRKs is a prerequisite for arrestin-mediated signaling [14]. To summarize, GPCRs interact with three classes of signal transducers – G proteins, GRKs, and arrestins – coupling to which determines the direction of ligand-induced signaling.
For a long time, GPCRs were believed to oscillate between two conformations, active and inactive, with agonists shifting the equilibrium towards the active state. The ability of a ligand to activate signaling upon binding to a receptor was characterized as “intrinsic efficacy”, which was believed to be an inherent property of the drug equally applicable to all signaling pathways initiated by a receptor. However, functional testing of several GPCRs for which numerous ligands are available revealed different orders of potency of these ligands depending on the downstream signaling monitored [15]. This pathway-specific signaling by a GPCR ligand is often referred to as functional selectivity or biased signaling. These observations led to the idea that GPCRs can assume multiple active conformations, which are conducive to coupling to different transducers, resulting in distinct signaling outcomes [16–18]. Indeed, biophysical studies of β2-adrenergic receptor (β2AR) and other GPCRs demonstrated that all receptors are flexible proteins existing in an ensemble of conformations in unliganded and liganded state [19–21]. It is widely believed that different ligands can stabilize distinct receptor conformations, resulting in the receptor coupling to signal transducer(s) that prefer a particular conformation [17, 22]. In recent years, attention in the field has focused on the ability of “biased” GPCR ligands to promote recruitment of G proteins or arrestins, thereby initiating G protein- or arrestin-dependent signaling. It has been suggested that in many cases G proteins or arrestins mediate the therapeutic effects of drugs, whereas the other signaling branch is responsible for deleterious side effects [5, 23].
A critical element in the concept of biased signaling is the assumption that the same receptor in different conformations preferentially couples to different transducers. Indeed, structures of several GPCRs with various ligands, some of which appear to be biased, do show differences in the receptor conformation depending on the bound ligand (reviewed in [24]). For example, G protein and arrestin –biased ligands induced different rearrangements in TM6, TM7, and H8 of β-adrenergic receptors [18, 25, 26]. The unbiased angiotensin-II induced on-axis rotation of TM3, resulting in outside flipping of N1113.35 side chain and a distinct configuration of the AT1 receptor polar network as compared to that in the AT1 receptor bound to arrestin-biased ligands (Fig. 1B,C) [27]. However, to signal, a GPCR must bind cytoplasmic transducers, and it is not evident that the distinct receptor conformations induced by different ligands would be retained in the receptor-transducer complex. Signal transducers bound to the cytoplasmic side of a GPCR often engage greater receptor surface than a ligand. Hence, the energy of the interaction is likely to be at least equal to, or even greater than, the energy of ligand binding. Therefore, the bound transducer is likely to affect GPCR conformation at least as much as the ligand, if not more. With this in mind, we focus exclusively on the available structures of GPCRs in complex with signaling partners [28–51].
Fig. 1. Comparison of the structures of muscarinic M1R and angiotensin AT1R in complex with Gq/11 or biased ligands.
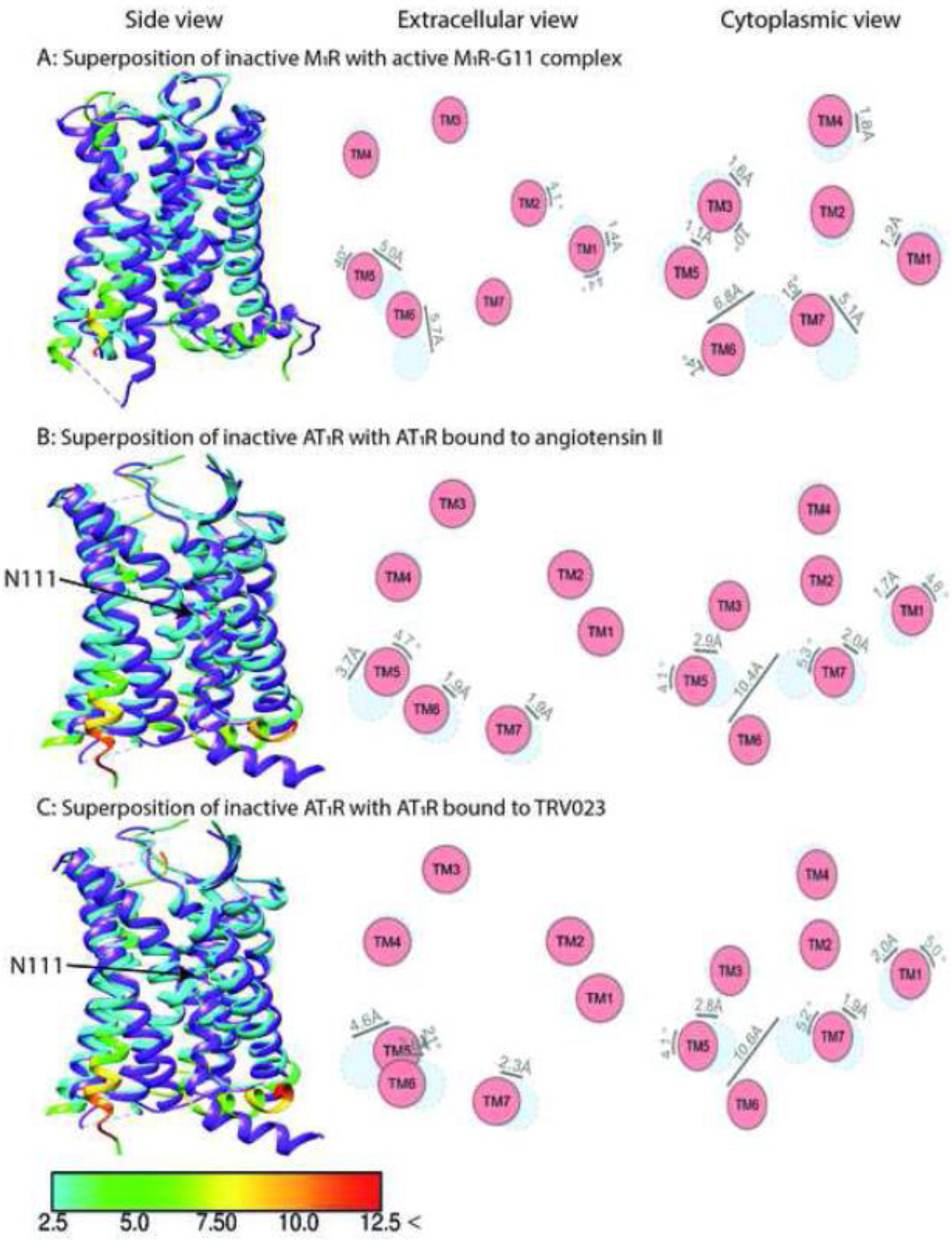
The structure of inactive M1R (PDB: 5CXV) [89] is superposed with activated M1R in complex with G11 (panel A, PDB: 6OIJ) [40]. The structure of inactive AT1R (PDB: 4YAY) [90] is superposed with activated AT1R in complex with its endogenous ligand angiotensin II (panel B, PDB: 6OS0) [27] and arrestin-biased TRV023 (panel C, PDB: 6OS1) [27]. Shown are side views (left panels), and schematic representation of helices from extracellular (middle panels) and cytoplasmic views (right panel). The positions of helices in the inactive state are shown in light blue, active in red. Inactive receptors are colored purple in left panels. The residues of the active receptors in complex with intracellular partner and balanced or biased agonists are color coded based on the RMSD of α-carbons from the matched residues in the inactive receptor with thresholds of 2.5 Å (cyan), 5 Å (green), 7.5 Å (yellow), 10 Å (orange), and 12.5 Å (red). The direction and extent of displacement of helices are also indicated by arrows. The critical AT1R residue N111 showing different orientation between structures is indicated by an arrow.
