

Abstract
Cholesterol biosynthesis occurs in the endoplasmic reticulum (ER). Its lego-like construction from water-soluble small metabolites via intermediates of increasing complexity to water-insoluble cholesterol requires numerous distinct enzymes. Dysfunction of the involved enzymes can cause several human inborn defects and diseases. Here, we review recent structures of three key cholesterol biosynthetic enzymes: Squalene epoxidase (SQLE), NAD(P)-dependent steroid dehydrogenase-like (NSDHL), and 3β-hydroxysteroid Δ8-Δ7 isomerase termed EBP. Moreover, we discuss structures of acyl-CoA:cholesterol acyltransferase (ACAT) enzymes, which are responsible for forming cholesteryl esters from cholesterol to maintain cholesterol homeostasis in the ER. The structures of these enzymes reveal their catalytic mechanism and provide a molecular basis to develop drugs for treating diseases linked to their dysregulation.
Keywords: cholesterol biosynthesis, SQLE, NSDHL, EBP, ACAT
Introduction
Cholesterol, a lipid component essential for growth and viability of mammalian cells, maintains membrane rigidity and permeability. It also serves as a precursor molecule for the biosynthesis of steroid hormones, bile acids, vitamin D, and a signaling transducer of the Hedgehog pathway (1–4). There are two major sources of cholesterol for humans: exogenous diet and endogenous biosynthesis. In cholesterol biosynthesis, 18 units of the fundamental building block acetyl-CoA are converted into lanosterol, the first sterol-like intermediate, in a series of reactions in the endoplasmic reticulum (ER) (5–7). Due to its low solubility, lanosterol synthesis occurs through several integral membrane enzymes and is eventually converted to cholesterol (Figure 1) (8,9).
Figure 1. Biosynthetic pathway of cholesterol and cholesteryl ester.
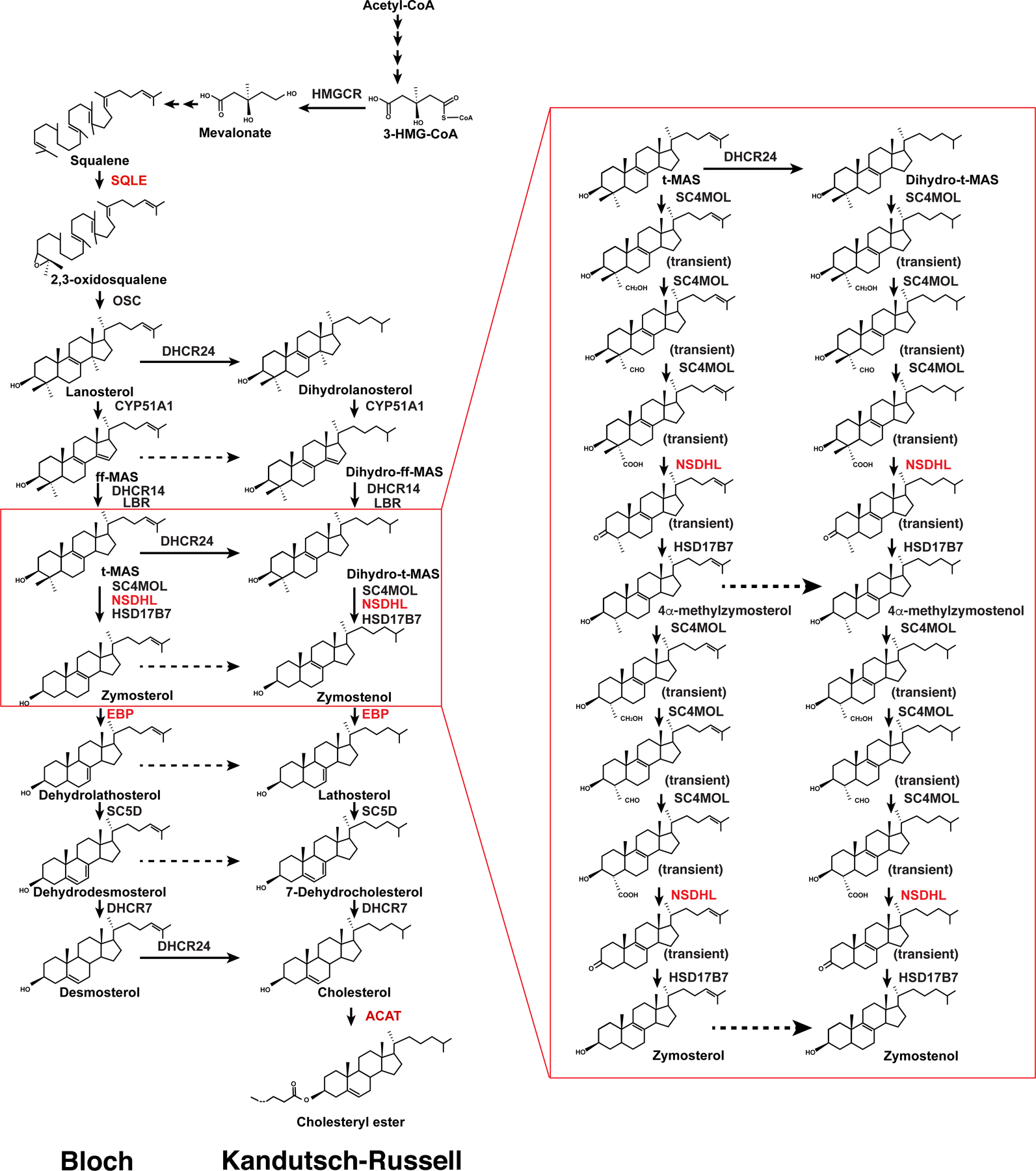
Enzymes that have been discussed in this review are colored in red. The left lane represents the Bloch pathway; the right lane represents the Kandutsch-Russell pathway.
