

Abstract
A novel organomediated cleavage of benzoyl group using ethane-1,2-diamine and acetic acid under neutral condition enables an efficient synthesis of 1-(6-nitropyridin-2-yl)thiourea, which previously has been challenging to prepare by conventional methods. The successful synthesis of 1-(6-nitropyridin-2-yl)thiourea as a synthon permits development of a variety of 18F labeled heterocycles as PET imaging ligands such as N-(pyridin-2-yl)thiazol-2-amine derivatives. The utility of this synthon is demonstrated with the synthesis of a 18F-labeled PET tracer for studying prion disease. In vitro autoradiography using this PET tracer on sagittal rat brain slices showed highest accumulation of radioactivity in the hippocampus, cortex, and striatum, in accordance with reported immunostaining of PrPc in rat brain.
Keywords: Cleavage reaction, Heterocycles, Organomediated reaction, ortho-Fluoropyridines, Positron emission tomography
1. Introduction
1-(Pyridin-2-yl)thioureas are versatile building blocks for synthesizing N-(pyridin-2-yl)thiazol-2-amine derivatives, which are used to construct a variety of heterocycles and bioactive compounds. These heterocycle compounds include anti-prion compound 1 [1–3], metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulator (PAM) 2 [4], anti-diabetic glucokinase activator 3 [5,6], anti-tubercular agent 4 [7], nerve growth factor TrKA inhibitor 5 [8], and antileishmanial agent 6 et al. (Fig. 1) [9].
Fig. 1.

Examples of bioactive N-(pyridine-2-yl)thiazol-2-amine derivatives.
Positron emission tomography (PET) is a technology currently available for in vivo assessment and quantification of specific biological and pharmacological processes in man and animal [10]. The application of PET relies on development of radiolabeled tracers possessing selectivity for targets of interest. Fluorine-18 (18F) has been predominately used for PET tracers in both clinic and research due to its favorable physical and nuclear characteristics, as well as its relatively long half-life compared to other positron emitting radioelements such as 11C, 13N, and 15O, allowing transportation of 18F-labeled compounds to off-site facilities.
Furthermore, fluorine is considered a classical bioisostere for replacement of univalent atoms and groups such as H, –CH3, and as a substitute for lone pairs of electrons in medicinal chemistry. Since fluorine and hydrogen share similar van der Waal’s radii, steric perturbations are of minimal concern, thus F-for-H substitutions are often well tolerated and useful in development of small molecule drugs and PET tracers [11]. Moreover, fluorine atom substitution increases metabolic stability [12]. An 18F may be introduced to the ortho position of the pyridine in structures 1–6 (Fig. 1) to generate PET tracers for studying these targets.
Pyridine derivatives are one of the most commonly studied heterocycles for incorporation of [18F]fluoride by SNAr reaction and thus the resultant ortho-[18F]fluoropyridines have emerged as a widely used functionality in PET tracers [13–15]. Pyridine has lower LUMO energy at ortho and para position than benzene, which allows the direct 18F-substitution by using either Br, I, Cl, NO2 or N+Me3 as a leaving group at ortho and para positions. It is reported that when 2-nitropyridine is subjected to [18F]KF/K222 in DMSO at 120 °C for 10 min 2-[18F]-fluoropyridine is obtained in 76% RCY, while using the trimethylammonium triflate precursor gives an 81% RCY at 120 °C for 5 min [16]. The 18F-substitution using Br, I, or Cl as leaving groups at ortho and para positions of pyridine requires elevated temperatures (150–180 °C). Although trimethylammonium triflate is a superior leaving group compared to the nitro group, there are some issues to limit its application in more complicated heterocycles and biomolecules: 1) The corresponding starting material is not commercially available which requires multistep syntheses; 2) It displays divergent reactivities under certain reaction conditions such as C-, O- & heteroatom arylation; O- & C-H methylation and a range of metal-catalyzed cross-coupling methodologies [17], which is preferably to introduce the trimethylammonium triflate at the late stage of the synthesis to avoid these conditions; 3) The orthoN+Me3−OTf pyridine analogues are prepared from the ortho-NMe2 pyridine analogues by using methyl triflate [18]. Since methyl triflate is a powerful methylating agent, it may react with many functional groups including very poor nucleophiles such as aldehydes, amides, and nitriles and some N-heterocycles. On the other hand, methyl triflate is also extremely hazardous. Therefore, due to the difficulty in preparation of the trimethylammonium triflate analogues we selected NO2 as the leaving group at ortho-pyridine position for the 18F-substitution in current approach. Because the 18F-substitution is preferably carried out at the last stage in a synthetic pathway, ortho-nitropyridine derivatives may offer better stability to different chemical reactions and transformations.
The ortho-nitropyridine derivative, 1-(6-nitropyridin-2-yl)thiourea 8, potentially can be a useful synthon for synthesizing precursor 9, thereby promoting development of 18F labeled heterocycles [18F]10 as PET tracers for numerous targets and applications (Scheme 1). To the best of our knowledge, there is no reported effective synthesis of 8. We disclose here an organomediated cleavage of benzoyl (Bz) group using ethane-1,2-diamine and acetic acid under neutral conditions enabling an efficient synthesis of 8a, which could be used to synthesize 18F-labeled heterocycles for PET. As an example of this application, 8a was used to prepare the PET radioligand [18F]10a for studying prion disease [1,3].
Scheme 1.

1-(6-Nitropyridin-2-yl)thioureas 8 could serve as a useful synthon for the development of 18F labeled heterocycles [18F]10.
