

Abstract
The bacterial surface display method was used to selectively screen for improved variants of carboxymethyl cellulase (CMCase). A library of mutated CMCase genes generated by DNA shuffling was fused to the ice nucleation protein (Inp) gene so that the resulting fusion proteins would be displayed on the bacterial cell surface. Some cells displaying mutant proteins grew more rapidly on carboxymethyl cellulose plates than controls, forming heterogeneous colonies. In contrast, cells displaying the nonmutated parent CMCase formed uniform tiny colonies. These variations in growth rate were assumed to result from altered availability of glucose caused by differences in the activity of variant CMCases at the cell surface. Staining assays indicate that large, rapidly growing colonies have increased CMCase activity. Increased CMCase activity was confirmed by assaying the specific activities of cell extracts after the expression of unfused forms of the variant genes in the cytoplasm. The best-evolved CMCases showed about a 5- and 2.2-fold increase in activity in the fused and free forms, respectively. Sequencing of nine evolved CMCase variant genes showed that most amino acid substitutions occurred within the catalytic domain of the enzyme. These results demonstrate that the bacterial surface display of enzyme libraries provides a direct way to correlate evolved enzyme activity with cell growth rates. This technique will provide a useful technology platform for directed evolution and high-throughput screening of industrial enzymes, including hydrolases.
Directed evolution is increasingly being used to improve biocatalysts (13). As more powerful combinatorial mutagenesis methods (e.g., combinatorial cassette mutagenesis, StEP and DNA shuffling [5]) become available, designing selection or screening strategies becomes the most critical step in the successful exploitation of generated molecular diversity (8, 13). Screening can be based on chromogenic substrates or easily observed colony phenotypes (16, 29), but the most direct methods of screening and selection link improved enzyme activity to the survival or growth rates of cells (12, 14, 21). Examples of this method include selection on plates containing increasing antibiotic concentrations (23, 28) and complementation selection with auxotrophs (1, 27). Unfortunately, these selection procedures are designed for specific enzyme activities, making the generalized identification of improved enzyme variants difficult (21). In addition, these methods work only if the activity of the target enzyme does not interfere with cellular metabolism and can be distinguished from the background of all other cell reactions (2).
An alternative selection method is to display libraries of mutated proteins on phage or microbial cell surfaces and then to select mutant enzymes having desirable properties (4, 25). Enzymes displayed on phage, for example, may be screened for improved affinity for desired substrates and the corresponding clones selected by panning techniques (22, 25). Recently, a library of surface protease OmpT was displayed, and clones showing improved substrate affinity were isolated by flow cytometry (18). In our study, we have used the ice nucleation protein (Inp)-based bacterial surface display system (10, 11) to selectively screen enzyme libraries for improved catalytic activity. Our model enzyme is carboxymethyl cellulase (CMCase). Because CMC (carboxymethyl cellulose) is a high-molecular-weight polymer, it is not transported into cells. Thus, cells that display CMCase on their surfaces only hydrolyze CMC in agar plates. Cell colonies that hydrolyze CMC can be easily recognized because they are surrounded by a clear halo after staining with Congo red (11). Because Escherichia coli cells displaying CMCases form tiny colonies on M9 minimal medium containing CMC as the sole carbon source, we reasoned that altered growth rates of transformants would be correlated with the activities of displayed CMCase variants. Thus, by selecting rapidly growing colonies, only the cells containing improved CMCase variants would be isolated and checked further, obviating laborious random plating and assay procedures. In this report, we describe a growth-based direct screening method for the identification of improved CMCases based on the display of an enzyme library on the bacterial cell surface. This technique may provide a useful technology platform for high-throughput selective screening of industrially important hydrolytic enzymes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
E. coli JM109 (recA1 supE44 endA1 hsdR17 gyrA96 relA1 thi Δ(lac-proAB) F′ [traD36 proAB+ lacIq lacZ ΔM15]) was used as a host cell for DNA manipulations and gene expression. pKK223-3 containing a tac promoter (Amersham Pharmacia Biotech, Uppsala, Sweden) was used so that the expression of the Inp fusion protein or foreign proteins could be induced with isopropyl-β-d-thiogalactoside (IPTG) for high-level gene expression in E. coli. pSSTS110 (10) was employed for surface display of CMCase. A CMCase gene (endo-β-1,4-glucanase, EC 3.2.1.4) from B. subtilis BSE616 (GenBank accession number D01057) was originated from plasmid pUBS101 (19). Recombinant E. coli cells were grown at 37°C in Luria-Bertani (LB) medium containing yeast extract, 5 g/liter; tryptone, 10 g/liter; and NaCl, 5 g/liter. When appropriated, ampicillin was added to a final concentration of 100 μg/ml. Cell growth was determined by measuring optical density of the culture at 600 nm (OD600) with an Ultraspec 2000 spectrometer (Amersham Pharmacia Biotech).