Existing structures of GPCR complexes with binding partners
Profound structural rearrangements in G proteins [52] and arrestins [53] induced by GPCR binding have been reviewed. Importantly, there are notable differences between the conformations of arrestins recruited to different receptors. The orientation of arrestin-2 relative to the seven transmembrane (TM) helix bundle present in all GPCRs is different in complex with muscarinic (M2R) [46], β1-adrenergic (β1AR) [54], and neurotensin (NTS1R) receptors [33, 49]. The extent of the interdomain twist also differs in arrestin complexes with different GPCRs [33, 46, 49, 51, 54]. The two non-visual subtypes, arrestin-2 and −3 (a.k.a. β-arrestin1 and 2), are not identical structurally and functionally [7, 55, 56], despite being highly homologous [57, 58]. Free arrestin-2 and −3 in the cytoplasm also form distinct oligomers, in which arrestin-3 assumes a receptor-bound-like (often called active) conformation, whereas arrestin-2 remains in the basal state [59]. Arrestin-3 is more often implicated in signaling [7]. It is possible that the conformation of GPCRs in complex with arrestin-3 is different from that with arrestin-2. As all non-visual GPCR structures solved thus far contain arrestin-2 [33, 46, 49, 54], the structures of the same receptor in complex with the two non-visual arrestins are necessary to address this issue.
Two aspects of the GRK interaction with a GPCR are important. One, GRKs are activated by docking to the receptor [60]. Two, the activated GRK phosphorylates the receptor it is bound to. Two recent structures of the rhodopsin-GRK1 complex [28] illuminate both these aspects. The αN helix of GRK1 docks into the interhelical cavity that appears in rhodopsin upon activation [61]. Compared to the α5 helix of the visual G protein transducin, the polarity is the opposite, and the angle differs by about 25° [28]. Upon receptor binding, this αN helix packs against the kinase domain, stabilizing its active conformation [28]. As these are the only GPCR-GRK structures available, they deserve more detailed analysis. Hydrophobic interactions between GRK1 and residues on the inner surface of rhodopsin TM3, TM5, and TM6 stabilize the complex [28]. Acidic residues of GRK1 interact with basic residues of rhodopsin (Lys3118.48, Arg3148.51, and Arg1353.50) [28]. However, only Leu6 is invariant in the αN helices of all GRKs [62], suggesting that ligand-dependent receptor conformations might favor particular GRKs, resulting in distinct phosphorylation patterns (“barcoding”) and, consequently, in different functional effects [63]. It is also informative that the target of GRK1, the rhodopsin C-terminus, is not resolved in the structures [28]. Most GPCRs have numerous GRK phosphorylation sites located in the C-terminus, in the third intracellular loop (ICL), or in both elements [64]. In rhodopsin, GRK targets are localized more compactly than in any other GPCR: six Ser and Thr residues (in mouse and human; seven in bovine) within a 10-residue stretch in the C-terminus [65]. However, for GRK1 to phosphorylate several targets (at least three rhodopsin-attached phosphates are needed for high affinity arrestin-1 binding [65, 66]), the rhodopsin C-terminus must have several different positions in the complex, which would preclude its resolution. Thus, the fact that it is not resolved suggests that it likely has several positions, “offering” different Ser and Thr residues to the catalytic site of bound GRK1. If the rhodopsin C-terminus were resolved in the structure, there would be only one Ser or Thr residue in the catalytic site, which would raise the question of how GRK1 manages to phosphorylate its other targets.
Do GPCRs have distinct conformations in complex with different partners?
As noted above, the cornerstone of the concept of biased signaling is the hypothesis that distinct receptor conformations favor particular partners: different G proteins, GRKs, and/or arrestins. Analysis of available structures of GPCRs in complex with intracellular partners does not support this hypothesis. Today we have structures of only one GPCR, rhodopsin, in complex with G protein [30, 34], arrestin-1 [35, 51], and GRK1 [28] (Fig. 2A,B,C). Because the full set of structures with all three types of interaction partners is unavailable for other receptors, we are forced to compare the structures of G protein and arrestin complexes of different GPCRs.
Fig. 2. Comparison of rhodopsin structure in complex with different partners.
