

Summary
Intracellular vesicles such as lysosomes contain micromolar to millimolar concentrations of Zn2+, and disturbing lysosomal Zn2+ homeostasis via lysosomal Zn2+ release leads to mitochondria damage and consequent lytic cell death. Methods have been developed to image cellular Zn2+ dynamics. Here, we present a protocol using GZnP3, a genetically encoded fluorescent Zn2+ indicator, to assess lysosomal Zn2+ release in cultured cells by fluorescence microscopy imaging.
For complete details on the use and execution of this protocol, please refer to Du et al. (2021) or Minckley et al. (2019).
Subject areas: Cell culture, Microscopy, Molecular/Chemical Probes
Graphical abstract
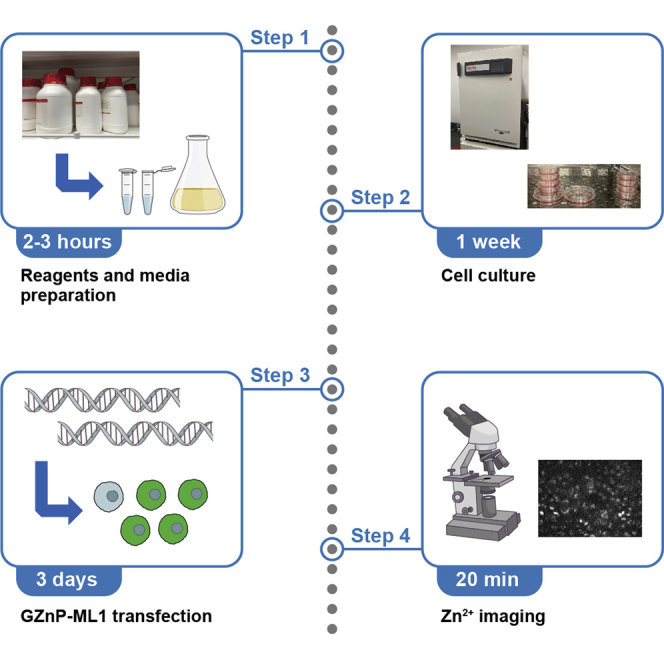
Highlights
-
•
Use green Zinc Probe (GZnP), a genetically encoded fluorescent Zn2+ sensor
-
•
Optical recording of real-time cellular Zn2+ dynamics
-
•
Use GZnP3-ML1 to monitor lysosomal Zn2+ release in live cell imaging
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Intracellular vesicles such as lysosomes contain micromolar to millimolar concentrations of Zn2+, and disturbing lysosomal Zn2+ homeostasis via lysosomal Zn2+ release leads to mitochondria damage and consequent lytic cell death. Methods have been developed to image cellular Zn2+ dynamics. Here, we present a protocol using GZnP3, a genetically encoded fluorescent Zn2+ indicator, to assess lysosomal Zn2+ release in cultured cells by fluorescence microscopy imaging.
Before you begin
TRPML1 (transient receptor potential mucolipin 1, or ML1), a protein that is mutated in type IV mucolipidosis (MLIV, a neurodegenerative lysosome storage disease), is a Ca2+ and Zn2+ permeable cation channel predominantly localized on the late endosome and lysosome membrane, and is central to lysosome functions, such as lysosome trafficking and biogenesis, and lysosomal ion homeostasis. For the first time, we reported that ML-SAs (ML-specific synthetic agonists) induced ML1 activation and subsequent lysosomal Zn2+ release via ML1, which causes a lysosomal Zn2+ release triggered, mitochondria-mediated non-apoptotic cell death in metastatic melanoma cells (Du et al., 2021).
GZnP3 is a genetically encoded fluorescent Zn2+-specific indicator which functions using a circularly permuted GFP fused with Zn2+-finger domains. Zn2+ binding induces the conformational change of the Zn2+-fingers, resulting in an increase in the fluorescent intensity of the GFP (Qin et al., 2016). GZnP3 (Zn2+-binding affinity, Kd = 1.3 nM) has approximately an 11-fold dynamic range from its apo state to Zn2+ saturation, making it the most sensitive protein-based Zn2+ sensor available for monitoring sub-nanomolar cellular Zn2+ dynamics. It has high specificity for Zn2+ over a range of other biologically relevant cations, including Ca2+ and Fe2+ (Minckley et al., 2019). As such, it is ideal for the detection of lysosomal Zn2+ release upon ML1 activation within individual cell. Here we are presenting a protocol of measuring lysosomal Zn2+ release upon ML1 activation that has a general application among various types of cells, e.g., MeWo, neuron, INS-1, COS-7 and HeLa cells.
