

Summary
CSNK2B has recently been implicated as a disease gene for neurodevelopmental disability (NDD) and epilepsy. Information about developmental outcomes has been limited by the young age and short follow up for many of the previously reported cases, and further delineation of the spectrum of associated phenotypes is needed. We present 25 new patients with variants in CSNK2B and refine the associated NDD and epilepsy phenotypes. CSNK2B variants were identified by research or clinical exome sequencing, and investigators from different centers were connected via GeneMatcher. Most individuals had developmental delay and generalized epilepsy with onset in the first two years. However, we found a broad spectrum of phenotypic severity, ranging from early normal development with pharmacoresponsive seizures to profound intellectual disability with intractable epilepsy and recurrent refractory status epilepticus. These findings suggest that CSNK2B should be considered in the diagnostic evaluation of patients with a broad range of NDD with treatable or intractable seizures.
Keywords: myoclonic seizures, Myoclonic Status in Nonprogressive Encephalopathy (MSNE), CSNK2A1, generalized epilepsy, casein kinase II (CK2)
INTRODUCTION
Protein kinase CK2 (formerly casein kinase II) is a ubiquitous protein serine/threonine kinase enriched in the brain. CK2 is a heterotetramer consisting of two catalytic subunits (α and/or α’) and two regulatory subunits (β).1, 2 De novo variants in CSNK2A1, encoding the α subunit, cause a syndromic neurodevelopmental disorder, Okur-Chung syndrome.3, 4 Recently, 14 individuals with NDD or epilepsy and de novo variants in CSNK2B, encoding the β subunit, have been reported.5–8 Here we present 25 additional patients and describe a broad spectrum of developmental and epilepsy phenotypes.
MATERIALS AND METHODS
CSNK2B variants were identified by research or clinical exome sequencing. Individuals were included if variants were predicted loss-of-function (LoF) and/or de novo. Investigators from different centers were connected via GeneMatcher.9 Epilepsy types and epilepsy syndromes were classified according to the International League Against Epilepsy 2017 classification.13 See Supplementary Information for additional information.
RESULTS
CSNK2B variants were identified in 25 previously unreported individuals with epilepsy and/or NDD, aged 19 months to 36 years, through an international collaboration. CSNK2B variants included six nonsense, three frameshift, three splice site, two start-loss, 10 missense and one in-frame duplication; grouping the nonsense, frameshift, splice site, and start-loss variants together, 14 predicted LoF variants were identified. Twenty-one variants were confirmed de novo. Four individuals for whom inheritance could not be determined carried a LoF variant: two canonical splice site, one stop-gain, and one frameshift. All variants were absent from gnomAD.10 While CSNK2B is intolerant to LoF variation (pLI = 0.92, o/e = 0.08, CI 0.03-0.38), two high confidence LoF variants affecting the canonical transcript (p.Glu173Ter and p.Pro146ThrfsTer100) were observed in gnomAD. However, each was from a disease cohort and absent from the “control” population.10 All variants identified met ACMG-AMP classification criteria as “pathogenic” or “likely pathogenic”.11
Seven missense variants were unique and one (p.Asp32Asn) occurred in three unrelated individuals (Fig. 1a). All eight missense variants were predicted deleterious by Polyphen-2 and CADD. Compared to “control” missense variants in gnomAD (n=16) and dbSNP (n=34), cohort missense variants were more likely to occur in the region of CK2β responsible for interfacing with the α or α’ subunit (4/8 versus 8/50, p=.049, Fisher exact test).12 The p.Cys137Arg variant disrupts a zinc binding site, the same residue affected in two previously reported individuals (p.Cys137Gly, p.Cys137Phe).7 Thus, in silico analyses suggest that missense variants in this cohort disrupt CK2β functional domains.
Figure 1.
