

Abstract
Small near-infrared (NIR) fluorescent proteins (FPs) are much needed as protein tags for imaging applications. We developed a 17 kDa NIR FP, called miRFP670nano3, which brightly fluoresces in mammalian cells and enables deep-brain imaging. By exploring miRFP670nano3 as an internal tag, we engineered 32 kDa NIR fluorescent nanobodies, termed NIR-Fbs, whose stability and fluorescence strongly depend on the presence of specific intracellular antigens. NIR-Fbs allowed background-free visualization of endogenous proteins, detection of viral antigens, labeling of cells expressing target molecules, and identification of double-positive cell populations with bispecific NIR-Fbs against two antigens. Applying NIR-Fbs as destabilizing fusion partners, we developed molecular tools for directed degradation of targeted proteins, controllable protein expression, and modulation of enzymatic activities. Altogether, NIR-Fbs enable the detection and manipulation of a variety of cellular processes based on the intracellular protein profile.
Keywords: nanobody, chromobody, iRFP, phytochrome, BphP, cyanobacteriochrome, CBCR
Optical imaging with near-infrared (NIR) fluorescent proteins (FPs) provides increased tissue penetration depths and a better signal-to-noise ratio due to reduced light-scattering, tissue absorption and autofluorescence in the NIR region (650–900 nm)1, 2. NIR FPs allow labeling of whole organisms, specific cell populations, organelles or individual proteins, and enable spectral multiplexing with FPs, biosensors and optogenetic tools active in visible range1.
The majority of available NIR FPs were engineered from bacterial phytochrome photoreceptors (BphPs)3 that use as a chromophore biliverdin IVα (BV), available in mammalian cells4, 5. However, for BV attachment BphP-based FPs require two PAS and GAF domains tightly interlinked by a complex “knot” structure and have a relatively high molecular weight of 35 kDa. In contrast, cyanobacteriochromes (CBCRs) can bind chromophores via GAF domain only and allow engineering of small 17 kDa NIR FPs. We recently reported a miRFP670nano, the first single-domain NIR FP, developed from CBCR6. Compared to BphP-derived NIR FPs, miRFP670nano is 2-fold smaller, naturally monomeric, tolerant to an acidic environment, denaturation conditions, cell fixation, and has significantly greater stability in mammalian cells6. Due to the compact single-domain fold with the N- and C- termini positioned close to each other, unlike BphP-based FPs, miRFP670nano performs well as the internal protein tag6.
While direct tagging of proteins with FPs ensures specificity and allows studying of protein dynamics in live cells, in some cases FP-fusion constructs behave differently from their endogenous analogs, due to altered expression level, turnover, and blocked by FP-tag functional domains7. Endogenous, not modified, proteins can be visualized by nanobodies (Nbs), which are single-domain 15 kDa antigen-binding fragments derived from camelid heavy-chain-only antibodies8. Despite their small size, Nbs bind antigens with high affinity and specificity9–11. The advantage of Nbs is their ability to recognize and bind a cognate (specific) antigen intracellularly12.
It has been shown that genetic fusions of Nbs with FPs of the GFP-like family, named chromobodies (Cbs), enabled visualization of protein dynamics in live cells13. However, imaging with Cbs often suffers from their excess inside a cell. Accumulation of fluorescent Nbs that remained unbound led to diffuse background signal, affecting the signal of antigen-bound Nbs13–15. To reduce the expression level of Cbs, low or inducible promoters could be selected14. Additionally, stable cell lines with a consistent Cb expression could be generated16, 17. These approaches allow to moderate the impact of background signal but do not allow studies of cell populations heterogeneous in antigen expression.
To control the level of intracellular Cbs, it was further suggested to use conditionally stable Nbs containing six point mutations in the Nb framework regions18. The diversity of Nbs is mainly determined by 3 complementarity determining regions (CDRs), whereas 4 framework regions are relatively conserved19. However, the introduction of mutations at the frameworks may affect the Nb antigen-binding properties20–23. The analysis of structures of the Nb-antigen complexes shows that a considerable number of contacts with antigen is mediated by the residues in the framework regions24, which limits this approach to reduce the intracellular level of Cbs.
Recently, an autorepression system based on the Krüppel associated box (KRAB) transcriptional repressor fused with antibody-like fibronectin-derived intrabodies (antigen binders functional in eukaryotic cells) that, in turn, fused to an FP, has been suggested25. This way the intrabodies expression depends on the antigen level because the unbound intrabodies fusions are translocated to the nucleus where the KRAB domain represses their transcription. This system can be also used with other intrabodies including Nbs23. However, the high accumulation of intrabodies in the nucleus produces a strong fluorescent signal23, 25, even if the cognate antigen is not expressed, limiting this approach to non-nuclear antigens and to homogeneously antigen-expressing cell populations.
In this work, we developed an enhanced CBCR-based single-domain NIR FP, called miRFP670nano3. We showed that it brightly fluoresces in mammalian cells without a supply of any cofactors and can be used for deep-tissue imaging in vivo. We then designed small 32 kDa fluorescent Nbs, internally tagged with the NIR FP, termed NIR-Fbs. NIR-Fbs efficiently recognize intracellular cognate antigens but are rapidly degraded in their absence. We first applied NIR-Fbs for re-coloring of cells expressing a visible-range FP into NIR, and then for NIR labeling of endogenous proteins. We next engineered bispecific NIR-Fb constructs for selective NIR labeling of only those cells that express two specific antigens. We then showed that NIR-Fbs can serve as the destabilizing fusion partner for regulatory proteins and peptides, enabling control of cellular processes with cell-selectivity mediated by the expression of specific intracellular epitopes. We finally applied NIR-Fbs for degradation of target proteins, regulation of protein expression, and kinase activities in an antigen-dependent manner.
Results
Molecular evolution of miRFP670nano protein.
A CBCR-based monomeric NIR FP miRFP670nano was used as a starting point for molecular evolution. We performed several rounds of random mutagenesis of miRFP670nano, followed by simultaneous saturating mutagenesis of the identified residues. In each round, initial screening was performed in E.coli co-expressing heme oxygenase for high BV production. However, the brightness of NIR FPs in mammalian cells (termed as effective brightness) may differ from the brightness in the heme oxygenase expressing bacteria. Since the NIR FP fluorescence is provided by BV chromophore, efficient binding of endogenous BV is essential for the NIR FP’s effective brightness in mammalian cells3, 26. Therefore, in each round of miRFP670nano mutagenesis, we screened the brighter clones not only in E.coli but also in transiently transfected HeLa cells. The 15 rounds of the molecular evolution resulted in a NIR FP, named miRFP670nano3, having 14 mutations relative to parental miRFP670nano (Supplementary Fig. 1).
Characterization of miRFP670nano3 protein in vitro.
An enhanced miRFP670nano3 protein exhibited fluorescence excitation and emission at 645 nm and 670 nm, respectively, which were similar to spectral properties of parental miRFP670nano (Fig. 1a–b and Supplementary Table 1). Molecular brightness (the product of a molar extinction coefficient and a quantum yield) of miRFP670nano3 was 2-fold higher than molecular brightness of miRFP670nano and 1.5-fold higher than widely used red FPs mCherry or mCardinal. Notably, the extinction coefficient (129,000 M−1cm−1) and fluorescence quantum yield (18.5%) of miRFP670nano3 are the highest among all monomeric NIR FPs with BV chromophore (Supplementary Table 1). Similar to parental miRFP670nano, miRFP670nano3 exhibited monomeric behavior in size exclusion chromatography. miRFP670nano3 with the polyhistidine tag and linker run as a monomer with the apparent molecular weight of 18.8 kDa (Fig. 1c and Supplementary Fig. 2).
Figure 1. Characterization of miRFP670nano3 protein.

(a) Absorbance spectra. (b) Fluorescence excitation spectrum recorded at 730 nm emission and emission spectrum recorded at 600 nm excitation. (c) Size exclusion chromatography of miRFP670nano3 and used molecular weight protein standards. (d) pH dependence of miRFP670nano3 fluorescence in comparison with parental miRFP670nano. (e) Effective (cellular) brightness of miRFP670nano3 and miRFP670nano in transiently transfected HeLa, N2A, U-2 OS, HEK293T, and NIH3T3 live cells. Fluorescence intensity was analyzed 72 h after transfection. The effective brightness of miRFP670nano was assumed to be 100% for each cell type. Data are presented as mean values ± s.d. for n = 3 transfection experiments. (f) Photobleaching kinetics of miRFP670nano3 in comparison with parental miRFP670nano in live HeLa cells. (g) Fluorescence intensity of live HeLa cells transiently transfected with miRFP670nano3, miRFP670nano, or EGFP before and after 4 h of incubation with 20 μg/ml cycloheximide. Data are presented as mean values ± s.d. for n = 3 transfection experiments. (h) Fluorescence intensity of live HeLa cells transiently transfected with miRFP670nano3, miRFP670nano, or EGFP before and after 4 h of incubation with 10 μM bortezomib. Data are presented as mean values ± s.d. for n = 3 transfection experiments. (i) Fluorescence intensity of live HeLa cells transiently transfected with miRFP670nano3, miRFP670nano, or EGFP 48 h and 120 h after transfection normalized to that at 48 h. Data are presented as mean values ± s.d. for n = 4 transfection experiments. Fluorescence intensity in (e, g, h, i) was measured by flow cytometry using a 640 nm excitation laser and a 670 nm LP emission filter for miRFP670nano and miRFP670nano3, and a 488 nm excitation laser and a 510/15 nm emission filter for EGFP. Gating was performed as shown in Supplementary Fig. 3.
Fluorescence of miRFP670nano3 was stable over a wide pH range of 4.0–11.0. (Fig. 1d). In contrast to parental miRFP670nano, which is more alkali sensitive, the developed miRFP670nano3 demonstrated high tolerance to both acidic and alkaline environments.