Preparation of ML-SA1 stock solution
Timing: 30 min
ML-SA1 stock solution
| Reagent | Final concentration (mM) | Amount |
|---|---|---|
| ML-SA1 | 10 | 3.62 mg |
| DMSO | n/a | 1 mL |
| Total | n/a | 1 mL |
Note: Aliquot ML-SA1 (Shen et al., 2012) stock solution and stored in -20 C freezer (up to 6 months) to avoid freeze-thaw cycles.
Preparation of imaging buffers
To monitor lysosomal Zn2+ release by GZnP3 imaging, cells will be imaged in the imaging buffer without Zn2+ and Ca2+ (Zn2+, Ca2+ free Hank’s Balanced Salt Solution (HBSS)) to minimize the possibility of extracellular ion effects on the GZnP3 signals. For neurons, INS-1, COS-7 and HeLa cells, we measured ML-SA1-evoked lysosomal Zn2+ release via ML1 in Zn2+, Ca2+ free HBSS. Here we are presenting a recipe for the preparation of Zn2+, Ca2+ free HBSS.
Zn2+ and Ca2+ free HBSS
| Reagent | Concentration | Amount to add for 1 L |
|---|---|---|
| NaCl | 137 mM | 8 g |
| KCl | 5.4 mM | 0.4 g |
| D-glucose | 16.8 mM | 3 g |
| HEPES | 30 mM | 7.14 g |
| MgCl2 | 1.1 mM | 1.1 mL of 1 M MgCl2 |
-
1.
Dissolve the above components in 900 mL ddH2O, once all dissolved, transfer the mixture to the clean 1 L graduated cylinder and adjust the volume to 1 L.
-
2.
Transfer the solution back to the 1 L beaker, and adjust the pH to 7.4 using 10 N NaOH.
-
3.
Filter the 1 L solution with a 0.22 μm bottle-top vacuum filter.
-
4.
Store long term (up to 1 month) at 4°C or short term (within 1 week) at room temperature (20°C–25°C).
Note: The ddH2O is made by Millipore Milli-Q® Integral 10 Water Purification System, which is deionized and filtered.
CRITICAL: However, for some types of cells, such as MeWo and HEK293 cells, Zn2+, Ca2+ free HBSS seems not the optimal imaging buffer as we barely detect any GZnP3 responses toward ML-SA1 application. Therefore, we used another imaging buffer containing Zn2+ and Ca2+, Tyrode’s solution, for GZnP3 imaging. If you choose to use this buffer for your cells in the imaging, you need additional control experiments, such as Glycyl-L-phenylalanine 2-naphthylamide (GPN) pretreatment to deplete lysosomal Zn2+ storage, to exclude the potential effects of extracellular ion on GZnP3 signals (please refer to step 5. Control experiments). Except the difference in the preparation of the two imaging buffers, all other steps, such as step 4. Image acquisition and step 6. Quantification and image analysis are similar for all tested cells. Here we are using MeWo cells as an example to demonstrate the use of this protocol and control experiments to image lysosomal Zn2+ release.
Tyrode’s solution
| Reagent | Final concentration (mM) | Amount |
|---|---|---|
| NaCl | 145 | 8.474 g |
| Glucose | 10 | 1.802 g |
| HEPES | 20 | 4.766 g |
| CaCl2 | 2 | 0.3803 g |
| MgCl2 | 1 | 0.406 g |
| KCl | 5 | 0.3728 g |
| ZnCl2 | 0.1 | 0.0136 g |
| ddH2O | n/a | To 1 L |
| Total | n/a | 1 L |
Adjust the pH to 7.4 with 10 N NaOH.