The activity of the cholesterol biosynthetic enzymes is closely related to human health. 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the first rate-limiting enzyme of cholesterol biosynthesis, is the target of statin that is widely used for lowering the blood cholesterol level to prevent cardiovascular diseases (10). Mutations of the membrane enzymes involved in the conversion of lanosterol to cholesterol lead to several human genetic diseases, caused by the toxic accumulation of sterol intermediates. For example, mutations in NSDHL cause congenital hemidysplasia (CHILD) syndrome and CK syndrome with ichthyosis and limb defects (11,12). Mutations in EBP can lead to Conradi-Hunermann syndrome, which commonly causes growth deficiency, short stature, and curvature of the spine (13). The deficiency of DHCR7 is associated with Smith-Lemli-Opitz syndrome (SLOS), a common inborn metabolic defect in Caucasians, causing developmental congenital disorder (14). Thus, structural and functional studies on these enzymes are critical to understand the molecular mechanism of cholesterol biosynthesis and may shed light on the molecular pathogenesis of the diseases caused by their dysfunction.
The structural analysis of cholesterol biosynthetic enzymes in the past 20 years has provided valuable insights into cholesterol biogenesis that has been reviewed elsewhere (8,15,16). This review focuses on the structural biology of three key cholesterol biosynthetic enzymes, SQLE, NSDHL, and EBP, published between 2019 and 2021. We also summarize recent structural observations on acyl-CoA:cholesterol acyltransferase (ACAT) enzymes that convert cholesterol to cholesteryl esters in the ER. References to original structural work on cholesterol biosynthetic enzymes have been denoted and provide important insights into cholesterol metabolism.
Structure of SQLE
Squalene epoxidase (SQLE), also known as squalene monooxygenase, catalyzes the first oxygenation step of cholesterol synthesis and is the second rate-limiting enzyme in cholesterol biosynthesis (Figure 1) (5). As a flavin adenosine dinucleotide (FAD)–containing epoxidase, it utilizes NADPH and molecular oxygen to oxidize squalene to 2,3-oxidosqualene. Human SQLE shows limited sequence similarity to the fungal form. Fungal SQLE inhibition interferes with cell membrane synthesis and fungal growth. Several fungal SQLE inhibitors, such as terbinafine (Lamisil) and naftifine (Naftin), exhibit significant specificity for the fungal SQLE without inhibiting the human homolog and are currently used for the treatment of fungal infections (17).
SQLE is a 64-kDa protein containing 574 amino acid residues with two domains: an N-terminal regulatory domain and a C-terminal catalytic domain. It belongs to group E flavin monooxygenases, which require external NADPH-cytochrome P450 reductase (P450R) as an electron donor for squalene epoxidation (18). Unlike other known flavin monooxygenases, SQLE catalyzes epoxidation but not hydroxylation of substrates. The N-terminal regulatory domain senses excess cholesterol in the ER membrane and regulates proteasomal degradation of SQLE in a cholesterol-dependent manner (19). Membrane-associated ring-CH type finger 6 (MARCH6), an E3 ubiquitin ligase, catalyzes the ubiquitination in the N-terminal regulatory domain (20). A recent study showed that squalene directly binds to the N-terminal regulatory domain of SQLE and reduces its ubiquitination by MARCH6. Thus, squalene serves a dual function towards SQLE as a substrate and as a regulator by increasing its metabolic capacity (21).
Crystal structures of the FAD-bound catalytic domain of SQLE and its complex with two different inhibitors were recently determined (22). The catalytic domain of SQLE consists of the FAD-binding domain, substrate-binding domain, and C-terminal α-helical domain (Figure 2a). The FAD-binding domain adopts a three-layer ββα sandwich architecture using the CATH nomenclature, which is alternatively referred to as a glutathione reductase (GR-2) Rossmann fold (18). The substrate-binding domain contains a two-layer βα sandwich with seven β-strands. The C-terminal α-helical domain mediates interaction with membranes. In both inhibitor-bound structures, a hydrogen bond between the tertiary amine of the inhibitor and the hydroxyl group of the conserved Y195 in the substrate-binding domain was observed (Figure 2b). This observation is consistent with all SQLE inhibitors containing a tertiary amine moiety and illustrates the importance of this interaction with Y195. In the FAD-only structure, Y195 exhibits a different conformation and forms a hydrogen bond with the side-chain amide of Q168 of the FAD-binding domain (Figure 2b). Mutations of Y195 inactivate SQLE, highlighting that Y195 is critical for enzymatic activity.