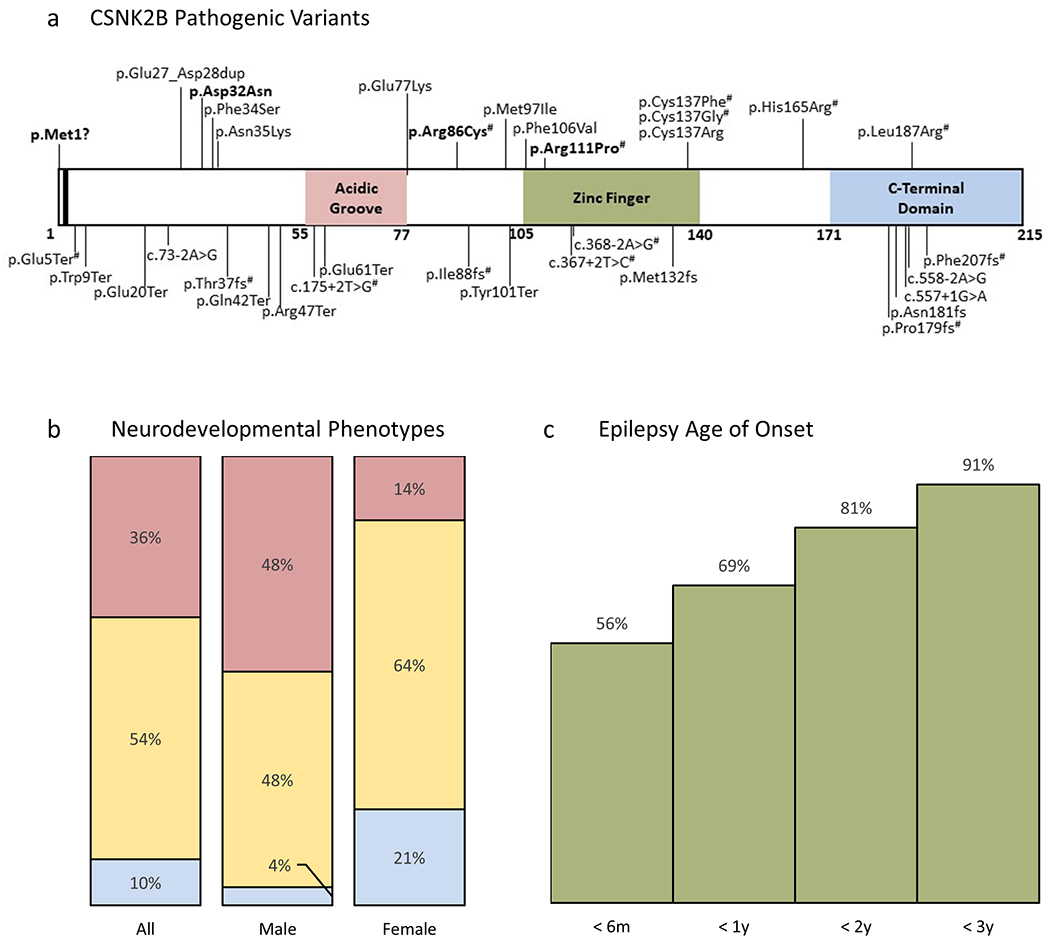
CSNK2B genotypes and phenotypes.
a) Mutational landscape of CSNK2B; boxes = functional domains; bar = autophosphorylation site; all 34 reported variants are shown (bold = recurrent variants; # = previously reported). Note p.Glu5Ter and p.Arg86Cys were reported in the same individual in Li et al.
b) Neurodevelopmental phenotypes across all reported CSNK2B patients (n = 39), males (n = 25), females (n = 14); blue = no delays to date, yellow = mild or not otherwise specified, red = moderate to profound.
c) Age of onset for all reported CSNK2B patients with epilepsy plotted as a cumulative frequency (n = 32).
Phenotypes are summarized by variant type and sex in Table 1 (see Table S1 in Supplementary Information for additional phenotypic details). Of the 25 individuals, 18 were male with females underrepresented (p=.02, binomial test). Thirteen had minor dysmorphic features. Other findings included cardiac comorbidities in four individuals, genitourinary problems in three, and endocrine abnormalities and hearing loss in two each. Most patients (23/25, 92%) presented with initial neurological symptoms prior to 2 years of age, with two typical patterns: seizures during the first year (10 patients) or language delay (in isolation or as part of global developmental delay) during the second year (11 patients).
Table 1.