Note: Store at 4°C (use within 1 month). The solutions contain glucose and may develop bacterial contamination after long-term storage.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DMEM high glucose medium | Thermo Fisher Scientific | Cat#11965092 |
| Phosphate Buffered Saline (PBS) | Thermo Fisher Scientific | Cat#20012027 |
| Poly-D-Lysine (PDL) | Gibco | Cat#A3890401 |
| Opti-MEM | Gibco | Cat#31985070 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668019 |
| Sodium chloride, NaCl | Sigma-Aldrich | S9888; CAS: 7647-14-5 |
| Calcium chloride, CaCl2 | Sigma-Aldrich | C8106; CAS: 10035-04-8 |
| Magnesium chloride, MgCl2 | Sigma-Aldrich | 208337; CAS: 7786-30-3 |
| Potassium chloride, KCl | Sigma-Aldrich | P9541; CAS: 7447-40-7 |
| Zinc chloride, ZnCl2 | Sigma-Aldrich | 208086; CAS: 7646-85-7 |
| Glucose | Sigma-Aldrich | G6152; CAS: 50-99-7 |
| HEPES | Sigma-Aldrich | H3375; CAS: 7365-45-9 |
| Glycyl-L-phenylalanine 2-naphthylamide (GPN) | Santa Cruz | sc-252858; CAS: 21438-66-4 |
| ML-SA1 | Tocris | 4746; CAS: 332382-54-4 |
| Pyrithione | Cayman | 29154; CAS: 13463-41-7 |
| TPEN | Cayman | 13340; CAS: 16858-02-9 |
| Experimental models: Cell lines | ||
| INS-1 | Sigma-Aldrich | SCC207 |
| COS-7 | ATCC | ATCC CRL-1651 |
| HeLa | ATCC | ATCC CCL-2 |
| MeWo | ATCC | ATCC HTB-65 |
| Recombinant DNA | ||
| GZnP3 | Addgene | Plasmid #161738 |
| GZnP3-ML1 | Minckley et al. (2019) | N/A |
| mCherry-ML1 | Du et al. (2021) | N/A |
| Software and algorithms | ||
| EasyRatioPro | HORIBA | https://www.horiba.com/ |
| OriginPro | OriginLab | https://www.originlab.com/ |
| Other | ||
| 18 mm Ø Cover glasses circular, #1.5 | Thermo Fisher Scientific | Cat#50-948-975 |
| 12-well Clear TC-treated Multiple Well Plates | Corning | 3513 |
| 0.2 μm pore size sterile syringe filter | Corning | 431229 |
| Metal perfusion chamber | Du et al. (2021) | N/A |
| 35 mm imaging dish | MatTek | P35GC-1.5-14-C |
Alternatives: All reagents can be obtained from different suppliers. High purity grades (>98%) for chemicals are recommended.
Step-by-step method details
Cell transfection
-
1.Preparation of PDL-coated 18 mm Ø coverslips.
-
a.Coat 18 mm Ø glass coverslips with 10 mg/mL PDL solution overnight (8–12 h) at 4°C.
-
b.Remove the PDL solution and wash the coverslips three times with PBS or sterilized water.
-
c.Store the coverslips in sterilized PBS or water at 4°C (use within 1 week) for future use.
-
a.
Note: Transfection approach varies between different cell types. You may use any transfection reagents that work best for your cells.
-
2.Check cell shape (adherent, fibroblast-like) and confluence (60%–70%). Transfect MeWo cells with 1 μg GZnP3-ML1 (conjugation of GZnP3 onto the N-terminus of ML1) or 1 μg mCherry-ML1 + 1 μg GZnP3 (given the GZnP3-ML1 plasmid is not available) using Lipofectamine 2000.
-
a.For each well:
-
i.Add 100 μL Opti-MEM each in two 1.5 mL tubes.
-
ii.Add 4 μL Lipofectamine 2000 to one tube and incubate the mixture at room temperature (20°C–25°C) for 5 min.
-
iii.Add 1 μg GZnP3-ML1 plasmid (or 1 μg mcherry-ML1 + 1 μg GZnP3) to the other tube and incubate the mixture at 20°C–25°C for 5 min.
-
iv.Mix the contents of the two tubes gently and incubate them at 20°C–25°C for 20 min.
-
v.Replace old culture medium of MeWo cells with 300 μL fresh culture medium.
-
vi.Gently drop the DNA & Lipofectamine mixture onto the cells.
-
vii.Incubate the cells in a 37°C 5% CO2 incubator for 4–6 h.
-
viii.Discard all medium (∼ 500 μL) from the well, wash twice with 1 mL PBS and add 1 mL fresh warmed cell culture medium.
-
i.
-
b.Cells are ready for imaging 48 h post transfection.
-
a.
Note: Both GZnP3 (the fluorescent Zn2+ indicator) and ML1 (the lysosomal Zn2+-release channel) are required here for imaging lysosomal Zn2+ release, thus the cells can be either transfected with GZnP3-ML1 or alternatively co-transfected with GZnP3 and ML1.
