

Abstract
The CNS regulates body weight; however, we still lack a clear understanding of what drives decisions about when, how much and what to eat. A vast array of peripheral signals provides information to the CNS regarding fluctuations in energy status. The CNS then integrates this information to influence acute feeding behaviour and long-term energy homeostasis. Previous paradigms have delegated the control of long-term energy homeostasis to the hypothalamus and short-term changes in feeding behaviour to the hindbrain. However, recent studies have identified target hindbrain neurocircuitry that integrates the orchestration of individual bouts of ingestion with the long-term regulation of energy balance.
Body weight (or more accurately body fat) is a homeo-statically regulated parameter similar to many other physiological processes. Changes in body fat are the product of the difference between energy intake and energy expended. This defence of body fat is orchestrated by the CNS. To accomplish this, minute-by-minute decisions on feeding must be integrated with long-term regulation of energy homeostasis. This regulation requires the integration of a wide range of different signals from the periphery that provide information on the status of various peripheral organs crucial to food absorption, energy storage or energy consumption. Historically, long-term energy balance regulation was thought to be directed by peripheral signals linked to energy storage in the form of adipose tissue, and short-term aspects of food intake regulation were thought to be controlled by signals originating from the gastrointestinal (GI) tract.
Recently, a more complex model is emerging. In particular, bariatric surgeries that manipulate the GI tract result in profound and sustained weight loss. This finding indicates that signals from the GI tract must impact not just decisions about the size of individual meals but also the long-term regulation of energy balance. As research has delved into the mechanisms that underlie the success of surgery, an expanding list of peripheral signals that act centrally to regulate energy homeostasis has been identified. In addition, new chemogenetic and optogenetic techniques are identifying target neurocircuitry that integrates the orchestration of individual bouts of ingestion with the long-term regulation of energy balance. This Review discusses the peripheral signals and nervous system (specifically the hypothalamus, the hindbrain and the vagus nerve) targets that have been implicated in the short-term and long-term regulation of energy homeostasis.
Vagus nerve.
The longest cranial nerve. It contains both motor and sensory fibres involved in the parasympathetic regulation of homeostatic processes.
Peripheral signals in feeding regulation
As a result of nutrient ingestion, the plasma levels of many gut peptides increase. The function of this increase is to regulate short-term satiety by altering the size and frequency of meals. Some of the more widely studied intestinal-secreted peptides include glucagon-like peptide 1 (GLP1), peptide YY (PYY) and cholecystokinin (CCK). These peptides are secreted by specialized enteroendocrine cells that have historically been defined by the peptides they secrete (the specifics on the regulation of secretion of these gut peptides are discussed in more detail in BOX 1). However, data that are more recent suggest that a distinction based on location within the intestine is more appropriate. For example, GLP1 cells within the proximal gut secrete both GLP1 and CCK, whereas the distal GLP1 cells secrete GLP1 and PYY but not CCK1.
Box 1 |. Gut peptide secretion.
Gut peptides are secreted from specialized intestinal endocrine cells called enteroendocrine cells. These peptides are secreted in response to nutrient ingestion and have widespread physiological effects. Depending on anatomical location, the various enteroendocrine cells release different peptides. For example, ghrelin is secreted from the stomach, whereas cholecystokinin (CCK) and gastric inhibitory polypeptide (GIP) are secreted from the proximal gut by I (REF. 130) and K cells, respectively. CCK reduces meal size, whereas GIP is thought of mostly for its ability to stimulate insulin secretion (see REF. 3 for review). By contrast, ghrelin is secreted from gastric X cells, is at its highest concentration during fasting and is suppressed by feeding3. The fasting-induced rise, but not the nutrient-induced suppression, of ghrelin is blocked by subdiaphragmatic vagotomy, indicating that two independent pathways regulate the nutrient-associated fluctuations in ghrelin131. In its active and acylated form, ghrelin increases appetite, gastric acid secretion and gastric motility132,133. Interestingly, compared with an isocalorific fat meal, protein was found to have a more potent suppressive effect on ghrelin levels, and both protein and fat have a more prolonged suppressive effect than carbohydrates134.
Glucagon-like peptide 1 (GLP1) and peptide YY (PYY) are secreted predominantly from enteroendocrine cells located in the distal intestine. Plasma GLP1 and PYY are co-secreted almost immediately after glucose ingestion2, and both peptides reduce food intake135. After protein and lipid ingestion, the secretion of GLP1 is slower and more sustained than after carbohydrate ingestion. However, in response to a mixed meal, GLP1 shows a biphasic response with peaks at 15 minutes and 2 hours postprandially (reviewed in REF. 3). Although GLP1-expressing cells are found throughout the small intestine1, PYY-secreting cells are found only in the distal gut1,3. Also in contrast to GLP1, enzymatic cleavage of PYY converts PYY1–36 to PYY3–36, its physiologically active form136. This cleavage process results in a slower postprandial rise in plasma PYY3.
Enteroendocrine cells.
Specialized endocrine cells within the intestine that secrete peptides important for regulating feeding and metabolism.