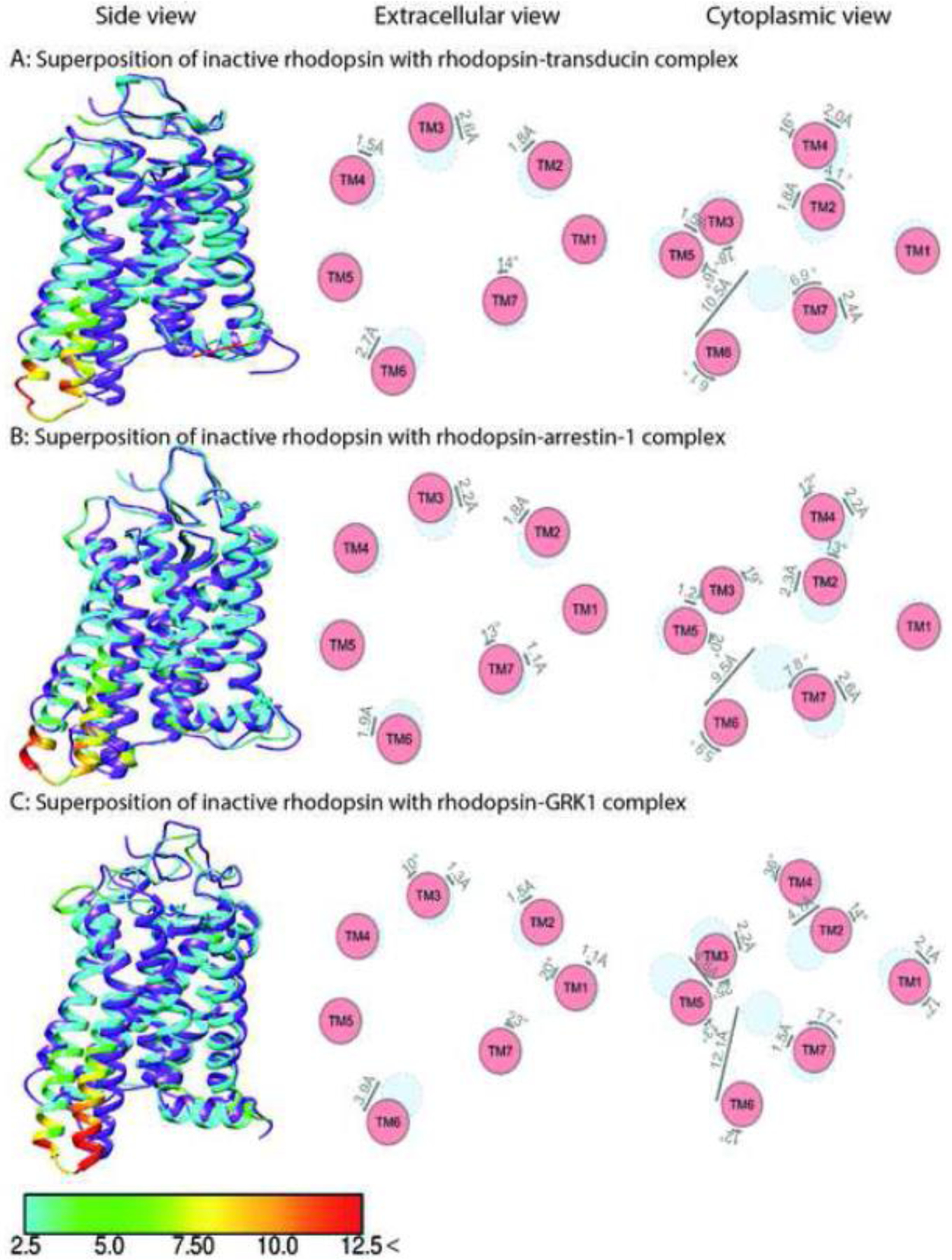
The structure of inactive rhodopsin (PDB: 1U19) [91] is superposed with activated rhodopsin in complex with transducin (panel A, PDB: 6OYA) [30], arrestin-1 (panel B, PDB: 4ZWJ) [35], and GRK1 (panel C, PDB: 7MT9) [28]. Shown are side view (left panels), and schematic representation of helices from extracellular (middle panels) and cytoplasmic views (right panel). The positions of helices in the inactive state are shown in light blue, active in red. Inactive receptor in the left panels is colored purple. The residues of the active rhodopsin in complex with intracellular partners are color coded based on the RMSD of α-carbons from the matched residues in the inactive receptor with thresholds of 2.5 Å (cyan), 5 Å (green), 7.5 Å (yellow), 10 Å (orange), and 12.5 Å (red). The direction and extent of displacement of helices are also indicated by arrows.
GPCR-G protein complexes
Comparison of the two earliest structures of GPCR-G protein complexes, rhodopsin-Gi [34] and β2AR-Gs [43] (Figs. 2A and 3A), revealed similar movements in TM2 and TM4 relative to the inactive state, but significantly different positions of TM6, TM1, and the connector between TM7 and intracellular helix 8 (H8). The outward movement of TM6 in the rhodopsin-Gi complex is about 8 Å smaller than in the β2AR-Gs complex. The outward movements of TM6 in the adenosine A1R-Gi2 [29], opioid μOR-Gi1 [36], serotonin 5-HT1BR-Go [32], muscarinic M2R-Go [40] (Fig. 4A), and rhodopsin-transducin [30] (Fig. 2A) complexes are also smaller than in the β2AR-Gs complex [43]. The Gs-coupled class B receptors, such as the human parathyroid receptor-1 or glucagon-like peptide 1 (GLP-1) receptor, display even larger movements of TM6 than the β2AR-Gs complex [38, 50, 67]. In contrast, two recently solved structures of class C GPCRs (metabotropic glutamate and GABA-B receptors) with Gi reveal minimal movement of TM6 (Fig. 4D), with ICL2, ICL3, and the C terminus serving as the key transducer binding receptor elements [39, 44, 45].
Fig. 3. Comparison of the structures of class A adrenergic β1AR and class B Glucagon GCGR in complex with Gs or arrestin-2.
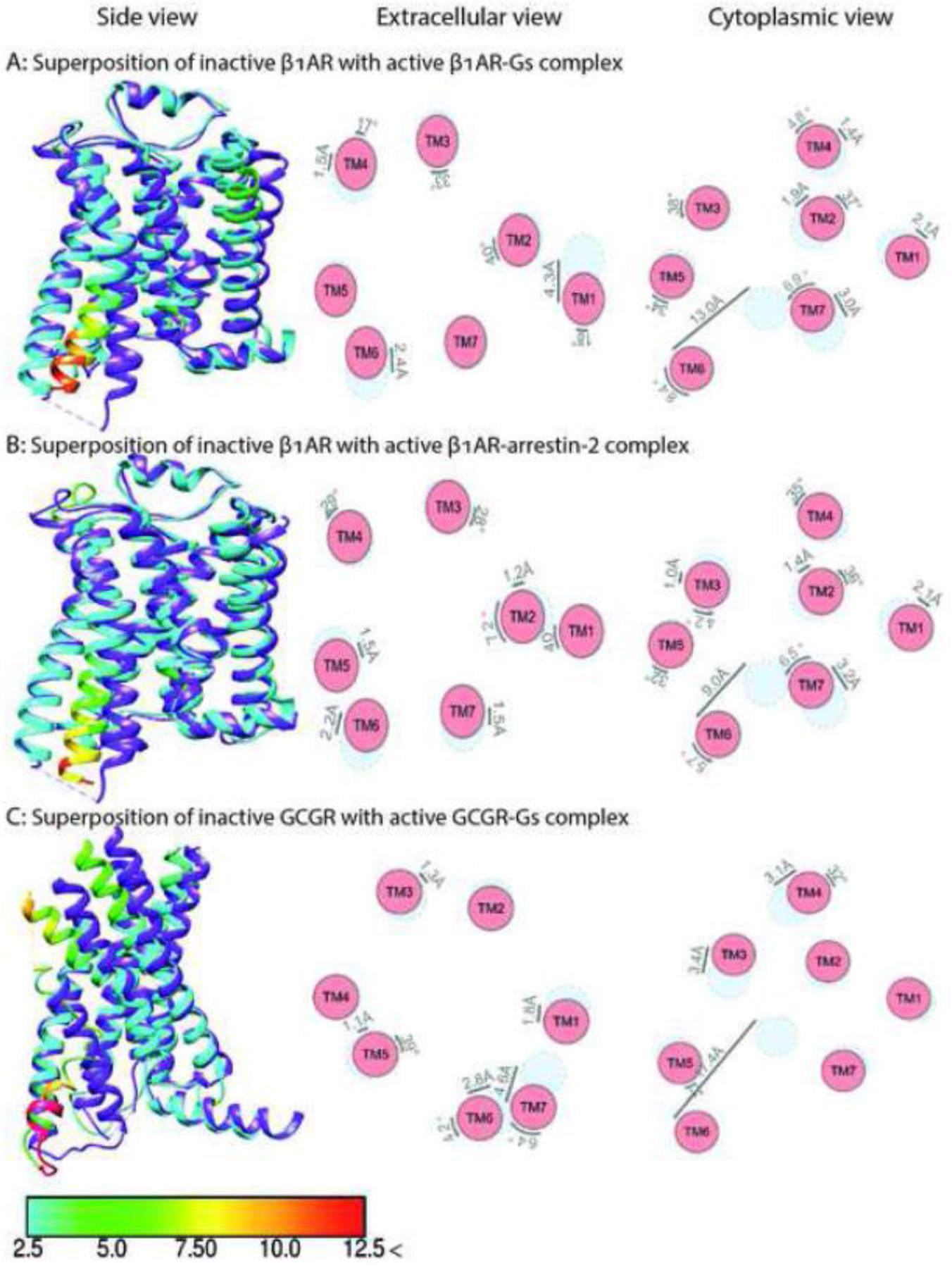