Performance of miRFP670nano3 in live mammalian cells.
miRFP670nano3 was brightly fluorescent in mammalian cells without adding exogenous BV. Its effective brightness was more than 4-fold higher than that of miRFP670nano in all tested mammalian cell lines (Fig. 1e, Supplementary Fig. 3 and Supplementary Table 1).
An important advantage of miRFP670nano over BphP-derived NIR FPs is high photostability. The enhanced miRFP670nano3 exhibited photostability close to that of miRFP670nano (Fig. 1f and Supplementary Table 1).
miRFP670nano3 was stable in mammalian cells. After 4 h incubation with cycloheximide, a protein synthesis inhibitor, the miRFP670nano3 expressing cells retained 85% of their fluorescence. Treatment with bortezomib, an inhibitor of proteasome-dependent protein degradation, increased the brightness of the miRFP670nano3 expressing cells only by 10%. Similar cellular stability was observed for miRFP670nano and EGFP (Fig. 1g,h and Supplementary Fig. 3). A fluorescence comparison of cells transiently expressing miRFP670nano3 48 h and 120 h after the transfection further confirmed its high cellular stability (Supplementary Fig. 4). Notably, 120 h after transfection the miRFP670nano3 expressing cells retained ~80% of their fluorescence, whereas the miRFP670nano- and EGFP-expressing cells only ~60% (Fig. 1i and Supplementary Fig. 3). miRFP670nano3 fluorescence was 85% at 48 h and reached a maximum 72 h after the transfection, which was similar to the miRFP670nano transfected cells (Supplementary Fig. 5).
To confirm that miRFP670nano3 is monomeric in mammalian cells, we performed an organized smooth endoplasmic reticulum (OSER) assay27. In this assay, FPs are fused to the cytoplasmic end of the endoplasmic reticular signal anchor (CytERM). The formation of dimers or oligomers should lead to the reorganization of ER architecture, which is observed as OSER whorl structures. OSER assay showed that miRFP670nano3 is monomeric in mammalian cells, with 87% of cells exhibiting normal phenotypes (Supplementary Fig. 6). Moreover, a ratio of the mean fluorescence intensities of the observed OSER structures and the nuclear envelope (NE) was 1.98±0.14, further indicating that miRFP670nano3 is a monomer27.
Due to its high brightness, pH-stability and compact single-domain fold, miRFP670nano3 performed well as C-, N- or internally inserted tags. In live mammalian cells, the fusions exhibited proper localization, including the fusions with proteins forming filaments, such as α-tubulin, β-actin and myosin, the fusions with transmembrane proteins, such as EGFR (epidermal growth factor receptor) and β2AR (β2-adrenergic receptor), and the fusions with proteins targeted to cellular compartments, such as clathrin, lysosome-associated membrane protein LAMP1 and histone 2B. As the internal tags, miRFP670nano3 was inserted between the helical and GTPase domains of the G-protein α-subunit (Gαs) and into the intracellular loop 3 of β2AR (Supplementary Fig. 7). Furthermore, primary rat cortical neurons expressing miRFP670nano3 exhibited bright homogenous fluorescence without the supply of exogenous BV (Supplementary Fig. 8).
Two-photon imaging in the brain and spinal cord in vivo.
To evaluate the utility of miRFP670nano3 for in vivo imaging, we sub-cloned it into an adeno-associated viral (AAV) vector carrying a synapsin promoter. The vector was then injected into the somatosensory cortex or spinal dorsal horn of Cx3cr1 GFP/+ mice with labeled microglia. Three to five weeks after injection, animals were prepared for in vivo two-photon imaging to determine protein expression and attainable imaging depth.
NIR FPs, in addition to their absorption peaks in the so-called Q band (i.e., 30–70 GM at 1200–1280 nm), exhibit high cross-sectional values in the so-called Soret band (180–450 GM at 890–950 nm)1, 28. This feature permits multiplex, subcellular resolution imaging of miRFP670nano3 and EGFP with single-wavelength two-photon excitation using a standard Ti:Sapphire laser. Our experiments used the excitation light powers that, based on our previous studies29–31, do not activate microglia.
We found that miRFP670nano3 brightly fluoresced in transduced neuronal cells without the need for external BV (Fig. 2). At the 920 nm wavelength optimally exciting EGFP, miRFP670nano3-positive cells were routinely visualized through the entire depth of the cortex, down to the entorhinal cortex level (~850 μm) that exceeded those with EGFP in Cx3cr1 GFP/+ mice with brightly EGFP-labeled microglia (Supplementary Videos S1–S3).
Figure 2. Multiplex two-photon imaging with miRFP670nano3 in the brain and spinal cord of live mice.
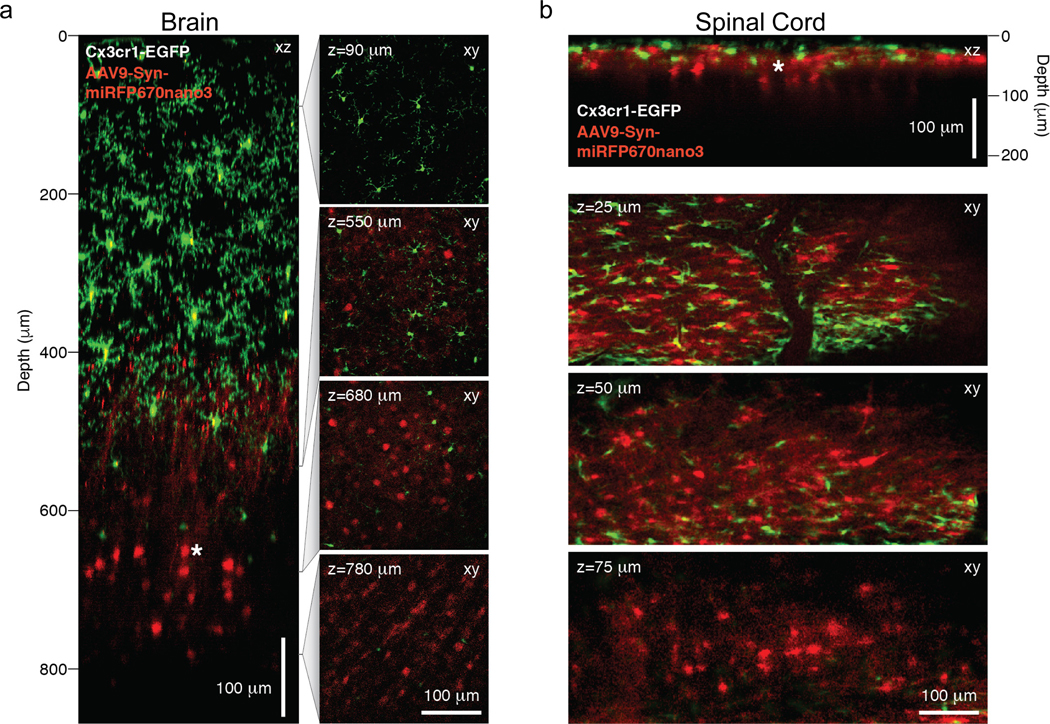
(a) Left, xz sub-projection from a xy fluorescence image stack showing neurons (red) and microglia (green) in the somatosensory cortex of a 14-weeks-old Cx3cr1 GFP/+ mouse five weeks after stereotactic AAV9-hSYN-miRFP670nano3 vector delivery into deep cortical layers. Right, four example images from the xy fluorescence image stack at the indicated depths (z) from the pial surface. (b) Top, xz sub-projection from a xy fluorescence image stack showing neurons (red) and microglia (green) in the lumbar spinal cord of a 12.5-weeks-old Cx3cr1 GFP/+ mouse five weeks after stereotactic AAV9-hSYN-miRFP670nano3 vector delivery into superficial dorsal horn laminae. Bottom, three example images from the xy fluorescence image stack at the indicated depths (z) from the pial surface. (a-b) GFP and miRFP670nano3 fluorescence images were acquired simultaneously using a 920 nm excitation light and emission filters 525/70 nm for EGFP and 645/75 nm for miRFP670nano3. No external BV was administered. Representative images of three experiments are shown. Scale bars, 100 μm.
It has been demonstrated that microglia respond to neuronal tissue perturbations with morphological changes and functional alterations29–32. Such changes were not observed in response to the long-term (3–5 weeks) miRFP670nano3 expression (Fig. 2 and Supplementary Videos S1–S3), indicating that miRFP670nano3 do not cause cytotoxicity and are well tolerated in vivo.
Construction of Nbs internally fused with miRFP670nano3.
We next designed internal fusions of miRFP670nano3 with a well-characterized Nb to GFP (NbGFP)20, 33. We tested three insertion positions in NbGFP (Fig. 3a and Supplementary Fig. 9) shown to be Nb split sites34. In cells co-expressing EGFP, all internal fusions exhibited NIR fluorescence, which varied depending on the insertion site and linkers length. Without EGFP, the fluorescence of internal fusions was not detected (Fig. 3b,c, Supplementary Fig. 3 and Supplementary Fig. 10), suggesting that fluorescence of fusions depended on the presence of a cognate antigen EGFP. In contrast, when miRFP670nano3 was fused to NbGFP as either N- or C-terminal tag, the fusions had NIR fluorescence in both the EGFP expressing and EGFP non-expressing cells (Supplementary Fig. 11). The brightest internal fusion had a miRFP670nano3 insertion between NbGFP residues Ser65 and Val66 with Gly4Ser linkers (Fig. 3b,c and Supplementary Fig. 10). This fusion variant was termed a NIR-FbGFP and subjected to further analysis. We expressed NIR-FbGFP in E.coli and found that, in contrast to mammalian cells, bacterial cells expressing NIR-Fb without antigen, exhibited NIR fluorescence (Supplementary Fig. 12a). NIR-FbGFP purified from E.coli recognized immobilized EGFP and bound it from solution in the dot-blot analysis (Supplementary Fig. 12b–e), suggesting that NIR-FbGFP expressed in E.coli retained both the miRFP670nano3 fluorescence and NbGFP antigen-binding properties.