HPLC analysis of CMC hydrolysates.
The products of CMC hydrolysis by CMCase were analyzed using high-pH anion-exchange chromatography (Dionex, Sunnyvale, Calif.). Separation of β-d-glucose (G1) and cellooligomers, such as cellopentaose (G5), cellotetraose (G4), cellotriose (G3), and cellobiose (G2), was accomplished by using a CarboPac PA1 analytical column (Dionex, 4 by 250 mm) and a CarboPac PA1 guard column (4 by 50 mm) with a mobile phase containing a mixture of eluent 1 (deionized water), eluent 2 (200 mM NaOH), and eluent 3 (200 mM NaOH, 1 M sodium acetate) at a flow rate of 1.0 ml/min. A PAD system with a gold electrode was used for detection of carbohydrates. A Dionex Advanced Computer Interface (ACI) model III was used for data acquisition with Dionex AI-450 software, version 3.32. For hydrolysis of cellopentaose (Sigma, St. Louis, Mo.), a reaction mixture containing 100 μl of 10 mg of cellopentaose per ml, 20 μl of purified CMCase (0.09 mg/ml), and 80 μl of 50 mM sodium phosphate buffer (pH 5.5) was incubated for 120 min at 37°C. To analyze the CMC hydrolysate, a reaction mixture containing 100 μl of 10 mg of CMC per ml, 20 μl of 3 × 108 cells displaying an evolved CMCase variant (2R52) and 80 μl of 50 mM sodium phosphate buffer (pH 5.5) were incubated for 180 min at 37°C. The cells were removed by centrifugation before high-pressure liquid chromatography (HPLC) analysis.
Random mutagenesis and enzyme library display.
Random mutagenesis of the CMCase gene as shown in Fig. 1 was performed by DNA shuffling as described previously (23, 30). Briefly, the substrates for the shuffling reaction were 1.3-kb double-stranded-DNA PCR products derived from pYSK3 by using a recombinant Taq DNA polymerase (TaKaRa Shuzo Co., Shiga, Japan) with two primers, YSK1 and YSK2, which are annealed to the outside of CMCase gene. PCR conditions were 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 60 s. After digestion of about 5 μg of the DNA substrates with DNase I (Boehringer Mannheim, Düsseldorf, Germany), 50- to 200-bp fragments were recovered on a 2% agarose gel and reassembled by PCR without primers by using a PCR program of 60 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 65 s. A 50-fold dilution of PCR assembled products was used for the final production of single PCR products of the correct size (1.3 kb) with 30 pmol of each primer and 30 additional PCR cycles (94°C for 60 s, 55°C for 60 s, and 72°C for 60 s). For this PCR amplification, two internal primers, YSK3 and YSK4, which anneal just inside of the first primer set, were used. After successful reassembly and amplification, reactions were verified by 0.8% agarose gel electrophoresis, and the shuffled products were purified with a Wizard PCR Prep Kit (Promega, Madison, Wis.), digested with terminal restriction enzymes, XmaI and HindIII, and subcloned into pYSK3. This process produced the plasmids containing the mutated CMCase genes that were fused to the 3′ end of the Inp gene. Plasmids were used to transform competent E. coli JM109 cells by a high-efficiency transformation method (9).