Image acquisition
The EasyRatioPro imaging system (HORIBA) is used for fluorescence imaging of live cells. As shown in Figure 1, this system consists of an inverted fluorescence microscope (Olympus IX71, Figure 1A) equipped with a CoolSnap HQ2 camera (pixel resolution 1392 × 1040, Photometrics, Figure 1B), a DeltaRam™ X monochromator (Photon Technology International, Figure 1C), and a LPS220B power supply (Photon Technology International). The DeltaRam™ X compact rapid switching monochromator allows virtually infinite excitation wavelengths from 250 to 650 nm. The system is automatically controlled by the EasyRatioPro software (ver 1.12.121.86). Acquisition rate could be set for continuous imaging (up to 250 frames/s for dual excitation or 1000 frames/s for dual emission measurements) or to specified intervals. The images are acquired using a 20× objective lens (Olympus). The experiment is usually carried out at room temperature, while a temperature control device could be used if needed.
Figure 1.

Hardware setup of the EasyRatio Pro Imaging system
(A–E) General view of the entire imaging system which consists of a microscope (A), a camera (B), a monochromator (C), a perfusion system (D) and a metal perfusion chamber (E).
A perfusion system (Figure 1D) is utilized for the solution application to a metal perfusion chamber (Figure 1E), which is customized to hold a Ø 18 mm glass coverslip. Alternatively, a 35 mm imaging dishes (MatTek) can be used without metal chamber. The perfusion system here is manually established by connecting the syringes as fluid reservoirs (10 mL or 60 mL, Corning), PE tubing (Warner Instruments, Cat. No. 64-0756), 3-way Luer locks (Baxter, Cat. No. 2C6241), roller clamps (Merit Medical, Cat. No. 700022001), and a perfusion manifold (Warner Instruments, Cat. No. 64-0211). This perfusion system uses gravity force to drive the solution flux, so the height level of the syringe bottom should be above of the perfusion manifold and the metal chamber. Manifold should be chosen with input ports to match the number of tubes to be connected. Input and output tubing are inserted with a friction fit. Roller clamps can be used to adjust the flow speed if needed. The metal perfusion chamber has two grooves to accommodate both the manifold tip (Figure 1E, on the left) and the outlet pipe (Figure 1E, on the right) to avoid direct flush onto the cells. The outlet pipe is connected with a liquid collection flask and a vacuum pump to collect waste solutions (also prevent the overflow within the perfusion chamber).
-
3.Preparation of the cells and the recording setup. Warm up the imaging buffer (Tyrode’s solution) to (20°C–25°C). Prepare ML-SA1 working solution (50 μM in Tyrode’s solution). Load all the solutions into the fluid reservoirs.
-
a.Put a coverslip with cells in the metal perfusion chamber, fill the chamber with the Tyrode’s solution (∼0.2 mL) and mount it on the inverted microscope.Note: Make sure the coverslip is aligned and the chamber is screwed on securely to prevent any liquid leakage.Note: The microscope must be capable of exciting and recording mCherry fluorescence (if the cells are transfected with mCherry-ML1; i.e., 561 nm excitation, 605 nm emission) and GZnP3 fluorescence (i.e., 488 nm excitation, 525 nm emission).
-
b.Prepare the perfusion system, adjust the inflow speed to a rate of 50–60 drops/min. Adjust the outflow speed accordingly to avoid overflow or dry out the cells.
-
c.Turn on the imaging system step by step as follows:
-
i.Turn on all the power switches.
-
ii.Turn on the DeltaRam-X monochromator.
-
iii.Turn on the camera.
-
iv.Turn on the computer/workstation, open the EasyRatioPro software (Figure 2A).
-
i.
-
d.Turn off the room lights. The experiment should be performed in a dark environment to reduce background noise signals.CRITICAL: The perfusion speed should be carefully controlled to avoid the washout of cells (Problem 1).Note: It is necessary to clean the perfusion system with ethanol to avoid the contamination of residual drugs from previous experiments.Note: Allow the solutions flow through entire perfusion system and ensure no air bubble arises. All the solution reservoirs (10 mL or 60 mL syringes) should be filled with a solution or ddH2O to avoid back-flow or air bubbles.Note: ML-SA1 is used to activate ML1 to release Zn2+ from the lysosomal lumen to the cytosol. Pyrithione, a Zn2+ ionophore, or TPEN, a Zn2+ chelator, can serve as the controls to confirm the availability of GZnP3 indicator (please refer to step 7. In situ response of GZnP3 to Zn2+ chelation and saturation for details).
