

Abstract

Polar molecular surface area provides a valuable metric when optimizing properties as varied as membrane permeability and efflux susceptibility. The EPSA method to measure this quantity has had a substantial impact in medicinal chemistry, providing insight into the conformational and stereoelectronic features that govern the polarity of small molecules, targeted protein degraders, and macrocyclic peptides. Recognizing the value of bioisosteres in replacing permeation-limiting polar groups, we determined the effects of common amide, carboxylic acid, and phenol bioisosteres on EPSA, using matched molecular pairs within the Merck compound collection. Our findings reinforce EPSA’s utility in optimizing permeability, highlight bioisosteres within each class that are particularly effective in lowering EPSA and others, which despite widespread use, offer little to no such benefit. Our method for matched-pair identification is generalizable across large compound collections and, thus, may constitute a flexible platform to study the effects of bioisosterism both in EPSA and other in vitro assays.
Keywords: EPSA, bioisosteres, matched molecular pairs, polar surface area
Valuable properties emerge from polarity displayed at the surface of a molecule. In synthetic chemistry, for instance, the purification of samples routinely relies on chromatographic methods to separate components of a mixture that differ with respect to exposed polarity. The aqueous solubility of compounds also depends strongly upon this property. In a predictive sense, calculated polar surface area (PSA) can correlate with the oral absorption and central-nervous-system penetrance of drug compounds.1,2 In antibiotics discovery, PSA as a fraction of total surface area is thought to influence the capacity of a molecule to gain entry to Gram-negative bacteria.3
As a means to assess the effects of intramolecular hydrogen bonding and steric shielding, the EPSA method for determining chromatographic apparent polarity has proved a valuable assay to medicinal chemistry teams and has become metonymous with the abstract quantity (exposed polar surface area) that it was designed to measure.4,5 As an experimental, rather than computational technique, EPSA offers particular advantages in the regime of large molecules beyond Lipinski’s Rule of Five,6 where convergence of conformational sampling methods used in three-dimensional PSA calculations is challenging, if not impossible. Thus, EPSA provides an empirical means to test, both qualitatively and quantitatively, the success of designs aimed at modulating exposed polarity through conformational and steric effects not captured with topological methods.7 The passive permeability of small molecules and macrocyclic peptides is widely known to correlate with EPSA, particularly when other permeability determinants, such as molecular weight and lipophilicity, are taken into account.8−57 Likewise, incorporating EPSA into models of blood–brain barrier penetrance has been shown to improve their predictive power.12 Medicinal chemists, therefore, largely rely on EPSA to evaluate designs aimed at reducing overall polarity, although modifications that increase EPSA can in some cases be desirable, for example, to improve aqueous solubility or to exclude distribution to the central nervous system. To aid in prospective molecular design, models have been developed to predict EPSA on the basis of structure.8,13
Constituent polar functional groups do not contribute equally to a molecule’s overall EPSA, as a result of differences in pKa, dipole moment, steric shielding, or intramolecular hydrogen bonding, among other considerations. Hence, when optimizing a scaffold, medicinal chemistry teams frequently focus their efforts on one or two groups whose properties present outsized obstacles to a desired physicochemical and pharmacokinetic profile. In so doing, these teams often seek to replace them with similarly sized groups capable of comparable interactions, that is to say, bioisosteres.14 The property effects of such replacements are most easily interpreted within the framework of matched molecular pairs (MMPs), where the group of interest represents the only change between molecules. As such, MMP analysis has figured prominently in the study of bioisosteres’ effects on bioactivity, lipophilicity,15 and membrane permeability,16 among other properties. Seeking to understand the effect of bioisosterism on EPSA, we investigated and here report the use of MMP analysis to quantify the impact of bioisosteres commonly used to replace permeation-limiting polar groups, namely, amides, carboxylic acids, and phenols.
To identify bioisosteric MMPs, we built a flexible protocol in Pipeline Pilot readily implemented by nonspecialists, using widely available components. This workflow is depicted schematically in Figure 1. Briefly, MMP identification began with a substructure search for molecules containing the bioisostere motif.17 An atom-mapped chemical reaction then transformed the hits from this search to the corresponding matched structures bearing the parent functional group. These transformed structures served as queries in a second, full-structure search identifying registered molecules satisfying a matched-pair relationship. Finally, MMPs were filtered for compound availability. Through adjustments to the substructure search and reaction scheme modules, this protocol identified >100 000 MMPs across >20 bioisostere types, with each search requiring 15 min or less of computation time in total.
Figure 1.