Figure 3. Design and evaluation of nanobodies (Nbs) internally fused with miRFP670nanos (NIR-Fbs) in live HeLa cells.

(a) Structure of Nb against GFP (NbGFP) (PDB ID: 3OGO) with indicated positions for insertion of miRFP670nano3. (b) The fluorescence intensity distribution of cells transfected with NbGFP, containing miRFP670nano3 inserted at G44/K45, S65/V66, or P90/E91 positions and co-expressed with or without EGFP. (c) Quantification of the data presented in (b). Data are presented as mean values ± s.d. for n = 3 transfection experiments. (d) Fluorescence intensity of cells transfected with the same amount of the pNIR-FbGFP plasmid and indicated ratios of the pEGFP-N1 plasmid to the pNIR-FbGFP plasmid. Fluorescence intensity of cells transfected with the pNIR-FbGFP and pEGFP-N1 plasmids with the ratio of 1:1 was assumed to be 100% for each FP. Data are presented as mean values ± s.d. for n = 3 transfection experiments. (e) Mean fluorescence intensity of cells transfected with NbGFP, containing miRFP670nano3 inserted at G44/K45, S65/V66 or P90/E91 sites via Gly4Ser linkers before and after 4 h of incubation with 10 μM bortezomib. Fluorescence intensity of cells co-transfected with pEGFP-N1 and NbGFP, containing miRFP670nano3 inserted at Ser65/Val66 was assumed to be 100%. Data are presented as mean values ± s.d. for n = 3 transfection experiments. (f) The fluorescence intensity distribution of cells transfected with NIR-Fbs to indicated antigens and co-expressed with or without cognate antigen. (g) Fluorescence images of cells transfected with NIR-Fbs to the indicated antigens and co-expressed with either mEGFP-labeled cognate antigens or with unfused EGFP as a control. Representative images of two experiments are shown. For imaging of miRFP670nano3 and EGFP, a 605/30 nm excitation and a 667/30 nm emission, and a 485/20 nm excitation and a 525/30 nm emission filter was used, respectively. In (b, c, d, e) fluorescence intensity was analyzed by flow cytometry using a 640 nm excitation laser and a 675/25 nm emission filter for miRFP670nano3, and a 488 nm excitation laser and a 510/15 nm emission filter for EGFP. Gating was performed as shown in Supplementary Fig. 3. In (g) scale bars, 10 μm.
To determine whether specificity to an antigen affected NIR-Fb behavior, we used EGFP bearing an N146I mutation, which has been shown to dramatically reduce binding with NbGFP35. Co-expression of NIR-FbGFP and EGFP/N146I caused a more than 10-fold decrease in NIR fluorescence of HeLa cells as compared to the cells co-expressed NIR-FbGFP and non-mutated EGFP (Supplementary Fig. 13), indicating that NIR-FbGFP performance depended on antigen specificity.
To assess whether an antigen concentration affects the protein level of NIR-Fb, we co-transfected cells with the fixed amount of a NIR-FbGFP-encoding plasmid and decreasing amounts of an EGFP-encoding plasmid. Despite we used the same amount of the NIR-FbGFP plasmid for cells transfection, the fluorescence intensity of NIR-FbGFP was different and depended on the EGFP expression level (Fig. 3d, Supplementary Fig. 3 and Supplementary Fig. 14), confirming that NIR-Fb exhibits the antigen-dependence in a dose-dependent manner. Finally, when we fused NIR-FbGFP to mTagBFP2 36, the mTagBFP2 fluorescence similarly depended on the presence of EGFP antigen (Supplementary Fig. 15).
These data suggested that a possible mechanism of NIR-FbGFP behavior in mammalian cells is the stabilization by antigen binding and degradation in the unbound state by the ubiquitin-proteasome system. To evaluate this hypothesis, we treated the NIR-FbGFP transfected cells with an inhibitor of proteasome-dependent protein degradation bortezomib. Whereas non-treated cells were non-fluorescent, the cells incubated with bortezomib demonstrated strong NIR fluorescence (Fig. 3e, Supplementary Fig. 3 and Supplementary Fig. 16) in agreement with this mechanism.
Engineering of NIR-Fbs using different nanobodies.
To study whether the NIR-Fb approach can be extended to other Nbs, we applied it to 9 more Nbs, such as Nb specific to actin37, BC2 Nb to β-catenin22, 38, 39, LaM4 Nb to mCherry40, Nb against ALFA-tag41, Nb ca1698 to E.coli DHFR (dihydrofolate reductase)42, Nb 59H10 and 2E7 to HIV (human immunodeficiency virus) antigens p2443 and gp4144, 45, and Nb 21 and m6 specific to spike SARS-Cov-2 antigen46, 47 (Supplementary Fig. 17).
Although these Nbs have a similar structural architecture, their mode of antigen binding is different. NbGFP employs all three CDR regions for antigen binding (PDB ID: 3OGO)20. Instead, in the LaM4 interaction with mCherry, the binding occurs via the CDR3 and only partly via CDR1 (PDB ID: 6IR1)48. Most of the contacting surfaces of the Nb6 and spike SARS-CoV-2 RBD (receptor binding domain) are contributed by the CDR1 and CDR247. The interactions in the BC2 Nb are formed by the CDR3 and framework regions 2 and 3 (PDB ID: 5IVN)22. The NbALFA binding with ALFA-peptide is mainly contributed by CDRs 2 and 3 but also involves framework region 2 (PDB ID: 6I2G)41. The Nb ca1698 binds to DHFR using residues from all three CDRs and framework 2 (PDB ID: 4EIG)42. To interact with the C-terminal domain of the HIV p24 antigen, the Nb 59H10 uses its framework region 2 and all three CDRs (PDB ID: 5O2U)43. The major contacts of the Nb 2E7 and HIV gp41 antigen are formed by residues in CDR1, CDR2 and framework region 2 (PDB ID:5HM1)45. The Nb 21 binds to spike SARS-CoV-2 RBD via all three CDRs and framework regions 2 and 346.
Despite differences in the structural basis of antigen binding, similar to NIR-FbGFP all engineered NIR-Fbs exhibited antigen-dependent stabilization in mammalian cells. The 8 engineered NIR-Fbs exhibited strong fluorescence in cells co-expressing their cognate antigens and were degraded in the absence of specific antigens (Fig. 3f,g and Supplementary Fig. 3). Only NIR-FbBC2 produced rather weak fluorescence that was consistent with the properties of the used Nb to β-catenin, which was reported to have low cellular expression22.
Re-coloring of fluorescent proteins with NIR-Fbs.
We next applied NIR-Fbs for re-coloring49 of EGFP- and mCherry-labeled proteins. HeLa cells were co-transfected with EGFP- or mCherry-tagged β-actin and α-tubulin and a 10-fold excess of NIR-FbGFP and NIR-FbmCherry encoding plasmids. Despite this, both NIR-FbGFB and NIR-FbmCherry strongly colocalized with the EGFP- or mCherry-labeled cytoskeletal proteins, and no background signal was observed (Fig. 4a,b and 4c,d).
Figure 4. Labeling of intracellular proteins with NIR-Fbs in live HeLa cells.

Re-coloring of EGFP and mCherry fluorescent proteins with NIR-Fbs: (a) EGFP-β-actin labeled with NIR-FbGFP, (b) EGFP-α-tubulin labeled with NIR-FbGFP, (c) mCherry-β-actin labeled with NIR-FbmCherry, and (d) mCherry-α-tubulin labeled with NIR-FbmCherry. (e) Schematic representation of NIR-FbALFA to ALFA tag. (f) ALFA-tagged α-tubulin, β-actin, myosin, and clathrin labeled with NIR-FbALFA. (g) Schematic representation of NIR-Fbactin to β-actin. (h) Endogenous β-actin labeled with NIR-Fbactin. (i) Schematic representation of non-fluorescent NIR-Fbactin/Y58C fused with EGFP for indirect EGFP-labeling of β-actin. (j) Endogenous β-actin labeled with NIR-Fbactin/Y58C–EGFP. In (a-d, f, h, j), scale bars, 10 μm. Representative images of two experiments are shown. For imaging of miRFP670nano3, EGFP, and mCherry, a 605/30 nm excitation and a 667/30 nm emission, a 485/20 nm excitation and a 525/30 nm emission, and a 560/25 nm excitation and a 607/36 nm emission filter was used, respectively.
The NIR re-coloring with NIR-FbGFP and NIR-FbmCherry can be applied to numerous available transgenic mice and cell lines expressing proteins of interest tagged with EGFP or mCherry50. NIR-Fbs exhibit antigen-dependence stabilization in a dose-dependent manner (Fig. 3d), which allows the re-coloring of cells heterogeneous for the EGFP or mCherry expression level.
Notably, NIR-FbALFA engineered with Nb specific to ALFA tag41 performed well in the labeling of various ALFA-tagged cellular proteins (Fig. 4e,f).
Furthermore, using NIR-Fbactin engineered with Nb specific for actin we showed that NIR-Fb could be fused with other FPs, like EGFP, for indirect labelling of proteins of interest (Fig. 4h–j). To avoid two-color (green and NIR) labelling in this application, we engineered a non-fluorescent miRFP670nano3 mutant, miRFP670nano3/Y58C, using the previously reported for CBCRs color-tuning mechanism in which the additional introduced tetrapyrrole-binding Cys residue caused the reduction of the BV chromophore3
Bispecific NIR-Fbs for labeling of double-positive cells.
We hypothesized that two fused NIR-Fbs for two different antigens would be stable only in the presence of both cognate antigens, forming a multi-antigen logic gate. To evaluate this hypothesis, we engineered a bispecific fusion construct containing NIR-Fbs to two different cognate antigens, EGFP and mCherry (Fig. 5a). Co-expression of both EGFP and mCherry resulted in miRFP670nano3 fluorescence, indicating that the bispecific NIR-FbGFP−NIR-FbmCherry fusion was stabilized by binding with both antigens. In contrast, when NIR-FbGFP−NIR-FbmCherry was co-expressed with either EGFP or mCherry alone the NIR fluorescence dropped >10-fold, indicating that the bispecific fusion was degraded (Fig. 5b,c). Thus, bispecific NIR-Fbs permit specific labeling of double-positive cell populations expressing both targeted antigens, efficiently acting as Boolean “and” elements. This property makes it very useful for the design of new signaling pathways and other synthetic biology applications in mammalian cells.