2. Results and discussion
Initially, syntheses of the pyridyl thiourea derivatives 8a and 12 were attempted using the reactions of pyridin-2-amines 7a and 11 with a thiocyanate that has often been applied to the synthesis of arylthioureas (Scheme 2) [19–24]. However, neither method A nor B gave the desired product 8a or 12. When the reactions were carried out by using method A, no products 8a or 12 were detected and both starting materials 7a and 11 remained in the reaction mixture (ESI, Table S1, entries 1 and 2). When acidic method B was used, no reaction occurred with 11 and NH4SCN (ESI, Table S1, entry 3). While more than 50% of 7a was consumed by using method B, no desired product 8a was detected and several unidentified by-products were observed in this reaction. Makam et al. also disclosed that the synthesis of pyridyl thioureas was not successful by using thiocyanate under con. HCl condition [25]. The failure for synthesizing thioureas from ortho-NO2 or ortho-F aminopyridine 7a and 11 might be due to the decreased nucleophilicity of the amino group on the electron deficient pyridine ring and the reactive nature of the ortho-nitro group.
Scheme 2.

Reactions of 7a and 11 with thiocyanates did not result in pyridyl thiourea 8a or 12.
An alternative 2-step reaction for the synthesis of pyridyl thioureas consists of formation of the N-(arylcarbamothioyl)benzamide derivative 15 via the reaction of pyridylamine 13 with benzoyl isothiocyanate 14, followed by cleavage of Bz group to give the pyridyl thiourea 16 (Scheme 3) [1,3,6,26]. We studied the synthesis of the ortho-NO2 and ortho-F pyridyl thiourea by using the 2-step reaction.
Scheme 3.

The 2-step reaction for the synthesis of the pyridyl thioureas.
The reaction of 4-chloroaniline 17 with benzoyl isothiocyanate 14 was accomplished within 5 min, giving the corresponding thiourea 18 in 99% yield (Table 1, entry 1). The reaction of 6-fluoropyridin-2-amine 11 with 14 was completed in 30 min to offer 19 in 98% yield (Table 1, entry 2), which indicates the lower reactivity of 11 compared to aniline 17. The reaction of 7a with 14 was much slower, and unreacted 7a could still be detected after 4 h under reflux in THF, (Table 1, entry 3). By extending the reaction time to 8 h, compound 20 was obtained in 97% yield. This further suggests that the strong electron-withdrawing nitro group significantly reduces the nucleophilicity of the amino group. Alternatively, compound 20 could be obtained in 95% yield within 10 min by refluxing 7a with 14 in toluene (Table 1, entry 5). Compound 20 precipitated out from the reaction mixture as light brown needle crystals when cooling down to room temperature (rt).
Table 1.
Synthesis of N-(arylcarbamothioyl)benzamide 18–20.
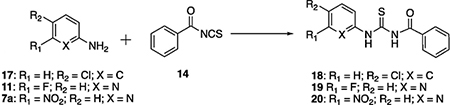
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Solvent | Compound | Time (min) | Temperature | Product (Yield) |
|
| |||||
| 1 | THF | 17 | <5 | reflux | 18 (99%) |
| 2 | THF | 11 | 30 | reflux | 19 (98%) |
| 3 | THF | 7a | 240 | reflux | 20 (90%) |
| 4 | THF | 7a | 480 | reflux | 20 (97%) |
| 5 | Toluene | 7a | 10 | reflux | 20 (95%) |
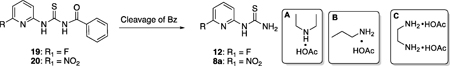
With intermediates 19 and 20, the cleavage of the Bz group was subsequently carried out under conventional basic conditions [3,8,25]. When 19 was treated in EtOH containing 5% aqueous sodium hydroxide solution (1.0 M), >95% of compound 12 (Table 2, entry 1) was obtained in 20 min. However, when 20 was treated in a similar condition, no desired product 8a was detected (Table 2, entry 2), and a large amount of red solid precipitated out of the solution in a few seconds. The collected solid showed very low solubility in DMSO and was later identified as oligomerization products by LC-MS analysis (ESI, Figure S1). Furthermore, when the reaction of 20 was carried out by using a weaker base such as K2CO3 or the organic base TEA (Table 2, entries 4 and 5), it also resulted in complicated reaction mixtures with red precipitate.
Table 2.
Cleavage of Bz group from N-(pyridin-2-ylcarbamothioyl)benzamides 19 and 20.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Entry | R | Reagents | solvent | T(°C) | Time (min) | Product (Yield) |
|
| ||||||
| 1 | F | NaOH | EtOH | reflux | 20 | 12 (99%) |
| 2 | NO2 | NaOH | EtOH | reflux | 10 | N.Dc |
| 3 | NO2 | NaOH | EtOH | rt | 10 | N.Dc |
| 4 | NO2 | K2CO3 | EtOH | reflux | 30 | tracec |
| 5 | NO2 | TEA | EtOH | reflux | 30 | tracec |
| 6 | NO2 | HBr | HOAc | rt | 600 | N.Dd |
| 7 | NO2 | HBr | HOAc | reflux | 60 | N.Dc |
| 8 | NO2 | HCl | water | reflux | 600 | tracec |
| 9 | NO2 | NH2NH2 | EtOH | reflux | 60 | N.Dc |
| 10a | NO2 | A | MeOH | rt | 1200 | N.Dd |
| 11a | NO2 | B | MeOH | rt | 1200 | N.Dd |
| 12a | NO2 | C | MeOH | rt | 1200 | 8a (17%) |
| 13a | NO2 | C | MeOH | reflux | 30 | 8a (55%) |
| 14b | NO2 | C | MeOH | reflux | 20 | 8a (97%) |
| 15b | F | C | MeOH | reflux | 20 | 12 (98%) |
One equiv of reagent was used.