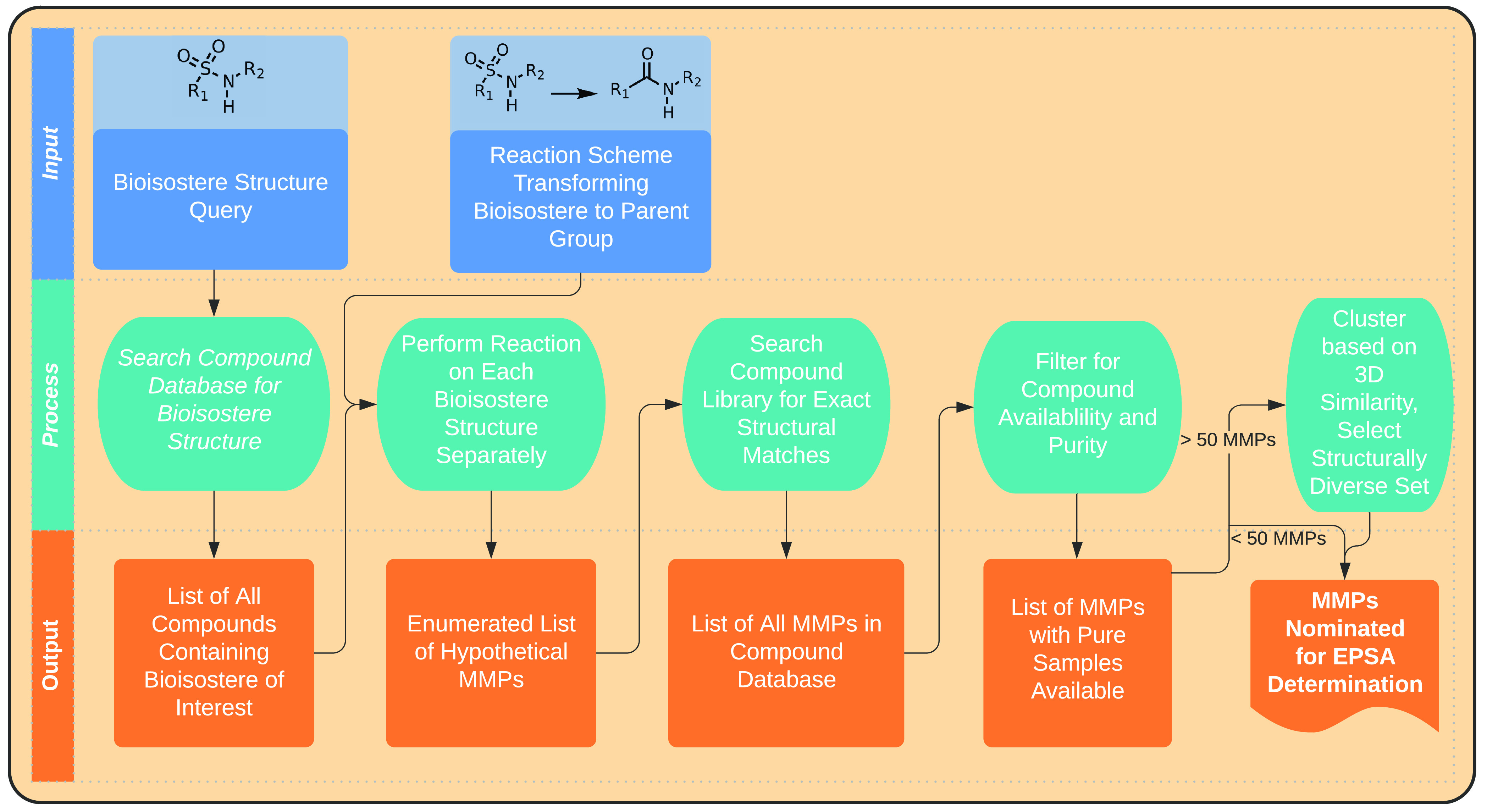
Schematic of the workflow used in this study to identify bioisosteric matched molecular pairs (MMPs).
MMPs identified in this way were clustered to ensure that the scaffolds selected for further study were maximally diverse with respect to three-dimensional shape. Within each MMP set, all bioisostere-containing structures were first expanded using OMEGA18,19 to generate up to 10 conformers. A 3D similarity matrix for all pairs of scaffolds was calculated with the FastROCS Toolkit,18,20 taking the highest similarity across conformers. Hierarchical density-based clustering21 of compounds based on these three-dimensional similarity scores afforded clusters of compounds, each representing one-half of a unique MMP. Final selections were made from these clustered arrays by visual inspection. Care was taken to match stereochemistry when appropriate because both pseudoenantiomeric and pseudodiastereomeric compounds would complicate subsequent analysis, which relies on a chiral chromatographic method to determine EPSA. In total, 23 bioisostere types were analyzed in this way, leading to the collection of EPSA data for of 499 MMPs or 998 individual compounds. From these measurements, ΔEPSA values were calculated for each MMP, reflecting the change in EPSA that resulted from bioisosteric replacement of the parent motif. The results from this analysis are presented below.
Amides are ubiquitous functional groups in medicinal chemistry, in large part for their ease of synthesis.22,23 Amide bonds can be common sites for hydrolytic metabolism, however, and the dual donor/acceptor character of secondary amide linkages can impart substantial polarity. Consequently, amide groups are proportionately less common among approved drugs targeting the central nervous system, where their polarity can hamper permeation and render molecules more susceptible toward transporter-mediated efflux.24 Correspondingly, by definition, macrocyclic peptides contain a multitude of amide linkages, which together with polar side chains and large size represent a major obstacle toward passive permeation.
Attempting to address problems arising from amide bond hydrolysis, polarity, or both, medicinal chemists have explored countless bioisosteres of this group.25 Within the Merck compound collection, the most common among these bioisosteres—defined by the number of MMPs identified prior to filtering and clustering—are N-methyl amides, esters, and sulfonamides, each comprising >10 000 MMPs. 1,2,3-Triazoles, 1,2,5-oxadiazoles, and α-trifluoromethylamines were well represented as well, each representing a category with >100 unique MMPs in our collection.
