

Abstract
The incidence of cutaneous melanoma continues to increase in pale skinned peoples in Europe and elsewhere. Epidemiological studies identified genetically determined phenotypes such as pale skin, freckles and red hair, and sunburn as risk factors for this cancer. The development of many melanocytic naevi is also genetically determined and a strong melanoma risk phenotype. Not surprisingly then, genome wide association studies have identified pigmentation genes as common risk genes, and to a lesser extent, genes associated with melanocytic naevi. More unexpectedly, genes associated with telomere length have also been identified as risk genes. Higher risk susceptibility genes have been identified, particularly >CDKN2A >as the most common cause, and very rarely genes such as >CDK4>, >POT1>, >TERT >and other genes in coding for proteins in the shelterin complex are found to be mutated. Familial melanoma genes are associated with an increased number of melanocytic naevi but not invariably and the atypical naevus phenotype is therefore an imperfect marker of gene carrier status. At a somatic level, the most common driver mutation is >BRAF>, second most common >NRAS>, third >NF1 >and increasing numbers of additional rarer mutations are being identified such as in >TP53>. It is of note that the >BRAF >and >NRAS >mutations are not C>T accepted as characteristic of ultraviolet light induced mutations.
Key words: susceptibility genes, somatic mutations, melanoma
Melanoma continues to increase in incidence in Europe; figures from the period 1995–2012 recently published showed increases in both in situ and invasive melanoma (1). IARC figures generated from data recorded up until 2012 were used to construct Fig. 1. It can be seen that the greater proportional rise in incidence in older men in the UK is mirrored in Australia albeit at a considerably higher incidence rate. Australia, however, appears to have achieved a decrease over time in incidence rates in the very young, probably related to the very active and long-standing public health activities in that country.
Fig. 1.

Incidence rates for melanoma in men in two genetically similar populations in England and in Australia. The figures were generated on line using the Globocan tool (gco.iarc.fr).
The common melanoma subtypes, superficial spreading melanomas (SSM), nodular melanomas (NM) and lentigo maligna melanomas (LMM) are essentially diseases of pale skinned individuals, and this observation along with the identification of reported sunburns as a significant risk factor led to the recognition that melanoma is caused by sun exposure. The comparison between rates in England and in Australia is useful as the sub-population of Australians who develop melanoma commonly claim UK ethnicity and previous genome-wide association studies confirmed inherited similarities (2): that is that this comparison in incidence therefore reflects the strong effect of sun exposure (in genetically similar people) on melanoma development.
SIGNIFICANCE
Melanoma continues to increase in incidence and therefore recognizing individuals at increased risk is especially important. This review discusses the associations between inherited genes which increase risk, and how the presence of those genes is manifest in family history or skin type. Environmental exposures, namely sun exposure leading to sunburn is aetiological in the genetically predisposed.
Fig. 2 shows a principal component analysis (PCA) from a genome-wide association study reported by the GenoMEL consortium (2). PCA analyses of inherited genetic variation effectively examines genome-wide genetic variation across the populations determining the underlying patterns. The first two components explain much of the overall pattern of variation; in this figure, each participant’s genome is represented by a “dot” reflecting on a 2 dimension plot the value of that person’s first two principal components – each of the principal components consists of many thousands of genetic variants across the genome. The dots in brown, orange, sky blue and dark green represent the genotype of blood samples from the UK, the Netherlands, Sydney and Brisbane respectively. The PCA did not consider the location of residence of the person or the laboratory that recruited them but when the two dimension graph is overlaid on the map of Europe, it is apparent that people recruited from the same location are together on the map and that the pattern of the geographical locations is also retained with the exception of the Australian populations which are superimposed on the map of Western Europe reflecting their ancestry. The map confirms that gene frequencies vary slowly and systematically across Europe reflecting the fact that local migration is the biggest determinant of change. For instance, one of the genes contributing to this pattern is the variation in the lactase gene reflecting the pattern of lactose intolerance across Europe. Thus the melanoma incidence curves in Fig. 1 reflect UK and Australian melanoma patients and this PCA suggests that these are more similar than populations sampled elsewhere in Europe.