Five equiv of reagent were used.
Complicated reaction mixtures.
No clear reaction was observed and 20 remained intact in the medium
The benzoyl group has been applied to protection of hydroxyl and amino groups in organic synthesis and is removed by bases, acids such as HCl or HBr-AcOH [27], and hydrazine [28,29]. Since the ortho-nitro group on pyridine is reactive and is not stable under the basic conditions, we studied other reported deprotection methods. Results showed that although 20 was relatively stable under acid conditions, no reaction was observed by using HBr-AcOH mixture (37% HBr in HOAC) at RT for 10 h (Table 2 entry 6). When 20 was heated to reflux in either HBr-HOAc or HCl (32 wt% in H2O), it led to complicated reaction mixtures (Table 2 entries 7 and 8), in which substitution of nitro group by Br or Cl was detected by LC-MS, respectively (ESI, Figures S2 and S3). By using the hydrazine method, 20 was completely consumed, however, no trace amount of product 8a could be detected (Table 2 entry 9 and Figures S4 and S5).
Inspired by our previous experience with organocatalyzed activation (primary amine-acid system) of carbonyl groups with low reactivity including alkyl and aryl ketones [30–32], α,β-unsaturated ketones [33,34], glycosides [32], and esters (furanones) [31,33], the organomediated cleavage of Bz group on the thiourea was investigated with different amine-acid systems (Table 2, entries 8–13). As the results showed, both the primary amine-acid A and the secondary amine-acid B with only one amino group did not promote this reaction at rt (Table 2, entries 10 and 11), although the starting material 20 was stable and was recovered under these neutral conditions. We then turned to ethane-1,2-diamine and acetic acid combination C, which had been successfully used for organocatalyzed domino reaction as well as Michael addition reaction [30,31]. Interestingly, the reaction of 20 with one equiv of C in MeOH at rt gave the desired product 8a in 17% yield with no by-product found (Table 2, entry 12). When the reaction was refluxed in methanol (65 °C) for 30 min, compound 8a was obtained in 55% yield (Table 2, entry 13). Finally, the cleavage of Bz group from 20 could be accomplished in 97% yield with 5 equiv of C by refluxing in methanol for 20 min (Table 2, entry 14). Product 8a crystalized from the reaction mixture as a light-yellow solid when cooling down. In a similar condition, the cleavage of Bz group from 19 gave 12 in 98% yield (Table 2, entry 15). These results demonstrate that the organomediated cleavage of Bz group from thioureas under neutral condition is a mild and general deprotection method specifically useful for basesensitive compounds.
This interesting 1,2-ethanediamine-AcOH mediated Bz-deprotection method prompted us to examine the reaction mechanism (Scheme 4). Since the mono primary amine did not promote the reaction, we propose that both amino groups of 1,2-ethanediamine synergistically participate in this reaction. Upon forming the iminium intermediate I by the reaction with an amino group, another amino group attacks the carbon of the iminium to form a 5-membered cyclic intermediate II and then the nucleophilic substitution by oxygen of water leads to the formation of the thiourea 8a and the intermediate III that releases the Bz group as the diamine adduct IV. The Bz-diamine adduct IV was isolated from the reaction mixture and confirmed by LC-MS (ESI Figure S6).
Scheme 4.

Proposed reaction mechanism for the deprotection of Bz from the thioureas 20.
The stability of 1-(6-nitropyridin-2-yl)thiourea 8a under basic conditions was then studied by in situ 1H NMR and LC-MS. In the 1H NMR study, 8a disappeared after addition of NaOH to the solution (Figure S7), wherein the soluble portion of the oligomerization products were revealed by the LC-MS analysis (ESI, Figure S1). This result clearly explains why compound 8a could not be prepared under the conventional conditions because the ortho-nitro group does not tolerate basic conditions.
As discussed previously, 8a is a useful synthon for synthesizing N-(6-nitropyridin-2-yl)thiazol-2-amine derivatives which can serve as precursors for direct 18F-radiolabeling to generate PET tracers. To demonstrate the feasibility, the reactions of 8a with 2-bromo-4′-phenylacetophenone 21 was carried out by refluxing in acetonitrile to give 9a in near quantitative yield (Scheme 5). In a similar reaction, 18F labeled compound 10a, which may have therapeutical effect for prion disease, was prepared [1,3]. The radioligand [18F]10a could serve as a useful tool for both disease imaging and drug development.
Scheme 5.

Synthesis of N-(pyridin-2-yl)thiazol-2-amine derivatives 9a and 10a
The radio-synthesis of [18F]10a was initially attempted via the direct 18F-fluorination of 9a (Scheme 6), however, the reaction resulted in only trace amount of [18F]10a and several unidentified by-products (Figure S8). Since the free amino group could interfere with the reaction, it should be protected to avoid side reactions. The Boc-protection of the amino group in 9a was achieved by using di-tert-butyl dicarbonate and NaI-DMAP co-catalyst in THF at rt to give 22 in >95% yield as a white solid.
Scheme 6.

The direct 18F-substitution of 9a gave a complicated reaction mixture.
Finally, the radiosynthesis of [18F]23 from 22 was accomplished by using either [18F]/K2CO3/K222 or [18F]TEAF in acetonitrile (Scheme 7), which gave [18F]23 in >85% yield along with 5–10% of [18F]10a in both reactions. The reaction mixture was treated with trifluoroacetic acid (TFA) at 25 °C and then purified by HPLC to obtain [18F]10a in 52% radiochemical yield (RCY) at the end of synthesis (decay corrected) with high purity (>95%). The radiolabeled compound [18F]10a was co-injected with the cold compound 10a to confirm the identity of the radiolabeled product (ESI Figure S9).