Analysis of a maximally diverse set of amide bioisostere MMPs, following shape-similarity clustering, revealed slight, yet significant, increases in EPSA accompanying the substitution of amides for sulfonamides (Δavg = 5 ± 2) and thioamides (Δavg = 4 ± 2) likely attributable to the combined size and HBD strengths of these groups (Figure 2). Amidine substitution—deployed historically to improve the pharmacokinetic properties of VLA-4 antagonists26 or to defeat glycopeptide antibiotic resistance27—led to greater EPSA as well, though the size of this effect varied greatly (Δavg = 22 ± 15), depending principally on the basicity of the amidine functional group.28,29 While sparsely represented in our compound collection, oxetanyl amine peptidomimetics30 provided no significant change in EPSA relative to their amide counterparts (Δ ranged from −5 to +3, n = 5).
Figure 2.

Changes in EPSA associated with common amide bioisosteres. Each MMP is plotted individually; bars and whiskers depict mean ± s.d. MMPs within the α-trifluoromethylamine set highlighted in orange (of which 1 → 2 is representative) all contain an α′-carboxy function responsible for their outlying ΔEPSA values.
Of the amide bioisosteres, we studied that preserve the parent group’s HBA capacity, while removing its donor function, all but one failed to reliably reduce EPSA—a trend consistent with analyses that illustrate HBD properties’ profound influence on EPSA and permeability.8 1,2,3-Triazoles, despite their weaker capacity for hydrogen bonding when compared to amides,31 did not offer consistent reductions in exposed polarity (Δavg = −2 ± 6), likely a consequence of their greater dipole moment.32 Accordingly, the lesser dipole embedded within 1,2,4-oxadiazoles led to reductions in EPSA on the order of Δavg = −12 ± 9 when compared to matched amide counterparts. Noting this, we investigated whether these bioisosteres’ differing reductions to EPSA corresponded to differences in cellular permeability as well. We compared the apparent permeability constants (Papp, measured using LLC-PK1 cells) of 10 amide → 1,2,4-oxadiazole and 5 amide → 1,2,3-triazole MMPs (Figure 3). We found that 1,2,4-oxadiazoles provided substantial boosts to permeability when incorporated into poorly permeable amide scaffolds (Papp < 10 × 10–6 cm s–1, 4 examples), and that these boosts corresponded to particularly negative MMP ΔEPSA values. Conversely, we found that in one instance, replacement of an amide group with a 1,2,3-triazole converted a freely permeable compound into a poorly permeable one (Papp 15 → 3 × 10–6 cm s–1, ΔEPSA = 3). We also found that N-methylation (Δavg = −12 ± 5) and ester substitution (Δavg = −15 ± 9) were among the simplest and most effective tactics to reduce the EPSA of amide-containing compounds (Figure 2). Both of these isosteres are abundant in naturally occurring permeable macrocyclic peptides33 and are widely recognized to improve passive permeability,34−37 further underscoring the connection of EPSA to membrane permeation and oral bioavailability.
Figure 3.

Comparison of the permeability (Papp, 10–6 cm s–1) and EPSA effects of amide → 1,2,4-oxadiazolone and amide → 1,2,3-triazole bioisosteric substitution. Transcellular permeability was assessed in LLC-PK1 cells at an analyte concentration of 1 μM. Gray dots denote MMPs for which ΔEPSA was not determined.
In some cases, the HBD role of amide groups is essential for bioactivity, and bioisosteres preserving this function while removing the acceptor carbonyl of the parent amide have been developed. α-Trifluoromethylamines represent one of the most successful such bioisosteres, having been deployed in the discovery of odanacatib, a peptidomimetic cathepsin K inhibitor, for instance.38 We found that, as with HBD-deleting bioisosteres, α-trifluoromethylamines afforded significant reductions in EPSA when incorporated in typical scaffolds (Δavg = −15 ± 8, Figure 2). Notably, within this category, we observed a set of outliers for which the associated reductions in EPSA were significantly and reproducibly greater (Δavg = −51 ± 3, n = 4). Each of these outlying points corresponded to molecular pairs possessing a free carboxylic acid positioned α′ to the amide (or trifluoromethylamine) nitrogen atom, a structural motif not present in any of the other 13 MMPs in this category. We postulate that this unusually large substitution effect arises through intramolecular hydrogen-bonding, which serves to mask the polarity of the carboxylic acid function, and whose effects are more pronounced in the case of α-trifluoromethylamines by virtue of their donor strength and compressed H–N–Cα′ bond angle (117° in amide 1 versus 109° in 2). Thus, from a design perspective, we note that in addition to representing an effective polarity-reducing amide bioisostere, the α-trifluoromethylamine motif also provides a particularly effective means by which to mask the EPSA contributions of adjacent acidic groups.
Carbamates are perhaps most commonly selected as amide bioisosteres for their improved proteolytic stability, though anecdotally they are known to improve membrane permeability as well.25,39 In our analysis of 56 MMPs describing amide → carbamate replacement, we found the resulting ΔEPSA measurements, followed a bimodal distribution, suggesting that the same bioisostere might exhibit divergent effects on EPSA (and thus on properties such as permeation) depending on structural context (Figure 4). We found that, while the extent of nitrogen-atom substitution (2° or 3°) had no significant effect when compared across subgroups (p = 0.41, see Supporting Information for details), the effect of molecular topology on ΔEPSA was pronounced. Namely, when embedded within a cyclic structure that forces the otherwise thermodynamically disfavored s-cis conformation, carbamates exhibited significantly greater EPSA than their cyclic amide counterparts (p < 0.001). Bioisosteric substitution of linear amides by contrast led to reductions in EPSA of approximately 5 units on average (Figure 4A). To better understand the physical basis for this finding, we studied the property effects of molecular topology in two closely related MMPs (Figure 4B). After exhaustive conformer generation40 and quantum-mechanical energy minimization of the resulting conformational ensembles, we computed Boltzmann-weighted average dipole moments41 and electrostatic potential values (describing the carbonyl HBA basicity and N–H HBD acidity)41,42 for linear molecules 3 and 4 and their cyclic congeners 5 and 6. When compared across these MMPs, the effects of bioisosteric substitution on computed HBA basicity and HBD acidity were mixed, confounding attempts to link EPSA effects to changes in individual atoms’ capacity for polar interaction. On the other hand, changes in molecular dipole, equal in magnitude but opposite in sign for 3 → 4 (Δμ = −1.1 D) and 5 → 6 (Δμ = +1.1 D), effectively accounted for the observed changes in EPSA. Thus, our findings indicate that, broadly, the EPSA effects of carbamate bioisosterism arise more through changes in molecular dipole than through atom-specific interactions and that the direction of such effects depends principally on conformational constraints imposed by the structure in which the group is embedded.