Figure 2. Structural mechanisms of three key enzymes in the cholesterol biosynthetic pathway.
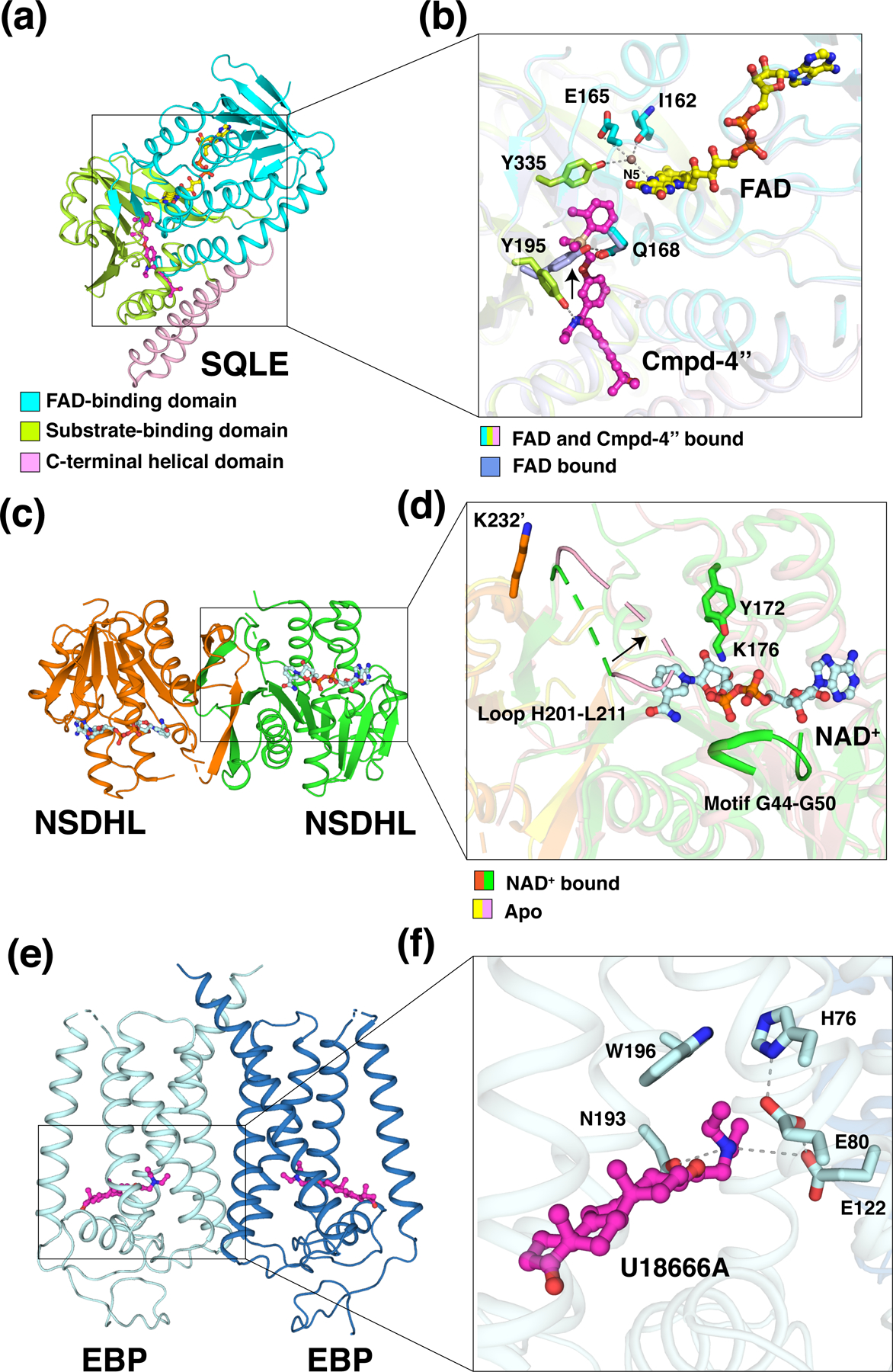
(a) Overall structure of the catalytic domain of SQLE bound to the inhibitor compound-4” (Cmpd-4”) (pdb: 6C6N). The FAD-binding domain, substrate-binding domain and C-terminal helical domain are colored in cyan, lime, and light pink, respectively. FAD (stick representation with carbon atoms in yellow) and Cmpd-4” (magenta) are shown. (b) The active site of SQLE. Y195 in the inhibitor-bound structure (lime) forms a hydrogen bond with the tertiary amine of Cmpd-4’’, while in the FAD-only structure (light blue, pdb: 6C6R), Y195 (light blue) interacts with Q168 (light blue) through a hydrogen bond. The black arrow indicates the conformational change of Y195. The side chains of Y335 and E165 and the main chain of I162 interact with N5 of FAD via a bridging water molecule (brown sphere). Y335, E165, and I162 are shown as sticks. (c) Overall structure of NSDHL complexed with NAD+ (pdb: 6JKH). NAD+ (pale cyan) is shown in stick representation. (d) The active site of NSDHL. The catalytic residues Y172 and K176 and motif G44-G50 are colored in green. The black arrow indicates the conformational change of loop H201-L211 from the NAD-bound structure (green and orange cartoon) to the apo structure (light pink and yellow cartoon, pdb: 6JKG). K232’ (orange) from the neighboring monomer interacts with loop H201-L211 at the dimer interface. (e) Overall structure of U18666A-bound EBP (6OHT). U18666A (magenta) is shown in stick representation. (f) The active site of EBP. The amine group of U18666A, which forms hydrophilic interactions with E122 and N193, is stabilized by W196 via a π-cation interaction. H76, E80, and E122 form a hydrogen-bonding network during enzymatic reaction. Residues are shown in pale cyan stick representation.