Summary of CSNK2B Clinical and Epilepsy Characteristics by Variant Type and Sex
| Clinical Characteristics | All n = 25 |
LoF* n = 14 |
Non-LoF n = 11 |
Males n = 18 |
Females n = 7 |
|---|---|---|---|---|---|
| Neurodevelopmental delay or disability | 24 | 14 | 10 | 18 | 6 |
| Speech delay or disability | 23 | 13 | 10 | 18 | 5 |
| Nonverbal | 5 | 4 | 1 | 5 | 0 |
| Motor delay or disability | 22 | 14 | 8 | 16 | 6 |
| Intellectual disability | 18 | 11 | 6 | 13 | 5 |
| Moderate to profound | 9 | 6 | 3 | 9 | 0 |
|
| |||||
| Autistic features or autism | 8 | 5 | 3 | 5 | 3 |
|
| |||||
| Problematic behavior | 13 | 9 | 4 | 8 | 5 |
|
| |||||
| Ataxia or impaired coordination | 9 | 7 | 2 | 7 | 2 |
|
| |||||
| Hypotonia | 13 | 8 | 5 | 12 | 1 |
|
| |||||
| Seizures | 21 | 13 | 8 | 15 | 6 |
|
| |||||
| Epilepsy | 19 | 12 | 6 | 13 | 5 |
|
| |||||
| Age of epilepsy onset† | |||||
| < 6 months | 8 | 5 | 3 | 7 | 1 |
| 6-12 months | 2 | 2 | 0 | 2 | 0 |
| 1-2 years | 3 | 2 | 1 | 1 | 2 |
| 2-3 years | 3 | 2 | 1 | 2 | 1 |
| > 3 years | 2 | 1 | 1 | 1 | 1 |
|
| |||||
| Generalized spike and wave | 14 | 10 | 4 | 10 | 4 |
|
| |||||
| Generalized tonic or tonic-clonic seizures | 11 | 8 | 3 | 9 | 2 |
| Absence seizures‡ | 6 | 2 | 4 | 5 | 1 |
| Myoclonic seizures | 6 | 5 | 0 | 5 | 1 |
| Atonic or myoclonic-atonic seizures | 4 | 4 | 0 | 2 | 2 |
| Focal-onset seizures | 4 | 2 | 2 | 2 | 2 |
|
| |||||
| Multiple seizure types | 10 | 6 | 4 | 9 | 1 |
|
| |||||
| Multiple anti-seizure medications | 13 | 9 | 4 | 10 | 3 |
|
| |||||
| Medically refractory | 8 | 5 | 3 | 7 | 1 |
All deletions, frameshift, nonsense and splice-site were considered predicted loss of function (LoF) for this table;
Age of onset could not be determined in one female LoF case;
Including typical absence, atypical absence and myoclonic-absences.
For 24/25 individuals (96%), there was an abnormal neurodevelopmental phenotype ranging from mild delays and/or learning disability to profound intellectual disability (ID). The only individual with normal development and epilepsy was a 3.5 year-old female with a missense variant (p.Arg86Cys; individual 22). Motor delays or impairment were observed in 22/25 (88%). Almost all (24/25, 96%) learned to walk, though some not until after five years of age. Hypotonia was common (13/25, 52%). Several patients (6/25, 24%) had an ataxic gait, and three others had impaired coordination. Language was almost universally affected (23/25, 92%), ranging from delayed speech to remaining nonverbal in adulthood. Of the 20 patients five years of age or older, five were non- or minimally verbal. A subset (8/25, 32%) had autistic features, and behavioral disturbances were common (13/25, 52%), including tantrums, aggression, and hyperactivity. Intellectual disability (ID) was diagnosed in 18/25 (72%) with differences in severity by sex; half of males (9/18) were reported to have moderate to profound ID while none of the seven female patients had more than mild cognitive impairment (p=.03, Fisher exact test).
Most patients (21/25, 84%) had a history of seizures. Individual 8 had a single seizure and individual 23 had febrile seizures only, leaving 19/25 (76%) to meet criteria for a diagnosis of epilepsy. Epilepsy was observed more often with LoF (13/14, 93%) than with other (6/11, 54%) variant types, but this finding was not significant (p=.056, Fisher exact test). Seizure and epilepsy phenotype details are provided in Table S1. Almost all individuals with epilepsy had onset of seizures in the first three years (16/19, 84%), with highest incidence (8/19, 42%) in the first six months. Generalized seizure types were common: 14/19 (74%) patients had generalized epilepsy, while two patients each had focal epilepsy, combined generalized & focal epilepsy, and unknown epilepsy. While generalized tonic-clonic seizures (GTC) were the most common seizure type reported, initial seizures in infants were often focal seizures or myoclonic seizures. Multiple seizure types were commonly reported in the same patient (e.g., myoclonic seizures and GTC for patient 4), occurring in 10/19 (53%).