Fig. 2.
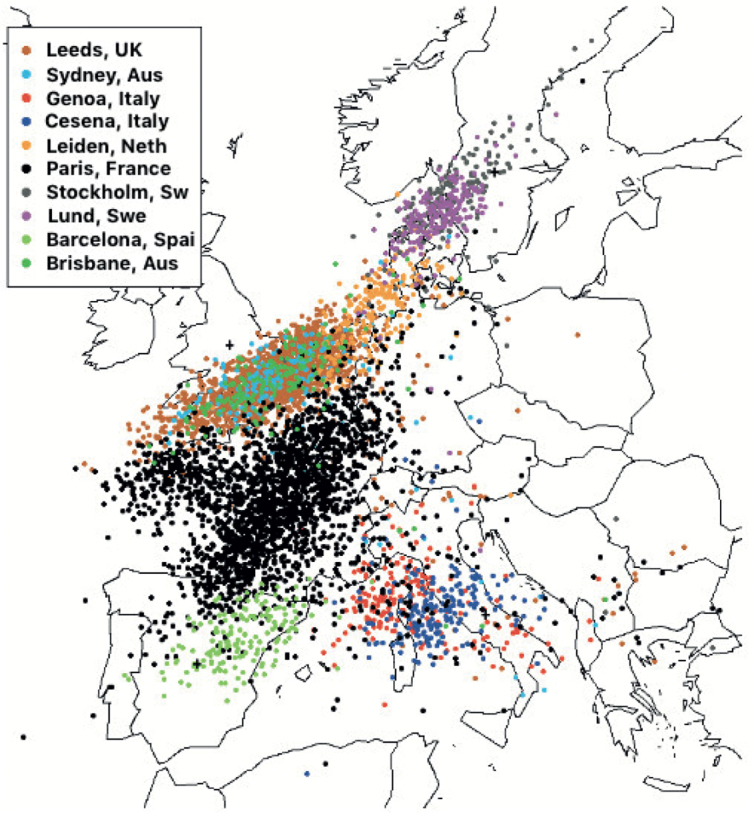
Principal components analysis (PCA) from a genome-wide association study reported by the GenoMEL consortium (2). The coloured dots represent a measure of the genetic inheritance of participants in a genetic study of melanoma. The superimposed blue, green and terracotta dots over the UK suggests that the participants from two sites in Australia (Sydney and Brisbane) were very similar genetically to those living in the UK. This was expected as many Australian melanoma patients report ethnicity as the UK. Comparing incidence in melanoma then between the UK and Australia is to some degree comparing incidence in two populations similar genetically but with very different sun exposure histories. Figure kindly prepared by Dr Mark Iles of the University of Leeds.
INHERITED (GERMLINE) GENOMIC VARIATION AND MELANOMA RISK
Skin colour genes
Although the markedly different incidence rates for genetically similar populations in the UK and Australia reflects the effects of very different patterns of sun exposure, cutaneous melanoma is a strongly genetically determined disease. Melanoma incidence is very strongly related to skin colour being predominantly a cancer of pale skinned individuals. Table I indicates that the most common melanoma subtypes, SSM, NM and the less common LMM and desmoplastic melanoma, are very much more common in fair skin, whereas the acral lentiginous melanoma (ALM) variety has approximately the same incidence in most ethnic groups. Table I reports incidence for different melanoma subtypes, SSM, NM, LMM and ALM. The ethnicity terms used are those used in North America: Non-Hispanic white (NHW), Hispanic white (HW), Asian and Black. The data show that the incidence of SSM, NM and LMM is highest in those with ethnicity associated with the palest skins, indeed there is some evidence for a gradation in incidence from typically palest to darkest skins. The data also show that the incidence of ALM does not differ with ethnicity and therefore inherited pigment genes.