To date, the reaction mechanism of SQLE has not been well resolved, owing in part to the lack of a structure with the substrate or a reaction intermediate. As a group E flavin monooxygenase, SQLE contains a loosely bound FAD and depends on an external NADPH-cytochrome P450 reductase (P450R) for the transfer of a reduced flavin to initialize the reaction (18). Subsequently, the reduced flavin reacts with molecular oxygen to form a highly reactive intermediate flavin hydroperoxide at the flavin C4a position (23), which is a critical region that controls oxygen transfer. This intermediate then transfers the terminal oxygen atom from the hydroperoxide to the substrate (24). After the reaction, a water molecule is released from the resulting hydroxyflavin to regenerate FAD. Recent research suggests that the highly conserved Y335 plays a critical role in the recruitment of reduced flavin and stabilization of the reactive intermediate to maintain catalytic efficiency, as the hydroxyl moiety of Y335 interacts with N5 via a bridging water molecule that is further engaged in hydrogen bonds with the carbonyl of I162 and the side chain of E165 (Figure 2b) (22).
Structure of NSDHL
NAD(P)-dependent steroid dehydrogenase-like (NSDHL) is a sterol-4α-carboxylate-3-dehydrogenase, which is an essential enzyme involved in cholesterol biosynthesis (25). NSDHL catalyzes the sequential removal of two methyl groups attached to atom C-4 through NAD+-dependent oxidative decarboxylation following their conversion into 4α-carboxylate groups present in meiosis-activating sterol (MAS) (26,27). NSDHL is anchored in the ER membrane through its C-terminal transmembrane helix. Bioinformatic analysis shows that NSDHL belongs to the short-chain dehydrogenase/reductase (SDR) family (28).
Crystallographic studies show that NSDHL with an N-terminal deletion (NSDHL-∆N) forms a homodimer, and each monomer is composed of five α-helices and nine β-strands (Figure 2c) (29). The NAD+-binding site is created by two βαβαβ Rossmann folds. Several hydrophilic and hydrophobic interactions were found between the surface residues of NSDHL and NAD+. The characteristic glycine-rich motif GGxGxxG (G44–G50) of the SDR family engages in forming hydrogen bonds as well as hydrophobic interactions with the adenine-ribose and pyrophosphate moieties (Figure 2d). In addition, the catalytic residues Y172 and K176, which belong to the conserved YxxxK motif in the SDR family, accommodate the nicotinamide-ribose moiety by hydrophilic interactions (Figure 2d). The nicotinamide ring of NAD+ is not clearly defined in the complex structure of NSDHL and NAD+, presumably due to the absence of a sterol substrate. In the apo form of the NSDHL structure, the loop H201–L211 occupies the position of the nicotinamide ring located in the active site and blocked the substrate binding site (Figure 2d). The structural significance of this loop was further confirmed by two NSDHL mutants (G205S and K232Δ), which are the most common causes of CHILD and CK syndromes, respectively (30). K232 is located near the loop of the other monomer in the dimeric interface (Figure 2d). These NSDHL mutants destabilize the protein fold, indicating the importance of the H201–L211 loop for the conformational change and the sequential binding of NAD+ and substrates. Thus, a specifically ordered and sequential mechanism is involved in the enzymatic reaction through NSDHL.
Structure of EBP
Emopamil-binding protein (EBP), which is the mammalian 3β-hydroxysteroid Δ8-Δ7 isomerase, shifts the double bond from the C8–C9 to the C7–C8 position at the B ring of https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/sterols in the postsqualene part of cholesterol biosynthesis (Figure 1). Certain cancer cells die through inhibition of EBP (31,32), implying that EBP is a potential target for cancer therapy.