The structure of inactive β1AR (PDB: 4GPO) [87] is superposed with activated β1AR in complex with Gs (panel A, PDB: 7JJO) [47] and arrestin-2 (panel B, PDB: 6TKO) [54]. The structure of inactive GCGR (PDB:5YQZ) [87] is superposed with activated GCGR in complex with Gs (panel C, PDB:6LMK) [42]. Shown are side views (left panel), and schematic representation of helices from extracellular (middle panels) and cytoplasmic views (right panel). The positions of helices in the inactive state are shown in light blue, active in red. Inactive receptors in left panels are colored purple. The residues of the active receptors in complex with intracellular partners are color coded based on the RMSD of α-carbons from the matched residues in the inactive receptor with thresholds of 2.5 Å (cyan), 5 Å (green), 7.5 Å (yellow), 10 Å (orange), and 12.5 Å (red). The direction and extent of displacement of helices are also indicated by arrows.
Fig. 4. Comparison of the structures of class A muscarinic M2R, class B Glucagon GCGR, and class C metabotropic glutamate mGlu2R in complex with Gi or arrestin-2.
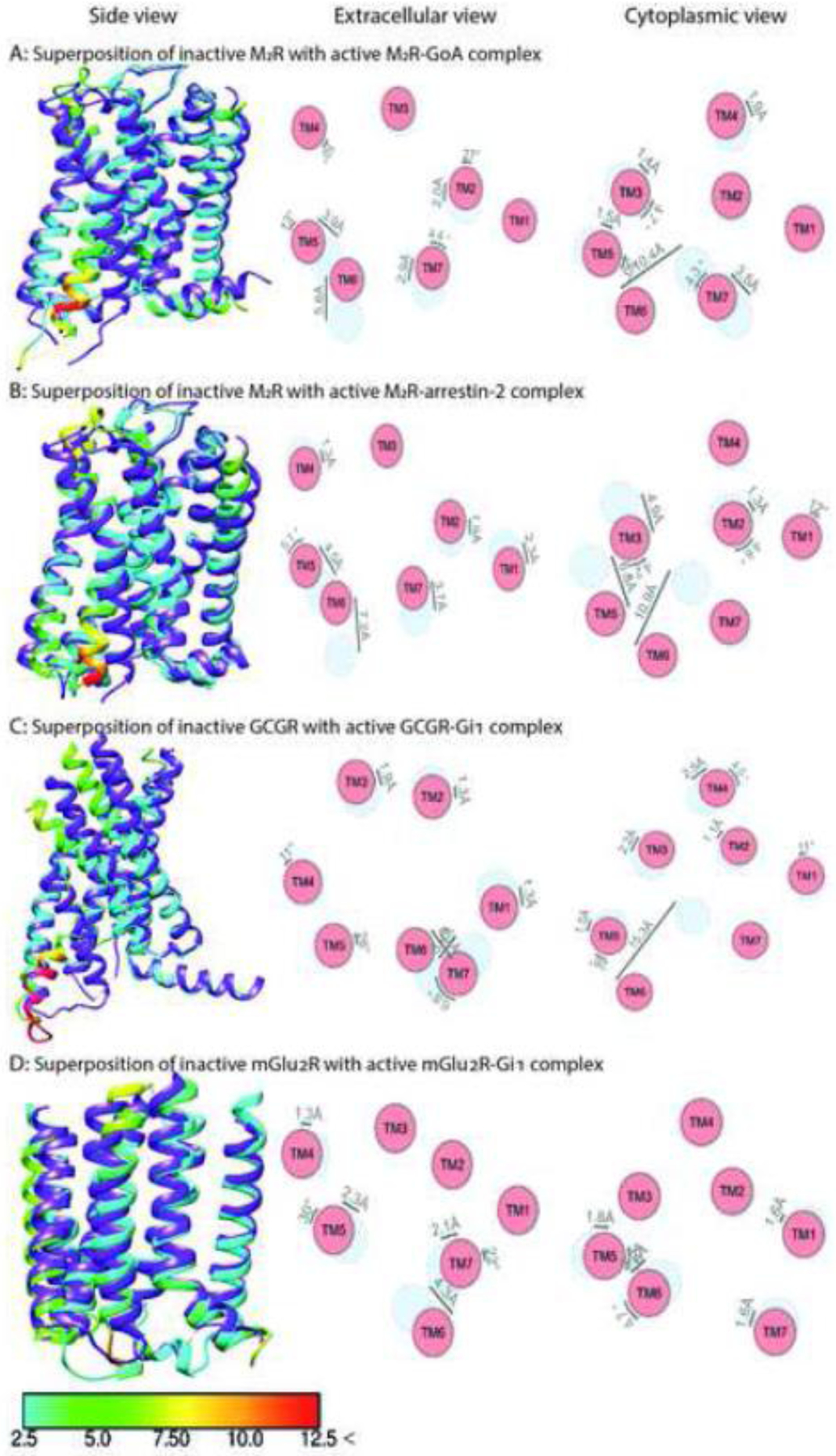
The structure of inactive M2R (PDB: 3UON) [88] is superposed with activated M2R in complex with GoA (panel A, PDB: 6OIK) [40] and arrestin-2 (panel B, PDB: 6U1N) [46]. The structure of inactive GCGR (PDB: 5YQZ) [87] is superposed with activated GCGR in complex with Gi1 (panel C, PDB: 6LML) [42]. The structure of inactive mGlu2R (PDB: 7MTQ) [44] is superposed with activated mGlu2R in complex with Gi1 (panel D, PDB: 7MTS) [44]. Shown are side views (left panels), and schematic representation of helices from extracellular (middle panels) and cytoplasmic views (right panel). The positions of helices in the inactive state are shown in light blue, active in red. Inactive receptors are colored purple in left panels. The residues of the active receptor in complex with intracellular partners are color coded based on the RMSD of α-carbons from the matched residues in the inactive receptor with thresholds of 2.5 Å (cyan), 5 Å (green), 7.5 Å (yellow), 10 Å (orange), and 12.5 Å (red). The direction and extent of displacement of helices are also indicated by arrows.