Figure 5. NIR-Fb fusions with antigen-dependent properties.

(a) Schematic representation of the bispecific NIR-FbGFP–NIR-FbmCherry fusion. If at least one cognate antigen (EGFP or mCherry) is not expressed in the cell, the whole fusion with the single attached antigen is degraded. (b) Fluorescence images of live HeLa cells transiently co-transfected with NIR-FbGFP–NIR-FbmCherry, EGFP cognate antigen and mCherry cognate antigen (top row), with NIR-FbGFP–NIR-FbmCherry and EGFP only (middle row), or with NIR-FbGFP–NIR-FbmCherry and mCherry only (bottom row). Scale bar, 10 μm. (c) Quantification of the data presented in (b). (d) Schematic representation of NIR-FbGFP–GAL4 and biNIR-FbGFP–GAL4 fusions for the antigen-dependent protein expression. In the presence of cognate antigen (EGFP), NIR-FbGFP–GAL4 or biNIR-FbGFP–GAL4 fusions drive expression of Gaussia luciferase (Gluc) reporter. Without cognate antigen, NIR-FbGFP–GAL4 and biNIR-FbGFP–GAL4 fusions are degraded, resulting in no Gluc expression. Bioluminescence signal of live HeLa cells co-transfected with 5xUAS Gluc, either EGFP or mTagBFP2, and (e) NIR-FbGFP–GAL4 or (f) biNIR-FbGFP–GAL4. (g) Schematic representation of NIR-FbGFP–NbALFA and biNIR-FbGFP–NbALFA fusions for antigen-dependent degradation of ALFA-tagged proteins. In the presence of cognate antigen (EGFP), ALFA-tagged GAL4 drives expression of Gluc reporter, whereas without EGFP the NIR-FB fusions are degraded together with bound ALFA-tagged GAL4. Bioluminescence signal of live HeLa cells co-transfected with ALFA-tagged GAL4, 5xUAS Gluc reporter, either EGFP or mTagBFP2, and (h) NIR-FbGFP–NbALFA or (i) biNIR-FbGFP–NbALFA. In (c, e, f, h, i) data are presented as mean values ± s.d. for n = 3 transfection experiments. For imaging of miRFP670nano3, EGFP, and mCherry, a 605/30 nm excitation and a 667/30 nm emission, a 485/20 nm excitation and a 525/30 nm emission, and a 560/25 nm excitation and a 607/36 nm emission filter was used, respectively.
Antigen-dependent gene regulation.
We next evaluated whether NIR-Fb could act as a destabilizing fusion partner for regulatory proteins. First, to confirm that NIR-Fb mediates antigen-dependent stability of its fusion partner, we constructed NIR-FbmCherry–EGFP fusion and evaluated its behavior in live cells co-transfected with mCherry or mTagBFP2, respectively (Supplementary Fig. 18a,b). EGFP fluorescence was observed only in the cells expressing mCherry (Supplementary Fig. 18b,c), suggesting that in the absence of cognate antigen both NIR-FbmCherry and its fusion-partner EGFP were degraded.
We then applied this approach to antigen-dependent gene expression. For this, we used a GAL4/UAS system in which the GAL4 transcription factor drives expression of the reporter gene downstream of its UAS (upstream activating sequence)’s DNA-binding site51. We fused GAL4 to NIR-FbGFP (Fig. 5d, right) and co-expressed it with the pUAS-Gluc reporter plasmid encoding Gaussia luciferase (Gluc) and with either EGFP or mTagBFP2. When mTagBFP2 was co-expressed instead of EGFP, the NIR-FbGFP–GAL4 was degraded, resulting in the 25-fold decrease of the bioluminescent signal as compared to cells co-transfected with EGFP (Fig. 5e). A biNIR-FbGFP–GAL4 construct, containing two NIR-FbGFP fused with GAL4 (Fig. 5d, left), exhibited even stronger antigen-dependence, providing more than the 50-fold decrease of the Gluc signal with co-expressed mTagBFP2 as compared to the EGFP-expressing cells (Fig. 5f).
NIR-Fbs for targeted degradation of cellular proteins.
We next studied whether NIR-Fb could drive the degradation of targeted proteins upon their binding. As a binding pair, we used NbALFA and its small antigen ALFA peptide (SRLEEELRRRLTE)41 fused to a protein. We tested the degradation of the GAL4 transcription factor. For this, we fused NIR-FbGFP and biNIR-FbGFP with NbALFA and GAL4 with ALFA peptide (Fig. 5g). Co-expression of ALFA-labeled GAL4 and pUAS-Gluc with NIR-FbGFP–NbALFA without EGFP resulted in ~75% reduction of the Gluc signal as compared to the EGFP-expressing cells (Fig. 5h). Moreover, with biNIR-FbGFP–NbALFA the ~85% reduction of the Gluc signal was observed in the absence of EGFP (Fig. 5i). These data suggested that intracellular proteins can be targeted for the directed antigen-dependent degradation mediated by binding with NIR-Fb fusions.
Control of kinases activity in antigen-dependent manner.
We next applied NIR-Fb for modulation of protein kinases activity. As for tagging kinases, we selected Protein Kinase A (PKA) and c-Jun N-terminal kinase (JNK). PKA is one of the key effectors of the cAMP-dependent pathway, while JNK regulates cellular responses to stress signals52. The activity of PKA can be specifically inhibited by a peptide comprising amino acids 14–22 (GRTGRRNAI) of the PKA inhibitor protein PKI53. This peptide binds to the catalytic subunit of PKA and acts as pseudosubstrate54. For inhibition of JNK activity, we chose the widely used inhibitory peptide composed of the amino acids 153–163 (RPKRPTTLNLF) of the JNK-interacting protein-1 JIP-155. The binding of this peptide induces rearrangement between the N- and C-terminal domains of JNK and distorts the ATP-binding pocket, inhibiting the catalytic activity56.
To develop antigen-dependent inhibitors for PKA and JNK kinases, we engineered constructs consisting of corresponding inhibitory peptides and non-fluorescent NIR-FbGFP/Y58C variant, containing non-fluorescent miRFP670nano3/Y58C mutant instead of miRFP670nano3 (Fig. 6a). The resultant NIR-FbGFP/Y58C–PKI and NIR-FbGFP/Y58C–JIP fusions were expressed in the presence and absence of EGFP in the reported earlier HeLa cell lines, expressing NIR FRET biosensors of PKA or JNK kinases6, respectively. The NIR FRET biosensors consisted of a miRFP670nano donor, a phosphoamino acid-binding domain, a peptide sequence of PKA or JNK kinases substrates, and a miRFP720 acceptor. Activated kinases phosphorylate the substrate peptides, resulting in a conformation rearrangement and FRET increase between the miRFP670nano donor and miRFP720 acceptor6. Kinase activity was determined after stimulation of PKA with 1 mM dibutyryl cyclic adenosine monophosphate (dbcAMP) and JNK with 1 μg/ml anisomycin. Co-transfection with NIR-FbGFP/Y58C–PKI and NIR-FbGFP/Y58C–JIP fusions with EGFP resulted in the inhibition of PKA and JNK kinases by ~80% and ~76%, respectively (Figs. 6b,d and 6c,e). These results demonstrated the ability of NIR-Fb inhibitory fusions to modulate kinases signaling in an antigen-dependent manner.
Figure 6. NIR-Fb fusions for modulation of protein kinase activity.

(a) Schematic representation of non-fluorescent NIR-FbGFP/Y58C-PKI fusions with kinase inhibitory peptides (PKIs). In the presence of the cognate antigen (in this case EGFP), the fusions inhibit the kinases activity. If the antigen is not expressed the fusions are degraded. (b) Live HeLa cells expressing NIR fluorescent PKA biosensor, transiently co-transfected with NIR-FbGFP/Y58C–PKI and EGFP cognate antigen (top row), or with NIR-FbGFP/Y58C–PKI and mTagBFP2 control (bottom row). Cells were imaged before and 45 min after the stimulation with dbcAMP. The FRET/donor ratio images are presented using intensity pseudocolor. Scale bar, 10 μm. (c) Quantification of the data presented in (b), control is cells not transfected with NIR-FbGFP/Y58C–PKI. (d) Live HeLa cells expressing NIR fluorescent JNK biosensor transiently transfected with NIR-FbGFP/Y58C–JIP and EGFP cognate antigen (top row), or with NIR-FbGFP/Y58C–JIP and mTagBFP2 control (bottom row). Cells were imaged before and 45 min after the stimulation with 1 μg/ml anisomycin. FRET/donor ratio images are presented using intensity pseudocolor. (e) Quantification of the data presented in (d), control is cells not transfected with NIR-FbGFP/Y58C–JIP. Scale bar, 10 μm. In (c, e) data are presented as mean values ± s.d. for n = 22 cells. For FRET imaging, a 605/30 nm excitation and a 667/30 nm (for miRFP670nano) and a 725/40 nm (for miRFP720) emission filters was used.
Discussion
By applying 15 rounds of molecular evolution to single-domain CBCR-based miRFP670nano, we have developed miRFP670nano3, the NIR FP with the highest quantum yield (Supplementary Table 1 and Fig. 1e). The molecular brightness of miRFP670nano3 is 2 or more times larger than that of the state-of-the-art two-domain BphP-based NIR FPs (Supplementary Table 1). miRFP670nano3 has a molecular weight of 17 kDa and exhibits high photostability (Supplementary Table 1 and Fig. 1d), protein stability in mammalian cells (Fig. 1g–i and Supplementary Fig. 4) and resistance to acidic and alkali environments (Fig. 1d–i and Supplementary Table 1). Size exclusion chromatography and OSER assay validated the monomeric status of miRFP670nano3 in vitro and mammalian cells (Fig. 1c and Supplementary Fig. 2,6). miRFP670nano3 performed well not only as the N- and C-terminal but also as an internal protein tag (Supplementary Fig. 7).