FIG. 1.

(A) Schematic diagram of plasmid construction and DNA shuffling procedure of the CMCase gene. The cel and mcel mean the CMCase and mature CMCase gene, and X and H show XmaI and HindIII, respectively. (B) Oligonucleotide primers used for PCR reactions. See the text for detail description.
Construction of plasmids for surface display and intracellular expression of CMCase.
For surface display of CMCase, the corresponding gene was subcloned into an Inp surface display vector, pSSTS110, as described below. The 1.3-kb PCR products derived from pUBS101 by using Pfu DNA polymerase (Stratagene, La Jolla, Calif.) with two primers (CMCF1 and CMCF2) were digested with XmaI and HindIII and then ligated with pSSTS110 which had been digested with the same enzymes, generating pYSK3. This plasmid contains only the gene encoding the mature form of CMCase from amino acids 31 to 499 and lacks the signal sequence required for secretion. To achieve intracellular expression of the free form of CMCase, 1.3-kb DNA fragments of the CMCase gene were obtained by 30 cycles of PCR (94°C for 30 s, 50°C for 30 s, and 72°C for 60 s) with two primers, YSK7 and YSK8. For correct translation of the CMCase gene in E. coli, the ATG start codon was added as indicated in boldface in Fig. 1B, and the XmaI and HindIII sites are underlined. PCR products were purified, digested with XmaI/HindIII, and ligated with pKK223-3 that had been digested with the same enzymes, resulting in a free-form expression vector, pYSK1.
Selection and screening.
Transformants displaying a library of CMCase variants on the surface of E. coli cells were spread on M9 minimal medium plates containing 0.5% (wt/vol) CMC (Sigma), 1 mM of IPTG (Sigma), 100 μg of thiamine per ml, and 50 μg of ampicillin per ml (M9-CMC plates). For the first positive selection, 150 larger colonies were picked up after a 72-h incubation at 37°C and subsequently transferred onto an LB plate containing 100 μg of ampicillin per ml and 1 mM IPTG (LB-Amp-IPTG). Halo-forming activities of the cells were analyzed by the Congo red method (11). After growth for 15 h at 37°C, bacterial colonies were overlaid with 10 ml of sterile top agar containing 0.5% CMC and then incubated at 37°C for 6 h to allow hydrolysis of CMC. After this incubation, the plates were flooded with 0.2% (wt/vol) Congo red. After 30 min, the Congo red solution was poured off, and the plates were washed with 10 ml of 1 M NaCl for 10 min. Colonies that hydrolyze CMC were identified by yellow halos, where Congo red staining is absent (20). During the first round of random mutagenesis and selection, 150 colonies were identified that showed higher growth rates and larger halos than control colonies containing the parent CMCase. These colonies were used for the next round of mutagenesis and selection; 1.3-kb fragments of evolved CMCases were amplified by colony PCR and then used as PCR templates for the next rounds. Colony PCR with YSK1 and YSK2 as primers was performed as described elsewhere (6) under PCR conditions of 30 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 60 s. Three rounds of random mutagenesis and screening were carried out, and 150 to 200 clones from each round were selected and characterized in detail.
CMCase assay.
Whole-cell and free-form CMCase activities were determined according to previous methods (11, 19). Enzymatic reactions were performed for 30 min at 37°C with mixing. One unit of enzyme was defined as the quantity of enzyme capable of releasing 1 μmol of glucose equivalent per min.
SDS-PAGE and Western blot analysis.
The expressed enzyme variants were analyzed by standard sodium dodecyl sulfate (SDS)–10% polyacrylamide gel electrophoresis (PAGE) and Western blot with rabbit anti-CMCase antibody. The soluble and insoluble fractions from total expressed CMCase were performed by centrifugation method. Briefly, 2.5 × 108 cells from 1 ml of culture (1.0 OD600) were pelleted by centrifugation at 12,000 rpm for 5 min. The cells were washed twice with 0.85% saline solution and resuspended in lysis buffer (50 mM Tris, 10 mM EDTA, 1 mM phenylmethylsulfonyl fluoride; pH 8.0). Then the cells were lyzed by ultrasonifier 450 (Branson, Danbury, Conn.). The soluble fraction was obtained from the supernatant after centrifugation at 12,000 rpm for 10 min, while the insoluble fraction was collected from the pellets.