-
a.
-
4.Image acquisition.
- a.
-
b.In the CHANNELS window, click the “R” and the “GFP” button at the bottom (Figure 2C).
-
c.In the IMAGE window, choose RAINBOW in PALETTE. Adjust gain and exposure time to obtain low but visible basal fluorescent signals. Set the read interval of each frame per 1–20 s, for MeWo cells, we used 3 s interval (Figure 2D).
-
d.Click the “RECORD” button in the TRANSPORT CONTROLS window, the monochromator will start working (Figure 2E).
-
e.Choose a ROI (Region of Interest) selector in the IMAGE window. Circle out the cell to show the intensity curves in Graph window (Figure 3, “E1”).
-
f.Click PLAY button in the TRANSPORT CONTROL window. The changes of GFP fluorescent signals will be recorded. Record baseline for at least 3 min, then add ML-SA1 (50 μM) to induce the lysosomal Zn2+ release. An example of an experimental setting showing the basal GZnP3 fluorescence and the fluorescence emission intensity trace of one MeWo cell upon ML-SA1 treatment (Figure 3, lower panel).
Note: The fluorescent stability of GZnP3 is great, so it could be used for long-term (hours) recording at different acquiring interval. However, cells under long-term exposure to ambient environment could become unhealthy, so additional temperature-controlled device and 5% CO2 environment may be required.
Figure 2.

EasyRatio Pro software setup
(A) General overview of the EasyRatio Pro software.
(B) “GRAPH” window.
(C) “CHANNELS” window.
(D) “IMAGE” window.
(E) “TRANSPORT CONTROLS” window.
Figure 3.

Software demonstration of the imaging of GZnP3 signal in MeWo cells expressing GZnP3-ML1
Upper panel, a representative image recorded from the cell (E1).
Lower panel, time series of fluorescent emission changes in the “GRAPH” window from the circled cell (E1).
After the recording is complete, remove the imaging chamber, discard the coverslip, wash the chamber thoroughly (to get rid of any residual chemicals), prepare the next coverslip, and repeat the imaging steps. Datasets are processed and analyzed with ImageJ (please refer to step 6. Quantification and image analysis for details).
-
5.Control experiments (optional).
-
a.Use FluoZin-3 AM (a cell permeable Zn2+-selective indicator, Kd=15 nM) and LysoTracker (a fluorescent dye broadly used to label lysosomes) to stain the cells, then challenge the cells with ML-SAs. Later, fluorescent images can be obtained and statistical analysis of the FluoZin-3 fluorescent intensity are performed to show ML-SA-induced lysosomal FluoZin-3 signal decrease, supporting lysosomal Zn2+ release into the cytosol upon ML1 activation.
-
b.Glycyl-L-phenylalanine 2-naphthylamide (GPN, a cathepsin C substrate that induces osmotic lysis of lysosomes) can be used to deplete lysosomal Zn2+ storage. GPN pre-treatment (200 μM) of cells largely abolishes or reduces the changes of GZnP3 signals upon ML-SA1 treatment, suggesting ML-SA-evoked GZnP3 signals are attributed to lysosomal Zn2+ release.
-
a.
Quantification and image analysis
-
6.Fluorescence should be normalized to the baseline fluorescence (F0, the baseline fluorescent intensity of GZnP3 prior to ML-SA1 treatment) indicated as ΔF/F0. This equation is defined below, where F is the fluorescence intensity of GZnP3.Plot the time on the x-axis and ΔF/F0 on the y-axis to observe changes in GZnP3 fluorescence over time.