Residues on the cytosolic face of TM6 tend to vary between Gs- and Gi/o-coupled receptors. Gi/o-coupled receptors carry polar or positively charged residues, stabilizing a straight TM6 conformation among the negatively charged lipid headgroups. In contrast, Gs-coupled receptors have many hydrophobic residues, favoring an outward TM6 movement, whereupon hydrophobic side chains bury inside the lipid bilayer [68]. Residues at TM6.36 in Gi-coupled receptors are often hydrophobic, supporting hydrophobic interactions with Leu (Leu353G.H5.25 by uniform residue numbering system [52]) in Gi [34]. This position is occupied by Thr/Ser in 72% of the Gs-coupled receptors, allowing hydrogen bonding with the carbonyl group of the residue preceding the TM6.36 by four positions, which stabilizes the bent TM6 conformation [68]. The higher flexibility of TM6 in Gs-coupled receptors is also attributed to the conserved Gly6.42, which prevents steric hindrance due to absence of side chain. Conserved Pro6.47 and Gly6.50 in Gs-coupled class B GPCRs allow bending of both the extracellular and intracellular ends of TM6, resulting in the largest TM6 movements [68]. Sequence alignment and MD simulations indicate a less dynamic and straighter conformation for TM6 of Gi-coupled receptors [34]. Yet there is significant variability: the movements of TM6 differ by about 4 Å among the Gi/o/t-coupled GPCRs [29, 30, 32, 36] (Figs. 2A, 4C,D).
While smaller TM6 movements are often observed upon Gi (as compared to Gs) binding, TM6 movement in the melatonin MT1R-Gi complex is very large (~15 Å), possibly due to the hydrophobic residues in TM6 of MT1R instead of the hydrophilic residues in many Gi-coupled receptors [41]. The issue becomes more complex when other G proteins are also considered. Upon coupling to their cognate Gq/11 proteins, TM6 of muscarinic M1R and histamine H1R move outward by ~6.8 Å and ~11.2 Å, respectively [40, 48] (Fig. 1A), i.e., in the range of complexes of other GPCRs with Gi. The TM6 movement is smaller than in class B (calcitonin, GLP-1, and parathyroid hormone receptor 1) [38, 50, 67] or class A (β1- and β2-adrenergic, adenosine A2A, and GPCR52) GPCRs [31, 43, 47, 69] with bound Gs. Our analysis of existing structures suggests that the conformation of the active receptor in complex with a partner is largely predetermined by the receptor sequence and that structural features of GPCRs that predispose them to a particular extent of TM6 movement remain invariable regardless of the bound ligand. This notion is supported by the same magnitude of TM6 movement in class B glucagon receptor bound to Gs and Gi (Figs. 3C, 4C) [42].
Thus, existing structures do not reveal a clear difference in the extent of TM6 movement that would predispose the receptor to coupling to G proteins of a particular subfamily. It is also worth noting that Gq proteins with the last few amino acids of the C-terminus of the Gα-subunit changed into those characteristic for Gs, Gi/o, or G12/13 subfamilies are routinely exploited to channel the signaling of all GPCRs to the Gq pathway, so that the easily detectable increase in intracellular Ca2+ can be used as a readout [70]. This means that every GPCR screened by this method can couple to Gq. A few residues at the C-terminus of the Gα-subunit are unstructured in free G proteins and do not change the overall structure of the G protein heterotrimer. In our view, apparent receptor versatility is hardly surprising considering the flexibility of GPCRs [19–21]. The switch of GPCR coupling by the exchange of several C-terminal residues of the Gα-subunit indicates that the residues lining the interhelical cavity, where this part of the Gα binds, rather than the overall conformation of the active receptor, largely determine G protein preference.
Comparing GPCR-G protein and -arrestin complexes
There are no discernible global structural differences in active GPCRs engaging G proteins versus arrestins, either. The extent of the movement of transmembrane helices, including TM6 that upon receptor activation moves more than others [61], is very similar in rhodopsin complex with arrestin-1 [35], Gi [34], and Gt [30] (Fig. 2A,B). Superposition of muscarinic M2R in complex with GoA [40] and arrestin-2 [46] reveals little difference in helix movement, including TM6 (Fig. 4A,B) [53].
Comparison of the rhodopsin structures in complexes with all three types of signaling partners – G proteins [30, 34], arrestin-1 [35, 51], and GRK1 [28] – revealed that in all cases, an α-helix of a partner inserts into the cavity between the cytoplasmic tips of transmembrane helices that opens upon GPCR activation [61]. This α-helix is the C-terminus of G protein α-subunit [34], an internal “finger loop” of arrestin-1 [35], and the N-terminus of GRK1 [28]. Rhodopsin structures in complexes with Gi/t are remarkably similar to its structure in complexes with other partners, arrestin-1 and GRK1 [28, 30, 34, 35, 51] (Fig 2A,B,C): the only notable difference is that in GRK1-bound rhodopsin TM6 displays ~3 Å greater upward movement at the cytoplasmic end compared to that in the rhodopsin-arrestin-1 or rhodopsin-G protein complexes [28, 30, 34, 35, 43, 51]. Notably, the activating “ligand” in all cases is the same, all-trans-retinal. Thus, the observed differences in the TM6 conformation appear to be induced by the binding partner, GRK1 on the one hand and G protein and arrestin on the other. However, rhodopsin is a highly specialized light receptor that has not been reported to initiate arrestin-mediated signaling.