An important advantage of miRFP670nano3 is its substantially increased quantum yield. miRFP670nano3 differs from parental miRFP670nano by 14 mutations (Supplementary Fig. 1). The overlay with the miRFP670nano’s crystal structure6 demonstrated that 3 mutations (V36T, V38S and V39H) are buried inside the protein close to the BV pocket and may form additional bonds with either BV chromophore or immediate chromophore environment, decreasing the chromophore flexibility. Other mutations may contribute to the overall protein rigidity. This makes the non-radiative chromophore transition less favorable, resulting in the increased quantum yield of miRFP670nano3.
Compared to miRFP670nano, miRFP670nano3 has twice increased molecular brightness but 4-fold higher brightness in mammalian cells, indicating the increased efficiency of the endogenous BV binding in mammalian cells.
miRFP670nano3 was imaged deeper in brain tissue that EGFP using two-photon excitation (Fig. 2). The possibility to excite miRFP670nano3 in the Soret band allows multiplexed imaging with the visible range FPs by a single two-photon laser. Altogether this makes miRFP670nano3 the FP of choice for imaging applications in the NIR.
Using miRFP670nano3 as the internal tag, we have developed a new antigen-stabilized type of NIR fluorescent Nbs, NIR-Fbs. NIR-Fbs are stabilized by the antigen-binding and are degraded in the absence of cognate antigens, such as endogenous proteins. Whereas the use of weak or inducible promoters allows only decreasing the Nb expression level14, the intracellular level of NIR-Fbs strongly depends on the amount of their cognate antigen (Fig. 3d), resulting in the specific labeling of cells heterogeneous in antigen expression. The NIR-Fb scaffold does not require the introduction of mutations into Nb framework regions15, often involved in the interaction with antigen20–23, and thus, do not perturb Nb antigen-binding properties.
It has been shown that antigen binding increases the stability of Nbs in mammalian cells23, which is consistent with the observed NIR-Fbs behavior. We hypothesize that the insertion of miRFP670nano3 could cause structural changes in Nb, which might explain the NIR-Fb antigen-dependent properties. Nbs have nine antiparallel β-strands forming two sandwiching β-sheets. Insertion of miRFP670nano3 at opposite sides of CDRs increases the distance between the adjacent β-strands, inevitably resulting in a deformation of the β-sheet surface, but does not preclude interacting with the antigen35. The binding of the antigen to NIR-Fb could either compensate for the deformation of the β-sheet or sterically hinder the approach of ubiquitination ligases to the Nb, effectively shielding it from the protein degradation machinery.
To demonstrate the generalizability of the NIR-Fb approach, we engineered 10 NIR-Fbs against different antigens including NIR-Fbs to FPs, mammalian endogenous proteins, small peptide tags, bacterial proteins, and viral antigens (Supplementary Fig. 17 and Fig. 3g,f). The antigen-dependent stabilization makes NIR-Fbs the valuable probes for labeling of mixed cell populations heterogenic by antigen expression and for specific detections of intracellular antigens. For example, engineered NIR-Fbs against gp41 protein of HIV envelope complex, HIV capsid protein p24, and SARS-CoV-2 spike protein can be used as fluorescent reporters of viral infection and for tracking of viral distribution.
NIR re-coloring with NIR-FbGFP and NIR-FbmCherry (Fig. 4a–d) could be applied for numerous transgenic mice and cell lines expressing proteins tagged with EGFP or mCherry. Double-antigen-dependent bispecific constructs consisting of two NIR-Fbs to different antigens (Fig. 5a–c) are useful probes to identify double-positive cells expressing both antigens.
Applying NIR-Fb as the destabilizing fusion partner for regulatory proteins allows controlling cellular processes exclusively in cells expressing the specific antigen. These processes include, but are not limited to, targeted protein degradation (Fig. 5g–i), control of gene expression (Fig. 5d–i), and regulation of enzymatic activity (Fig. 6). The NIR-Fb scaffold allows combining Nbs of different specificity with various regulatory molecules. Further enhancement of degradation ability can be achieved by the multiplication of the number of NIR-Fb copies (Fig. 5f and Fig. 5i).
NIR-Fbs can be used for making protein fusions, re-coloring of cellular structures labeled with other FPs, monitoring endogenous protein levels and localization, manipulating endogenous protein levels at desired locations, conditional manipulation of protein activity in certain cells or locations, and controlling expression of genes in certain cells, among other applications.
The small size and universal design of NIR-Fb, together with several thousand available Nbs57 of high structural similarity58, should allow detecting and/or manipulating functionality of a wide range of cellular proteins and diverse signaling pathways based on the intracellular protein profile. Several approaches for Nb selection from libraries59 and for their rational design60 should make the NIR-Fb constructs available for almost any cellular protein. Further developments building upon this work should expand our ability to rapidly generate NIR-Fb-based biosensors and effectors against a diversity of intracellular epitopes, for cell- or protein-specific applications.
Methods
Mutagenesis and directed molecular evolution.
The genes encoding miRFP670nano3 protein and its mutants were amplified by polymerase chain reaction (PCR) and inserted into the pBAD/His-B vector (Invitrogen/Thermo Fisher Scientific) at KpnI/EcoRI sites. All oligonucleotide PCR primers were purchased from Biomers. For BV synthesis, E.coli host cells were cotransformed with a pWA23h plasmid encoding heme oxygenase from Bradyrhizobium ORS278 under rhamnose promoter61, 62. Random mutagenesis of genes encoding miRFP670nano3 was performed with a GeneMorph II random mutagenesis kit (Agilent Technologies). Site-specific mutagenesis and saturated mutagenesis were performed by overlap-extension PCR. For library construction, a mixture of mutated genes was cloned into pBAD/His-B vector and electroporated into LMG194 host cells (Invitrogen/Thermo Fisher Scientific), containing a pWA23h plasmid. Typical mutant libraries contained 107- 108 clones. Flow cytometry screening of mutant libraries was performed using an Influx cell sorter (BD Biosciences). 640 nm laser for excitation and 670/30 nm emission filters were used for the selection of positive clones. Before sorting, cells were grown in LB/ampicillin/kanamycin medium supplemented with 0.02% rhamnose and 0.005% arabinose for 5 h at 37°C and then for 20 h at 22°C. The next day bacterial cells were pelleted, washed and diluted with phosphate-buffered saline (PBS) to an optical density of 0.03 at 600 nm. Cells collected after sorting were incubated in SOC medium for 1 h at 37°C and then plated on LB/ampicillin/kanamycin Petri dishes supplemented with 0.005% arabinose and 0.02% rhamnose overnight at 37°C. Screening of brightest clones was performed with Leica M205 fluorescence stereomicroscope equipped with CCD camera (Tucsen), using filter set 650/45 nm excitation and 710/50 nm emission. About 30 selected clones were subcloned into a pcDNA3.1 plasmid (Invitrogen/Thermo Fisher Scientific) and evaluated in transiently transfected HeLa cells.
Protein expression and characterization.
Proteins were expressed in LMG194 bacterial cells, cotransformed with pWA23h plasmid, encoding heme oxygenase. Bacterial cells were grown to an optical density of 0.5–0.7 at 600 nm in LB/ampicillin/kanamycin medium supplemented with 0.02% rhamnose and, then, to induce miRFP670nano3 or NIR-Fb expression, 0.005% arabinose was added. Bacteria were cultured for 5 h at 37°C and, then, at 22°C for 20 h. Protein purification was performed with Ni-NTA agarose (Qiagen). Proteins were eluted with PBS containing 100 mM EDTA.
To perform size exclusion liquid chromatography a 2 ml volume of purified miRFP670nano3 was applied on the HiLoad 16/600 Superdex-200 column (GE Healthcare) equilibrated with 20 mM Tris-HCl, 300 mM NaCl buffer, pH 8.0, containing 50 μM EDTA, 10% glycerol, 150 mM NaCl, 1 mM DTT, 0.2 mM PMSF, 0.01% EP-40 and 0.2 mM benzodiazepine. A 1 ml/min flow rate was used. The column was calibrated using the gel filtration standards from Bio-Rad.
Fluorescence spectra were recorded with a Cary Eclipse fluorimeter (Agilent Technologies). Absorbance measurements were performed with a Hitachi U-2000 spectrophotometer. The extinction coefficient of miRFP670nano3 was determined as a ratio between the absorbance value of the main peak at the Q-band and the value of the peak at the Soret band, assuming the latter to have the extinction coefficient of free BV of 39,900 M−1cm−1 63. The fluorescence quantum yield of miRFP670nano3 was determined using a Nile blue dye as standard. All measurements were performed in PBS (pH 7.4). The pH stability was studied using a series of Hydrion buffers (Micro Essential Laboratory).
Distance between the adjacent β-strands of an Nb after insertion of miRFP670nano3 was estimated with a Coot software (Crystallographic Object-Oriented Toolkit)64 using an Nb structure from the crystallized GFP:antiGFP-Nb complex (PDB ID: 3OGO).
Dot-blot assay.
The 2 μl of purified EGFP or NIR-FbGFP and control antigen bovine serum albumin were spotted in PBS (proteins concentration 1 μg/ml) to the nitrocellulose membranes. The membranes were air-dried and incubated with 10% non-fat skim milk in PBS at 37°C for 1 h. Then membranes were washed with PBS and incubated with EGFP or NIR-FbGFP (1:100 dilution, PBS, 0.05% Tween-20), respectively, at 37° C for 1 h. After incubation membraned were washed and imaged using the Leica M205 fluorescence stereomicroscope, using two filter sets: 650/45 nm excitation and 710/50 nm emission for NIR-FbGFP and 480/40 nm excitation and 535/50 nm emission for EGFP.