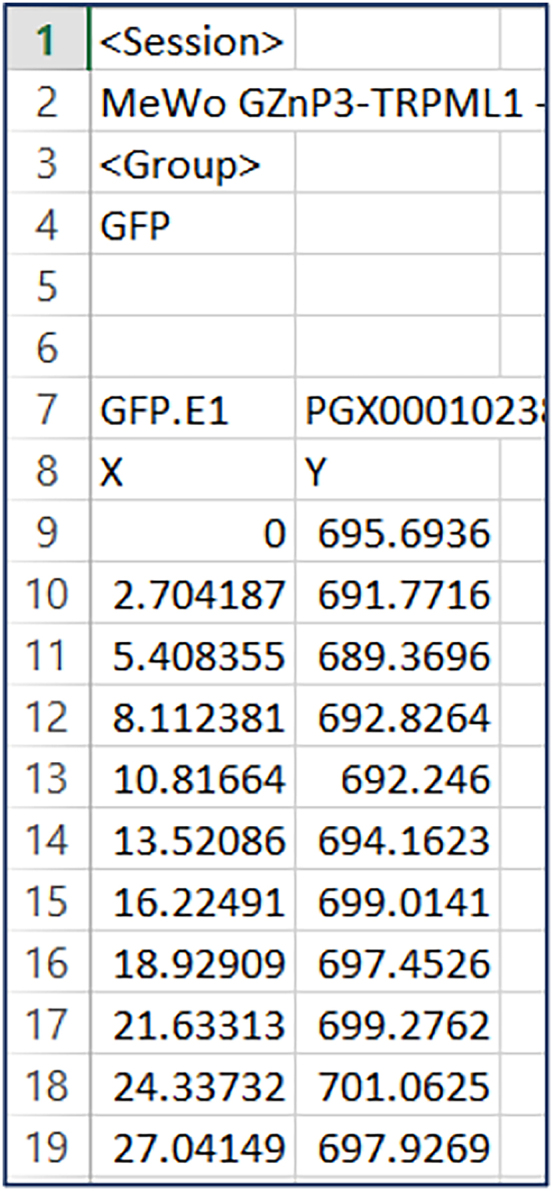 Here we include a detailed image analysis of a MeWo cell as an example:
Here we include a detailed image analysis of a MeWo cell as an example:-
a.Use ImageJ to analyze GZnP3 fluorescence. Export the session data as an Excel file (Table above is an example for one MeWo cell. X is time in second. Y is GZnP3 fluorescence intensity (F)).
-
b.The dataset contains the F at every recording time-point for the cell. Average the F from the time points before the application of ML-SA1 to get the baseline fluorescent intensity of GZnP3 (F0) for the cell. Use the equation (step 6) to calculate the ΔF/F0 for individual time point. Plot the time points on the x-axis and their corresponding ΔF/F0 on y-axis to reveal the changes of GZnP3 signals over time.Average ΔF/F0 from all recorded cells to generate an average time series of ΔF/F0. In OriginPro, plot the time on the x-axis and the average ΔF/F0 on the y-axis to observe the GZnP3 responses of all the recorded cells over time (Figure 4B).
-
a.
-
7.In situ response of GZnP3 to Zn2+ chelation and saturation (optional).Note: For most of the cells that are transfected with either GZnP3-ML1 alone or co-transfected with mCherry-ML1 and GZnP3 together, 48 h should be enough for the exogenous protein expression (Figure 5). Here, we are providing a step-by-step method to validate the GZnP3 indicator expression in the transfected cells. This section can be also used as a control template to test your microscope system and GZnP3 responses in your cells.Preparation of imaging buffer with 100 μM Zn2+Timing: 15–30 min
-
a.Make a 1 M intermediate stock solution of ZnCl2 with ddH2O in a 1.5 mL Eppendorf tube.CRITICAL: 1 M ZnCl2 will start to precipitate quickly (∼1 h), so it is important to make this solution fresh and use it immediately.
-
b.Add 5 μL 1 M ZnCl2 to 50 mL imaging buffer (Zn2+, Ca2+ free HBSS) to get 100 μM Zn2+ HBSS.
-
c.Store long term (1 month) at 4°C and short term (1 week) at room temperature (20°C–25°C).Preparation before imaging.Timing: 5 min
-
d.Cells can be either transfected with GZnP3-ML1 alone or co-transfected with mCherry-ML1 and GZnP3. 48 h post-transfection, wash the cells three times with 1 mL Zn2+, Ca2+ free HBSS.
-
e.Prepare the image chamber for imaging.Image acquisition and drug treatment.
-
f.Mount the imaging chamber on the inverted microscope.
-
i.The microscope must be capable of exciting and recording GZnP3 fluorescence (i.e., 488 nm excitation, 525 nm emission).
-
i.
-
g.Acquire images using a CCD camera, every 10–20 s.