Table I.
Incidence rates reported by the North American SEER registry by ethnicity. Modified from Wang et al. (3)
| Non-Hispanic white | Hispanic white | Asian | Black | Total | |
|---|---|---|---|---|---|
| Superficial spreading melanoma | 9.05 (8.96–9.13) | 1.12 (1.05–1.19) | 0.31 (0.27–0.35) | 0.15 (0.12–0.18) | 6.18 (6.13–6.24) |
| Nodular melanoma | 1.80 (1.76–1.84) | 0.49 (0.44–0.54) | 0.14 (0.12–0.17) | 0.06 (0.04–0.08) | 1.30 (1.28–1.33) |
| Lentigo maligna melanoma | 1.87 (1.83–1.90) | 0.23 (0.19–0.27) | 0.06 (0.05–0.08) | 0.02 (0.01–0.04) | 1.37 (1.35–1.40) |
| Acral lentiginous melanoma | 0.21 (0.20–0.22) | 0.24 (0.21–0.28) | 0.17 (0.14–0.20) | 0.19 (0.16–0.23) | 0.20 (0.19–0.22) |
Age-adjusted incidence rates per 100,000 person-years.
Melanocytic naevi genes
The second risk phenotype is the presence of greater numbers of melanocytic naevi (4), both of the “common” or banal type and the presence of larger naevi described clinically as atypical naevi and histologically as dysplastic naevi. Twin studies have reported evidence for high hereditability for this phenotype in the order of around 65% (5, 6). Thus the two phenotypes most predictive of melanoma risk (pale skin and the presence of many naevi) are shown to be predominantly genetically determined.
New low-medium penetrance loci
Genome wide association studies have increased steadily in power to identify larger numbers of inherited genetic variation associated with increased risk of the common subtypes of melanoma (7, 8) and indeed with the risk phenotypes as a result of collaboration between multiple research groups. The role of inherited pigmentation genes in melanoma susceptibility is clear but there are also a number of genetic loci associated with increased numbers of melanocytic naevi and with telomere length. Telomeres are nucleotide repeat sequences which protect the ends of chromosomes, from excessive shortening and becoming tangled during cell division. Genes such as that coding for telomerase and additional genes coding for proteins in the so-called shelterin complex play an important role in maintaining telomeres. A number of inherited genetic variants are reported to determine telomere length and a genetic score predicting longer telomeres has been shown to strongly predict melanoma risk (9). In short, common genes associated with paler skin and in particular skin which tends to burn in the sun (predominantly the gene coding for the melanocortin receptor 1, MC1R); others which are associated with having more naevi; and genes associated with longer telomeres are melanoma risk genes, and to a large degree explain variation in melanoma incidence in different populations worldwide. Further genes associated with risk will certainly be found and other pathways may therefore be identified: a recent genome wide association study of naevi reported some evidence of pathways not previously supposed to be associated with naevus pathogenesis (8).
The low to medium penetrance (risk) genes identified in genome wide association studies each increase risk a little and melanoma occurs essentially in individuals who have inherited several risk alleles and who like the sun, in particular intermittent sun exposure. The likelihood is that risk of melanoma increases progressively with higher numbers of the risk alleles.
RARE INHERITED MUTATIONS
Rarer inherited mutations are associated with a high risk of melanoma (high penetrance) so that clustering of cases occurs in families. The definition usually used to define a melanoma family is at least 3 cases in close relatives. The commonest high penetrance susceptibility gene is CDKN2A which notably codes for two quite distinct proteins: p16INK4a and p14ARF. P16INK4a is a cyclin-dependent kinase inhibitor in the RB1 cell cycle control pathway, and p14ARF binds the p53-stabilizing protein MDM2 in the p53 signalling pathway. The CDKN2A gene is therefore involved in the regulation of two critical cell cycle regulatory pathways. A very small number of melanoma families have causal mutations in the gene which codes for CDK4 to which p16 binds and these families appear to have a very similar phenotype to those with CDKN2A mutations (10).