From the distal gut, GLP1 and PYY are co-secreted postprandially from the same enteroendocrine cells1–3. Genetic disruption and/or pharmacological manipulation of GLP1 and PYY reveal that these peptides do not just regulate the size of individual meals but also influence long-term energy balance. PYY-knockout (KO) mice are hyperphagic and obese; these effects are reversed by exogenous PYY administration4. Although whole-body GLP1 receptor (GLP1R)5 and preproglucagon (Gcg, the gene that codes for GLP1)6 KO mice have levels of adiposity similar to those of their wild-type littermates, exogenous intravenous administration of GLP1 has been found to reduce food intake in healthy-weight, obese and type 2 diabetes mellitus (T2DM) subjects7,8. Furthermore, long-acting agonists for GLP1R have been approved in the United States and Europe for the treatment of obesity. These data indicate that at least pharmacological manipulation of GLP1 influences long-term regulation of body mass.
In contrast to the above primarily distally secreted peptides, CCK, also an appetite suppressant, is secreted from the duodenum. Although CCK has two known receptors, CCK-A receptor (CCK-AR) and CCK-B receptor (CCK-BR), the satiety effect of CCK seems to be due to activation of CCK-AR rather than CCK-BR. The fact that CCK-AR antagonists increase feeding supports a role for endogenous CCK in the regulation of feeding3. However, when administered over time, CCK reduces meal size but increases meal number, leading to no net change in food intake9. These data have been cited as evidence of the traditional model of gut signals being involved in the regulation of meals but not in long-term energy homeostasis. Despite this, CCK does appear to be important for long-term regulation of body weight as CCK-AR polymorphisms have been reported to be associated with obesity10,11. Thus, CCK is another example of a short-term regulator of feeding that can contribute to long-term energy balance.
In contrast to the above-mentioned gut-secreted peptides that are thought to regulate individual meals, leptin, secreted from and circulating in proportion to adiposity, is an indicator of long-term energy stores. Insulin, a pancreatic hormone crucial for blood glucose regulation, also circulates in proportion to body fat content12,13; thus, insulin is also thought to serve as an adiposity signal. Direct administration of both leptin and insulin has been found to reduce food intake14,15; conversely, insulin or leptin deficiency has been found to increase food intake14,16. The predominance of literature that has explored the site of action for leptin and insulin in the regulation of energy homeostasis has focused on a model in which these hormones first act within the hypothalamus, which then activates the hindbrain and in turn lead to changes in feeding behaviour.
Ghrelin is secreted from enteroendocrine cells of the stomach. However, circulating ghrelin is highest in a fasted state, and levels are suppressed by feeding. In contrast to the above satiety peptides, central or peripheral administration of ghrelin (thought to be one of the only orexigenic hormones secreted from the gut) leads to rapid but short-lived increases in food intake17. However, there is also a case to be made for a role of ghrelin in long-term regulation of body weight. Like insulin and leptin, ghrelin circulates in proportion to adiposity18. Chronic administration of ghrelin increases body mass17, and disruption of ghrelin signalling makes mice resistant to weight gain in response to obesogenic diets19.
Altogether, the data discussed in this section highlight the traditional ‘players’ involved in the regulation of meals and in long-term energy homeostasis. Although there may be some credence in distinguishing signals by short-term and long-term regulators of energy status, there is also evidence for integration of these two processes as deficiency in some short-term signals leads to adiposity. Interestingly, with the exception of ghrelin, these peptides have clear anorectic effects, but only GLP1 has been successfully targeted for the treatment of obesity. Clinical targeting of CCK was ceased owing to the impact of pharmacological activation of CCK-AR on increasing pancreatitis. Leptin is very potent at suppressing feeding; however, owing to diet-induced or obesity-induced changes to leptin signalling, its clinical efficacy is limited to patients with genetic leptin deficiency. The anorectic effect of insulin is isolated to its CNS actions, and this is at odds with its potent anabolic actions in the periphery. Thus, to effectively utilize any of these peptides to treat obesity, understanding the underlying neurocircuitry that drives their efficacy is critical.
Neural circuitry in regulating energy homeostasis
The discovery of leptin and subsequent work defining the hypothalamic neurocircuitry (FIG. 1) through which leptin regulates energy homeostasis revolutionized the way we think about body-weight regulation. This approach was hypothalamic centric and focused on the descending projections from the hypothalamus to the hindbrain. At that time, the role of the hindbrain was thought to be more important for regulating acute feeding behaviour rather than long-term energy homeostasis. More complex genetic strategies have allowed for more direct manipulation of expression and now even activity of specific neurons and/or specific regions of the CNS. These technologies have led to the development of a more complex but integrated model whereby peripheral signals activate both descending (hypothalamus to hindbrain) and ascending (hindbrain to hypothalamus) neurocircuits that are essential for regulation of energy homeostasis. This section discusses the historical role of the hypothalamus but also highlights recent data delineating the complex neurocircuitry linking the hindbrain, specifically the nucleus of the solitary tract (NTS), to higher brain centres in the regulation of energy homeostasis.
Figure 1 |. Peripheral-to-CNS signals of energy status.