-
h.Record baseline GZnP3 fluorescence for 3–5 min to ensure stable sensor fluorescence, then add either ML-SA1 to open lysosomal ML1 channel (cause lysosomal Zn2+ release), or 100 μM TPEN to chelate Zn2+ from the GZnP3 indicator, bringing it to its minimum-fluorescent apo-state (Figures 5A–5D).
-
i.As shown in Figure 5F, the cells are incubated with TPEN for 5 min, then washed 3 times with Zn2+, Ca2+ free HBSS.
-
j.Add 100 μM Zn2+ HBSS (with 2.5 μM pyrithione) to influx Zn2+ and saturate the GZnP3 indicator to its fluorescent maximum.
-
k.After the recording is complete, use ImageJ to analyze GZnP3 fluorescence (please refer to step 6. Quantification and image analysis for the details).
-
a.
Figure 4.

Lysosomal Zn2+ release imaged in MeWo cells expressed GZnP3-ML1
(A) Scheme diagram of the lysosome-targeted genetically encoded Zn2+ sensor, GZnP3-ML1, in which GZnP3 is linked to the N terminus of ML1.
(B) Average time series of fluorescence intensity recorded from the MeWo cells in response to ML-SA1. (Figure reprinted with permission from Du et al., 2021).
Figure 5.

Lysosomal Zn2+ release in various cell types measured by GZnP3
(A–D) ML1-mediated Zn2+ signals measured by GZnP3 responses in (A) primary rat hippocampal neurons, (B) INS-1 cells, (C) COS-7 cells, and (D) HeLa cells expressing GZnP3 and mCherry-ML1. Cells were treated with 50 μM ML-SA1 at 0 s and 100 μM TPEN at 300 s.
(E) Bar graph of maximum GZnP3 signal (ΔF/F0) for each of the four cell types. Mean GZnP3 signal maximum (±SEM) 300 s after ML-SA1 addition. (Figure reprinted with permission from Minckley et al. [2019].)
(F) In situ response of GZnP3 to Zn2+ chelation and saturation. Representative GZnP3 signals in HeLa cells treated with 100 μM TPEN at 0 s, washed three times with Zn2+, Ca2+ free HBSS, then treated with 100 μM ZnCl2 and 2.5 μM pyrithione at 300 s.
Expected outcomes
The expression of either GZnP3 together with mCherry-ML1 or the fusion construct GZnP3-ML1 in the cells enables the acquisition of continuous, reproducible, and relatively stable fluorescence signals that allows direct tracking of lysosomal Zn2+ release via ML1 activation at the cellular level. As for the control, we have used GZnP3-Rab7 (Rab7 is used as a late endosome marker) in our previous work to demonstrate the requirement of ML1 for the GZnP3 imaging (Du et al., 2021). We expect that our protocol will provide a feasible and reproducible method to monitor lysosomal Zn2+ dynamics upon ML1 activation (see Du et al., 2021; Minckley et al., 2019).
Limitations
This protocol is most suitable for monitoring lysosomal Zn2+ release upon lysosomal ML1 activation (the main Zn2+ permeable channels reside on lysosomes). One major limitation of using the current protocol is the requirement of exogenous expression of ML1 and GZnP3 (or the fusion construct GZnP3-ML1) in the cells. We hope that the development of new ML-SAs with higher potency can bypass the requirement of ML1 overexpression. Another limitation of using this method is the availability of the entire setup, e.g., the perfusion system. This could be solved by performing live cell imaging under a spinning-disk or laser-scan confocal microscope (at least equipped with the bandpass emission filters for GFP and mCherry). Without the perfusion system, manually drop-in drugs or solutions can be optimized for the imaging. The third limitation is that the current protocol only works for adherent cells. Although the coverslips are treated with PDL to enhance cell attachment, non-adherent cells are not suitable for live cell imaging. Finally, the sensitivity of the GZnP3 indicator is not optimal to respond to subtle Zn2+ changes that are far off the detection level. Vesicular Zn2+ concentration varies among different cells, for example, the labile Zn2+ in the lysosomes of HEK 293 cells is relatively low (data not shown), thus the GZnP3 responses are barely detectable upon ML-SA1 treatment in Zn2+, Ca2+ free HBSS imaging buffer. To enhance the GZnP3 signals, HEK293 cells can be cultured with the growth medium containing high Zn2+ (supplemented with 100 μM ZnCl2) 24 h before imaging (to increase the intracellular Zn2+ storage; the amount and time course of ZnCl2 supplementation were determined based on our preliminary FluoZin-3 AM staining assay). Afterward, performing the imaging in Tyrode’s solution can help to obtain GZnP3 responses upon ML-SA1 treatment.