Construction of mammalian plasmids.
To construct plasmids encoding miRFP670nano3 or its mutants, the respective genes were inserted into a pcDNA3.1 plasmid (Invitrogen/ThermoFisher Scientific) at KpnI/EcoRI sites.
To engineer plasmids for protein tagging and labeling of intracellular structures, the miRFP670nano3 gene was swapped with miRFP703 either as C- (for LAMP1 and H2B) or N-terminal fusions (for α-tubulin, β-actin, myosin and clathrin) and with miRFP670nano as internal fusion (β2AR and Gαs). To construct the EGFR-miRFP670nano3 fusion, miRFP670nano3 was swapped with PATagRFP (Addgene #31950). To construct the pCytERM-miRFP670nano3 plasmid for the OSER assay, the miRFP670nano3 gene was swapped with mGFP at AgeI/NotI sites of the pCytERM-mGFP cloning vector (Addgene #62237).
Nanobodies internally fused with miRFP670nano3 were generated by overlap PCR, and the resulted constructs were inserted into the pcDNA plasmid at the KpnI/EcoRI sites. Nb to GFP was amplified from a pcDNA3.1-NSlmb-vhhGFP4 plasmid (Addgene #35579). Nb to actin was amplified from a plasmid commercially available from ChromoTek. Nb to mCherry was amplified from a pGEX6P1-mCherry-Nanobody plasmid (Addgene #70696). Nb to ALFA-tag was amplified from a pET51b(+)-EGFP-NbALFA plasmid (Addgene #136626). Genes of Nb 2E7 to HIV gp41, Nb21 and Nbm6 to SARS-CoV-2 to spike protein were synthesized by GenScript. A NIR-Fbactin/Y58C–EGFP fusion was generated by overlap PCR and then a Tyr58Cys mutation was introduced to miRFP670nano3 by site-directed mutagenesis.
Gene encoding RBD (333–529) of SARS-CoV-2 spike protein was amplified from a pDONR223-SARS-CoV-2 S plasmid (Addgene #149329). Human β-catenin was amplified from a pCI-neo-β-catenin plasmid (Addgene #16518). HIV p24 was amplified from a pET51b(+)-SNAP-p24 plasmid (Addgene #130718). DHFR was amplified from a pET22b-ecDHFR plasmid (Addgene #109055). HIV gp41 was amplified from a pLAI-Env plasmid (Addgene #133996). The PCR-amplified genes were then fused with mEGFP (Addgene #62237) by SOE-PCR and inserted into a pcDNA3.1 plasmid. ALFA-fused α-tubulin, β-actin, myosin, and clathrin were generated by PCR with primer encoding ALFA-tag.
To generate NIR-Fb fusions, a 20 residue linker (Gly4Ser)4 was inserted into a pcDNA plasmid by the BamHI/NotI sites. For the construction of NIR-FbGFP fused to mTagBFP2, NIR-FbGFP was inserted into a pcDNA3.1 plasmid, containing the (Gly4Ser)4 linker at the KpnI/BamHI sites, and mTagBFP2 was inserted at the NotI/XbaI sites. To generate bispecific NIR-Fbs, NIR-FbGFP was inserted into the pcDNA3.1 plasmid, containing the (Gly4Ser)4 linker at the KpnI/BamHI sites, and NIR-FbmCherry was inserted at the NotI/XbaI sites. To generate a NIR-FbmCherry–EGFP fusion, EGFP was inserted into the pcDNA3.1 plasmid, containing a linker (Gly4Ser)4 and NIR-FbmCherry at NotI/XbaI site. For the construction of N- and C-terminal fusions of Nb to GFP and miRFP670nano3, their genes were inserted into a pcDNA3.1 plasmid, containing the (Gly4Ser)4 linker at the KpnI/BamHI or at the NotI/XbaI sites, respectively.
To engineer a NIR-FbGFP –GAL4 fusion, the GAL4-VP16 sequence was PCR-amplified from a pGV-ER plasmid (Systasy) and inserted at the NotI/XbaI sites into the pcDNA3.1 plasmid containing NIR-FbGFP and linker (Gly4Ser)4. To generate a NIR-FbGFP–NbALFA, the NIR-FbGFP was inserted into the pcDNA-3.1 plasmid, containing the (Gly4Ser)4 linker at the KpnI/BamHI sites, and NbALFA was inserted at the NotI/XbaI sites. To generate biNIR-FbGFP–GAL4 and biNIR-FbGFP–NbALFA fusions, two NIR-FbGFP genes were joined by SOE-PCR and swapped with NIR-FbGFP. The ALFA-tag was added to the N-terminus of GAL4-VP16 with oligonucleotide primer and inserted into the pcDNA3.1 plasmid, at the KpnI/XbaI site. To generate fusions with inhibitory peptides, the peptide sequences were introduced to the NIR-FbGFP with the (Gly4Ser)4 linker by PCR, and a Y58C mutation was introduced to miRFP670nano3 by overlap PCR.
Mammalian cells and transfection.
HeLa (CCL-2), N2A (CCL-131), U-2 OS (HTB-96), HEK293T (CRL-3216) and NIH3T3 (CRL-1658) cells were obtained from the ATCC. Cells were cultured in a DMEM medium supplemented with 10% FBS, 0.5% penicillin-streptomycin and 2 mM glutamine (Invitrogen/ThermoFisher Scientific) at 37°C. For live-cell fluorescence microscopy, cells were plated in 35 mm glass-bottom Petri dishes (Greiner Bio-One). Transient transfections were performed using polyethylenimine65 or Effectene reagent (Qiagen).
To study protein stability, HeLa cells were incubated for 4 h with 10 μM bortezomib (Santa Cruz) or 20 μg/ml cycloheximide (Santa Cruz).
An effective FP brightness was determined as a NIR fluorescence of transiently transfected live HeLa cells 72 h after the transfection, without a supply of exogenous BV. The effective brightness of parental miRFP670nano was assumed to be 100%.
Neuronal culture and transfection.
Primary rat cortical neurons were prepared in the Neuronal Cell Culture Unit, University of Helsinki. All animal work was performed under the ethical guidelines of the European convention and regulations of the Ethics Committee for Animal Research of the University of Helsinki. Cells were plated at a density of 500,000–700,000 per 35 mm glass-bottom dish, coated with Poly-L-Lysine (0.01 mg/ml) (Merck). Neurons were grown at 37°C and 5% CO2 in neurobasal medium (Gibco) supplemented with B27 (Invitrogen/Thermo Fisher Scientific), L-glutamine (Invitrogen/Thermo Fisher Scientific), and penicillin-streptomycin (Lonza). Cultured neurons were transfected with pcDNA plasmids encoding respective miRFP670nano3 at 4–5 days in vitro (DIV) using Effectene reagent (Qiagen) and imaged 48–72 h after transfection.
Wide-field fluorescence microscopy.
Live cells were imaged with an Olympus IX81 inverted epifluorescence microscope, equipped with a Lambda LS Xenon light source (Sutter). An ORCA-Flash4.0 V3 camera (Hamamatsu) was used for image acquisition. Cells were imaged using either a 20× 0.75 NA air or a 60× 1.35 NA oil immersion objective lens (UPlanSApo, Olympus). During imaging, HeLa cells were incubated in a cell imaging solution (Invitrogen/Life Technologies) and kept at 37°C. The microscope was operated with a SlideBook v.6.0.8 software (Intelligent Imaging Innovations). For imaging of miRFP670nano3, 605/30 nm excitation and 667/30 nm emission filters (Chroma) were used. For EGFP, 485/20 nm excitation and 525/30 nm emission filters (Chroma) were used. For mCherry, 560/25 nm excitation and 607/36 nm emission filters (Chroma) were used. The data were analyzed using a SlideBook v. 6.0.8 (Intelligent Imaging Innovations) and a Fiji v.1.50b software.
Photobleaching measurements were performed in live HeLa cells 48 h after transfection using a 60× 1.35 NA oil immersion objective lens (UPlanSApo, Olympus) and a 605/30 nm excitation filter. The light power density measured at the back aperture of the objective lens was 8.25 mW cm−2, which gives an estimate of 4.3 W cm−2 at the focal objective plane.
For the OSER assay, U-2 OS cells were transfected with a pCytERM-miRFP670nano3 plasmid and imaged 48 h after the transfection using an Olympus IX81 inverted epifluorescence microscope. The percentage of cells with normal reticular-shaped ER and without incorrect localization was determined. A ratio of mean intensity of the whorls structure (OSER) to mean intensity of the nuclear envelope (NE) measurement was quantified using an ImageJ v.1.51u.
To obtain FRET images of HeLa cells expressing NIR FRET biosensors, a 605/30 excitation filter and two emission filters (667/30 nm for miRFP670nano and 725/40 nm for miRFP720) were used. Emission ratios were obtained by calculating background-subtracted FRET intensities divided by background-subtracted miRFP670nano intensities for JNK and PKA NIR biosensors. FRET measurements were quantified using ImageJ v.1.51u. Intensity-modulated display mode was generated with a full-spectrum lookup table. HeLa cells expressing JNK and PKA NIR-biosensors were starved for 6 h with DMEM medium (Gibco/ThermoFisher Scientific) before imaging.
Flow cytometry.
Flow cytometry analysis was performed using an Accuri C6 (BD Biosciences) and a NovoCyte Quanteon 4025 (Agilent) flow cytometers. Before analysis, live cells were washed and diluted in cold PBS to a density of 500,000 cells per ml. At least 30,000 cells per sample were recorded. Using Accuri C6 flow cytometer, the fluorescence intensity of miRFP670nano and miRFP670nano3 expressing cells was analyzed using the 640 nm excitation laser and 675/25 nm or 670 nm LP emission filters. The fluorescence intensity of EGFP was analyzed using a 488 nm laser for excitation, and its fluorescence was detected with a 510/15 nm emission filter. Using a NovoCyte Quanteon 4025 flow cytometer, the fluorescence intensity of miRFP670nano3 expressing cells was analyzed using a 637 nm excitation laser and a 667/30 emission filter. The fluorescence intensity of EGFP was analyzed using a 488 nm excitation laser, and a 525/45 nm emission filter. The fluorescence intensity of mTagBFP2 was analyzed using a 405 nm excitation laser, and a 445/45 nm emission filter. The fluorescence intensity of mCherry was analyzed using a 561 nm excitation laser, and a 615/20 nm emission filter. Supplementary Fig. 3 exemplifies the gating strategy. The data were analyzed using a FlowJo v.7.6.2 software.