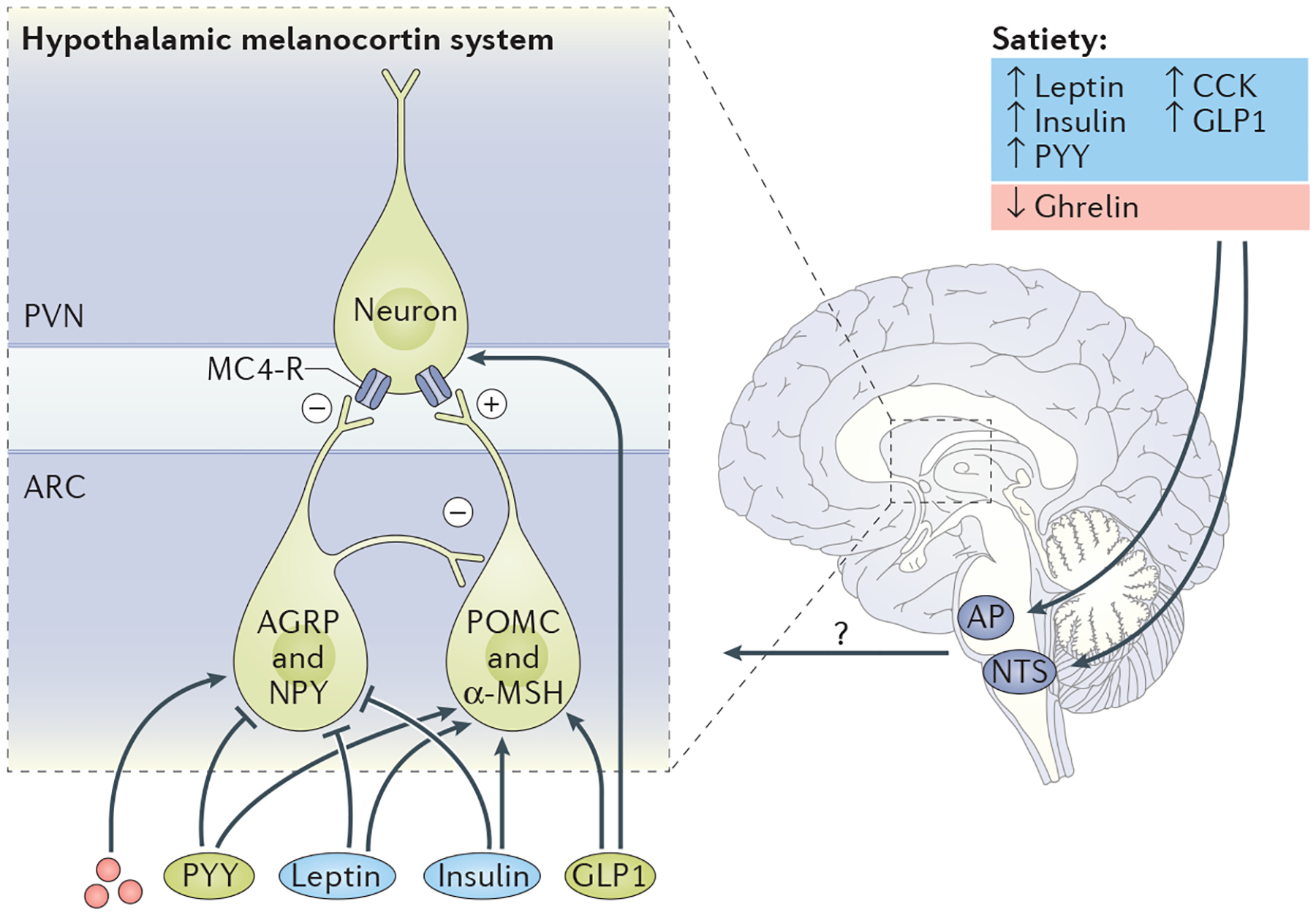
Gut-secreted peptides, such as glucagon-like peptide 1 (GLP1), cholecystokinin (CCK) and peptide YY (PYY), are secreted from the gastrointestinal tract in response to nutrient ingestion. Conversely, ghrelin levels are highest during fasting and are suppressed during a meal. Leptin and insulin, secreted from adipose and pancreatic cells, respectively, circulate in proportion to adiposity and therefore represent signals of long-term energy storage. The ‘hypothalamic-centric’ view has held that long-term energy homeostasis is regulated by the hypothalamic melanocortin system (agouti-related protein (AGRP)/neuropeptide Y (NPY) neurons and pro-opiomelanocortin (POMC) neurons). Indeed, leptin and insulin regulate the activity of these neuronal populations, and at least some gut peptides may also regulate the hypothalamic melanocortin system. However, there is an increasingly appreciated role of the ascending circuits from the hindbrain to the hypothalamus that are critical in regulating both short-term and long-term energy homeostasis. ?, unidentified peptides or circuits that regulate energy homeostasis; α-MSH, α-melanocyte-stimulating hormone; AP, area postrema; ARC, arcuate nucleus; MC4-R, melanocortin receptor 4; NTS, nucleus of the solitary tract; PVN, paraventricular nucleus.