Figure 4.
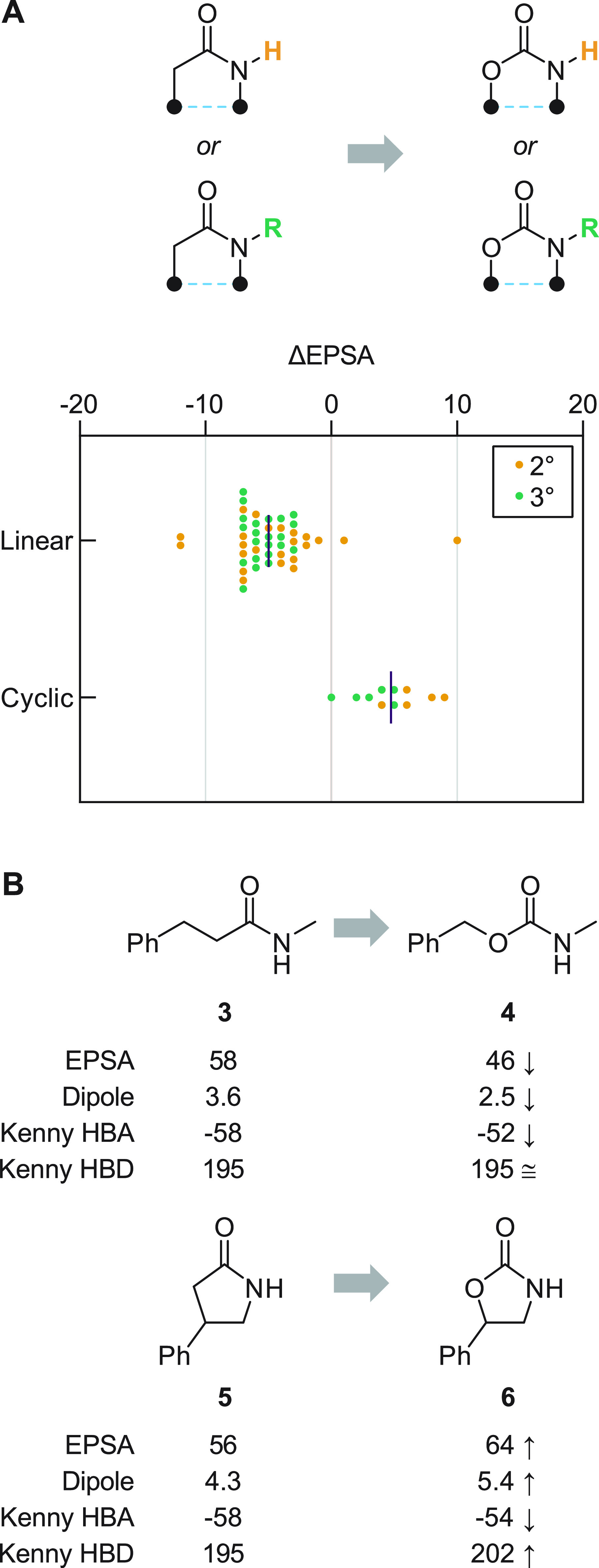
EPSA effects of amide → carbamate replacement. (A) Carbamates show reduced EPSA relative to their amide counterparts when embedded in linear substructures; the converse is true of cyclic amide → carbamate MMPs. The effect of N substitution (2° versus 3°) is insignificant. Each MMP is plotted individually; bars depict mean values. (B) Computed properties of two representative MMPs suggest that changes to overall dipole, rather than to individual atoms’ capacity for hydrogen bonding, are responsible for the observed ΔEPSA effects. Dipole units are debye; HBA acidity and HBD basicity calculated using Kenny electrostatic potential method41,42 (units are kcal/mol; see Supporting Information for details).
Among the functional groups most commonly encountered in medicinally relevant molecules, carboxylic acids contribute substantially to poor membrane permeability and high EPSA alike. Consequently, compounds bearing this group are rarely orally bioavailable and are often ill-suited for interacting with targets that reside within the cell or beyond the blood-brain barrier. Beyond limiting absorption, carboxylic acids also carry a pronounced susceptibility toward phase-II metabolism (glucuronidation), further impacting the pharmacokinetic and toxicologic risk profiles of compounds incorporating them.43 Nonetheless, owing to their prevalence in screening libraries and capacity to form anchoring electrostatic interactions with cationic residues (e.g., arginine), carboxylic acids are widespread among hits identified by high-throughput screens, including by mRNA display and other peptide-screening platforms. Striving to address the pharmacokinetic limitations of carboxylic acids, medicinal chemistry teams have explored a variety of bioisosteric groups,16,44 seven of which we elected to study with respect to their impact on EPSA (Figure 5).