Generalized epileptiform discharges, including generalized spike-and-wave and polyspike-and-wave, were reported in 14/19 (74%). Photic stimulation was not reported to induce a true photoparoxysmal response, though the abundance of discharges was reported to increase during photic stimulation for individual 9. Slowing of the EEG background was reported in 9/19 (47%) patients with epilepsy. EEG tracings were available for direct inspection in 6/19 patients with epilepsy (individuals 1, 2, 4, 5, 6, and 24). In each of these individuals, epileptiform abnormalities most often consisted of high-amplitude generalized or lateralized polyspikes that occurred most often during sleep. The incidence of epileptiform abnormalities varied by individual from rare to abundant. Ictal EEG recorded diffuse electrographic seizure onset for patients 2, 4 and 24. Representative tracings are shown in Fig. S1.
Diagnoses included epilepsy with myoclonic-absences (individual 22) and epilepsy with myoclonic-atonic seizures (individuals 1 and 2). The concurrent use of multiple anti-seizure medications (ASMs) was common (13/19, 68%) and many patients (8/19, 42%) were medically refractory. The ASMs most often reported to be helpful were valproic acid (6 cases), lamotrigine (4 cases) and zonisamide (4 cases), though these were commonly used in combination with at least one other ASM (see Supplementary Table S1 for additional details). No ASM was reported to exacerbate seizures. The course of the epilepsy tended to improve over time. However, several patients had increased seizure frequency from 7-12 years of age, including three patients with LoF variants who had particularly severe epilepsy with recurrent episodes of refractory status epilepticus (individuals 4, 9 and 24).
Combining our cohort with previously published individuals, 35/39 (90%) had NDD and 14/39 (36%) had moderate to profound impairment (Fig. 1b). In the combined cohort, females (14/39, 36%) were no longer underrepresented (p=.055, binomial test), but phenotypic differences by sex persisted, where 12/25 (48%) of males were more severely affected, compared with 2/14 (14%) of females (p=.04, Fisher exact test). A total of 32/39 individuals (82%) had epilepsy, 22/32 (69%) with onset in the first six months of life (Fig. 1c).
DISCUSSION
We describe the genotype and clinical phenotype of 25 patients with variants in CSNK2B, the largest cohort to date, bringing the total number of reported individuals to 39. Patients usually presented with seizures and/or developmental delay within the first two years of life and we observed high variability in the severity of the phenotype. Most individuals had delays spanning multiple domains, but were primarily affected in terms of language and cognition. While motor delays were common, almost all patients became ambulatory; in contrast, one-fifth of patients remained non-verbal, and over two-thirds had cognitive impairment that ranged from mild to profound. The majority of patients had epilepsy, most often generalized, which ranged from pharmacoresponsive seizures to medically intractable epilepsy with recurrent episodes of refractory SE.
The spectrum of phenotypic severity was not readily explained by variant type. However, males were more likely than females to have more severe ID, suggesting that sex may be one factor influencing outcome. Indeed, the underrepresentation of females in our cohort may reflect ascertainment bias. The association remained significant when our cohort was pooled with other published individuals, but ultimately larger numbers will be necessary to confirm the observation.
Whereas we identified a neurodevelopmental phenotype in 24/25 individuals, a recent report described normal development in 3/9 individuals.7 This difference is likely accounted for by the young age of their cohort, as all three individuals were two years of age or younger. Since mild ID and learning disabilities may not be evident until later, the rates of mild NDD may be underestimated. Similarly, while we noted that 24/25 individuals learned to walk, 4/9 of their patients were not walking – three of whom were two years or younger. As we noted acquisition of ambulation as late as five years of age, we speculate that some of the younger patients may ultimately learn to walk.