Mutation carriers are more likely to have multiple primaries than those without such mutations (11), a little earlier age of onset and pancreatic cancer occurs in some CDKN2A families reported from mainland Europe and the USA. Studies in specific founder CDKN2A mutation families from Sweden (12) and the Netherlands have reported increased rates of cancers associated with smoking (13) but the risks of cancers other than melanoma and pancreatic cancer are not yet sufficiently well established to infer screening requirements, see https://www.ncbi.nlm.nih.gov/books/NBK7030/. That risks remain unclear to some extent reflects bias of ascertainment: in order to identify new high risk inherited cancer genes, researchers typically tested families who had multiple cases of the same cancer. Work is ongoing currently within GenoMEL (www.genomel.org) to address this deficiency.
Familial melanoma has been recognised for many years and between 1994 (14) and 2013, only CDKN2A and CDK4 mutations were recognised as familial melanoma genes. These mutations were identified not least because the majority of affected families are at increased risk of only melanoma, sometimes also with some pancreatic cancer and families were ascertained for investigation on the basis essentially of multiple melanoma cases. There was, in essence, a deliberate bias, in that families with multiple cases of melanoma were selected for invitation to participate in research. This was the usual method for the identification of highly penetrant genes using genetic linkage studies where co-segregation of genetic variants with the cancer was required. Now that whole exomic or genomic sequencing and “panels” of cancer genes are used to identify high risk genes in families, genes are being identified with association with melanoma and an increased number of various other cancer types. As a result, rarer mutations in additional melanoma susceptibility genes have been identified. These have been seen in less than 2% of UK families with 3 or more melanoma cases. They are predominantly genes which are involved in telomere function/maintenance, first the gene named Protection of Telomeres I (POT1) which was described simultaneously in two groups in melanoma families (15, 16). Additional mutations were described in other genes in the shelterin telomere protection complex of which POT1 is a subunit (17), and in TERT (18, 19). Telomere function is therefore clearly important in melanoma pathogenesis. Finally inherited mutations in the BAP1 gene, which were originally reported as an inherited cause of uveal melanoma but were quickly then associated additionally with a risk of lung cancer and meningiona (20) are now recognised also to increase the risk of cutaneous melanoma (21). Subsequently the mutations were recognised as also associated with renal cancer and mesotheliomas. Unusual but generally benign “spitzoid” melanocytic lesions of the skin were reported to be part of the syndrome in 2011 (21).
The role of gene testing and screening is therefore in the process of change. As the penetrance of these genes which increase the risk of melanoma and other cancers, becomes clearer then appropriate screening should be possible and gene testing/counselling likely to be increasingly performed.
Families with inherited melanoma susceptibility to melanoma often also have more melanocytic naevi than is usual in that population. This phenotype, called the atypical mole syndrome or the dysplastic naevus syndrome was originally thought to be a key component of the Familial Melanoma “Syndrome” (22). Indeed, there is certainly an association: mutations are more likely to have larger numbers of naevi (23). However, it is recognised now that some families with the same mutation may or may not have many naevi, so that family members with normal naevi may yet be found to carry the susceptibility gene. It has been postulated that the rather variable association between inherited high risk melanoma genes and naevi may be complicated by the variable co-inheritance of common lower risk melanoma susceptibility genes (23). In the dermatology or melanoma clinic, then the factors which should alert the medical team to the possibility of inherited high-risk melanoma susceptibility are, the atypical naevus syndrome, multiple primaries, relatively early onset and the co-occurrence of pancreatic cancer in some populations at least. The single most important factor, however, is family history of cancer. So, only 2% of apparently sporadic melanomas even with 2 primaries have inherited CDKN2A mutations (24), but in our own studies > 50% of families with 4 or more melanoma cases have such mutations. In the dermatology or melanoma clinic then, the presence of many naevi or more than one primary should alert the team to the possibility of a higher risk but family history is the strongest evidence for highly penetrant melanoma susceptibility genes. A review published by Sancy Leachman and GenoMEL (25) made recommendations for genetic counselling, but the identification of genes such as POT1 and TERT which increase the risk of cancers other than melanoma means that these recommendations will be revised as more data become available.