Figure 5.

Changes in EPSA associated with common carboxylic acid bioisosteres. Each MMP is plotted individually; bars and whiskers depict mean ± s.d. α,α-Difluoro and α,α-dimethyl acids were matched with their unsubstituted (−CH2CO2H) counterparts.
We found that the magnitude of EPSA changes accompanying isosteric substitution of carboxylic acid groups was, broadly speaking, much greater than those observed for amides. This finding is consistent with the general principle that charged groups contribute greater overall polarity than do neutral ones, as well as with prior findings regarding the disproportionate impact of carboxylic acid groups on experimentally determined EPSA values.4,45 For instance, replacement with the neutral, isoelectronic nitro group afforded dramatic reductions in EPSA (Δavg = −73 ± 36), a finding useful for benchmarking, though there are widely known safety risks surrounding the nitro group that limit its incorporation into drugs. Accordingly, we found that EPSA depended strongly on relative acidity, with α,α-difluorinated carboxylic acids showing greater EPSA than their nonfluorinated counterparts (Δavg = 129 ± 35; pKa,avg = 1.0),28 while α,α-dimethylation of aliphatic acids led to modest decreases in EPSA (Δavg = −19 ± 7; pKa,avg = 4.4). Differences in acidity likewise account for the observation that oxazolidinediones (Δavg = −25 ± 15, pKa,avg = 5.6) and 1,3,4-oxadiazolones (Δavg = −47 ± 29, pKa,avg = 7.6), but not 1,2,4-oxadiazolones (Δavg = −1 ± 15, pKa,avg = 5.0), serve as excellent EPSA-lowering carboxylic acid bioisosteres.46
By contrast, we found that tetrazole compounds, despite marginally higher pKa and logD when compared to carboxylic acid counterparts,16 nonetheless showed greater EPSA values (Δavg = 34 ± 13, n = 25) in our study. Tetrazoles have come to constitute perhaps the single most popular carboxylic acid bioisostere (>750 such MMPs were present in our compound collection) owing to their resistance toward metabolism47 and successful deployment in the discovery of leukotriene receptor 1 antagonist48 and angiotensin II receptor blocker49 drugs. Our results suggest that this replacement may come at significant and underappreciated cost to membrane permeability, consistent with findings from the pioneering work of Huryn, Ballatore, and co-workers.16 Thus, in cases where carboxylic acid groups must be replaced to address poor drug permeation or transporter-mediated efflux, our results do not support the use of the tetrazole bioisostere.
When incorporated within the amino acid tyrosine, the phenol group plays a critical role in molecular recognition both at protein–protein50 and protein–drug interfaces.51 Consequently, tyrosyl residues are common among hits emerging through peptide-screening methods, including mRNA display. Indeed, owing to their chemical ubiquity, conformational rigidity, overall lipophilicity, and capacity to form strong hydrogen bonds with biological targets, phenols are often found in small-molecule leads as well. This is true despite phenols’ liability toward glucuronidation (phase-II metabolism) in vivo, which frequently limits the pharmacokinetic developability—most notably the oral bioavailability—of drugs that incorporate them. A wide array of bioisosteric replacements for the phenol group has been advanced, often with an explicit eye toward boosting oral availability.52,53 However, many such bioisosteres are more polar than the phenol groups they are designed to replace, and the impact of these bioisosteres on polarity-dependent properties such as permeability (itself a determinant of oral bioavailability) are rarely described.
In an effort to identify scaffolds that preserve a degree of phenols’ capability for polar interaction while reducing overall polarity when compared across MMPs, we studied the impact of six bioisosteres on EPSA (Figure 6). Of these, difluoromethyl-substituted benzenes provided the greatest reduction in EPSA on average (Δavg = −22 ± 8), consistent with reports of this group’s greater lipophilicity and reduced capacity for hydrogen bonding when compared to phenols.54,55 Pyridines, whose basic nitrogen might mimic the HBA or HBD role of hydroxyl groups depending on local pH in the binding pocket, elicited similar reductions in EPSA, though the effects were more varied in magnitude—in some cases where intramolecular hydrogen bonding present in the phenol was disrupted through bioisosteric substitution, the direction of ΔEPSA was positive, for instance. We found that indoles led to modest yet consistent increases in EPSA across MMPs (Δavg = 5 ± 4), and that the same was true for indazoles, albeit to a lesser extent (Δavg = 4 ± 5). Perhaps most notably, despite their increased heteroatom and HBD count, 2-aminopyridines and 2-aminopyrimidines both provided considerable reductions in EPSA when used in place of phenols (Δavg = −5 ± 8 and −9 ± 8 respectively), an observation likely attributable to electrostatic interaction between nNδ– and N–Hδ+ that masks both the HBA and HBD motifs’ effect on overall polarity–a permeability-boosting phenomenon commonly observed in ortho-fluorinated anilines as well.56
Figure 6.

Changes in EPSA associated with common phenol bioisosteres. Each MMP is plotted individually; bars and whiskers depict mean ± s.d.