Troubleshooting
Problem 1
Cells detach during imaging. This problem may arise in step 4. Image acquisition.
Potential solution
Glass coverslips should be coated optimally with PDL or other appropriate coating reagents to strengthen the cell attachment. The optimal coating reagent used for different cell types could be variable, so a prior optimization step is required to determine an appropriate coating reagent for each cell type. Also, the inflow/outflow speed of perfused solutions should be carefully controlled to avoid the cell washout.
Problem 2
Slow GZnP3 response upon ML-SA1 treatment. This problem may arise in step 4. Image acquisition.
Potential solution
The perfusion system should be checked for potential problems. The potential problems include, but not limited to, leakage (e.g., PE tubing broke), clogged (air bubbles clog the fluid flow), slow flow etc. Efficient solution exchange is essential for cells to access drugs of predefined concentration. Adjust the flow speed to allow efficient solution exchange to obtain predicted observation in positive control tests (refer to step 7. In situ response of GZnP3 to Zn2+ chelation and saturation for details). All chemicals (e.g., ML-SA1) should be prepared freshly from stock solutions or powders to ensure the chemicals of expected efficiency or strength will be applied to the cells.
Problem 3
GZnP3 fluorescence signal is undetectable in step 4. Image acquisition.
Potential solution
This issue is very likely due to not enough GZnP3 indicator expression in cells. Adjust the transfection protocol by increasing the amount of plasmids and/or Lipofectamine 2000 to achieve the optimal protein expression. Excess Lipofectamine 2000 usually makes cells unhealthy, so other transfection reagents such as FuGENE HD (Roche) could be tried alternatively. Here, we suggest to refer to step 7. In situ response of GZnP3 to Zn2+ chelation and saturation for validation of the expression of GZnP3 indicators in cells.
Problem 4
Unexpected fluorescence oscillation seen after solution switch or accidently. This may arise in step 4. Image acquisition.
Potential solution
Prior to the experiment, make sure that perfusion system has been washed thoroughly with ethanol and ddH2O to avoid the contamination of residual chemicals. Any interruption to the light pathway of the recording system, including but not limited to, light contamination, leakage of chamber, movement of coverslip, vibration, could induce unexpected light signals to the camera, so a constant dark environment is required, in addition, an air table that isolates microscope settings and samples from ambient vibration is necessary.
Problem 5
Background noise during recording in step 4. Image acquisition.
Potential solution
The imaging process is light sensitive therefore a constant dark environment is required. Background noises may result from outside light contamination.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Wanlu Du (wanludu@umich.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by NIH project grants (R21CA252428 to W.D. and R01NS110590 to Y.Q.). Additional support was provided by an M-cubed grant from the University of Michigan (MICHR Diamond Award to W.D.).
Author contributions
Conceptualization, W.D. and H.X.; methodology, M.G., T.M., Y.Q., and W.D.; investigation, M.G., M.H., T.M., P.P., and W.D.; writing – original draft, M.G., M.H., and W.D.; writing – review & editing, M.G., M.H., T.M., P.P., Y.Q., and W.D.; funding acquisition, Y.Q. and W.D.; supervision, W.D.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate/analyze datasets/code.
References
- Du W., Gu M., Hu M., Pinchi P., Chen W., Ryan M., Nold T., Bannaga A., Xu H. Lysosomal Zn(2+) release triggers rapid, mitochondria-mediated, non-apoptotic cell death in metastatic melanoma. Cell Rep. 2021;37:109848. doi: 10.1016/j.celrep.2021.109848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minckley T.F., Zhang C., Fudge D.H., Dischler A.M., LeJeune K.D., Xu H., Qin Y. Sub-nanomolar sensitive GZnP3 reveals TRPML1-mediated neuronal Zn(2+) signals. Nat. Commun. 2019;10:4806. doi: 10.1038/s41467-019-12761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y., Sammond D.W., Braselmann E., Carpenter M.C., Palmer A.E. Development of an optical Zn(2+) Probe based on a single fluorescent protein. ACS Chem. Biol. 2016;11:2744–2751. doi: 10.1021/acschembio.6b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D., Wang X., Li X., Zhang X., Yao Z., Dibble S., Dong X.P., Yu T., Lieberman A.P., Showalter H.D., Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.