The pharmacological properties of EBP are similar to the so-called σ1 receptor, which serves as a modulator of diverse signaling-pathway effectors (33). Thus, EBP is of high pharmacological interest because it binds a wide range of structurally diverse pharmacologically active compounds, including antidepressants, antipsychotics, opioid analgesics, sterol biosynthesis inhibitors and anti-tumor reagent (34,35). Although these compounds contain few shared features, all the EBP ligands contain a positively charged amine group, which mimics the carbocationic intermediate, representing a minimal EBP-binding pharmacophore. Together with 7-dehydrocholesterol reductase (DHCR7), EBP forms the microsomal anti-estrogen (e.g. tamoxifen) binding site (AEBS) (36), which is different from the canonical nucleolar tamoxifen binding site in the estrogen receptor α.
Despite shared properties with the σ1 receptor, sequence analysis shows that the mammalian EBP shares no structural similarity with the σ1 receptor. By contrast, the TM6SF2 protein, which is correlated to nonalcoholic fatty liver disease (37), shares significant sequence homology with EBP; and σ2 receptor, which is highly expressed in a number of diverse tumor cells (38), shares a similar membrane topology with EBP (39). The crystal structure of an EBP complex with its inhibitor U18666A shows that EBP forms a homodimer, with each monomer containing five transmembrane helices (TMs) (Figure 2e) (40). These TMs exhibit a novel fold to generate a large cavity. This cavity contains numerous aromatic residues that provide a hydrophobic environment to host a lipophilic ligand. Two polar residues N193 and E122 in the cavity recognize the amine group of the ligand, which can additionally be stabilized by W196 through a π-cation interaction (Figure 2f). For the EBP-mediated isomerization, an acid-base catalytic mechanism was proposed, analogous to that of ketosteroid isomerase (41). The proton donor H76 initiates the reaction by protonating Δ8-sterol at C9, thus creating a carbenium ion at C8, which is then neutralized by elimination of the C7 proton. The removal of this proton from the carbocationic sterol intermediate would be achieved by E80 that would in turn be stabilized by E122. Similar to the amine group of EBP ligands, the short-lived carbocationic sterol intermediate would be stabilized through a π-cation interaction with W196. With the release of the Δ7-isomer product, the proton would be shuttled back to H76 through the hydrogen-bonding network (40). Due to the reversibility of sterol ∆8-∆7 isomerization, H76 and E80 could both act as proton donors and acceptors, respectively, depending on the direction of the enzymatic reaction.
Cholesterol esterification by ACATs
The acyl-CoA:cholesterol acyltransferase (ACAT) enzymes, also named sterol O-acyltransferase (SOAT), including ACAT1 and ACAT2, convert cholesterol to cholesteryl esters that become incorporated into lipoproteins or stored in cytosolic lipid droplets to maintain the homeostasis of cellular cholesterol (2,42). ACAT1 is ubiquitously expressed, while expression of ACAT2 is limited to hepatocytes and intestinal cells (43). ACAT1 is associated with Alzheimer’s disease and several cancers (44,45). Specific knockout of ACAT2 in the intestine or liver can prevent the accumulation of dietary hepatic cholesteryl esters and hypercholesterolemia in vivo (46). Recently, the structures of ACAT1 and ACAT2 have been determined by cryo-electron microscopy (47–50). These two enzymes share a similar membrane protein topology, containing nine TMs, and both form a tetramer (Figure 3a). The structures reveal endogenous acyl-CoA in the active-site cavity created by TMs 4–9, which is accessible for the cholesterol substrate through the lipid bilayer to contact the acyl-CoA in the center of the enzyme (Figure 3b). Nevanimibe, an ACAT inhibitor, directly interacts with the catalytic histidine to prevent the esterification (47,50).
Figure 3. Mechanism of ACAT-mediated cholesterol esterification.