The hypothalamus
One of the most intensively studied populations of hypothalamic neurons that regulate energy homeostasis is within the arcuate nucleus (ARC) of the hypothalamus and is composed of the agouti-related protein (AGRP) /neuropeptide Y (NPY) and pro-opiomelanocortin (POMC) neurons. These neurons serve to stimulate and inhibit feeding, respectively. There is extensive research that has focused on the CNS action of leptin and insulin on these neuronal populations. Both POMC and AGRP/NPY neurons express leptin and insulin receptors. In addition, both insulin and leptin have been found to activate the POMC anorexic pathways and, conversely, to inhibit the AGRP/NPY orexigenic pathways20, suggesting that these neurons are critical for leptin action. However, specific deletion of hypothalamic leptin receptors on POMC neurons or within discrete areas of the hypothalamus fails to replicate the phenotype of the whole-body leptin-receptor KO21–23, leading to the speculation that extrahypothalamic leptin receptors are more critical in leptin action.
Despite being thought of primarily as regulators of satiety, both AGRP/NPY and POMC neuronal populations also contain receptors for PYY and ghrelin24,25, but GLP1Rs are found only on a small population of POMC neurons20. Assuming that circulatory factors can penetrate the ARC, these neurons could then directly sense changes in any of these circulating factors and, in turn, regulate food intake. Certainly, this region is responsive to exogenous administration of these peptides. Indeed, direct intra-ARC administration of GLP1 (REF. 26) and PYY27 reduced feeding, whereas intra-ARC ghrelin administration28 increased feeding. However, for GLP1, the anorexic effect is more potent when directly injected into the paraventricular nucleus versus the ARC of the hypothalamus26,29,30. Although GLP1Rs are found on POMC neurons, deletion of these specific receptors using the Cre–LoxP system results in no metabolic phenotype, and these mice are equally responsive to a long-acting GLP1 agonist26. These data suggest that POMC GLP1Rs are not necessary for the anorexic effect of GLP1. By contrast, delivery of PYY directly into the ARC suppressed food intake, reduced NPY expression and increased FOS expression (the latter being a marker for neuronal activity)27, and PYY increased the electrical activity of isolated POMC neurons27. Altogether, these data suggest that both AGRP/NPY and POMC neurons are potential regions of the CNS for which PYY can regulate feeding. Conversely, the orexigenic effect of ghrelin is thought to be via activation of receptors located on NPY rather than POMC neurons31. All of these data demonstrate that exogenous administration of GLP1, PYY and ghrelin can act via POMC and/or AGRP/NPY neurons to regulate feeding. However, whether endogenous sources of these peptides are able to reach these neurons is less clear. The median eminence, located just below the ARC, is a CNS region with a leaky blood–brain barrier, suggesting the possibility that these circulating gut peptides reach the ARC; however, the circulating half-life, certainly of GLP1, is limited (<2 minutes), which leaves this possibility debatable.
In the past few years, new technologies, including optogenetics and DREADDs (designer receptors exclusively activated by designer drugs), have revealed a more complicated role for these neurons. The basis for these technologies is discussed in greater detail in BOX 2. These technologies were initially applied to AGRP and POMC neurons, likely initially as a proof of concept. However, the data have revealed a more in-depth understanding of how these neuronal populations interact. Both optogenetic and chemogenetic activation of AGRP neurons immediately evoked voracious food intake whether the animals were fed or fasted32,33. By contrast, POMC neurons in the ARC need to be chronically stimulated in order to decrease feeding32,34. These data underscore the role of POMC neurons in the regulation of long-term energy homeostasis but also suggest that AGRP neurons can regulate meal patterning. Stimulation of POMC neurons causes the release of α-melanocyte-stimulating hormone, which binds to downstream receptors in the paraventricular hypothalamus, a process necessary for its satiety effect. The ability of photoactivation of POMC neurons to suppress feeding was found to be dependent upon melanocortin receptor activity32. Although AGRP inhibits POMC neuronal activity, photostimulation of AGRP neurons evoked a feeding response that is independent of the melanocortin system32. Further clarification of the circuit has revealed that stimulation of AGRP neurons activates not only neurons within the paraventricular nucleus of the hypothalamus but also neurons in the lateral hypothalamus and dorsal medial neurons in the bed nucleus of the stria terminalis to regulate feeding35. Thus, AGRP regulates feeding in both melanocortin-dependent and melanocortin-independent pathways. Together, these studies have expanded what we know about the hypothalamic neurocircuitry stemming from AGRP and POMC neurons in the regulation of energy homeostasis. The results of the study emphasize the role of the hypothalamic AGRP neurons as a broader homeostatic centre that regulates not only long-term homeostatic control of feeding but also short-term control of meal initiation.
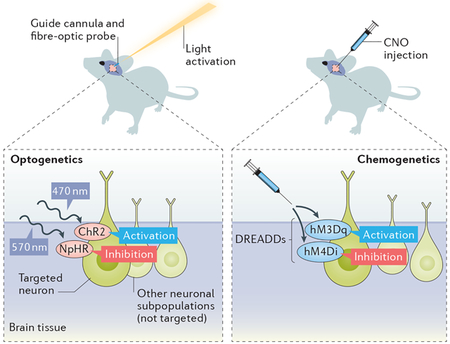
Box 2 |. Optogenetic and chemogenetic activation of CNS neurons.
The increasing availability of two particular techniques, optogenetics and chemogenetics, that allow for acute regulation of nerve activity has provided a new frontier in our understanding of CNS-induced regulation of energy homeostasis.
Optogenetics.