Melanoma is an uncommon second malignancy in inherited retinoblastoma (26) and there are reports of a possible small increase of risk in carriers of BRCA2 mutations (27) and possibly Lynch syndrome susceptibility genes although the evidence for the latter is not at this time convincing.
SOMATIC MELANOCYTIC NAEVUS GENOMICS
Melanocytic naevi are both markers of melanoma risk and precursors of melanoma. They are benign proliferations which arise progressively starting in the first year of life, but which stop appearing at the age of 40 years or so. The proliferation of melanocytes sufficient to produce detectable naevi results from the development of mutations in genes predominantly in the RAS/RAF/MEK/ERK pathway. The most common mutation is BRAFV600E but NRAS, and less commonly KRAS mutations occur. The prevalence of such mutations differs between naevi of different types, recently reviewed by Roh et al. (28). Roh et al. estimated that BRAF mutations drive 78% of common acquired naevi, 60% of dysplastic naevi, 7% of blue and 6% of Spitz naevi. Similarly, they estimated that NRAS mutations drive 95% of giant pigmented congenital naevi, 70% of small/medium naevi and 2% blue and Spitz naevi. GNAQ mutations occur in 84% of blue naevi.
Neither BRAF nor NRAS mutations have the classical genetic signature of mutagenesis as a result of ultraviolet (UV) light exposure: C>T mutations (29), but as described above, there is clear epidemiological evidence of a relationship between naevus number and sun exposure. The precise pathogenesis of such mutations remains as yet unclear but these observations suggest a complex relationship between intermittent sun exposure and naevogenesis. It has been queried whether BRAF mutations might actually result from DNA damage consequent upon exposure to UVA (30).
Whatever the route, the activation of the RAS/RAF/MEK/ERK pathway appears to drive the proliferation of naevi but the mutations eg in BRAF also induce senescence and therefore in the majority of naevus proliferation eventually ceases, resulting in growth cessation and ultimately clinical involution. Where this senescence is overcome as a result of additional mutations, then dysplastic naevi may develop and evolve into superficial spreading melanomas. As reported by Shain et al. (31), as melanoma evolves from benign naevi through to invasive tumours, then the proportion of lesions with loss of the CDKN2A gene, increased expression of TERT, increased numbers of additional mutations and copy number changes steadily increases resulting in more aggressive tumours. An on-line data source https://www.mycancergenome.org/content/disease/melanoma/ estimates the frequency of the driver mutations in melanoma as BRAF in 37–50%, CTNNB1 (2–4%), GNA11 (1%), GNAQ (1%), KIT (2–8%), MEK1 (6–7%), NF1 (12%) and NRAS (13–25%). The proportion of each in different melanoma subtypes differs, so the same data source reported that in melanomas arising on for example the trunk 50% have BRAF, 20% RAS compared with melanomas arising in skin with sun damage, RAC1, SNX31, TACC1, STK19 and ARID2), 3 of which: RAC1, PPP6C and STK19 were recurrent. Hayward et al. (33) reported in addition significant mutation of TP 53 in cutaneous melanoma and that the significant mutations were BRAF, NRAS and NF1 in acral melanoma and SF 3B1 in mucosal melanoma.
Large mutation burden in melanomas
Although, the classic driver mutations of naevi do not have C>T mutations, melanomas were shown by the Sanger Institute to have the greatest number of mutations of any cancer and that these mutations were predominantly C>T (29). Mutations are not surprisingly more frequent in tumours which arose on chronically sun exposed skin and the probability is that these mutations are predominantly passenger mutations: that is that they don’t play a key role in tumour progression. However, overall mutation rates were reported to be highest in lung cancer and melanoma (29), both of which have good responses to checkpoint blockade and the supposition is that this is at least in part attributable to mutation derived neoantigens capable of stimulation immune responses to the melanoma cells.