DNA sequencing.
The 1.3-kb DNA fragment encoding the evolved CMCase and its flanking regions was sequenced in both forward and reverse directions by using a BigDye Terminator Ready-Reaction kit and an ABI Prism 377 DNA Sequencer (Perkin-Elmer/Applied Biosystems, Foster City, Calif.).
RESULTS AND DISCUSSION
Generation of glucose by surface-displayed CMCases and growth of E. coli.
The B. subtilis BSE616 CMCase (19) used in this work is similar to that of Bacillus sp. D04 (7), having both endo- and exoglucanase activities. Our preliminary HPLC analysis showed that hydrolysis of CMC by this enzyme produced glucose, cellobiose, and oligosaccharides. The CMCase also cleaved cellopentaose (G5) to cellotetraose (G4), cellotriose (G3), cellobiose (G2), and glucose (G1). In order to determine if Inp-CMCase fusion proteins have the same catalytic activity as free-form CMCase, the products of hydrolysis were analyzed by using a Dionex high-pH anion-exchange ion chromatography system (Fig. 2). Treatment of CMC with surface-displayed CMCases (Fig. 2C) and purified unfused CMCases (Fig. 2B) generated the same products, including glucose, cellobiose, cellotriose, and cellotetraose. The glucose liberated by hydrolysis of CMC can be used as a carbon source for cell growth. As expected, E. coli cells displaying the nonmutated parent CMCase formed uniform tiny colonies on M9-CMC minimal medium plates, reflecting their homogeneous growth rates (Fig. 3A).
FIG. 2.

HPLC chromatogram of CMC hydrolysates. (A) Standard chromatogram of glucose (G1), cellobiose (G2), cellotriose (G3), cellotetraose (G4), and cellopentaose (G5) at a concentration of 2 g/liter. (B) Chromatogram of cellopentaose hydrolysate produced by treatment with the purified parent CMCase. (C) Chromatogram of CMC hydrolysate produced by treatment with E. coli whole cells displaying Inp-CMCase fusion proteins.
FIG. 3.

Colonies of E. coli JM109 displaying CMCase on their surfaces (magnification, ×2.5). Parent CMCase (A) and the shuffled CMCase library (B) from the first round of mutagenesis are shown. Colonies were formed after 72 h of growth on M9 minimal agar plate at 37°C containing 0.5% (wt/vol) CMC as the sole carbon source, 50 μg of ampicillin per ml, 100 μg of thiamine per ml, and 1 mM IPTG. (C) Transformants of the shuffled CMCase library were spread and grown for 24 h on M9 glucose agar plate at 37°C.
E. coli cells displaying a CMCase library form heterogeneous colonies on CMC plates.
Following random mutagenesis of the 1.3-kb CMCase gene by DNA shuffling (Fig. 1), mutated DNA fragments were subcloned into the Inp surface display vector, pYSK3. This CMCase library was transformed into E. coli. Transformed cells were spread on M9-CMC minimal medium agar plates and incubated for 72 h at 37°C, producing 100 to 300 colonies per plate. The library size was more than 1.0 × 106 colonies per transformation. E. coli colonies displaying CMCase variants showed heterogeneity in colony size, reflecting differences in growth rates (Fig. 3B). Such differences in colony size, which are presumably due to differences in the hydrolytic activity of CMCase variants, were the basis of screening for putative improved CMCase variants. Colony size did not vary along with varying levels of expression of CMCase variants. Total cellular level of expressed Inp-CMCase variant fusions in the cell was considered to be constant, which was confirmed by SDS-PAGE (data not shown). This might result from the fact that the initiation of transcription and translation of them was started from the completely same genetic elements, e.g., the tac promoter and Shine-Dargarno sequence, and mutated CMCase constitutes only one-third of fusion proteins from C-terminal end. Moreover, colonies did not vary in size directly after recovery from transformation. When the transformants were spread on M9 glucose agar plate, colonies were formed more homogeneously in size (Fig. 3C), indicating that transformation did not affect the colony size. It is notable that most colonies on plates containing mutant libraries were smaller than those containing the parent CMCase, indicating that most mutations occurred were deleterious (Fig. 3B).