(see the figure, right) In optogenetics, light-responsive opsin proteins that serve as ion channels are genetically incorporated into targeted neuronal populations using cell-type-specific promoters that drive Cre recombinase. This incorporation enables the light-manipulated activation (channelrhodopsin 2 (ChR2)) or inhibition (halorhodopsin (NpHR) or archaerhodopsin) of neuronal activity. To do this, a guide cannula targeted to the region of interest is surgically implanted into the rodent’s brain. An ultrafine fibre-optic probe that emits light-emitting diode (LED) light is inserted into the guide cannula, and modulation of the light intensity and frequency then regulates the activity of the opsin-expressing neurons so that the resulting behaviour can be assessed. This method enables the manipulation of neuronal activity within a very short time frame, and the intensity of the light can be adjusted to increase or decrease neuronal activity.
Chemogenetics.
(see the figure, left) Although optogenetics utilizes the expression of light-sensitive ion channels, chemogenetics uses the same cell-type-specific promoters to drive Cre recombinase to incorporate chemically engineered receptors called DREADDs (designer receptors exclusively activated by designer drugs) into neurons of interest. These receptors bind to a specific physiologically inert drug, clozapine N-oxide (CNO). Depending on which receptors are introduced, these G-protein-coupled muscarinic receptors either excite (hM3Dq) or inhibit (hM4Di) the neurons of interest when exposed to CNO.
Strengths and weaknesses of these techniques.
Both chemogenetics and optogenetics enable researchers to regulate the specific neuronal activity and to monitor behavioural changes or to identify downstream circuits. Unlike optogenetics, which provides millisecond precision, chemogenetic manipulation can take several hours. However, the advantage of chemogenetics is that it can be applied for an extended period of time to understand the long-term impact of activating or inhibiting the neurons of interest137. Both can be limited by the availability or specificity of the Cre-driven promoters available. Surgical equipment and expertise are necessary if using adeno-associated viruses to incorporate the optogenetic or chemogenetic proteins and also for inserting the guide cannula that will hold the fibre-optic cable for optogenetics. Additionally, light penetration into the tissue can be a limitation of optogenetics, and this technique cannot be applied if the cells of interest have dispersed neuroanatomy. With chemogenetics, an assumption critical for the success of the technique is that CNO is physiologically inert in control animals. However, a recent study found that a metabolite of CNO, clozapine, has off-target metabolic effects138, highlighting the importance of using CNO-injected control groups.
The peripheral nervous system
Because of the rapidity of changes in feeding behaviour in response to peripherally derived signals, the role of the peripheral nervous system has been hypothesized to be essential for short-term meal regulation. Vagal afferent neurons innervate the intestinal crypts and villi throughout the intestine36, the stomach and the portal vein37 (the vein that receives blood flow from the intestine). All of these nerve endings are optimally placed to receive incoming signals from ingested food. Evidence suggests that these neurons sense not only the type but also (via mechanical feedback) the volume of ingested nutrients. Some evidence suggests that gut peptides such as GLP1 and CCK also regulate feeding via vagal afferents. Thus, it is possible that nutrient signals indirectly activate the peripheral nervous system by increasing gut peptides but could also directly activate nutrient sensors expressed within the vagal neurons themselves (FIG. 2).