Scheme 7.

Radiosynthesis of [18F]10a from precursor 22.
In vitro autoradiography experiments of [18F]10a (93 kBq/mL, 1.0–2.0 nM) were performed on sagittal rat brain slices (20 μm). Fig. 2 depicts a heterogeneous distribution of radioactivity in rat brain sections at the baseline with highest accumulation of radioactivity in the hippocampus, neocortex, striatum and thalamus, the regions known to express high levels of PrPc, in accordance with the reported immunostaining of rat brain.[35,36] The rat brain slices that were coincubated with [18F]10a and 10 μM of cold compound 10a displayed a substantially reduced and homogeneous accumulation of radioactivity (Fig. 2, self-blocking). These results suggested that ligand [18F]10a had excellent in vitro binding specificity and further in vitro and in vivo evaluation of the compound is underway.
Fig. 2.

In vitro autoradiography of [18F]10a on sagittal rat brain slices (20 μm). Anatomical regions are detailed in: St, Striatum; Cx, Cortex; Th, Thalamus; Hi, Hippocampus; Cr, Cerebellum.
3. Conclusion
Compound 8a and its derivatives could be used to construct a series of bioactive heterocycles such as N-(pyridin-2-yl)thiazol-2-amine derivatives, which could be utilized for PET tracer development toward different imaging targets. However, synthesis of 8a from 6-nitropyridin-2-amines 7a was challenging due to the reactive nature of the ortho-nitro group and significantly reduced nucleophilicity of the amino group. Our study found that the cleavage of Bz group from thiourea 20 was not achievable by conventional methods since the ortho-nitropyridine was not stable under basic conditions. Therefore, a novel organomediated method was developed using an ethane-1,2-diamine and acetic acid system to remove the Bz group under neutral condition. By using this method, 8a was efficiently synthesized in high yield under mild conditions. To further demonstrate the feasibility for its application on PET tracer development, the coupling of 8a with 2-bromo-4′-phenylacetophenone 21 was carried out to afford 9a (>98%). Synthesis of the radioligand [18F]10a was successfully accomplished and was further applied to the in vitro autoradiography on sagittal rat brain slices, which showed highest accumulation of radioactivity in the hippocampus, cortex, and striatum in agreement with the reported immunostaining of PrPc in rat brain.
4. Experimental section
4.1. General methods
All reagents and starting materials were obtained from the commercial sources including Sigma-Aldrich (St. Louis, MO), Thermo Fisher Scientific, Oakwood Products, Inc., Matrix Scientific and used as received. The reactions were monitored by TLC using a UV lamp monitored at 254 nm. If necessary, the reactions were also checked by LC − MS using the Agilent 1200 Series HPLC system coupled with a multiwavelength UV detector and a model 6310 ion trap mass spectrometer (Santa Clara, CA) equipped with a Luna C18 column (Phenomenex, 100 × 2 mm, 5 μm, 100 Å). The RP-HPLC was carried out by using a 7 min gradient method (LC-MS Method): the mobile phase A was water with 0.1% Formic acid (FA) added; mobile phase B was acetonitrile with 0.1% FA added; gradient: 5% B to 95% B from 0 to 3 min, 95% B from 3 to 4.5 min, 95% to 5% B from 4.5 to 5 min, 5% B from 5 to 7 min; flow rate at 0.7 mL/min. The silica gel used in flash column chromatography was from Aldrich (Cat. 60737, pore size 60 Å, 230–400 mesh). The products were identified by 1H NMR and 13C NMR using a Varian 500 MHz spectrometer. Chemical shifts were expressed as ppm and calculated downfield or upfield from the NMR signal of the reference standard. J was expressed as Hz, and its splitting patterns were reported as s, d, t, q, or m. Unless otherwise specified, the purity of all new compounds was over 95% determined by HPLC.
4.2. Radiochemistry procedure
[18F]Fluoride was generated by a GE PETtrace 16.5 Mev cyclotron (GE Healthcare, Waukesha, WI, USA) using 18O enriched water (Isoflex Isotope, San Francisco, CA) with proton bombardment. Fluorine-18 labeling of [18F]10a was accomplished in one pot via two steps. First, [18F]fluoride in 18O-enriched water was passed through a QMA Sep-Pak cartridge (Waters, Milford, MA) to trap [18F]fluoride ions, which was washed off by a mixture of acetonitrile (0.9 mL) and water (0.1 mL) solution containing 5.0 mg of tetraethylammonium bicarbonate. And the solvents were evaporated at 115 °C in a stream of nitrogen. To remove water completely, 1.0 mL of acetonitrile was added and evaporated in a stream of nitrogen three more times. To the residue containing [18F]fluoride was added 22 (2.0 mg) in 0.7 mL of acetonitrile. The resulting solution was heated to 85 °C for 10 min and then cooled to room temperature, and the solvent was removed by a stream of nitrogen (2 mins). Then 0.5 mL of TFA was added to the reactor for 5 mins at room temperature. The resulting mixture was diluted with water and neutralized by NaOH then purified by a semipreparative HPLC (Waters 4000 system equipped with an Xbridge BEH C18 OBD column: 130 Å, 5 μm, 10 × 250 mm) by eluting with a solution of water and acetonitrile (20:80) at a flow rate of 4 mL/min to give the fractions containing [18F]10a. The combined fraction was diluted with water to 40 mL and loaded on a C18 Sep-Pak column to give the final formulation of [18F]10a in saline containing 10% ethanol. The purity of [18F]10a was over 99% that was analyzed by an analytical HPLC (Waters 2487 series equipped with a UV detector and a BIOSCAN radioactivity detector and an ACE 5 C18-AR column: 250 × 10 mm, 5 μm). The identity of [18F]10a was confirmed by co-injection of the cold compound 10a in HPLC analysis.