Rapidly growing colonies produce larger halos on CMC plates.
The 150 to 200 colonies originally selected for rapid growth on M9-CMC plates were transferred to LB agar plates containing 0.5% CMC (LBC). These colonies were assayed for CMCase activity by checking their ability to form halos detected by staining with Congo red. Congo red reacts with certain polysaccharides, including β-d-glucans and substituted cellulose, to form colored dye-polysaccharide complexes, but it does not bind with glucose or cellooligosaccharides such as cellobiose, cellotriose, or cellotetraose. Thus, after staining with Congo red, a clear yellow zone (halo) forms around colonies that hydrolyze CMC (26). All rapidly growing colonies on the M9-CMC plates produced larger halos on LBC plates than those displaying the parent CMCase, indicating improved activity of surface-displayed CMCases (Fig. 4A). In contrast, Congo red staining assays showed that transformants that were selected randomly from normal LB-Amp plates produced either no halos or halos of similar size to those produced by cells displaying the parent CMCase (Fig. 4B). This observation demonstrates that isolating cells from outgrowing colonies is an essential step in the selection process.
FIG. 4.
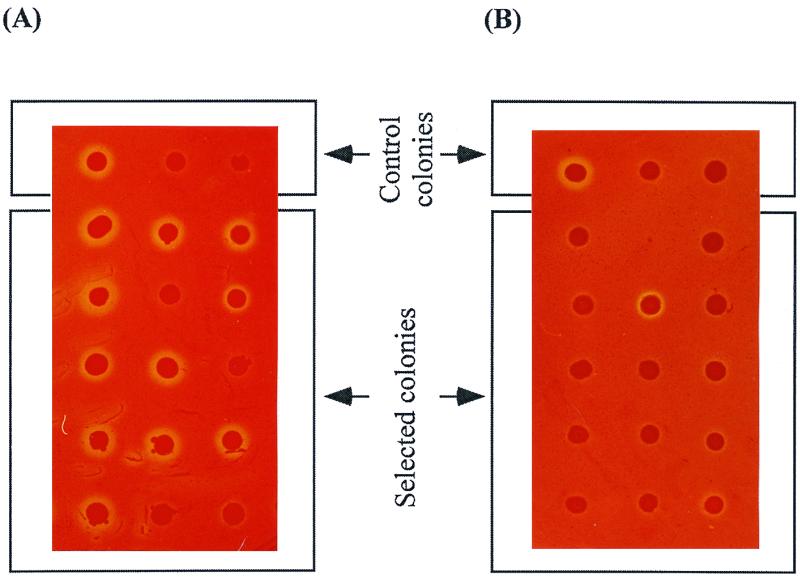
Congo red staining of E. coli JM109 colonies displaying evolved CMCase variants on LB-Amp-IPTG agar plates topped with 0.5% (wt/vol) CMC. (A) Colonies selected from M9-CMC plates showing outgrowth. (B) Colonies randomly chosen from a library of transformants grown on LB-Amp plates. Control colonies are shown in each photograph. The first control colony is JM109(pYSK3), the second is JM109(pEIN229), and the third is JM109(pKK223-3); all other colonies were selected as CMCase variants.
Activities of evolved CMCases.
To further improve enzyme activity, two more rounds of random mutagenesis, display, and selection were performed. The surface enzyme activities of the evolved CMCase variants obtained through three rounds of DNA shuffling were characterized (Fig. 5A). All the clones obtained through these selection and screening steps showed higher hydrolytic activities than clones containing the parent CMCase. Three clones from the first round (1R86, 1R169, and 1R186) showed 1.2- to 1.8-fold increases in activity, while three clones from the second (2R29, 2R33, and 2R59) and the third round (3R38, 3R139, and 3R256) showed 2.2- to 3.8-fold and 3.2- to 5-fold increased activities, respectively. These results demonstrate that colony outgrowth on M9-CMC plates and formation of larger halos on LBC agar plates were indeed due to the increased activities of surface-displayed CMCases.