Gaussia luciferase assay.
For an antigen-dependent GAL4 stabilization, HeLa cells were co-transfected with plasmids encoding NIR-FbGFP–GAL4 or biNIR-FbGFP–GAL4, reporter plasmid pUAS-Gluc (5x UAS), and with either EGFP or mTagBFP in a 1:10:89 ratio. HeLa cells were co-transfected with plasmids encoding ALFA-tagged GAL4-VP16, reporter plasmid pUAS-Gluc (5x UAS), NIR-FbGFP–NbALFA or biNIR-FbGFP–NbALFA, and with either EGFP or mTagBFP2 in the 1:10:44.5:44.5 ratio. As controls, cells transfected with reporter plasmid pUAS-Gluc and with or without ALFA-tagged GAL4-VP16 were used. To measure Gluc activity, the co-transfected HeLa cells were grown in 24-well plates. 10 h after the transfection mixture was removed, a fresh growth medium was added, and cells were incubated for 48 h. Then, 5 μl of culture media was mixed with 100 μl of 2 μM coelenterazine (Invitrogen/Thermo Fisher Scientific) in PBS in wells of a 96-well half-area white plate (Costar). Bioluminescence was measured using a Victor X3 multilabel plate reader (PerkinElmer).
Virus production and stereotactic injection.
The miRFP670nano3 gene was PCR amplified and subcloned into an AAV transfer vector downstream of the human synapsin promoter and upstream of WPRE and hGHpA sequences. This vector was co-transfected into HEK293-AAV cells (Vector Biolabs) along with a pAdeno-helper vector and a pRC-AAV9 rep-cap plasmid. Recombinant AAV9 production was then carried out using a protocol developed by the Byungkook Lim laboratory at the University of California at San Diego. The recombinant AAV9-hSYN-miRFP670nano3-WPRE-hGHpA was titered by qPCR using primers designed to the hGHpA sequence. The titer of the virus was 1.4E+12 GC ml-1.
0.4 μl of undiluted AAV was injected into the cortex (coordinates: AP −0.8 - (−1.75) mm, ML 1.45–1.65 mm, DV 0.25–1.1 mm) or dorsal horn of the L3-L4 spinal cord (ML 0.3, DV 0.3–0.5 mm). Surgical procedures closely followed previously established protocols30, 66. Briefly, thin-wall glass pipettes were pulled on a Sutter Flaming/Brown micropipette puller (model P-97). Pipette tips were cut at an acute angle under 10x magnification using sterile techniques. Tip diameters were typically 15–20 μm. Pipettes that did not result in sharp bevels nor had larger tip diameters were discarded. Millimeter tick marks were made on each pulled needle to measure the virus volume injected into the brain or spinal cord.
Mice were anesthetized with isoflurane (4% for induction; 1%−1.5% for maintenance) and positioned in a computer-assisted stereotactic system with digital coordinate readout and atlas targeting (Leica Angle Two). Body temperature was maintained at 36–37 °C with a direct current (DC) temperature controller and ophthalmic ointment was used to prevent eyes from drying. A small amount of depilator cream (Nair) was used to remove hair over the dorsal areas of the injection site thoroughly. The skin was cleaned and sterilized with a two-stage scrub of betadine and 70% ethanol.
For brain injections, a midline incision was made beginning just posterior to the eyes and ending just past the lambda suture. The scalp was pulled open and periosteum cleaned using scalpel and forceps to expose the desired hemisphere for calibrating the digital atlas and coordinate marking. Once reference points (bregma and lambda) were positioned using the pipette needle and entered into the program, the desired target was set on the digital atlas. The injection pipette was carefully moved to the target site (using AP and ML coordinates). Next, the craniotomy site was marked and an electrical micro-drill with a fluted bit (0.5 mm tip diameter) was used to thin a 0.5–1 mm diameter part of the bone over the target injection site. Once the bone was thin enough to flex gently, a 30G needle with an attached syringe was used to carefully cut and lift a small (0.3–0.4 mm) segment of bone.
For spinal cord injections, surgical scissors were used to make a small (around 10 mm) incision along the midline. Fascia connecting the skin to the underlying muscle was removed with forceps. The skin was held back by retractors. Using blunt dissection, lateral edges of the spinal column were isolated from connective tissue and muscle. Tissue from the vertebra of interest and one vertebra rostral and caudal to the site of spinal cord exposure was removed with forceps. The spine was then stabilized using Cunningham vertebral clamps and any remaining connective tissue on top of the exposed vertebrae was removed with a spatula. Using a small sterile needle, an approximately 0.3 mm opening was made in the tissue overlying the designated injection site.
For injection, a drop of AAV was carefully pipetted onto parafilm (1–2 μl) for filling the pulled injection needle with the desired volume. Once loaded with sufficient volume, the injection needle was slowly lowered into the brain or spinal cord until the target depth was reached. Manual pressure was applied using a 30-ml syringe connected by shrink tubing and 0.4 μl of the virus was slowly injected over 5–10 min. Once the virus was injected, the syringe’s pressure valve was locked. The position was maintained for approximately 10 min to allow the virus to spread and avoid backflow upon needle retraction. Following the injection, head or spinal cord clamps were removed, muscle approximated, and the skin sutured along the incision. Mice were given subcutaneous Buprenex SR (0.5 mg per kg) and allowed to recover before placement in their cage.
Animal preparation for imaging.
All live animal procedures were performed following the guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at the Salk Institute. We used male Cx3cr1 GFP/+ mice (stock #005582, Jackson Laboratories) with an age of 8–14 weeks at the time of imaging (6–9 weeks at the time of stereotactic injection). 20 mice were used in this study.
Surgical procedures closely followed established protocols29, 30, 66. Briefly, mice were anesthetized with isoflurane (4–5% for induction; 1%−1.5% for maintenance) and implanted with a head or spinal plate on a custom surgical bed (Thorlabs). Body temperature was maintained at 36°C–37°C with a DC temperature control system and ophthalmic ointment was used to prevent eyes from drying. Depilator cream (Nair) was used to remove hair above the imaging site. The skin was thoroughly cleansed and disinfected with a two-stage scrub of betadine and 70% ethanol.
For brain imaging, a scalp portion was surgically removed to expose frontal, parietal, and interparietal skull segments. Scalp edges were attached to the lateral sides of the skull using a tissue-compatible adhesive (3M Vetbond). A custom-machined metal plate was affixed to the skull with dental cement (Coltene Whaledent, cat. no. H00335), allowing the head to be stabilized with a custom holder. An approximately 3 mm diameter craniotomy was made over the AAV injection site. A 1.5% agarose solution and coverslip were applied to the exposed tissue. The coverslip was affixed to the skull with dental cement to control tissue motion. Imaging commenced immediately after optical window preparation.
For spinal cord imaging, a laminectomy was performed over the AAV injection site. The dura mater overlying the spinal cord was kept intact. A custom-cut #0 coverslip was placed over the exposed tissue and fixed to the spine with dental cement to control tissue motion. The depth of anesthesia was monitored throughout the experiment and adjusted as needed to maintain a breath rate of approximately 55–65 breaths per minute. Saline was supplemented as needed to compensate for fluid loss.
Two-photon in vivo microscopy.
Live animal imaging was performed 3–5 weeks after AAV injection, as previously described29, 30, 66. Briefly, a Sutter Movable Objective Microscope equipped with a pulsed femtosecond Ti:Sapphire laser (Chameleon Ultra II, Coherent) with two fluorescence detection channels was used for imaging (green emission filter: ET-525/70M (Chroma); near-infrared emission filters: ET645/75M (Chroma) in conjunction with FF01–720/SP (Semrock); dichroic beamsplitter: 565DCXR (Chroma); photomultiplier tubes: H7422–40 GaAsP (Hamamatsu)). The laser excitation wavelength was set between 880–930 nm. The average laser power was <10–15 mW at the tissue surface and adjusted with depth as needed to compensate for signal loss due to scattering and absorption. An Olympus 20× 1.0 NA water immersion objective was used for light delivery and collection. Z-stacks included up to 900 images, acquired at 1–2 μm axial step size, used a two- to four-frame average, 256 or 512 × 512-pixel resolution, and 1.0–2.5× zoom (corresponding to 701–282 μm fields of view).
Image data processing.
Data were processed in ImageJ or Fiji software (SciJava; https://fiji.sc). xz projections were created from xy fluorescence image stacks using the ‘Reslice’ and ‘3D Project’ functions. Motion artifacts in spinal cord images were reduced using the ‘Image Stabilizer’ plugin (transformation: affine). Supplemental Videos S1–S3 were created using Igor Pro (version 8.04; WaveMetrics).
Reproducibility.
The experiments were not randomized. The investigators were not blinded to allocation during the experiments and outcome assessment. No sample-size estimation was performed to ensure adequate power to detect a pre-specified effect size.
Supplementary Material
Supplementary Video S1 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the neocortex of a 14-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).
Supplementary Video S2 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the neocortex of a 13-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).
Supplementary Video S3 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the lumbar spinal dorsal horn of a 12.5-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).
Acknowledgments
We thank N. Peitsaro and N. Aarnio from the Flow Cytometry Core Facility and staff of the Neuronal Cell Culture Unit of the University of Helsinki for the technical assistance. This work was supported by grants NS108034, NS112959, NS103522 and GM122567 from the US National Institutes of Health (NIH), 322226 from the Academy of Finland, and the Magnus Ehrnrooth Foundation. S.P. was supported in part by the NIH Intramural Research Program for the Vaccine Research Center of the National Institute of Allergy and Infectious Diseases.