4.3. In vitro autoradiography
Rat brains were cut into sections 20 μm-thick in a cryostat, mounted on Histobond adhesion slides and used for [18F]10a phosphor screen autoradiography. Brain slices were pre-incubated with Tris-HCl buffer (50 mM), MgCl2 (1.2 mM) and CaCl2 (2 mM) solution for 10 min at ambient temperature, followed by incubation in 10% neutral buffered formalin for another 10 min. The brain slices were then washed with Tris-HCl buffer followed by incubation with [18F]10a (93 kBq/mL, 1–2 nM) for another 45 mins. For the blocking studies, the rat brain slices were incubated with [18F]10a containing cold compound 10a (10 μM) to determine the specificity of radioligand binding. After incubation, brain slices were rinsed with ice-cold buffer three times for 2 min and then were dipped in cold distilled water for 10 s. The brain sections were allowed to air dry before transfer of the slides to a storage phosphor screen (BAS-MS2025, GE Healthcare, NJ, USA) that had been photobleached immediately prior by exposure on a white light box for a minimum of 15 min. Autoradiograms were obtained by a GE Typhoon FLA 9000 Imager and regions of interest (ROIs) were carefully drawn.
4.4. General synthetic procedures
General procedure A for the synthesis of N-(arylcarbamothioyl) benzamides. To a solution of the corresponding pyridin-2-amine (3.0 mmol) in THF (5.0 mL) was added benzoyl isothiocyanate (1.2 equiv). The reaction mixture was refluxed until pyridin-2-amine was consumed. After cooling to rt, the reaction mixture was subjected to silica gel column chromatography directly using ethyl acetate and hexanes as eluents to give pure products
General procedure B for the synthesis of 20. To a solution of 6-nitropyridin-2-amine (3.0 mmol) in toluene (5.0 mL) was added benzoyl isothiocyanate (1.2 equiv). The reaction mixture was refluxed for 10 min. After cooling, needle crystals were collected by filtration.
General procedure C for the synthesis of N-(pyridin-2-ylcarbamothioyl)benzamides. To a solution of N-(arylcarbamothioyl) benzamide (2.0 mmol) in MeOH (5.0 mL) was added ethane-1,2-diamine (10.0 mmol) and acetic acid (20.0 mmol). The reaction mixture was refluxed and monitored by HPLC until N-(arylcar-bamothioyl)benzamide was consumed. After cooling to room temperature, the products were collected by filtration. The filtrates were further subjected to silica gel column chromatography directly using ethyl acetate and hexanes as eluent to give pure products.
General procedure D for the synthesis of N-(pyridin-2-yl)thiazol-2-amine derivatives. To a solution of N-(pyridin-2-ylcarbamothioyl) benzamides (0.7 mmol) in acetonitrile (5.0 mL) was added 2-bromo-4′-phenylacetophenone (0.7 mmol). The reaction mixture was refluxed and a large amount of solid precipitated out within 2 min. The solid was collected by simple filtration as pure products.
General procedure E for the Boc protection. To a solution of 9a or 10a (0.5 mmol) in THF (5 mL) was added Di-tert-butyl dicarbonate (2.5 mmol), DMAP (2.5 mmol) and NaI (0.5 mmol). The reaction mixture was stirred at room temperature for 30 s and was subjected to silica gel column chromatography using ethyl acetate and hexanes as eluents to give pure product.
4.5. Synthesis
Compound 18 was prepared according to the general procedure A in 99% yield as white solid. 1H NMR (500 MHz, Chloroform-d) δ 9.10 (s, 1H), 7.97–7.80 (m, 2H), 7.77–7.60 (m, 3H), 7.54 (t, J = 7.8 Hz, 2H), 7.48–7.31 (m, 2H).
Compound 19 was prepared according to the general procedure A in 98% yield as white solid. 1H NMR (500 MHz, Chloroform-d) δ 9.06 (s, 1H), 8.78 (dd, J = 7.9, 2.1 Hz, 1H), 7.93–7.88 (m, 2H), 7.86 (q, J = 8.0 Hz, 1H), 7.75–7.61 (m, 1H), 7.59–7.45 (m, 2H), 6.79 (dd, J = 8.0, 2.7 Hz, 1H). 13C NMR (126 MHz, Chloroform-d) δ 177.3, 166.5, 162.0 (d, J = 242.6 Hz), 149.5 (d, J = 14.3 Hz), 142.7 (d, J = 7.5 Hz), 134.0, 131.5, 129.4, 127.7, 112.5 (d, J = 4.4 Hz), 106.6 (d, J = 35.2 Hz).
Compound 20 was prepared according to the general procedure A in 90% yield as light yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.24 (d, J = 6.9 Hz, 1H), 9.15 (s, 1H), 8.23–8.01 (m, 2H), 7.91 (d, J = 7.6 Hz, 2H), 7.67 (t, J = 7.4 Hz, 1H), 7.56 (t, J = 7.7 Hz, 2H). 13C NMR (126 MHz, Chloroform-d) δ 178.2, 166.6, 155.1, 150.5, 141.4, 134.2, 131.3, 129.4, 127.7, 121.5, 115.0.