Our analysis of existing structures puts in doubt the feasibility of biasing GPCR signaling towards a particular G protein subfamily or GRKs/arrestins by ligands stabilizing distinct global receptor conformations. A possibility exists that subtle local structural alterations caused by different ligands like the side chain rearrangements [18, 25, 26] might bring about distinct signaling consequences. These fine differences can be captured in the structures of the GPCR-effector complexes. In the structures of sphingosine-1-phosphate (S1P) receptor 1 bound to Gi activated by unbiased S1P and two arrestin-biased agonists, the intermediate flipping of W2696.48 and the retained interaction between F2656.44 and N3077.49 appear associated with arrestin biased ligands [71]. However, it remains unclear whether these structural features confer signaling bias, since in all cases the SP1 receptor was bound to Gi. Three structures of Gi-bound chemokine receptor CCR1 were recently solved, where the receptor had no ligand (CCR1 has constitutive ligand-independent activity), bound unbiased shorter or arrestin-biased longer form of CCL15 [72]. These structures suggest that the position of the side chain of Y2917.43 differs upon activation by unbiased and arrestin-biased agonist [72]. Here again, the CCR1 receptor was in complex with Gi in all cases, suggesting that all side chain positions induced by differentially biased ligands are conducive to Gi coupling. Unfortunately, no structures of these receptors with either non-visual arrestin are available. Conceivably, receptor affinity for G protein and/or arrestin might depend on the nature of bound agonist, but we don’t have experimental evidence supporting this hypothesis.
Screening for signaling bias
Bias in signaling by different ligands of the same GPCR is often documented in cells exogenously expressing the receptor of interest. This is usually interpreted as proof that different ligands induce distinct changes in the conformational ensemble of the receptor, shifting it to conformations conducive to coupling to particular transducers. However, these data are open to alternative interpretations. Every cell expresses 10–12 thousand different proteins [73], including many GPCR subtypes. Even at low concentrations, virtually every ligand binds several different GPCRs (e.g., see https://pdsp.unc.edu/databases/kidb.php). Naturally, different ligands bind distinct sets of receptors. Thus, their action on the receptor of interest occurs on different backgrounds, which might lead to distinct outcomes even when they act on that receptor similarly. It is technically challenging to account for this kind of artefact. Cells that are not transfected with the receptor of interest and cells expressing it treated by saturating antagonist concentration are used as controls. The caveat is that in both cases the signal from the receptor of interest is absent. Hence, other receptors binding the ligands used have nothing to modulate. The off-target effects of the ligands can be monitored in untransfected cells by a label-free method, such as dynamic mass redistribution (DMR) [74]. For example, DMR showed significant effects of many neuropeptide S receptor ligands on HEK293 cells that did not express this receptor [75]. Similarly, some nociception/orphanin receptor agonists produced DMR signal in HEK293 cells that did not express this receptor [76]. Importantly, these are one-way experiments: DMR signal in the absence of GPCR of interest shows that the ligand acts via other receptors and/or non-receptor-mediated mechanisms, but the lack of detection does not prove anything, as it might reflect insufficient sensitivity.
Another problem is selective screening for G protein- and arrestin-mediated signaling. While many cellular events are attributed to Gα and Gβγ signaling, the issue of arrestin-mediated signaling independent of G proteins remains controversial [77, 78]. For example, ERK1/2 phosphorylation was assumed to be a G protein-independent signature of arrestin-mediated signaling [79]. Subsequent studies revealed that arrestins and receptor internalization are dispensable for GPCR signaling to ERK1/2 [80], and that G proteins, not arrestins, often determine the level of ERK1/2 phosphorylation [77]. Importantly, while G protein-mediated signaling is usually measured directly by the production of second messengers, arrestin-mediated signaling is not: arrestin recruitment to GPCRs is often measured in lieu of signaling [81]. Considering that the functional outcome of arrestin binding to a GPCR is expected to be different depending on the pattern of receptor phosphorylation (“barcode hypothesis” [63]), this does not appear to be an adequate substitution. Moreover, there is always a signaling consequence of arrestin recruitment: suppression of G protein-mediated signaling via the well-established mechanism of homologous desensitization [6]. In addition, problems with bias quantification even in cultured cells can lead to misinterpretation of reduced agonist efficacy as G protein bias [82].
Even biasing the receptor by mutagenesis does not yield unambiguous results. E.g., the DRY mutants of angiotensin AT1R and histamine H1R previously believed to signal exclusively via arrestins were shown to activate G proteins [83]. Thus, “biased” GPCR mutants may not be as biased as one would wish. Discovery of GPCR megaplexes in which the receptor interacts with G protein and arrestin simultaneously adds another level of complexity [84]. Moreover, Gαi family members were found to bind arrestin-2 and form noncanonical scaffolds, which may have their own signaling signature [85].
Concluding remarks
The observed ligand-dependent bias of signaling by the same receptor needs to be explained. Solved structures of GPCRs activated by different ligands bound to different G proteins, arrestins, and GRK1 do not yield clear clues, extrapolatable to all members of the GPCR superfamily, regarding the conformational basis of signaling bias. One cannot tell which signal transducer a GPCR is coupled to by looking at its conformation in the complex. However, there are important caveats in focusing exclusively on the available structures. First, the structures of too few receptors were solved with more than one transducer. Second, GPCR complexes with G proteins, GRKs, and arrestins show the final result, but do not reveal the sequence of events in the process of each transducer’s binding to the receptor. This information might be critical for assessing the feasibility of biasing ligands [86]. We know too little about the dynamics of partner binding to GPCRs, where different paths might reveal the basis of bias better than the final complexes used for structure determination (see Outstanding Questions).
Outstanding questions.
GPCRs are remarkably flexible. To what extent can a ligand shift conformational landscape of a GPCR? Is this extent sufficient to direct signaling to a specific downstream pathway?
How do GPCRs select a particular G protein? Does the receptor structure predispose it to couple to a specific G protein? Does a receptor choose a G protein, or is it the G protein that choses the receptor? Is this interaction “lock and key” or “induced fit”?
Are GPCR complexes with arrestin-2 and arrestin-3 different? What determines which arrestin the receptor prefers?
Is there arrestin-mediated signaling totally independent of G proteins? Or are G proteins required for arrestin-mediated signaling, either in a sequence (G proteins act first, arrestins come next) or a complex (G proteins and arrestins bind the receptor simultaneously, or interact independently of GPCRs).
Last, but not least: to what extent is GPCR preference for a particular signal transducer determined by the complement of available transducers in a cell or by the dynamics of their interaction?
Highlights.