FIG. 5.

Comparisons of CMCase activities and expression levels of improved variants and the parent CMCase. (A) Enzyme activities of the surface-displayed enzymes. (B) Enzyme activities of their corresponding free-form enzymes. (C) Western blot of the soluble fractions of corresponding enzyme variants in same order.
To determine if increased enzyme activity was affected by fusing CMCase to Inp, evolved CMCase genes were excised from Inp and expressed in the cytoplasm. The activities of each of the unfused CMCases from cell extracts were assayed. Figure 5B shows that specific activities of free-form CMCase variants were much higher than for the parent enzyme. For example, 2R59 from the second round and 3R256 from the third round showed about 1.8- and 2.2-fold higher activities, respectively, compared to the parent CMCase (Fig. 5B). It is probable that the extent of improvement of free-form enzymes is lower than the improvement of Inp-CMCase fusions because the CMCases were selected in the Inp-fused form during the evolution experiments. Nevertheless, the trend toward improved activities of evolved enzymes was well correlated in Inp-CMCase fusion proteins and unfused CMCases (Fig. 5). Additionally, Western blot analysis showed that very similar levels of the evolved and parent CMCases were produced (Fig. 5C), indicating that changes in the activities of the variants were not due to the changes in protein expression levels. In order to determine whether mutations affected solubility of enzyme variants, soluble and insoluble fractionation of total cellular proteins and subsequent Western blotting were performed. The results showed that more than 98% of the expressed enzyme variants were soluble (Fig. 5C). Even though the insoluble form of enzymes were detected only in the case of variants, 1R186 and 3R256, the amount was less than 2% of total expressed enzymes (data not shown).
These results demonstrate that potential structural constraints caused by fusing CMCase to a large Inp molecule did not significantly hamper the display of a functional CMCase. In screening systems involving secreted enzymes, special devices are often required to prevent the diffusion of enzymes to other cells (17). Assays of secreted enzymes require expression-normalization before the evaluation of changes in specific enzyme activities (15). Expression levels did not change when enzyme libraries were displayed by fusion to the anchoring motif, Inp.
Sequence analysis.
Complete DNA sequences for nine evolved CMCase genes were obtained and their mutation sites were identified. Table 1 summarizes the positions of amino acid changes in each gene and the number of silent mutations with respect to the parent CMCase gene. Mutated proteins had between 1 and 8 amino acid substitutions in the 469 amino acids that make up the mature CMCase. Meanwhile, they also contained silent mutations for up to 8 base changes. All the nucleotide substitutions were randomly distributed throughout the CMCase gene. Exceptionally, 3R38 was a truncated form containing only amino acids 31 to 368 of CMCase. It is possible that use of the surface display method may affect the process of directed evolution since selections are made on fusion proteins. This may explain why Inp fusions of CMCase variants showed consistently higher activities than the corresponding free enzymes. Although it is premature to comment on the evolved CMCase variants without structural information, it should be interesting to determine whether directed evolution of enzymes in their fused form generates a characteristic set of variants.
TABLE 1.
Amino acid substitution and the number of silent mutations of selected CMCase variants
| Mutant | Amino acid substitutions | No. of silent mutations |
|---|---|---|
| 1R86 | K45E, K55R, R83W, I191T, N223Y, T256A, Q350R, N381D | 2 |
| 1R169 | F91L, I188V, S335L, T342A, T481S | 8 |
| 1R186 | G123E, I339V, N415D | 2 |
| 2R29 | A222V, S308P, L315M, S335L, A360S, N368Y, A397T | 3 |
| 2R33 | N39I, K60R, N244S, G267D, Q357R, I370V, T382I | 2 |
| 2R59 | D317N, K347N | 0 |
| 3R38 | K109N, truncated form containing amino acids 31 to 368 | 5 |
| 3R139 | K109N, G123E, S335L, Q357R | 2 |
| 3R256 | V90I, T260A, T285I, K347R, Y358H, H424Y, A430P | 7 |
Conclusion.