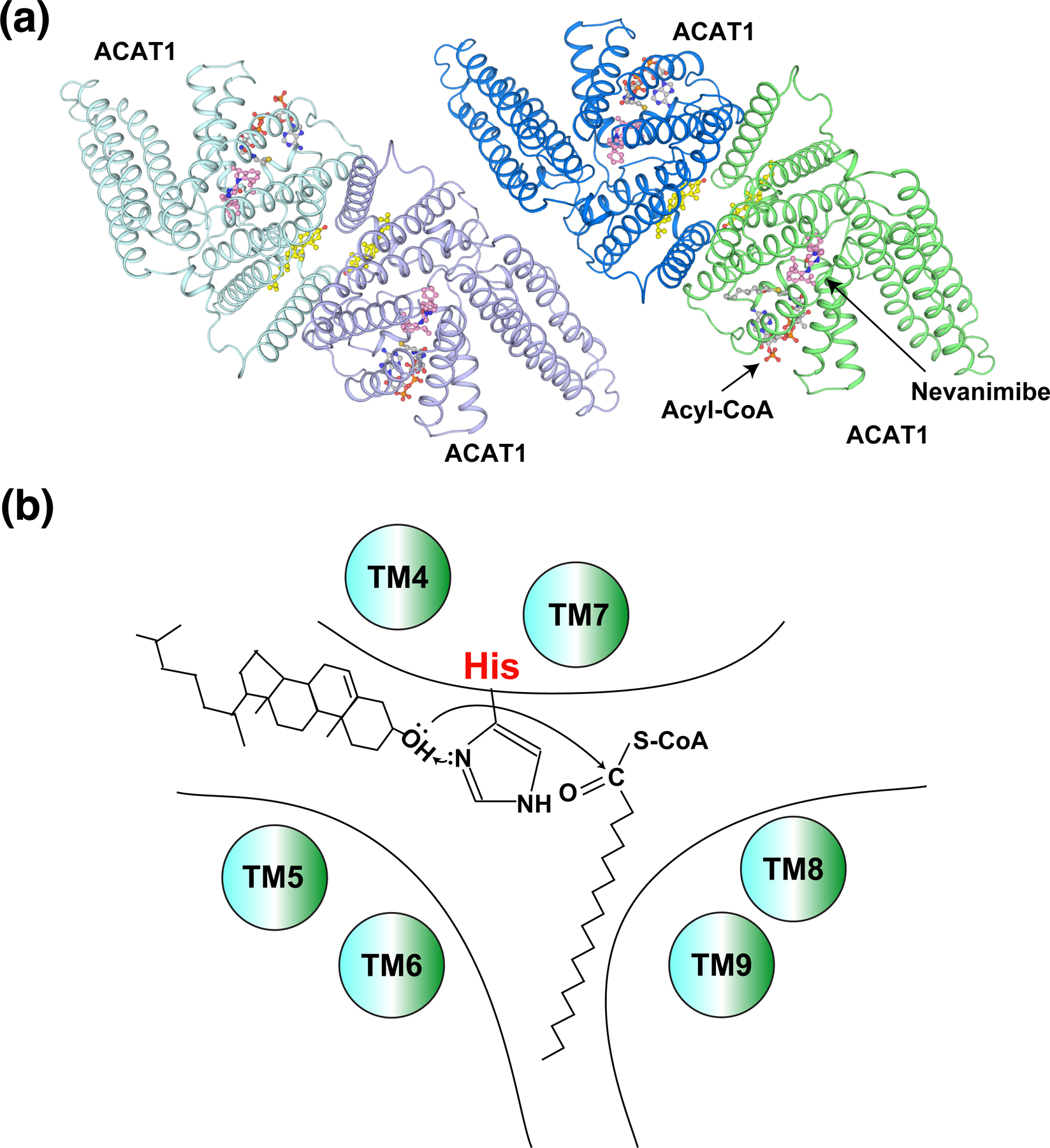
(a) Overall structure of the ACAT1 tetramer (pdb: 6VUM). Cholesterol (yellow), acyl-CoA (grey), and nevanimibe (light pink) are shown as sticks. (b) Working model of ACAT enzymes, showing the catalytic histidine, cholesterol, and acyl-CoA.
Conclusions
The structural studies of the cholesterol biosynthetic enzymes combined with biochemical investigations have revealed major insights into their catalytic mechanism. Recent atomic structures of SQLE, NSDHL, EBP, and ACAT1/2 demonstrate how these enzymes recruit their substrates and facilitate the chemical reactions. The structures of SQLE, EBP, and ACAT1/2 have been determined with inhibitors, accelerating the development of specific inhibitors. While these structures provide important insights into cholesterol biosynthesis and metabolism, the mechanism of how cholesterol synthesis is regulated by the sterol concentration via HMGCR in the ER demands further investigation (10). Moreover, the mechanisms of how these membrane enzymes aptly avoid the accumulation of sterol intermediates and their diffusion into the ER during the conversion of lanosterol to cholesterol, which would be highly toxic to cells, remain unclear.
Acknowledgement
We apologize to our colleagues whose work could not be cited due to space limitations. This work was supported by NIH grant R21 AI154191 (E.W.D), R01 GM135343 and Welch Foundation (I-1957) (to X.L.). X.L. is a Damon Runyon-Rachleff Innovator supported by the Damon Runyon Cancer Research Foundation (DRR-53S-19) and a Rita C. and William P. Clements Jr. Scholar in Biomedical Research at UT Southwestern Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no conflict of interest.