Our delineation of the epilepsy phenotype highlights the early age of onset, most commonly in infancy, and the predominantly generalized seizures. Generalized discharges were reported for the majority of patients with epilepsy, and we noted on direct review of EEG tracings that sleep-potentiated generalized polyspike bursts were a common feature (Fig. S1). The abundance of epileptiform abnormalities and presence of background slowing, like the severity of the epilepsy and NDD, were variable. While in a few cases a specific epilepsy syndrome could be diagnosed, this was the exception. In marked contrast to a prior report,7 we did not find the epilepsy universally easy to control. Most patients with epilepsy required multiple ASMs, with valproic acid the most often reported to be effective. Many had refractory seizures, including three males with LoF variants (patients 4, 9 and 24) and particularly severe and intractable epilepsy with recurrent episodes of SE. All three patients were refractory to numerous combinations of ASMs, and patients 4 and 24 to ketogenic diet and vagal nerve stimulator as well.
Limitations of this study include the heterogeneity of the clinical data. Data were not collected in a standardized prospective manner and we relied heavily on the clinical judgement of treating physicians. Our results should be seen as provisional, and further refinement of phenotypes and clarity on genotype effects and other phenotypic determinants are expected with the report of increasing numbers of patients.
How alteration of CSNK2B results in NDD and epilepsy is not understood. The heterozygous predicted LoF variants in our patients suggest a haploinsufficiency mechanism. As β subunit dimerization is required for CK2 holoenzyme formation,1 reduced expression of CSNK2B may result in fewer complexes. Impaired formation of heterotetramers may also explain the pathogenicity of some missense variants, as 4/8 were in a region that interfaces with the α or α’ subunit. Decreased CK2 holoenzyme activity may represent a unifying disease mechanism.
CK2 is abundant in brain, specifically in neurons and neuroepithelia,14 where it is involved in the generation and growth of neuronal processes.15 Moreover, CK2 is enriched in post-synaptic densities,16 CK2 activity increases during long-term potentiation (LTP),17 and CK2-selective inhibitors block NMDA receptor dependent synaptic transmission and LTP.18 Clarity on the disease mechanism in CSNK2B-related NDD and epilepsy will require additional work in model systems.
CSNK2B is a developmental disease gene associated with a range of severity in NDD and epilepsy phenotypes. Language delays, cognitive impairment, and early-onset generalized epilepsy are the most common features with the severe end of the spectrum including nonverbal patients with profound intellectual disability, intractable epilepsy, and recurrent refractory SE. In addition, a range of non-cerebral malformations may be seen. In our cohort, the severity of neurological impairments was greater than reported to date, serving as a call for further studies into the mechanisms of CSNK2B-related NDD and epilepsy in hopes that more specific approaches to treatment may be sought in the future.
Supplementary Material
Supplementary Figure. Electroencephalography (EEG) in CSNK2B-associated epilepsy.
a) Interictal EEG during wakefulness at age 6 years (individual 1), showing a brief generalized discharge arising from a mildly slow background. b) Status epilepticus at age 12 years (individual 4), characterized electrographically by generalized polyspikes and polyspike and wave at ~2 Hz, and clinically by altered mental status with intermittent periods of rhythmic jerking of the arms and/or legs. Seizure lasted over one hour and 43 minutes and the image is from an epoch approximately 1 hour after onset. c) Interictal EEG during sleep at age 6 years (individual 2), showing frontally predominant generalized polyspikes. d) Interictal EEG during sleep at age 6 years (individual 1), showing abundant multifocal and generalized spikes and polyspikes. e) Ictal EEG from sleep at age 6 years (individual 2), characterized electrographically by an initial generalized polyspike discharge corresponding clinically to a myoclonic jerk, and then by polyspike and wave at 2-3 Hz accompanied by arousal and non-rhythmic limb movements. f) Interictal EEG during sleep at age 9 years (individual 5), showing generalized polyspikes.
Acknowledgements
We are very appreciative of all the families who enrolled in the various studies involved. We thank pSERG (Pediatric Status Epilepticus Research Group) for connecting the Boston Children’s group with the study. This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. Funding for the Undiagnosed Diseases Program-Victoria was provided by philanthropic donation and the Murdoch Children’s Research Institute. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program; sequencing and analysis was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure of Conflicts of Interest
Several authors have industry relationships as follows: Weimin Bi has a relationship with Baylor College of Medicine (BCM); BCM and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories (BMGL), which performs clinical exome sequencing. Lindsay Burrage is employed by the Department of Molecular and Human Genetics at Baylor College of Medicine, which received financial support from Baylor Genetics during the conduct of the study. David B. Goldstein is founder and holds equity with Q State – Pairnomix; is founder and holds equity with Praxis Therapeutics; holds equity with Apostle Inc.; and has received personal fees from AstraZeneca, Gilead Sciences, and GoldFinch Bio. Kristin G. Monaghan is an employee of GeneDx. Nicholas Stong is an employee of, and has equity in, Bristol Myers Squibb. The remaining authors have no conflicts of interest.