In summary, we report the EPSA changes associated with bioisosteres used in the optimization of polar groups, using data collected across MMPs identified within the Merck compound collection. As benchmarks, these data serve to illustrate not only the average impact of each bioisosteric replacement but also the relative variability of this change, as the surveyed MMPs span a range of scaffolds representative of a large corporate collection, and were selected with structural diversity as an explicit aim. Our results showcase the sensitivity of the EPSA method toward pKa effects and identify bioisosteres within each category that offer significant polarity reduction when used in place of the parent group. We therefore expect these findings to aid teams engaged in the property optimization of drug molecules, by offering a means to prioritize—or disqualify—designs aimed at improving membrane permeability or efflux susceptibility, for instance. Moreover, we hope that the method we describe for MMP identification, similarity clustering, and selection may be useful to others studying the effects of bioisosteres on pertinent molecular properties.
Acknowledgments
This report is dedicated to the memory of our colleague, David M. Tellers. M.J.M. is indebted to Michael D. Altman and Xavier Fradera for their exceptional mentorship, scientific input, and assistance building automated workflows for MMP identification and similarity scoring. Sebastian E. Schneider is gratefully acknowledged for his assistance in the hierarchical clustering of 3D similarity matrices. The authors thank Karin M. Otte, Michael J. Hafey, Tony Pereira, Andrew M. Haidle, Derun Li, Kaustav Biswas, Chunhui Huang, James A. Baker, and Shawn P. Walsh for helpful discussions.
Glossary
Abbreviations
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- log D
logarithm of the distribution coefficient, a measure of a compound’s lipophilicity
- MMP
matched molecular pair
- PSA
polar surface area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00114.
Author Contributions
M.J.M. conceived the project and performed molecular modeling. D.A.L. and D.A.V. collected the EPSA measurements. A.E. and M.J.M. chose bioisostere categories to study, identified MMPs, selected compounds for EPSA measurement, analyzed the results, and wrote the manuscript.
This work was supported in part by the Merck Future Talent Program.
A.K.E., D.A.L, and M.J.M. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. D.A.V. is an employee of Eurofins PSS Insourcing Solutions.
The authors declare the following competing financial interest(s): A.K.E., D.A.L, and M.J.M. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. D.A.V. is an employee of Eurofins PSS Insourcing Solutions.
Supplementary Material
References
- Palm K.; Stenberg P.; Luthman K.; Artursson P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. 10.1023/A:1012188625088. [DOI] [PubMed] [Google Scholar]
- Kelder J.; Grootenhuis P. D. J.; Bayada D. M.; Delbressine L. P. C.; Ploemen J.-P. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. 10.1023/A:1015040217741. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Farrell W.; Shalaeva M.; Sciabola S.; Anderson D.; Yan J.; Philippe L.; Shapiro M. J. High Throughput Method for the Indirect Detection of Intramolecular Hydrogen Bonding. J. Med. Chem. 2014, 57, 2920–2929. 10.1021/jm401859b. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Philippe L.; Shapiro M. J. EPSA: A Novel Supercritical Fluid Chromatography Technique Enabling the Design of Permeable Cyclic Peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. 10.1021/ml500239m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doak B. C.; Over B.; Giordanetto F.; Kihlberg J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Shalaeva M.; Caron G.; Ermondi G.; Philippe L. Relationship between Passive Permeability and Molecular Polarity Using Block Relevance Analysis. Mol. Pharmaceutics 2017, 14, 386–393. 10.1021/acs.molpharmaceut.6b00724. [DOI] [PubMed] [Google Scholar]
- Mathiowetz A. M.Design Principles for Intestinal Permeability of Cyclic Peptides. In Methods in Molecular Biology: Cyclic Peptide Design; Goetz G., Ed.; Humana Press: New York, NY, USA, 2019: pp 1–16. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Defining optimum lipophilicity and molecular weight ranges for drug candidates—Molecular weight dependent lower logD limits based on permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- Furukawa A.; Townsend C. E.; Schwochert J.; Pye C. R.; Bednarek M. A.; Lokey R. S. Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem. 2016, 59, 9503–9512. 10.1021/acs.jmedchem.6b01246. [DOI] [PubMed] [Google Scholar]
- Boehm M.; Beaumont K.; Jones R.; Kalgutkar A. S.; Zhang L.; Atkinson K.; Bai G.; Brown J. A.; Eng H.; Goetz G. G.; Holder B. R.; Khunte B.; Lazzaro S.; Limberakis C.; Ryu S.; Shapiro M. J.; Tylaska L.; Yan J.; Turner R.; Leung S. S. F.; Ramaseshan M.; Price D. A.; Liras S.; Jacobson M. P.; Earp D. J.; Lokey R. S.; Mathiowetz A. M.; Menhaji-Klotz E. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide–Peptoid Hybrid CXCR7 Modulators. J. Med. Chem. 2017, 60, 9653–9663. 10.1021/acs.jmedchem.7b01028. [DOI] [PubMed] [Google Scholar]
- Russo G.; Barbato F.; Grumetto L.; Philippe L.; Lynen F.; Goetz G. H. Entry of therapeutics into the brain: Influence of exposed polarity calculated in silico and measured in vitro by supercritical fluid chromatography. Int. J. Pharm. 2019, 560, 294–305. 10.1016/j.ijpharm.2019.02.008. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Shalaeva M. Leveraging chromatography based physicochemical properties for efficient drug design. ADMET 2018, 6, 85–104. 10.5599/admet.529. [DOI] [Google Scholar]
- Meanwell N. A. Synopsys of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Landry M. L.; Crawford J. J. LogD contributions of Substituents Commonly Used in Medicinal Chemistry. ACS Med. Chem. Lett. 2020, 11, 72–76. 10.1021/acsmedchemlett.9b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalas P.; Gay B.; Lasfargeas C.; James M. J.; Tran V.; Vijayendran K. G.; Brunden K. R.; Kozlowski M. C.; Thomas C. J.; Smith A. B. III; Huryn D. M.; Ballatore C. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59, 3183–3203. 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As isosteres were typically less common than the corresponding parent structures, this approach provided fewer hits in the first stage of the protocol, making the MMP search more efficient overall.
- OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- Hawkins P. C. D.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant J. A.; Gallardo M. A.; Pickup B. J. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. . [DOI] [Google Scholar]
- McInnes L.; Healy J.; Astels S. hdbscan: Hierarchical density based clustering. Journal of Open Source Software 2017, 2, 205. 10.21105/joss.00205. [DOI] [Google Scholar]
- Schneider N.; Lowe D. M.; Sayle R. A.; Tarselli M. A.; Landrum G. A. Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem. 2016, 59, 4385–4402. 10.1021/acs.jmedchem.6b00153. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Altmann E.; McKenna J. M. The most Common Functional Groups in Bioactive Molecules and How Their Popularity has Evolved Over Time. J. Med. Chem. 2020, 63, 8408–8418. 10.1021/acs.jmedchem.0c00754. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. Defining Desirable Central Nervous System Drug Space through the Alignment of Molecular Properties, in Vitro ADME, and Safety Attributes. ACS Chem. Neurosci. 2010, 1, 420–434. 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S.; Carmona A. V.; Tiwari A. K.; Trippier P. C. Amide Bond Isosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. 10.1021/acs.jmedchem.0c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann W. K.; Durette P. L.; Lanza T.; Kevin N. J.; de Laszlo S. E.; Kopka I. E.; Young D.; Magriotis P. A.; Li B.; Lin L. S.; Yang G.; Kamenecka T.; Chang L. L.; Wilson J.; MacCoss M.; Mills S. G.; Van Riper G.; McCauley E.; Egger L. A.; Kidambi U.; Lyons K.; Vincent S.; Stearns R.; Colletti A.; Teffera J.; Tong S.; Fenyk-Melody J.; Owens K.; Levorse D.; Kim P.; Schmidt J. A.; Mumford R. A.. The discovery of sulfonylated dipeptides as potent VLA-4 antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 2709–2713. [DOI] [PubMed] [Google Scholar]
- Wu Z.-C.; Boger D. L. Maxamycins: Durable Antibiotics Derived by Rational Redesign of Vancomycin. Acc. Chem. Res. 2020, 53, 2587–2599. 10.1021/acs.accounts.0c00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- pKa values were calculated using ACD Classic: ACD/Percepta, ver. 2018.2.1; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2021; www.acdlabs.com. [Google Scholar]
- Amidines with moderate basicity (pKa ∼ 5–7) typically exhibited EPSA values no greater than 10 units above the matched amides, while with increased basicity, ΔEPSA could reach as high as + 44. See Supporting Information for more details.
- McLaughlin M.; Yazaki R.; Fessard T. C.; Carreira E. M. Oxetanyl Peptides: Novel Peptidomimetic Modules for Medicinal Chemistry. Org. Lett. 2014, 16, 4070–4073. 10.1021/ol501590n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doiron J. E.; Le C. A.; Bacsa J.; Breton G. W.; Martin K. L.; Aller S. G.; Turlington M. Structural Consequences of the 1,2,3-Triazole as an Amide Bioisostere in Analogues of the Cystic Fibrosis Drugs VX-809 and VX-770. ChemMedChem. 2020, 15, 1720–1730. 10.1002/cmdc.202000220. [DOI] [PubMed] [Google Scholar]
- Rečnik L.-M.; Kandioller W.; Mindt T. L. 1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear peptides with Biological Activity. Molecules 2020, 25, 3576. 10.3390/molecules25163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlbach C. L.; Lexa K. W.; Bockus A. T.; Chen V.; Crews P.; Jacobson M. P.; Lokey R. S. Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Future Med. Chem. 2015, 7, 2121. 10.4155/fmc.15.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia O.; Greenberg S.; Chatterjee J.; Laufer B.; Opperer F.; Kessler H.; Gilon C.; Hoffman A. The Effect of Multiple N-Methylation on Intestinal Permeability of Cyclic Hexapeptides. Mol. Pharmaceutics 2011, 8, 479–487. 10.1021/mp1003306. [DOI] [PubMed] [Google Scholar]
- Wang C. K.; Northfield S. E.; Colless B.; Chaousis S.; Hamernig I.; Lohman R.-J.; Nielsen D. S.; Schroeder C. I.; Liras S.; Price D. A.; Fairlie D. P.; Craik D. J. Rational design and synthesis of an orally bioavailable peptide guided by NMR amide temperature coefficients. Proc. Nat. Acad. Sci. U.S.A. 2014, 111, 17504–17509. 10.1073/pnas.1417611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono Y.; Morimoto J.; Townsend C.; Kelly C. N.; Naylor M. R.; Lee H.-W.; Lokey R. S.; Sando S. Amide-to-Ester Substitution Improves Membrane Permeability of a Cyclic Peptide Without Altering Its Three-Dimensional Structure. ChemRxiv 2020, 10.26434/chemrxiv.12272861.v1. [DOI] [Google Scholar]; This content is a preprint and has not been peer-reviewed.