Ligand-specific GPCR signaling in cells and structures of ligand-specific receptor conformations suggested the possibility of selective conformation-dependent coupling to particular transducers resulting in biased signaling outcomes
However, available structures of GPCRs with different signal transducers do not reveal transducer-specific conformations
Dynamics of binding and/or complement of signal transducers, rather than the final structure of the receptor-transducer complex, may determine transducer preference
Acknowledgements
Supported in part by NIH grants R35 GM122491, RO1 EY011500, and Cornelius Vanderbilt Endowed Chair from Vanderbilt University (to V.V.G.).
Glossary
- Arrestins
Four-member family of proteins in most vertebrates. Arrestins are involved in desensitization of G protein-mediated GPCR signaling, but they have also been shown to participate in cell signaling beyond classical desensitization. The systematic nomenclature of these proteins is based on the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin).
- Biased ligand
A ligand selectively activating one/some of all available signaling pathways downstream of the receptor.
- Biased signaling
Selective ligand-specific signaling of the same receptor due to activation of a subset of downstream pathway(s).
- GPCRs
G protein coupled receptors; a superfamily of receptors described initially as conveying signals across biological membranes via the activation of G proteins. Later the signaling via arrestins was also reported. All GPCRs have an extracellular N-terminus, seven transmembrane α-helices (TMs) connected by three extracellular (ECL) and three intracellular (ICL) loops, and an intracellular C-terminus. Based on structure analysis, about 4% of protein coding sequences in the human genome encode 831 different GPCRs. More than 85% of GPCRs belong to class A (rhodopsin-like) with the orthosteric ligand binding site between transmembrane helices. Humans have 25 Class B (secretin receptor-like) GPCRs that have a much larger extracellular N-terminus with internal disulfide bonds that participates in ligand (peptide in this class) binding. Humans have even fewer class C GPCRs. These receptors are obligatory dimers, where each protomer has a very large N-terminal “venus flytrap” (VFT) domain related to bacterial periplasmic transporters of amino acids and ions. VFTs are attached to the seven transmembrane helix bundle, which is a structural hallmark of all GPCRs. In class C VFTs bind orthosteric ligands. The interhelical site where orthosteric ligands bind in class A GPCRs often binds allosteric modulators in class C.
- G proteins
Guanine nucleotide-binding proteins that act as molecular switches inside the cells. Their activation is based on GTP/GDP exchange. Activated GPCR catalyzes the exchange of GDP bound to inactive αβγ heterotrimer for GTP, which activates G proteins. GTP-liganded G proteins dissociate from the receptor and separate into Gα-GTP and Gβγ, both of which regulate various effectors. G proteins are divided into four subfamilies based on their α subunits. G proteins with Gαs/olf activate adenylyl cyclase, thereby increasing intracellular cAMP. Gαi/o/z inhibit adenylyl cyclase; transducin (Gt) in this subfamily activates cGMP phosphodiesterase in photoreceptors. Gαq/11 activate phospholipase C, which cleaves phosphatidylinositol-4,5-biphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 binds IP3 receptors in the endoplasmic reticulum, which allows calcium ions to escape to the cytoplasm. DAG activates protein kinase C in cooperation with cytoplasmic calcium. Gα12/13 target RhoGEFs containing an amino-terminal RGS homology domain.
- GRKs
GPCR kinases, belonging to the family of AGC protein kinases. GRKs use ATP to phosphorylate serine and threonine residues in cytoplasmic GPCR elements, making them “attractive” for arrestins. GRKs of the 1/7 (visual) and 2/3 subfamilies exclusively target activated GPCRs, whereas GRKs of the 4/5/6 subfamily, although preferring active GPCRs, can phosphorylate some inactive receptors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fredriksson R et al. (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63, 1256–72. [DOI] [PubMed] [Google Scholar]
- 2.Masuho I et al. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal 8 (405), ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hermans E (2003) Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol Ther 99 (1), 25–44. [DOI] [PubMed] [Google Scholar]
- 4.Holze J et al. (2020) Ligand-Specific Allosteric Coupling Controls G-Protein-Coupled Receptor Signaling. ACS Pharmacol Transl Sci 3 (5), 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seyedabadi M et al. (2019) Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol Ther 200, 148–178. [DOI] [PubMed] [Google Scholar]
- 6.Carman CV and Benovic JL (1998) G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol 8, 335–344. [DOI] [PubMed] [Google Scholar]
- 7.Peterson YK and Luttrell LM (2017) The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev 69 (3), 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurevich EV and Gurevich VV (2006) Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esmaeeli A et al. (2020) Low-dose angiotensin AT1 receptor beta-arrestin-biased ligand, TRV027, protects against cisplatin-induced nephrotoxicity. Pharmacol Rep 72 (6), 1676–1684. [DOI] [PubMed] [Google Scholar]
- 10.Gurevich VV and Gurevich EV (2019) Plethora of functions packed into 45 kDa arrestins: biological implications and possible therapeutic strategies. Cell Mol Life Sci 76 (22), 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurevich VV and Gurevich EV (2020) Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi M et al. (2018) G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the beta2-adrenergic receptor. Sci Signal 11 (544), 544. [DOI] [PubMed] [Google Scholar]
- 13.Ballesteros JA and Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in neurosciences, pp. 366–428, Elsevier. [Google Scholar]
- 14.Gurevich VV and Gurevich EV (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol Sci 25, 105–111. [DOI] [PubMed] [Google Scholar]
- 15.Urban JD et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- 16.Vaidehi N and Kenakin T (2010) The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr Opin Pharmacol 10 (6), 775–81. [DOI] [PubMed] [Google Scholar]
- 17.Wootten D et al. (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19 (10), 638–653. [DOI] [PubMed] [Google Scholar]
- 18.Lamichhane R et al. (2020) Biased Signaling of the G-Protein-Coupled Receptor beta2AR Is Governed by Conformational Exchange Kinetics. Structure 28 (3), 371–377 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manglik A et al. (2015) Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 161 (5), 1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elgeti M and Hubbell WL (2021) DEER Analysis of GPCR Conformational Heterogeneity. Biomolecules 11 (6), 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J et al. (2019) Conformational Complexity and Dynamics in a Muscarinic Receptor Revealed by NMR Spectroscopy. Mol Cell 75 (1), 53–65.e7. [DOI] [PubMed] [Google Scholar]
- 22.Rajagopal S et al. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80 (3), 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rankovic Z et al. (2016) Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26 (2), 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilger D (2021) The role of structural dynamics in GPCR-mediated signaling. FEBS J 288 (8), 2461–2489. [DOI] [PubMed] [Google Scholar]
- 25.Liu JJ et al. (2012) Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335 (6072), 1106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warne T et al. (2012) Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20 (5), 841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wingler LM et al. (2020) Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367 (6480), 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q et al. (2021) Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Draper-Joyce CJ et al. (2018) Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 558 (7711), 559–563. [DOI] [PubMed] [Google Scholar]