Figure 2 |. Endocrine and neuronal pathways to the CNS.
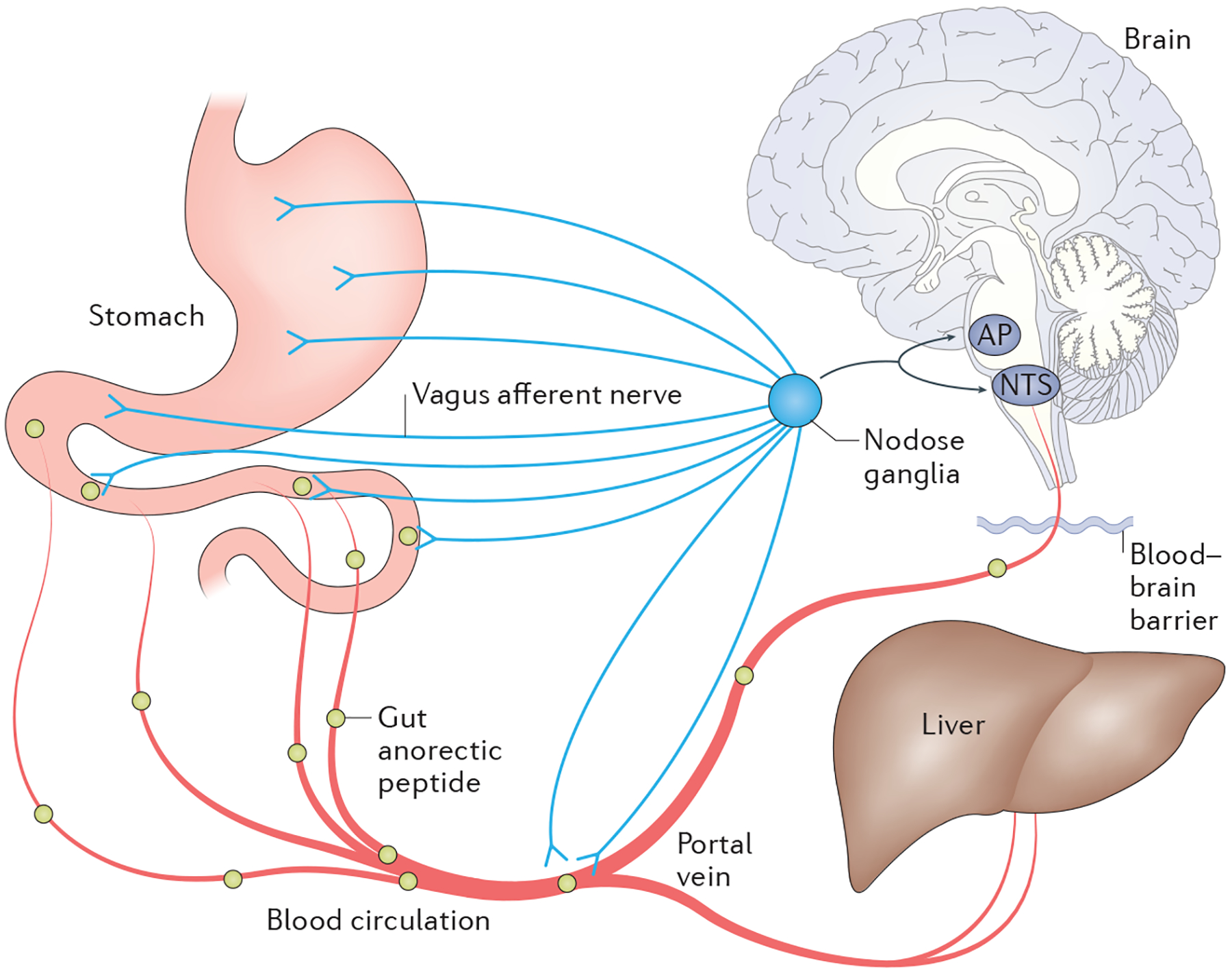
The nodose ganglion (cell bodies of the vagus nerve) connects peripheral innervation of the stomach, intestine and portal vein to the hindbrain. These afferent nerve endings have receptors that can bind to many hormones and nutrients. Furthermore, blood flow from the intestine dumps first into the portal vein and then into the general circulation. Thus, hormones and nutrients that are increased postprandially can have endocrine or direct neural pathways to signal the CNS about the changes in nutrient status. AP, area postrema; NTS, nucleus of the solitary tract.
Afferent neurons.
Peripheral sensory neurons that carry ascending nerve impulses from peripheral organs to the brain and spinal cord.
Before the discovery of the many gut and other peripherally secreted hormones that act via the peripheral and/or CNS to regulate feeding, it was hypothesized that ingested nutrients themselves were responsible for changes in feeding behaviour38. Since then, physiological studies have demonstrated that vagal innervation is necessary for nutrient-induced regulation of feeding and glucose homeostasis39, that vagal neurons are directly stimulated by specific types of nutrients40–42 and that the specific intracellular machinery located within these neuronal populations is necessary for regulation of energy homeostasis43,44. Although drugs that inhibit both glucose and fatty acid metabolism increase feeding, glucoprivic agents are hypothesized to directly activate the CNS, whereas lipoprivic agents work via vagal activation45. Such work supports the hypothesis that nutrients regulate feeding, at least partially, through vagal signals. However, until recently, the mechanism(s) underlying direct activation were unknown.
Nutrient receptors and transporters are expressed within the vagus nerve. In regards to lipid sensing, the stimulatory effect of mercaptoacetate, a lipoprivic agent, is dependent upon a specific G-protein-coupled receptor (GPCR), GPCR 40 (GPR40; also known as FFAR1), that is activated by long-chain fatty acids46,47. Further support that these neurons have the necessary intracellular machinery to sense lipids is demonstrated by data in which several nuclear transcription factors that are activated by nutrients have been identified within the nodose ganglion, the cell bodies of the vagus nerve. These include liver X receptor-α (LXRA; also known as NR1H3), LXRB, peroxisome proliferator-activated receptor-α (PPARα) and PPARγ43. Additionally, nodose ganglion expression of PPARγ43 and LXRA and/or LXRB44 are both necessary for diet-induced weight gain. In both cases, the impact on body weight was due to stimulation of dietary-induced thermogenesis rather than altered feeding, demonstrating that the gut–brain axis does more than just regulate acute feeding patterns. These data provide clues into the specific intracellular machinery that is important for nutrient sensing, but how those nutrients (in this case lipids) come to interact with this intracellular machinery remains unknown. For example, whether PPARγ is translocated to the terminals or whether it is downstream of fatty acid-induced activation of terminal receptors is an open question.
G-Protein-coupled receptor (GPCR).
Seven-transmembrane receptor that is coupled to, and activates, a heterotrimeric G protein, which subsequently activates a series of downstream signalling cascades; this receptor class is large and diverse and binds to a variety of nutrients and hormones.
Nodose ganglion.
Contains the cell bodies of neurons of the vagus nerve.
Nutrient sensing.
A process by which nutrients or their by-products directly activate cell signalling cascades that, in turn, regulate metabolism.