Copy number changes
Copy number changes have been elucidated to some extent. Hodis et al. (32) described a low prevalence of amplifications in melanoma overall: 11% CCND1, 6% KIT, 3% CDK4, 13% TERT, and 4% MITF. The deletions were dominated by those in CDKN2A (38%) and PTEN (25%). Overall the data support the view that copy number changes are more common in acral lentiginous melanomas than in those in sun-exposed sites. In Table II we have summarised some of the recent reports of copy number variation in acral lentiginous melanomas, and by comparison with the proportions reported by Hodis et al. (32) it can be seen that with this albeit limited data, copy number changes are more frequent in acral lentiginous melanoma than in melanomas arising in sun-exposed sites.
Table II.
The recently reported data looking at copy number changes in acral lentiginous melanoma
| Copy number alteration | Reference | n (%) |
|---|---|---|
| Amplification AURKA | Yan et al. 2018 (34) | 472 (25) |
| Amplification GAB2/PAK1 | Chernoff et al. 2009 (35) Yeh et al. 2019 (36) |
122 (22) |
| Amplification CCND1 | Sauter et al. 2002 (37) Yeh et al. 2019 (36) |
122 (21) |
| Amplification CDK4 | Curtin et al. 2005 (38) Yeh et al. 2019 (36) |
122 (22) |
| Deletion NF1 | Liang et al. 2017 (39) | 34 (12) |
| Yeh et al. 2019 (36) | 122 (15) | |
| Inactivation NF1 cooperating factor SPRED1 | Yeh et al. 2019 (36) | 122 (7) |
| Deletion CDKN2A | Liang et al. 2017 (39) | 34 (35) |
| Amplification or point mutation TERT promoter | Liang et al. 2017 (39) | 34 (35) |
In conlusion, cutaneous melanoma is a good example of gene environment interaction, in that it is largely (but not exclusively) a cancer of genetically determined pale skinned peoples, when they experience sun burn or sun damage. The identification of genes associated with risk from low to high risk has led to the identification of biological processes involved in tumourigenesis. The genetic changes occuring in the tumours adds more to what is known about tumourigenesis but also has lead to the evolution of treatment options for advanced disease.
REFERENCES
- 1.Sacchetto L, Zanetti R, Comber H, Bouchardy C, Brewster DH, Broganelli P, et al. Trends in incidence of thick, thin and in situ melanoma in Europe. Eur J Cancer 2018; 92: 108–118. [DOI] [PubMed] [Google Scholar]
- 2.Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet 2009; 41: 920–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Zhao Y, Ma S. Racial differences in six major subtypes of melanoma: descriptive epidemiology. BMC Cancer 2016; 16: 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang YM, Newton-Bishop JA, Bishop DT, Armstrong BK, Bataille V, Bergman W, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer 2009; 124: 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Easton D, Cox G, Macdonald A, Ponder B. Genetic susceptibility to naevi – a twin study. Br J Cancer 1991; 64: 1164–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wachsmuth RC, Gaut RM, Barrett JH, Saunders CL, Randerson-Moor JA, Eldridge A, et al. Heritability and geneenvironment interactions for melanocytic nevus density examined in a U.K. adolescent twin study. J Invest Dermatol 2001; 117: 348–352. [DOI] [PubMed] [Google Scholar]
- 7.Macgregor S, Montgomery GW, Liu JZ, Zhao ZZ, Henders AK, Stark M, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet 2011; 943: 1114–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duffy DL, Zhu G, Li X, Sanna M, Iles MM, Jacobs LC, et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat Commun 2018; 9: 4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iles MM, Bishop DT, Taylor JC, Hayward NK, Brossard M, Cust AE, et al. The effect on melanoma risk of genes previously associated with telomere length. J Natl Cancer Inst 2014. Sep 17106. pii: dju267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puntervoll HE, Yang XR, Vetti HH, Bachmann IM, Avril MF, Benfodda M, et al. Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet 2013; 50: 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet 2007; 44: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helgadottir H, Hoiom V, Jonsson G, Tuominen R, Ingvar C, Borg A, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet 2014; 51: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Snoo FA, Bishop DT, Bergman W, van Leeuwen I, van der Drift C, van Nieuwpoort FA, et al. . Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res 2008; 14: 7151–7157. [DOI] [PubMed] [Google Scholar]