- 30.Gao Y et al. (2019) Structures of the Rhodopsin-Transducin Complex: Insights into G-Protein Activation. Mol Cell 75 (4), 781–790 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.García-Nafría J et al. (2018) Cryo-EM structure of the adenosine A(2A) receptor coupled to an engineered heterotrimeric G protein. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García-Nafría J et al. (2018) Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558 (7711), 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W et al. (2020) Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579 (7798), 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang Y et al. (2018) Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558 (7711), 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang Y et al. (2015) Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 523 (7562), 561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koehl A et al. (2018) Structure of the micro-opioid receptor-Gi protein complex. Nature 558 (7711), 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y et al. (2020) Molecular basis of β-arrestin coupling to formoterol-bound β(1)-adrenoceptor. Nature 583 (7818), 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang YL et al. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546 (7656), 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin S et al. (2021) Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594 (7864), 583–588. [DOI] [PubMed] [Google Scholar]
- 40.Maeda S and Qu Q (2019) Structures of the M1 and M2 muscarinic acetylcholine receptor/ G-protein complexes. 364 (6440), 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okamoto HH et al. (2021) Cryo-EM structure of the human MT1-Gi signaling complex. Nat Struct Mol Biol 28 (8), 694–701. [DOI] [PubMed] [Google Scholar]
- 42.Qiao A et al. (2020) Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 367 (6484), 1346–1352. [DOI] [PubMed] [Google Scholar]
- 43.Rasmussen SG et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seven AB et al. (2021) G-protein activation by a metabotropic glutamate receptor. Nature 595 (7867), 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen C et al. (2021) Structural basis of GABAB receptor-Gi protein coupling. Nature 594 (7864), 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staus DP et al. (2020) Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579 (7798), 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su M et al. (2020) Structural Basis of the Activation of Heterotrimeric Gs-Protein by Isoproterenol-Bound beta1-Adrenergic Receptor. Mol Cell 80 (1), 59–71 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia R et al. (2021) Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat Commun 12 (1), 2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin W et al. (2019) A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res 29 (12), 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y et al. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546 (7657), 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou XE et al. (2017) Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell 170 (3), 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flock T et al. (2015) Universal allosteric mechanism for Galpha activation by GPCRs. Nature 524 (7564), 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seyedabadi M et al. (2021) Receptor-Arrestin Interactions: The GPCR Perspective. Biomolecules 11 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Min K et al. (2020) Crystal Structure of β-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 28 (9), 1014–1023.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhan X et al. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol 406, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lymperopoulos A et al. (2019) Not all arrestins are created equal: Therapeutic implications of the functional diversity of the β-arrestins in the heart. World J Cardiol 11 (2), 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Indrischek H et al. (2017) Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol 17 (1), 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attramadal H et al. (1992) Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem 267, 17882–17890. [PubMed] [Google Scholar]
- 59.Chen Q et al. (2021) An Eight Amino Acid Segment Controls Oligomerization and Preferred Conformation of the two Non-visual Arrestins. J Mol Biol 433 (4), 166790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palczewski K et al. (1991) Mechanism of rhodopsin kinase activation. J Biol Chem 266, 12949–12955. [PubMed] [Google Scholar]
- 61.Farrens DL et al. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274 (5288), 768–770. [DOI] [PubMed] [Google Scholar]
- 62.Gurevich EV et al. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 133 (1), 40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nobles KN et al. (2011) Distinct Phosphorylation Sites on the {beta}2-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of {beta}-Arrestin. Sci Signal 4(ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gurevich EV and Gurevich VV (2020) GRKs as Modulators of Neurotransmitter Receptors. Cells 10 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendez A et al. (2000) Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28, 153–164. [DOI] [PubMed] [Google Scholar]
- 66.Vishnivetskiy SA et al. (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem 282, 32075–32083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao LH et al. (2019) Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364 (6436), 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou XE et al. (2019) Structural biology of G protein-coupled receptor signaling complexes. Protein Sci 28 (3), 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin X et al. (2020) Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 579 (7797), 152–157. [DOI] [PubMed] [Google Scholar]
- 70.Coward P et al. (1999) Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal Biochem 270 (2), 242–8. [DOI] [PubMed] [Google Scholar]
- 71.Xu Z et al. (2022) Structural basis of sphingosine-1-phosphate receptor 1 activation and biased agonism. Nat Chem Biol, in press. [DOI] [PubMed] [Google Scholar]
- 72.Shao Z et al. (2022) Identification and mechanism of G protein-biased ligands for chemokine receptor CCR1. Nat Chem Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilhelm M et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509 (7502), 582–7. [DOI] [PubMed] [Google Scholar]
- 74.Fang Y et al. (2006) Resonant waveguide grating biosensor for living cell sensing. Biophys. J 91, 1925–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruzza C et al. (2018) Pharmacological profile of the neuropeptide S receptor: Dynamic mass redistribution studies. Pharmacol Res Perspect 6 (6), e00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malfacini D et al. (2018) NOP receptor pharmacological profile - A dynamic mass redistribution study. PLoS One 13 (8), e0203021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grundmann M et al. (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 9 (1), 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luttrell LM et al. (2018) Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shenoy SK et al. (2006) beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281 (2), 1261–73. [DOI] [PubMed] [Google Scholar]
- 80.O’Hayre M et al. (2017) Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci Signal 10 (484). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hauge Pedersen M et al. (2021) A novel luminescence-based β-arrestin recruitment assay for unmodified receptors. J Biol Chem 296, 100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gillis A et al. (2020) Intrinsic Efficacy of Opioid Ligands and Its Importance for Apparent Bias, Operational Analysis, and Therapeutic Window. Mol Pharmacol 98 (4), 410–424. [DOI] [PubMed] [Google Scholar]
- 83.Pietraszewska-Bogiel A et al. (2020) Not So Dry After All: DRY Mutants of the AT1A Receptor and H1 Receptor Can Induce G-Protein-Dependent Signaling. ACS Omega 5 (6), 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thomsen ARB et al. (2016) GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166 (4), 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith JS et al. (2021) Noncanonical scaffolding of Galphai and beta-arrestin by G protein-coupled receptors. Science 371 (6534). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gusach A et al. (2020) Beyond structure: emerging approaches to study GPCR dynamics. Curr Opin Struct Biol 63, 18–25. [DOI] [PubMed] [Google Scholar]
- 87.Huang J et al. (2013) Crystal structure of oligomeric beta1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol 20 (4), 419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haga K et al. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482 (7386), 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thal DM et al. (2016) Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531 (7594), 335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang H et al. (2015) Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 161 (4), 833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Okada T et al. (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol 342 (2), 571–83. [DOI] [PubMed] [Google Scholar]