Compound 12 was prepared according to the general procedure C in 98% yield as white solid. 1H NMR (500 MHz, Chloroform-d) δ 10.20 (s, 1H), 9.46 (s, 1H), 7.74 (q, J = 8.0 Hz, 1H), 7.14 (s, 1H), 6.81 (d, J = 7.8 Hz, 1H), 6.60 (dd, J = 8.0, 2.0 Hz, 1H). 19F NMR (470 MHz, Chloroform-d) δ −68.5. 13C NMR (126 MHz, Chloroform-d) δ 181.0, 161.4 (d, J = 244.5 Hz), 151.0 (d, J = 14.0 Hz), 143.4 (d, J = 8.4 Hz), 108.8 (d, J = 4.8 Hz), 103.0 (d, J = 34.1 Hz).
Compound 8a was prepared according to the general procedure C in 97% yield as light brown solid. 1H NMR (500 MHz, DMSO-d6) δ 11.05 (s, 1H), 10.02 (d, J = 3.7 Hz, 1H), 9.24 (s, 1H), 8.12 (t, J = 8.0 Hz, 1H), 7.92 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 181.2, 153.4, 152.23, 143.1, 119.4, 112.2. LC-MS (LC-MS Method): tR = 2.97 min, m/z: [M+H]+ Calcd forC6H7N4O2S 199.0; Found 199.1.
Compound 9a was prepared according to the general procedure D in 98% yield as light brown solid. 1H NMR (500 MHz, DMSO-d6) δ 8.07 (t, J = 8.0 Hz, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.82 (dd, J = 7.8, 2.7 Hz, 1H), 7.75–7.66 (m, 4H), 7.65 (d, J = 3.0 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 7.34 (t, J = 7.3 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 159.5, 154.9, 151.7, 149.2, 141.9, 140.2, 139.7, 134.1, 129.5, 128.1, 127.5, 127.2, 127.0, 126.8, 117.8, 110.2, 108.1. LC-MS (LC-MS Method): tR = 4.50 min, m/z: [M+H]+ Calcd for C20H15N4O2S 375.09; Found 375.1.
Compound 10a was prepared according to the general procedure D in 99% yield as white solid. 1H NMR (500 MHz, Chloroform-d) δ 7.87 (dd, J = 8.0, 5.6 Hz, 3H), 7.72 (d, J = 8.2 Hz, 2H), 7.63–7.54 (m, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.40–7.32 (m, 1H), 7.16 (d, J = 7.7 Hz, 1H), 7.00 (s, 1H), 6.73 (dd, J = 8.1, 2.0 Hz, 1H). 13C NMR (126 MHz, Chloroform-d) δ 162.7, 161.9 (d, J = 246.9 Hz), 147.0 (d, J = 13.0 Hz), 143.8 (d, J = 8.2 Hz), 139.6, 139.6, 129.1, 128.3, 128.2, 127.1, 126.4, 125.7, 109.5 (d, J = 4.7 Hz), 104.4, 104.3 (d, J = 33.9 Hz). LC-MS (LC-MS Method, ESI): tR = 4.01 min, m/z: [M+H]+ Calcd for C20H15FN3S 348.1; Found 348.1.
Compound 22 was prepared according to the general procedure E in 99% yield as light yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 8.33 (d, J = 8.0 Hz, 1H), 8.21 (t, J = 7.9 Hz, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.60–7.54 (m, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.41 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.24 (s, 1H), 1.47 (s, 10H). 13C NMR (126 MHz, Chloroform-d) δ 160.4, 155.9, 151.5, 151.5, 149.9, 141.7, 140.8, 140.7, 133.1, 129.9, 128.9, 127.4, 127.3, 127.0, 126.4, 117.5, 108.95, 85.1, 28.1. LC-MS (LC-MS Method): tR = 4.71 min, m/z: [M+H]+ Calcd for C25H23N4O4S 475.1; Found 475.0.
Compound 23 was prepared according to the general procedure E in 99% yield as white solid. 1H NMR (500 MHz, Chloroform-d) δ 7.96 (q, J = 7.9 Hz, 1H), 7.74–7.66 (m, 2H), 7.60–7.55 (m, 2H), 7.55–7.50 (m, 2H), 7.41 (t, J = 7.6 Hz, 2H), 7.33 (dd, J = 7.9, 1.9 Hz, 2H), 7.21 (s, 1H), 7.02 (dd, J = 8.1, 2.8 Hz, 1H), 1.48 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 162.8 (d, J = 244.1 Hz), 160.7, 151.9, 150.3 (d, J = 13.5 Hz), 149.9, 142.7 (d, J = 7.4 Hz), 140.84, 140.51, 133.6, 128.9, 127.4, 127.3, 127.0, 126.5, 121.1 (d, J = 4.9 Hz), 109.2, (d, J = 34.0 Hz), 108.6, 84.3, 28.1. LC-MS (LC-MS Method): tR = 4.89 min, m/z: [M+H]+ Calcd for C25H23FN3O2S 448.1; Found 448.0.
Supplementary Material
Acknowledgments
The studies were supported by the NIH grants R01NS100164, K25AG061282 and S10OD025234.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2022.105804.