Reference and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Brown MS, Radhakrishnan A and Goldstein JL (2018) Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annual review of biochemistry, 87, 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo J, Yang H and Song BL (2019) Mechanisms and regulation of cholesterol homeostasis. Nature reviews. Molecular cell biology [DOI] [PubMed]
- 3.Qi X and Li X (2020) Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends in biochemical sciences, 45, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radhakrishnan A, Rohatgi R and Siebold C (2020) Cholesterol access in cellular membranes controls Hedgehog signaling. Nat Chem Biol, 16, 1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerqueira NM, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN, Ramos MJ and Fernandes PA (2016) Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry, 55, 5483–5506. [DOI] [PubMed] [Google Scholar]
- 6.Porter FD and Herman GE (2011) Malformation syndromes caused by disorders of cholesterol synthesis. Journal of lipid research, 52, 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitsche MA, McDonald JG, Hobbs HH and Cohen JC (2015) Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. eLife, 4, e07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Roberti R and Blobel G (2015) Structure of an integral membrane sterol reductase from Methylomicrobium alcaliphilum. Nature, 517, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhu AV, Luu W, Li D, Sharpe LJ and Brown AJ (2016) DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Progress in lipid research, 64, 138–151. [DOI] [PubMed] [Google Scholar]
- 10.Schumacher MM and DeBose-Boyd RA (2021) Posttranslational Regulation of HMG CoA Reductase, the Rate-Limiting Enzyme in Synthesis of Cholesterol. Annual review of biochemistry, 90, 659–679. [DOI] [PubMed] [Google Scholar]
- 11.König A, Happle R, Bornholdt D, Engel H and Grzeschik KH (2000) Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet, 90, 339–346. [PubMed] [Google Scholar]
- 12.McLarren KW, Severson TM, du Souich C, Stockton DW, Kratz LE, Cunningham D, Hendson G, Morin RD, Wu D, Paul JE et al. (2010) Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am J Hum Genet, 87, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L et al. (1999) Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet, 22, 291–294. [DOI] [PubMed] [Google Scholar]
- 14.Porter FD (2008) Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. European journal of human genetics : EJHG, 16, 535–541. [DOI] [PubMed] [Google Scholar]
- 15.Istvan ES and Deisenhofer J (2001) Structural mechanism for statin inhibition of HMG-CoA reductase. Science, 292, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 16.Thoma R, Schulz-Gasch T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M and Ruf A (2004) Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature, 432, 118–122. [DOI] [PubMed] [Google Scholar]
- 17.Ryder NS (1992) Terbinafine: mode of action and properties of the squalene epoxidase inhibition. Br J Dermatol, 126 Suppl 39, 2–7. [DOI] [PubMed] [Google Scholar]
- 18.Huijbers MM, Montersino S, Westphal AH, Tischler D and van Berkel WJ (2014) Flavin dependent monooxygenases. Arch Biochem Biophys, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 19.Gill S, Stevenson J, Kristiana I and Brown AJ (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab, 13, 260–273. [DOI] [PubMed] [Google Scholar]
- 20.Zelcer N, Sharpe LJ, Loregger A, Kristiana I, Cook EC, Phan L, Stevenson J and Brown AJ (2014) The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol Cell Biol, 34, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshioka H, Coates HW, Chua NK, Hashimoto Y, Brown AJ and Ohgane K (2020) A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate. Proc. Natl. Acad. Sci. U. S. A, 117, 7150–7158. • The authors reported that squalene, the substrate of human SQLE, can allosterically stabilize the enzyme through directly binding to the N terminal regulatory domain, thereby reducing interaction with and ubiquitination by MARCH6.
- 22. Padyana AK, Gross S, Jin L, Cianchetta G, Narayanaswamy R, Wang F, Wang R, Fang C, Lv X, Biller SA et al. (2019) Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat Commun, 10, 97. • • This study describes the crystal structures of the catalytic domain of FAD bound human SQLE and its complex with two distinct inhibitors and provides insights into the function and inhibition mechanism of SQLE.
- 23.Chaiyen P, Fraaije MW and Mattevi A (2012) The enigmatic reaction of flavins with oxygen. Trends Biochem Sci, 37, 373–380. [DOI] [PubMed] [Google Scholar]
- 24.Abe I, Seki T and Noguchi H (2000) Potent and selective inhibition of squalene epoxidase by synthetic galloyl esters. Biochem Biophys Res Commun, 270, 137–140. [DOI] [PubMed] [Google Scholar]
- 25.Caldas H and Herman GE (2003) NSDHL, an enzyme involved in cholesterol biosynthesis, traffics through the Golgi and accumulates on ER membranes and on the surface of lipid droplets. Hum Mol Genet, 12, 2981–2991. [DOI] [PubMed] [Google Scholar]
- 26.Baudry K, Swain E, Rahier A, Germann M, Batta A, Rondet S, Mandala S, Henry K, Tint GS, Edlind T et al. (2001) The effect of the erg26–1 mutation on the regulation of lipid metabolism in Saccharomyces cerevisiae. J Biol Chem, 276, 12702–12711. [DOI] [PubMed] [Google Scholar]
- 27.Mo C, Valachovic M, Randall SK, Nickels JT and Bard M (2002) Protein-protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc. Natl. Acad. Sci. U. S. A, 99, 9739–9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orengo CA, Jones DT and Thornton JM (1994) Protein superfamilies and domain superfolds. Nature, 372, 631–634. [DOI] [PubMed] [Google Scholar]
- 29. Kim DG, Cho S, Lee KY, Cheon SH, Yoon HJ, Lee JY, Kim D, Shin KS, Koh CH, Koo JS et al. (2021) Crystal structures of human NSDHL and development of its novel inhibitor with the potential to suppress EGFR activity. Cell Mol Life Sci, 78, 207–225. • • The structures of human NSDHL in apo form and in complex with NAD reported in this paper reveal the structural determinants for recognition of NAD by NSDHL and the unique conformational change upon the binding of NAD.