- Klein V. G.; Bond A. G.; Craigon C.; Lokey R. S.; Ciulli A. Amide-to-Ester Substitution as a Strategy for Optimizing PROTAC Permeability and Cellular Activity. J. Med. Chem. 2021, 64, 18082–18101. 10.1021/acs.jmedchem.1c01496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Black W. C.; Bayly C. I.; Davis D. E.; Desmarais S.; Falgueyret J.-P.; Léger S.; Li C. S.; Massé F.; McKay D. J.; Palmer J. T.; Percival M. D.; Robichaud J.; Tsou N.; Zamboni R. Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 2005, 15, 4741–4744. 10.1016/j.bmcl.2005.07.071. [DOI] [PubMed] [Google Scholar]; b Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J.-P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V.-L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger. MacroModel, release 2020-1; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger. Jaguar, release 2020-1; Schrödinger, LLC: New York, NY, USA, 2020.
- Kenny P. W.; Montanari C. A.; Prokopczyk I. M.; Ribeiro J. F. R.; Sartori G. R. Hydrogen Bond Basicity Prediction for Medicinal Chemistry Design. J. Med. Chem. 2016, 59, 4278–4288. 10.1021/acs.jmedchem.5b01946. [DOI] [PubMed] [Google Scholar]
- Skonberg C.; Olsen J.; Madsen K. G.; Hansen S. H.; Grillo M. P. Metabolic activation of carboxylic acids. Expert Opin. Drug Metab. Toxicol. 2008, 4, 425–438. 10.1517/17425255.4.4.425. [DOI] [PubMed] [Google Scholar]
- Ballatore C.; Huryn D. M.; Smith A. B. III Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem. 2013, 8, 385–395. 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The dominance of carboxylic acids’ contribution to chromatographic interaction likely arises through correlated hydrogen bonding with the urea motif embedded in the stationary phase (Chirex 3014), an interaction that each of the carboxylic acid bioisosteres studied here are capable of mimicking, to varying extent.
- Reutershan M. H.; Machacek M. R.; Altman M. D.; Bogen S.; Cai M.; Cammarano C.; Chen D.; Christopher M.; Cryan J.; Daublain P.; Fradera X.; Geda P.; Goldenblatt P.; Hill A. D.; Kemper R. A.; Kutilek V.; Li C.; Martinez M.; McCoy M.; Nair L.; Pan W.; Thompson C. F.; Scapin G.; Shizuka M.; Spatz M. L.; Steinhuebel D.; Sun B.; Voss M. E.; Wang X.; Yang L.; Yeh T. C.; Dussault I.; Marshall C. G.; Trotter B. W. Discovery of MK-4688: an Efficient Inhibitor of the HDM2–p53 Protein–Protein Interaction. J. Med. Chem. 2021, 64, 16213. 10.1021/acs.jmedchem.1c01524. [DOI] [PubMed] [Google Scholar]
- Herr R. J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002, 10, 3379–3393. 10.1016/S0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- Juby P. F.3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones. U.S. Patent US 4,122,274, Oct. 24, 1978.
- Chiu A. T.; McCall D. E.; Price W. A.; Wong P. C.; Carini D. J.; Duncia J. V.; Wexler R. R.; Yoo S. E.; Johnson A. L.; Timmermans P. B. M. W. M. Nonpeptide Angiotensin II Receptor Antagonists. VII. Cellular and Biochemical Pharmacology of DuP 753, an Orally Active Antihypertensive Agent. Journal of Pharmacology and Experimental Therapeutics 1990, 252, 711–718. [PubMed] [Google Scholar]
- Bogan A. A.; Thorn K. S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Khazanov N. A.; Carlson H. A. Exploring the Composition of Protein-Ligand Binding Sites on a Large Scale. PloS Comput. Biol. 2013, 9, e1003321. 10.1371/journal.pcbi.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.-L.; Burnett D. A.; Spring R.; Greenlee W. J.; Smith M.; Favreau L.; Fawzi A.; Zhang H.; Lachowicz J. E. Dopamine D1/D5 Receptor Antagonists with Improved Pharmacokinetics: Design, Synthesis, and Biological Evaluation of Phenol Bioisosteric Analogues of Benazepine D1/D5 Antagonists. J. Med. Chem. 2005, 48, 680–693. 10.1021/jm030614p. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Angell R. M.; Bhamra I.; Brown D.; Bull J.; Christopher J. A.; Cooper A. W. J.; Fazal L. H.; Giordano I.; Hind L.; Patel V. K.; Ranshaw L. E.; Sims M. J.; Skone P. A.; Smith K. J.; Vickerstaff E.; Washington M. N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of Lck: Indazoles as phenol isosteres with improved pharmacokinetics. Bioorg. Med. Chem. Lett. 2007, 17, 4363–4368. 10.1016/j.bmcl.2007.04.029. [DOI] [PubMed] [Google Scholar]
- Zafrani Y.; Sod-Moriah G.; Yeffet D.; Berliner A.; Amir D.; Marciano D.; Elias S.; Katalan S.; Ashkenazi N.; Madmon M.; Gershonov E.; Saphier S. CF2H, a Functional Group-Dependent Hydrogen-Bond Donor: Is it a More or Less Lipophilic Bioisostere of OH, SH, and CH3?. J. Med. Chem. 2019, 62, 5628–5637. 10.1021/acs.jmedchem.9b00604. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; McLoughlin J. I. Hydrogen Bond Donor Properties of the Difluoromethyl Group. J. Org. Chem. 1995, 60, 1626–1631. 10.1021/jo00111a021. [DOI] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.