References
Uncategorized References
- [1].Li Z, Silber BM, Rao S, Gever JR, Bryant C, Gallardo-Godoy A, Dolghih E, Widjaja K, Elepano M, Jacobson MP, Prusiner SB, Renslo AR, 2-Aminothiazoles with improved pharmacotherapeutic properties for treatment of prion disease, ChemMedChem 8 (5) (2013) 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, Li Z, Jacobson MP, Huang Y, Benet LZ, Prusiner SB, Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice, Pharm. Res. 30 (4) (2013) 932–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR, 2-Aminothiazoles as therapeutic leads for prion diseases, J. Med. Chem. 54 (4) (2011) 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hong S-P, Liu KG, Ma G, Sabio M, Uberti MA, Bacolod MD, Peterson J, Zou ZZ, Robichaud AJ, Darío Doller, Tricyclic thiazolopyrazole derivatives as metabotropic glutamate receptor 4 positive allosteric modulators, J. Med. Chem. 54 (14) (2011) 5070–5081. [DOI] [PubMed] [Google Scholar]
- [5].Hinklin RJ, Baer BR, Boyd SA, Chicarelli MD, Condroski KR, DeWolf WE, Fischer J, Frank M, Hingorani GP, Lee PA, Neitzel NA, Pratt SA, Singh A, Sullivan FX, Turner T, Voegtli WC, Wallace EM, Williams L, Aicher TD, Discovery and preclinical development of AR453588 as an anti-diabetic glucokinase activator, Bioorg. Med. Chem. 28 (1) (2020) 115232, 10.1016/j.bmc.2019.115232. [DOI] [PubMed] [Google Scholar]
- [6].Hinklin RJ, Boyd SA, Chicarelli MJ, Condroski KR, DeWolf WE, Lee PA, Lee W, Singh A, Thomas L, Voegtli WC, Williams L, Aicher TD, Identification of a new class of glucokinase activators through structure-based design, J. Med. Chem. 56 (19) (2013) 7669–7678. [DOI] [PubMed] [Google Scholar]
- [7].Kesicki EA, Bailey MA, Ovechkina Y, Early JV, Alling T, Bowman J, Zuniga ES, Dalai S, Kumar N, Masquelin T, Hipskind PA, Odingo JO, Parish T, Seleem MN, Synthesis and Evaluation of the 2-Aminothiazoles as Anti-Tubercular Agents, PLoS ONE 11 (5) (2016) e0155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim S-H, Tokarski JS, Leavitt KJ, Fink BE, Salvati ME, Moquin R, Obermeier MT, Trainor GL, Vite GG, Stadnick LK, Lippy JS, You D, Lorenzi MV, Chen P, Identification of 2-amino-5-(thioaryl)thiazoles as inhibitors of nerve growth factor receptor TrkA, Bioorg. Med. Chem. Lett. 18 (2) (2008) 634–639. [DOI] [PubMed] [Google Scholar]
- [9].Bhuniya D, Mukkavilli R, Shivahare R, Launay D, Dere RT, Deshpande A, Verma A, Vishwakarma P, Moger M, Pradhan A, Pati H, Gopinath VS, Gupta S, Puri SK, Martin D, Aminothiazoles: Hit to lead development to identify antileishmanial agents, Eur. J. Med. Chem. 102 (2015) 582–593. [DOI] [PubMed] [Google Scholar]
- [10].Paans AM, van Waarde A, Elsinga PH, Willemsen AT, Vaalburg W, Positron emission tomography: the conceptual idea using a multidisciplinary approach, Methods 27 (3) (2002) 195–207. [DOI] [PubMed] [Google Scholar]
- [11].Kilbourn MR, Fluorine-for-hydrogen: a strategy for radiolabeling, not a replacement, Nucl. Med. Biol. 40 (8) (2013) 956–958. [DOI] [PubMed] [Google Scholar]
- [12].Pettersson M, Hou X, Kuhn M, Wager TT, Kauffman GW, Verhoest PR, Quantitative Assessment of the Impact of Fluorine Substitution on P-Glycoprotein (P-gp) Mediated Efflux, Permeability, Lipophilicity, and Metabolic Stability, J. Med. Chem. 59 (11) (2016) 5284–5296. [DOI] [PubMed] [Google Scholar]
- [13].Dolle F, Fluorine-18-labelled fluoropyridines: advances in radiopharmaceutical design, Curr. Pharm. Des. 11 (25) (2005) 3221–3235. [DOI] [PubMed] [Google Scholar]
- [14].Gobbi LC, Knust H, Körner M, Honer M, Czech C, Belli S, Muri D, Edelmann MR, Hartung T, Erbsmehl I, Grall-Ulsemer S, Koblet A, Rueher M, Steiner S, Ravert HT, Mathews WB, Holt DP, Kuwabara H, Valentine H, Dannals RF, Wong DF, Borroni E, Identification of Three Novel Radiotracers for Imaging Aggregated Tau in Alzheimer’s Disease with Positron Emission Tomography, J. Med. Chem. 60 (17) (2017) 7350–7370. [DOI] [PubMed] [Google Scholar]
- [15].Sergeev ME, Morgia F, Lazari M, Wang C, van Dam RM, Titania-catalyzed radiofluorination of tosylated precursors in highly aqueous medium, J. Am. Chem. Soc. 137 (17) (2015) 5686–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dolci L, Dolle F, Jubeau S, Vaufrey F, Crouzel C, 2-[18F]fluoropyridines by no-carrier-added nucleophilic aromatic substitution with [18F]FK-K222 - a comparative study, J. Labelled Compd. Radiopharm. 42 (10) (1999) 975–985. [Google Scholar]
- [17].Washington JB, Assante M, Yan C, McKinney D, Juba V, Leach AG, Baillie SE, Reid M, Trialkylammonium salt degradation: implications for methylation and cross-coupling, Chem. Sci. 12 (20) (2021) 6949–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM, Cuthbertson A, One Step Radiosynthesis of 6-[18F]Fluoronicotinic Acid 2,3,5,6-Tetrafluorophenyl Ester ([18F]F-Py-TFP): A New Prosthetic Group for Efficient Labeling of Biomolecules with Fluorine-18, J. Med. Chem. 53 (4) (2010) 1732–1740. [DOI] [PubMed] [Google Scholar]