Footnotes
Competing interests
The authors declare no competing interests.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability. The data supporting the findings of this study are available within the article and its Supplementary Information. All other data that support the findings of the study are available from the corresponding author upon reasonable request. The major plasmids constructed in this study, their maps, and sequences are deposited to Addgene. The nucleotide sequence in GenBank for miRFP670nano3 is MW627294.
Editor’s Summary
miRFP670nano3 offers improved near-infrared imaging and was used to develop fluorescent nanobodies whose stability and fluorescence strongly depend on antigen binding, with broad implications for detecting and manipulating cellular targets.
References
- 1.Shcherbakova DM, Stepanenko OV, Turoverov KK & Verkhusha VV Near-Infrared Fluorescent Proteins: Multiplexing and Optogenetics across Scales. Trends Biotechnol 36, 1230–1243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chernov KG, Redchuk TA, Omelina ES & Verkhusha VV Near-Infrared Fluorescent Proteins, Biosensors, and Optogenetic Tools Engineered from Phytochromes. Chem Rev 117, 6423–6446 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Oliinyk OS, Chernov KG & Verkhusha VV Bacterial Phytochromes, Cyanobacteriochromes and Allophycocyanins as a Source of Near-Infrared Fluorescent Probes. Int J Mol Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shcherbakova DM, Baloban M. & Verkhusha VV Near-infrared fluorescent proteins engineered from bacterial phytochromes. Curr Opin Chem Biol 27, 52–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran MT et al. In vivo image analysis using iRFP transgenic mice. Exp Anim 63, 311–319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliinyk OS, Shemetov AA, Pletnev S, Shcherbakova DM & Verkhusha VV Smallest near-infrared fluorescent protein evolved from cyanobacteriochrome as versatile tag for spectral multiplexing. Nature Communications 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snapp E. Design and use of fluorescent fusion proteins in cell biology. Curr Protoc Cell Biol Chapter 21, 21 24 21–21 24 13 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bathula NV, Bommadevara H. & Hayes JM Nanobodies: The Future of Antibody-Based Immune Therapeutics. Cancer Biother Radiopharm (2020). [DOI] [PubMed] [Google Scholar]
- 9.Akiba H. et al. Structural and thermodynamic basis for the recognition of the substrate-binding cleft on hen egg lysozyme by a single-domain antibody. Sci Rep 9, 15481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deschaght P. et al. Large Diversity of Functional Nanobodies from a Camelid Immune Library Revealed by an Alternative Analysis of Next-Generation Sequencing Data. Front Immunol 8, 420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Sapienza G, Sofia MAR & Rosa T.-d. Single-Domain Antibodies As versatile Affinity Reagents for Analytical and Diagnostic Applications. Frontiers in Immunology 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheloha RW, Harmand TJ, Wijne C, Schwartz TU & Ploegh HL Exploring cellular biochemistry with nanobodies. J Biol Chem 295, 15307–15327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traenkle B. & Rothbauer U. Under the Microscope: Single-Domain Antibodies for Live-Cell Imaging and Super-Resolution Microscopy. Front Immunol 8, 1030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilar G, Vigano MA, Affolter M. & Matsuda S. Reflections on the use of protein binders to study protein function in developmental biology. Wires Dev Biol 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schnell U, Dijk F, Sjollema KA & Giepmans BN Immunolabeling artifacts and the need for live-cell imaging. Nat Methods 9, 152–158 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Keller BM et al. A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells. Antibodies (Basel) 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maier J, Traenkle B. & Rothbauer U. Real-time analysis of epithelial-mesenchymal transition using fluorescent single-domain antibodies. Sci Rep 5, 13402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang JC et al. Detection and manipulation of live antigen-expressing cells using conditionally stable nanobodies. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem 82, 775–797 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Kirchhofer A. et al. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol 17, 133–138 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Fanning SW & Horn JR An anti-hapten camelid antibody reveals a cryptic binding site with significant energetic contributions from a nonhypervariable loop. Protein Sci 20, 1196–1207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun MB et al. Peptides in headlock--a novel high-affinity and versatile peptide-binding nanobody for proteomics and microscopy. Sci Rep 6, 19211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keller BM et al. Chromobodies to Quantify Changes of Endogenous Protein Concentration in Living Cells. Mol Cell Proteomics 17, 2518–2533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zavrtanik U, Lukan J, Loris R, Lah J. & Hadzi S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J Mol Biol 430, 4369–4386 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Gross GG et al. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78, 971–985 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shemetov AA, Oliinyk OS & Verkhusha VV How to Increase Brightness of Near-Infrared Fluorescent Proteins in Mammalian Cells. Cell Chem Biol 24, 758–766 e753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costantini LM, Fossati M, Francolini M. & Snapp EL Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic 13, 643–649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piatkevich KD et al. Near-infrared fluorescent proteins engineered from bacterial phytochromes in neuroimaging. Biophys. J. 113, 2299–2309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nimmerjahn A, Kirchhoff F. & Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Tufail Y. et al. Phosphatidylserine Exposure Controls Viral Innate Immune Responses by Microglia. Neuron 93, 574–586 e578 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fourgeaud L. et al. TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nimmerjahn A. Monitoring neuronal health. Science 367, 510–511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caussinus E, Kanca O. & Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol 19, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Yu D. et al. Optogenetic activation of intracellular antibodies for direct modulation of endogenous proteins. Nat Methods 16, 1095–1100 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Kubala MH, Kovtun O, Alexandrov K. & Collins BM Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci 19, 2389–2401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subach OM, Cranfill PJ, Davidson MW & Verkhusha VV An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 6, e28674 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panza P, Maier J, Schmees C, Rothbauer U. & Sollner C. Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development 142, 1879–1884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traenkle B. et al. Monitoring interactions and dynamics of endogenous beta-catenin with intracellular nanobodies in living cells. Mol Cell Proteomics 14, 707–723 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Virant D. et al. A peptide tag-specific nanobody enables high-quality labeling for dSTORM imaging. Nat Commun 9, 930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fridy PC et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods 11, 1253–1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gotzke H. et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat Commun 10, 4403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyen D, Wechselberger R, Srinivasan V, Steyaert J. & Barlow JN Mechanistic analysis of allosteric and non-allosteric effects arising from nanobody binding to two epitopes of the dihydrofolate reductase of Escherichia coli. Biochim Biophys Acta 1834, 2147–2157 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Gray ER et al. Unravelling the Molecular Basis of High Affinity Nanobodies against HIV p24: In Vitro Functional, Structural, and in Silico Insights. ACS Infect Dis 3, 479–491 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Strokappe NM et al. Super Potent Bispecific Llama VHH Antibodies Neutralize HIV via a Combination of gp41 and gp120 Epitopes. Antibodies (Basel) 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traenkle B, Segan S, Fagbadebo FO, Kaiser PD & Rothbauer U. A novel epitope tagging system to visualize and monitor antigens in live cells with chromobodies. Sci Rep 10, 14267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang Y. et al. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 370, 1479–1484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoof M. et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 370, 1473–1479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z. et al. Structural insights into the binding of nanobodies LaM2 and LaM4 to the red fluorescent protein mCherry. Protein Sci 30, 2298–2309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prole DL & Taylor CW A genetically encoded toolkit of functionalized nanobodies against fluorescent proteins for visualizing and manipulating intracellular signalling. BMC Biol 17, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abe T. & Fujimori T. Reporter mouse lines for fluorescence imaging. Dev Growth Differ 55, 390–405 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Caygill EE & Brand AH The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol Biol 1478, 33–52 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Gerits N, Kostenko S, Shiryaev A, Johannessen M. & Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal 20, 1592–1607 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Yi JJ, Wang H, Vilela M, Danuser G. & Hahn KM Manipulation of endogenous kinase activity in living cells using photoswitchable inhibitory peptides. ACS Synth Biol 3, 788–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu C, Ke P, Zhang J, Zhang X. & Chen X. Protein Kinase Inhibitor Peptide as a Tool to Specifically Inhibit Protein Kinase A. Front Physiol 11, 574030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barr RK, Kendrick TS & Bogoyevitch MA Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem 277, 10987–10997 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Heo YS et al. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J 23, 2185–2195 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilton EE, Opyr MP, Kailasam S, Kothe RF & Wieden HJ sdAb-DB: The Single Domain Antibody Database. ACS Synth Biol 7, 2480–2484 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Mitchell LS & Colwell LJ Comparative analysis of nanobody sequence and structure data. Proteins 86, 697–706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen TD et al. Engineering a growth sensor to select intracellular antibodies in the cytosol of mammalian cells. J Biosci Bioeng 124, 125–132 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Hussain M, Angus SP & Kuhlman B. Engineering a Protein Binder Specific for p38alpha with Interface Expansion. Biochemistry 57, 4526–4535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shcherbakova DM & Verkhusha VV Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Methods 10, 751–754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piatkevich KD, Subach FV & Verkhusha VV Far-red light photoactivatable near-infrared fluorescent proteins engineered from a bacterial phytochrome. Nat Commun 4, 2153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filonov GS & Verkhusha VV A near-infrared BiFC reporter for in vivo imaging of protein-protein interactions. Chem Biol 20, 1078–1086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P, Lohkamp B, Scott WG & Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Longo PA, Kavran JM, Kim MS & Leahy DJ Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol 529, 227–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sekiguchi KJ et al. Imaging large-scale cellular activity in spinal cord of freely behaving mice. Nat. Commun. 7, 11450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video S1 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the neocortex of a 14-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).
Supplementary Video S2 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the neocortex of a 13-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).
Supplementary Video S3 Z-stack of two-photon images showing miRFP670nano3-labeled neurons (red) in the lumbar spinal dorsal horn of a 12.5-weeks-old anesthetized Cx3cr1 GFP/+ mouse with labeled microglia (white).