Footnotes
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Gietz RD, Graham KC, Litchfield DW. Interactions between the subunits of casein kinase II. J Biol Chem. 1995;270(22):13017–21. [DOI] [PubMed] [Google Scholar]
- 2.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66(11–12):1817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ceglia I, Flajolet M, Rebholz H. Predominance of CK2alpha over CK2alpha’ in the mammalian brain. Mol Cell Biochem. 2011;356(1-2):169–75. [DOI] [PubMed] [Google Scholar]
- 4.Okur V, Cho MT, Henderson L, Retterer K, Schneider M, Sattler S, et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum Genet. 2016;135(7):699–705. [DOI] [PubMed] [Google Scholar]
- 5.Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat. 2017;38(8):932–41. [DOI] [PubMed] [Google Scholar]
- 6.Sakaguchi Y, Uehara T, Suzuki H, Kosaki K, Takenouchi T. Truncating mutation in CSNK2B and myoclonic epilepsy. Hum Mutat. 2017;38(11):1611–2. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Gao K, Cai S, Liu Y, Wang Y, Huang S, et al. Germline de novo variants in CSNK2B in Chinese patients with epilepsy. Sci Rep. 2019;9(1):17909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima M, Tohyama J, Nakagawa E, Watanabe Y, Siew CG, Kwong CS, et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet. 2019;64(4):313–22. [DOI] [PubMed] [Google Scholar]
- 9.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018;25(1):154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mestres P, Boldyreff B, Ebensperger C, Hameister H, Issinger OG. Expression of casein kinase 2 during mouse embryogenesis. Acta Anat (Basel). 1994;149(1):13–20. [DOI] [PubMed] [Google Scholar]
- 15.Ulloa L, Diaz-Nido J, Avila J. Depletion of casein kinase II by antisense oligonucleotide prevents neuritogenesis in neuroblastoma cells. EMBO J. 1993;12(4):1633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soto D, Pancetti F, Marengo JJ, Sandoval M, Sandoval R, Orrego F, et al. Protein kinase CK2 in postsynaptic densities: phosphorylation of PSD-95/SAP90 and NMDA receptor regulation. Biochem Biophys Res Commun. 2004;322(2):542–50. [DOI] [PubMed] [Google Scholar]
- 17.Charriaut-Marlangue C, Otani S, Creuzet C, Ben-Ari Y, Loeb J. Rapid activation of hippocampal casein kinase II during long-term potentiation. Proc Natl Acad Sci U S A. 1991;88(22):10232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura R, Matsuki N. Protein kinase CK2 modulates synaptic plasticity by modification of synaptic NMDA receptors in the hippocampus. J Physiol. 2008;586(13):3195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. Electroencephalography (EEG) in CSNK2B-associated epilepsy.
a) Interictal EEG during wakefulness at age 6 years (individual 1), showing a brief generalized discharge arising from a mildly slow background. b) Status epilepticus at age 12 years (individual 4), characterized electrographically by generalized polyspikes and polyspike and wave at ~2 Hz, and clinically by altered mental status with intermittent periods of rhythmic jerking of the arms and/or legs. Seizure lasted over one hour and 43 minutes and the image is from an epoch approximately 1 hour after onset. c) Interictal EEG during sleep at age 6 years (individual 2), showing frontally predominant generalized polyspikes. d) Interictal EEG during sleep at age 6 years (individual 1), showing abundant multifocal and generalized spikes and polyspikes. e) Ictal EEG from sleep at age 6 years (individual 2), characterized electrographically by an initial generalized polyspike discharge corresponding clinically to a myoclonic jerk, and then by polyspike and wave at 2-3 Hz accompanied by arousal and non-rhythmic limb movements. f) Interictal EEG during sleep at age 9 years (individual 5), showing generalized polyspikes.