- 14.Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 1994; 8: 22–26. [DOI] [PubMed] [Google Scholar]
- 15.Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 2014; 46: 478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong K, Robles-Espinoza CD, Rodriguez D, Rudat SS, Puig S, Potrony M, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol 2019; 155: 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aoude LG, Pritchard AL, Robles-Espinoza CD, Wadt K, Harland M, Choi J, et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst 2014; 107. pii: dju408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013; 339: 959–961. [DOI] [PubMed] [Google Scholar]
- 19.Harland M, Petljak M, Robles-Espinoza CD, Ding Z, Gruis NA, van Doorn R, et al. Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer 2016; 15: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 2011; 48: 856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011; 43: 1018–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bale SJ, Chakravarti A, Greene MH. Cutaneous malignant melanoma and familial dysplastic nevi: evidence for autosomal dominance and pleiotropy. Am J Hum Genet 1986; 38: 188–196. [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor NJ, Mitra N, Goldstein AM, Tucker MA, Avril MF, Azizi E, et al. Germline Variation at CDKN2A and Associations with Nevus Phenotypes among Members of Melanoma Families. J Invest Dermatol 2017; 137: 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harland M, Cust AE, Badenas C, Chang YM, Holland EA, Aguilera P, et al. Prevalence and predictors of germline CDKN2A mutations for melanoma cases from Australia, Spain and the United Kingdom. Hered Cancer Clin Pract 2014; 12: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leachman SA, Carucci J, Kohlmann W, Banks KC, Asgari MM, Bergman W, et al. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol 2009; 61: 677e1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traboulsi EI, Zimmerman LE, Manz HJ. Cutaneous malignant melanoma in survivors of heritable retinoblastoma. Arch Ophthalmology 1988; 106: 1059–1061. [DOI] [PubMed] [Google Scholar]
- 27.Breast Cancer Linkage C . Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 1999; 91: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 28.Roh MR, Eliades P, Gupta S, Tsao H. Genetics of melanocytic nevi. Pigment Cell Melanoma Res 2015; 28: 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas NE, Berwick M, Cordeiro-Stone M. Could BRAF mutations in melanocytic lesions arise from DNA damage induced by ultraviolet radiation? J Invest Dermatol 2006; 126: 1693–1696. [DOI] [PubMed] [Google Scholar]
- 31.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med 2015; 373: 1926–1936. [DOI] [PubMed] [Google Scholar]
- 32.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell 2012; 150: 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017; 545: 175–180. [DOI] [PubMed] [Google Scholar]
- 34.Yan J, Yu J, Wu X, Xu T, Yu H, Dai J, et al. Increased AURKA gene copy number correlates with poor prognosis and predicts the efficacy of high-dose interferon therapy in acral melanoma. J Cancer 2018; 9: 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chernoff KA, Bordone L, Horst B, Simon K, Twadell W, Lee K, et al. GAB2 amplifications refine molecular classification of melanoma. Clin Cancer Res 2009; 15: 4288–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh I, Jorgenson E, Shen L, Xu M, North JP, Shain AH, et al. Targeted genomic profiling of acral melanoma. J Natl Cancer Inst 2019; 111: 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauter ER, Yeo UC, von Stemm A, Zhu W, Litwin S, Tichansky DS, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res 2002; 62: 3200–3206. [PubMed] [Google Scholar]
- 38.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353: 2135–2147. [DOI] [PubMed] [Google Scholar]
- 39.Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res 2017; 27: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]