The surface display of hydrolytic industrial enzyme libraries could provide a high-throughput screening environment according to the ability of the cells to grow on nonutilizable substrates as described here. The bacterial surface display method would be a technology platform for selection of improved enzymes produced by directed evolution, if Inp-enzyme variants, for example, were functionally displayed. In addition, enzymes such as proteases and lipases, which are toxic to the cells when expressed in the cytosol, can be selected for improved characteristics by using these techniques. While most directed evolution experiments have attempted to improve binding of non-natural substrates and/or enzyme stability in non-natural environments (2), we have shown here that overall catalytic efficiency of cellulase toward its natural substrate (cellulose) can be improved under physiological conditions.
ACKNOWLEDGMENTS
This work was supported in part by grant NB0470 from the Ministry of Science and Technology of Korea.
We thank S. H. Koh for his help with the HPLC analysis of CMC hydrolysates, E. S. Choi for critical reading of the manuscript, and S. H. Park for the CMCase gene and for helpful discussion.
REFERENCES
- 1.Asanuma S, Yamagishi A, Tanaka N, Oshima T. Serial increase in the thermal stability of 3-isopropylmalate dehydrogenase from Bacillus subtilis by experimental evolution. Protein Sci. 1998;7:698–705. doi: 10.1002/pro.5560070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold F H, Volkov A A. Directed evolution of biocatalysts. Curr Opin Chem Biol. 1999;3:54–59. doi: 10.1016/s1367-5931(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 3.Cowan D A. Industrial enzymes. In: Moses V, Cape R E, editors. Biotechnology: the science and the business—1991. Chur, Switzerland: Harwood Academic Publishers; 1991. pp. 311–340. [Google Scholar]
- 4.Georgiou G, Stathopoulos C, Daugherty P S, Nayak A R, Iverson B L, Curtiss R., III Display of heterologous proteins on the surface of microorganisms: from the screening of combinatorial libraries to live recombinant vaccines. Nat Biotechnol. 1997;15:29–34. doi: 10.1038/nbt0197-29. [DOI] [PubMed] [Google Scholar]
- 5.Giver L, Arnold F H. Combinatorial protein design by in vitro recombination. Curr Opin Chem Biol. 1998;2:335–338. doi: 10.1016/s1367-5931(98)80006-9. [DOI] [PubMed] [Google Scholar]
- 6.Gussow D, Clackson T. Direct clone characterization from plaques and colonies by the polymerase chain reaction. Nucleic Acids Res. 1989;17:4000. doi: 10.1093/nar/17.10.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han S J, Yoo Y J, Kang H S. Characterization of a bifunctional cellulase and its structural gene: the cel gene of Bacillus sp. D04 has exo- and endoglucanase activity. J Biol Chem. 1995;270:26012–26019. doi: 10.1074/jbc.270.43.26012. [DOI] [PubMed] [Google Scholar]
- 8.Harayama S. Artificial evolution by DNA shuffling. Trends Biotechnol. 1998;16:76–82. doi: 10.1016/s0167-7799(97)01158-x. [DOI] [PubMed] [Google Scholar]
- 9.Inoue H, Jojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 10.Jung H C, Lebeault J M, Pan J G. Surface display of Zymomonas mobilis levansucrase by using the ice-nucleation protein of Pseudomonas syringae. Nat Biotechnol. 1998;16:576–580. doi: 10.1038/nbt0698-576. [DOI] [PubMed] [Google Scholar]
- 11.Jung H C, Park J H, Park S H, Lebeault J M, Pan J G. Expression of carboxymethylcellulase on the surface of Escherichia coli using Pseudomonas syringae ice nucleation protein. Enzyme Microb Technol. 1998;22:348–354. doi: 10.1016/s0141-0229(97)00224-x. [DOI] [PubMed] [Google Scholar]