- [19].Hammoud H, Elhabazi K, Quillet R, Bertin I, Utard V, Laboureyras E, Bourguignon J-J, Bihel F, Simonnet G, Simonin F, Schmitt M, Aminoguanidine Hydrazone Derivatives as Nonpeptide NPFF1 Receptor Antagonists Reverse Opioid Induced Hyperalgesia, ACS Chem. Neurosci. 9 (11) (2018) 2599–2609. [DOI] [PubMed] [Google Scholar]
- [20].Chikhale R, Thorat S, Choudhary RK, Gadewal N, Khedekar P, Design, synthesis and anticancer studies of novel aminobenzazolyl pyrimidines as tyrosine kinase inhibitors, Bioorg. Chem. 77 (2018) 84–100. [DOI] [PubMed] [Google Scholar]
- [21].Chikhale R, Menghani S, Babu R, Bansode R, Bhargavi G, Karodia N, Rajasekharan MV, Paradkar A, Khedekar P, Development of selective DprE1 inhibitors: Design, synthesis, crystal structure and antitubercular activity of benzothiazolylpyrimidine-5-carboxamides, Eur. J. Med. Chem. 96 (2015) 30–46. [DOI] [PubMed] [Google Scholar]
- [22].Rothweiler U, Stensen W, Brandsdal BO, Isaksson J, Leeson FA, Engh RA, Svendsen JSM, Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules, J. Med. Chem. 59 (21) (2016) 9814–9824. [DOI] [PubMed] [Google Scholar]
- [23].Hay MP, Turcotte S, Flanagan JU, Bonnet M, Chan DA, Sutphin PD, Nguyen P, Giaccia AJ, Denny WA, 4-Pyridylanilinothiazoles that selectively target von Hippel-Lindau deficient renal cell carcinoma cells by inducing autophagic cell death, J. Med. Chem. 53 (2) (2010) 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chikhale R, Thorat S, Pant A, Jadhav A, Thatipamula KC, Bansode R, Bhargavi G, Karodia N, Rajasekharan MV, Paradkar A, Khedekar P, Design, synthesis and pharmacological evaluation of pyrimidobenzothiazole-3-carboxylate derivatives as selective L-type calcium channel blockers, Bioorg. Med. Chem. 23 (20) (2015) 6689–6713. [DOI] [PubMed] [Google Scholar]
- [25].Makam P, Kannan T, 2-Aminothiazole derivatives as antimycobacterial agents: Synthesis, characterization, in vitro and in silico studies, Eur. J. Med. Chem. 87 (2014) 643–656. [DOI] [PubMed] [Google Scholar]
- [26].Bollenbach M, Salvat E, Daubeuf F, Wagner P, Yalcin I, Humo M, Letellier B, Becker LJ, Bihel F, Bourguignon JJ, Villa P, Obrecht A, Frossard N, Barrot M, Schmitt M, Phenylpyridine-2-ylguanidines and rigid mimetics as novel inhibitors of TNFalpha overproduction: Beneficial action in models of neuropathic pain and of acute lung inflammation, Eur. J. Med. Chem. 147 (2018) 163–182. [DOI] [PubMed] [Google Scholar]
- [27].Ben-Ishai D, Altman J, Peled N, The synthesis of p-substituted D, L-phenylglycines by the amidoalkylation of benzylchloride and N-benzylbenzamide, Tetrahedron 33 (20) (1977) 2715–2717. [Google Scholar]
- [28].Boger DL, Machiya K, Total synthesis of (+)-duocarmycin SA, J. Am. Chem. Soc. 114 (25) (1992) 10056–10058. [Google Scholar]
- [29].Boger DL, McKie JA, Nishi T, Ogiku T, Total Synthesis of (+)-Duocarmycin A and epi-(+)-Duocarmycin A and Their Unnatural Enantiomers: Assessment of Chemical and Biological Properties, J. Am. Chem. Soc. 119 (2) (1997) 311–325. [Google Scholar]
- [30].Wang J, Li Q, Qi C, Liu Y, Ge Z, Li R, Primary 1,2-diamine catalysis III: an unexpected domino reaction for the synthesis of multisubstituted cyclohexa-1,3-dienamines, Org. Biomol. Chem. 8 (19) (2010) 4240–4242. [DOI] [PubMed] [Google Scholar]
- [31].Wang W, Wang J, Zhou S, Sun Q, Ge Z, Wang X, Li R, An efficient organocatalytic enantioselective Michael addition of aryl methyl ketones with 2-furanones: highly functionalized chiral 3,4-substituted lactones, Chem. Commun. (Camb.) 49 (13) (2013) 1333–1335. [DOI] [PubMed] [Google Scholar]
- [32].Wang J-F, Lei M, Li Q, Ge Z-M, Wang X, Li R-T, Runtao Li A novel and efficient direct aldol condensation from ketones and aromatic aldehydes catalyzed by proline–TEA through a new pathway, Tetrahedron 65 (25) (2009) 4826–4833. [Google Scholar]
- [33].Wang J, Qi C, Ge Z, Cheng T, Li R, Efficient direct asymmetric vinylogous Michael addition reactions of gamma-butenolides to chalcones catalyzed by vicinal primary-diamine salts, Chem. Commun. (Camb.) 46 (12) (2010) 2124–2126. [DOI] [PubMed] [Google Scholar]
- [34].Wang J, Wang X, Ge Z, Cheng T, Li R, Highly enantioselective Michael addition of cyclopentanone with chalcones via novel di-iminium mechanism, Chem. Commun. (Camb.) 46 (10) (2010) 1751–1753. [DOI] [PubMed] [Google Scholar]
- [35].Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB, Regional mapping of prion proteins in brain, Proc Natl Acad Sci U S A 89 (16) (1992) 7620–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR, Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease, Proc. Natl. Acad. Sci. U S A 105 (29) (2008) 10238–10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.