More recent data have identified the genotypes of populations of vagal neurons that are distinct in innervation, CNS projections and function. In vivo calcium imaging of GLP1R-expressing vagal neurons found that the majority of these neurons increased calcium in response to gastric and intestinal distention but not in response to nutrient ingestion, suggesting that these neurons are mechano-sensitive and not nutrient sensitive48. By contrast, in vivo calcium imaging of vagal neurons expressing a GPCR called GPR65 indicates that these neurons increase calcium levels in response to nutrients but not mechanical stimuli. GPR65-expressing neurons have a much greater innervation of intestinal villi and project to the NTS commissural zone, whereas GLP1R-expressing neurons innervate the stomach and intestinal muscle and project to the medial NTS. Altogether, more and more data are accumulating to indicate that the nodose ganglion is home to several distinct populations of neurons that regulate different physiological processes. Furthermore, several nutrient sensors are expressed within the apical membrane of gut epithelia, including sodium-gated glucose transporters (Na+/glucose cotransporter 1 (SGLT1; also known as SLC5A1)), glucose transporter 2, sweet and bitter taste receptors, many other GPCRs (GPR40 and GPR119) and so forth. The extent to which these sensors are linked to direct vagal activation will likely be an important direction of future work delineating the function of the gut–brain axis in the regulation of feeding.
Although we have much to learn regarding if and how nutrients are directly sensed by the vagus nerve to regulate feeding, it is clear that nutrients indirectly regulate vagal activity by initiating the release of gut peptides and neurotransmitters from neighbouring enteroendocrine cells49, which we know regulate feeding. For example, glucose has been found to directly stimulate the release of serotonin from enterochromaffin cells50 and to modulate vagal activity by regulating the trafficking of serotonin receptors to the neuronal membrane, enhancing exposure to the released serotonin51.
The role of the vagus nerve in mediating the impact of gut-secreted peptides has been extensively studied but primarily through chemical or surgical ablation approaches. Although some have shown that portal vein infusion of GLP1 regulated feeding patterns52, general food intake was not suppressed in rats53,54. Total vagotomy attenuates the inhibition of food intake by both PYY and GLP1 in rodents55 and in patients who have had vagotomy plus pyroplasty56. However, a confounding issue with these types of studies is that vagotomy ablates both vagal afferent and efferent neurons and causes alterations in gut motility that could independently regulate feeding. An alternative surgical ablation approach has been subdiaphragmatic deafferentation, which removes all afferent and about 50% of efferent fibres. Using this more specific approach, feeding responses to GLP1 (REF. 54), PYY27,55 and long-acting GLP1 (REF. 57) and PYY58 agonists are also blunted, suggesting that afferent fibres are necessary for these L cell-secreted peptides to suppress feeding. However, this effect could be overcome with higher doses of PYY59 and GLP1 (REF. 57). Whether this latter effect reflects an impact of these higher doses of GLP1 and/or PYY on the CNS, rather than vagal receptors, or the lack of importance of vagal afferents on the action of these two peptides remains unknown. Lastly, although some studies do not demonstrate a role for the vagus nerve in mediating the satiating effect of CCK60,61, CCK does regulate vagal activity. A recent study using injection of a neurotoxin specific to CCK into the nodose ganglion abolished the anorexic effect of exogenous CCK62. However, given that these studies also use exogenous administration of the peptide of interest, it remains unknown whether these gut peptides play a physiological role in regulating feeding through the vagus nerve.
Efferent neurons.
Peripheral motor neurons that carry descending nerve impulses from the CNS to peripheral organs.
A more specific way to target these sensory neurons is via genetic manipulation. Interestingly, the long-term energy homeostasis signal, leptin, may act via the vagus nerve to regulate body weight. Leptin receptor expression has been found in the nodose ganglion, and genetic ablation of these receptors using tissue-specific genetic strategies resulted in increased body mass and food intake with no impact on energy expenditure and reduced sensitivity to CCK-induced anorexia63.
By contrast, mice with a genetic knockdown of GLP1Rs within the nodose ganglion have no significant changes in long-term body mass, food intake or gastric-emptying rate compared with control animals64. In addition, they have normal weight loss in response to a long-acting GLP1 agonist64. Consistent with these latter findings, long-acting GLP1 agonists do not activate vagal afferents innervating the stomach48. Although lentiviral-mediated knockdown of GLP1R specifically within the nodose ganglion resulted in no long-term changes in body weight, it did result in increased meal size, accelerated gastric-emptying rate and increased postprandial glucose levels65. Taken as a whole, the data indicate that GLP1 certainly regulates vagal activity, feeding patterns and GI function but is not necessary for long-term energy homeostasis. The bottom line from all of this is that as technologies improve, researchers may be able to get a grasp on the confounding variables that limit the ability to dissociate the physiological versus the pharmacological impact of these gut peptides on the vagus nerve and, consequently, a better understanding of the factors, be they nutrient or hormonal, that impact the role of the vagus nerve in regulating meal intake.
The hindbrain
Vagal afferent neurons terminate within the NTS66. Again, historically, this region of the brain was thought to be more specifically involved in regulating acute feeding behaviour. However, recent work has highlighted its role in long-term energy balance as well.