- 12.Kast P, Hilvert D. 3D structural information as a guide to protein engineering using genetic selection. Curr Opin Struct Biol. 1997;7:470–479. doi: 10.1016/s0959-440x(97)80109-1. [DOI] [PubMed] [Google Scholar]
- 13.Kuchner O, Arnold F H. Directed evolution of enzyme catalysts. Trends Biotechnol. 1997;15:523–530. doi: 10.1016/S0167-7799(97)01138-4. [DOI] [PubMed] [Google Scholar]
- 14.MacBeath G, Kast P, Hilvert D. Redesigning enzyme topology by directed evolution. Science. 1998;279:1958–1961. doi: 10.1126/science.279.5358.1958. [DOI] [PubMed] [Google Scholar]
- 15.Miyota Y, Komada S, Momose H, Taguchi S. Quantitative assay system for specific enzyme activity using antibody: the case of protease, subtilisin BPN′. J Biotechnol. 1998;66:157–163. doi: 10.1016/s0168-1656(98)00148-5. [DOI] [PubMed] [Google Scholar]
- 16.Moore J C, Arnold F H. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- 17.Naki D, Paech C, Ganshaw G, Schellenberger V. Selection of a subtilisin-hyperproducing Bacillus in a highly structured environment. Appl Microbiol Biotechnol. 1998;49:290–294. [Google Scholar]
- 18.Olsen M, Georgiou G, Iverson B. Display of enzyme libraries on E. coli: screening of combinatorial libraries by FACS. San Diego, Calif: IBC's Second International Symposium on Directed Evolution of Industrial Enzymes; 1998. [Google Scholar]
- 19.Park S H, Kim H K, Pack M Y. Characterization and structure of the cellulase gene of Bacillus subtilis BSE616. Agric Biol Chem. 1991;55:441–448. [PubMed] [Google Scholar]
- 20.Park S H, Pack M Y. Cloning and expression of a Bacillus cellulase gene in Escherichia coli. Enzyme Microb Technol. 1986;8:725–728. [Google Scholar]
- 21.Skandalis A, Encell L P, Loeb L A. Creating novel enzymes by applied molecular evolution. Chem Biol. 1997;4:889–898. doi: 10.1016/s1074-5521(97)90297-0. [DOI] [PubMed] [Google Scholar]
- 22.Soumillion P, Jespers L, Bouchet M, Marchand-Brynaert J, Sartiaux P, Fastrez J. Phage display of enzymes and in vitro selection for catalytic activity. Appl Biochem Biotechnol. 1994;47:175–189. doi: 10.1007/BF02787933. [DOI] [PubMed] [Google Scholar]
- 23.Stemmer W P. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 24.Tomme P, Warren R A J, Gilkes N R. Cellulose hydrolysis by bacteria and fungi. Adv Microb Physiol. 1995;37:2–81. doi: 10.1016/s0065-2911(08)60143-5. [DOI] [PubMed] [Google Scholar]
- 25.Wang C I, Yang Q, Craik C S. Phage display of proteases and macromolecular inhibitors. Methods Enzymol. 1996;267:52–68. doi: 10.1016/s0076-6879(96)67005-0. [DOI] [PubMed] [Google Scholar]
- 26.Wood P J, Erfle J D, Teather R M. Use of complex formation between Congo red and polysaccharides in detection and assay of polysaccharide hydrolases. Methods Enzymol. 1988;60:59–74. [Google Scholar]
- 27.Yano T, Oue S, Kagamiyama H. Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc Natl Acad Sci USA. 1998;95:5511–5515. doi: 10.1073/pnas.95.10.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaccolo M, Gherardi E. The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 beta-lactamase. J Mol Biol. 1999;285:775–783. doi: 10.1006/jmbi.1998.2262. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J H, Dawes G, Stemmer W P. Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc Natl Acad Sci USA. 1997;94:4504–4509. doi: 10.1073/pnas.94.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Arnold F H. Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res. 1997;25:1307–1308. doi: 10.1093/nar/25.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]