- 30.Morimoto M, Souich C, Trinh J, McLarren KW, Boerkoel CF and Hendson G (2012) Expression profile of NSDHL in human peripheral tissues. J Mol Histol, 43, 95–106. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Theodoropoulos PC, Eskiocak U, Wang W, Moon YA, Posner B, Williams NS, Wright WE, Kim SB, Nijhawan D et al. (2016) Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci Transl Med, 8, 361ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segala G, David M, de Medina P, Poirot MC, Serhan N, Vergez F, Mougel A, Saland E, Carayon K, Leignadier J et al. (2017) Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat Commun, 8, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maurice T and Su TP (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther, 124, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moebius FF, Reiter RJ, Bermoser K, Glossmann H, Cho SY and Paik YK (1998) Pharmacological analysis of sterol delta8-delta7 isomerase proteins with [3H]ifenprodil. Mol Pharmacol, 54, 591–598. [DOI] [PubMed] [Google Scholar]
- 35.Laggner C, Schieferer C, Fiechtner B, Poles G, Hoffmann RD, Glossmann H, Langer T and Moebius FF (2005) Discovery of high-affinity ligands of sigma1 receptor, ERG2, and emopamil binding protein by pharmacophore modeling and virtual screening. J Med Chem, 48, 4754–4764. [DOI] [PubMed] [Google Scholar]
- 36.Pavlik EJ, Nelson K, Srinivasan S, Powell DE, Kenady DE, DePriest PD, Gallion HH and van Nagell JR Jr. (1992) Resistance to tamoxifen with persisting sensitivity to estrogen: possible mediation by excessive antiestrogen binding site activity. Cancer Res, 52, 4106–4112. [PubMed] [Google Scholar]
- 37.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, Vogt TF, Hobbs HH and Cohen JC (2014) Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet, 46, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kayed H, Kleeff J, Ding J, Hammer J, Giese T, Zentgraf H, Büchler MW and Friess H (2004) Expression analysis of MAC30 in human pancreatic cancer and tumors of the gastrointestinal tract. Histol Histopathol, 19, 1021–1031. [DOI] [PubMed] [Google Scholar]
- 39.Alon A, Lyu J, Braz JM, Tummino TA, Craik V, O’Meara MJ, Webb CM, Radchenko DS, Moroz YS, Huang XP et al. (2021) Structures of the σ(2) receptor enable docking for bioactive ligand discovery. Nature, 600, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Long T, Hassan A, Thompson BM, McDonald JG, Wang J and Li X (2019) Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat Commun, 10, 2452. • • This paper contains an extensive structural study on human EBP, including the structures of complex with two different inhibitors. Structural bases for catalytic and inhibition mechanisms by the enzyme are described.
- 41.Pollack RM (2004) Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg Chem, 32, 341–353. [DOI] [PubMed] [Google Scholar]
- 42.Chang TY, Li BL, Chang CC and Urano Y (2009) Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab, 297, E1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee RG, Willingham MC, Davis MA, Skinner KA and Rudel LL (2000) Differential expression of ACAT1 and ACAT2 among cells within liver, intestine, kidney, and adrenal of nonhuman primates. Journal of lipid research, 41, 1991–2001. [PubMed] [Google Scholar]
- 44.Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E, Seidah NG, Oddo S, LaFerla FM, Spencer TA et al. (2010) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proceedings of the National Academy of Sciences of the United States of America, 107, 3081–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C et al. (2016) Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature, 531, 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohshiro T, Matsuda D, Sakai K, Degirolamo C, Yagyu H, Rudel LL, Omura S, Ishibashi S and Tomoda H (2011) Pyripyropene A, an acyl-coenzyme A:cholesterol acyltransferase 2-selective inhibitor, attenuates hypercholesterolemia and atherosclerosis in murine models of hyperlipidemia. Arteriosclerosis, thrombosis, and vascular biology, 31, 1108–1115. [DOI] [PubMed] [Google Scholar]
- 47.Long T, Sun Y, Hassan A, Qi X and Li X (2020) Structure of nevanimibe-bound tetrameric human ACAT1. Nature, 581, 339–343. • • [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian H, Zhao X, Yan R, Yao X, Gao S, Sun X, Du X, Yang H, Wong CCL and Yan N (2020) Structural basis for catalysis and substrate specificity of human ACAT1. Nature, 581, 333–338. • • [DOI] [PubMed] [Google Scholar]
- 49. Guan C, Niu Y, Chen SC, Kang Y, Wu JX, Nishi K, Chang CCY, Chang TY, Luo T and Chen L (2020) Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nature communications, 11, 2478. • • The cryo-EM structures of human ACAT1 provides important observations for understanding the catalytic and inhibition mechanisms of ACAT enzymes.
- 50.• Long T, Liu Y and Li X (2021) Molecular structures of human ACAT2 disclose mechanism for selective inhibition. Structure The cryo-EM structures of human ACAT2 complex with distinct inhibitors provide further insight into ACAT mediated cholesterol esterification.