Several gut peptides have receptors located throughout the hindbrain region and have been found to activate this region. For example, peripherally administered PYY has been demonstrated to activate neurons within the area postrema (AP) and NTS59, and CNS administration of GLP1 suppresses feeding when administered into various CNS regions, including the NTS67. The fact that GLP1 is also made in the NTS suggests an interesting possibility that gut and CNS GLP1 systems are integrated in the regulation of energy homeostasis. However, GLP1Rs are not colocalized to GCG neurons, nor do isolated GCG neurons respond to GLP1 (REF. 68). Currently, it remains unclear whether their is redundancy between the peripheral and CNS sources of GLP1, whether they act on distinct targets and have distinct functions. In fact, GCG neurons seem to be more important for regulation of long-term body weight. For example, chronic downregulation of GCG within the NTS increased body mass and food intake in rats69, and chemogenetic stimulation of GCG neurons in mice reduced food intake, but this was dependent upon the metabolic state of the animal70. A caveat to this is that mice with genetic downregulation of the CNS GLP1R had normal body mass whether fed chow or a high-fat diet, suggesting that these receptors are not necessary for normal body-weight regulation64. Comparisons across these studies are difficult in that some are in rats and some are in mice. Although there are few studies that compare rats and mice, one study did find that GLP1R signalling was necessary for the aversive response to lithium chloride in rats but not in mice71. Thus, whether these discordant results are due to species differences, to developmental compensation in the mice or to some other methodological issues is an open question. Regardless, these data highlight that at least one set of hindbrain neurons that express GCG are important in the regulation of long-term energy homeostasis.
Although the role of leptin in regulating energy homeostasis has historically focused on the hypothalamus as a central locale for mediating this effect, targeted deletion of leptin receptors within specific hypothalamic nuclei has failed to initiate the degree of metabolic impairments seen in the diabetic (db/db) mouse21–23. Leptin receptors are also found within the AP and the NTS72,73, and leptin injections directly into the NTS suppressed feeding74. However, neurons within the hindbrain region that contain the leptin receptor are diverse. For example, leptin receptors within the NTS are colocalized with distinct subpopulations of GCG-expressing versus CCK-expressing neurons75. Interestingly, both leptin68 and CCK76 have been shown to depolarize isolated GCG neurons, suggesting that leptin directly, although CCK indirectly, activates GCG neurons. Interestingly, another population of leptin receptors are located on POMC neurons that are also expressed in the NTS. To distinguish between the functions of ARC and NTS POMC neurons, one study injected a Cre-dependent adeno-associated virus expressing the activating DREADD into the ARC or the NTS and then administered clozapine N-oxide (CNO)34. The authors found that activation of POMC neurons within the NTS suppressed feeding within hours, whereas it took repeated administrations of CNO for at least 4 days to reduce feeding when POMC neurons within the ARC were activated34. Although these data suggest the interesting possibility that distinct populations of POMC neurons differentially regulate short-term versus long-term feeding, the expression of POMC within the NTS is thought to be very low75, suggesting that these effects in the NTS are pharmacological.
Once activated, NTS neurons activate other brain regions, which in turn regulate feeding behaviour and energy homeostasis. One specific neuronal circuit receiving NTS projections is the lateral parabrachial nucleus (PBN). The PBN has been identified as a critical nucleus for regulating feeding responses to leptin, PYY and GLP1 (REFS 77–79). However, the identities of the neurons important for this effect have only recently been discovered. Calcitonin gene-related peptide (CGRP; also known as CALC)-expressing neurons within the PBN are activated by feeding, and chemogenetic activation initiated meal termination and suppressed subsequent meal initiation80. Although silencing of these neurons using Cre-dependent expression of tetanus toxin light chain (tetanus toxin chain L) increased meal size and duration, it also decreased meal frequency such that overall body mass was not affected. Interestingly, these mice also did not suppress feeding in response to CCK and leptin and had a blunted response to a long-acting GLP1 agonist (exendin 4). The authors went on to define two distinct sets of upstream neurons that, when chemogenetically stimulated, excited PBN CGRP neurons (as indicated by FOS) and suppressed feeding: one set expressing CCK and another expressing the noradrenergic neurotransmitter dopamine β-hydroxylase81. These data highlight the complexity of the neural networks that regulate feeding behaviour and may shed light on why obesity, and presumably disruptions to these neural networks, is so difficult to treat.
Although we have separated the discussion of the hypothalamus and NTS in this Review, it is important to highlight some of the neural networks that link the NTS to the hypothalamus, and specifically to POMC and AGRP neurons. With regards to the PBN, there are dense projections from the AGRP to the lateral PBN that are not colocalized with CGRP neurons, indicating that AGRP neurons inhibit CGRP neurons indirectly through other PBN circuits82.
One other hindbrain nucleus that receives neuronal inputs from the NTS is the dorsal raphe nucleus (DRN). The DRN has been implicated in the control of feeding83 and has reciprocal connections with numerous hypo thalamic nuclei84. Photogenetic and chemogenetic activation of DRN neurons expressing vesicular GABA transporter (vGAT; also known as SLC32A1), an inhibitory neurotransmitter, and glutamate, an excitatory neurotransmitter, increased and suppressed feeding, respectively85. Chemogenetic inhibition of DRN vGAT neurons in obese (ob/ob) mice reduced food intake and caused weight loss, suggesting that the action of these neurons is independent of leptin. Further work is needed to clarify the projections from these neurons that are critical in mediating their effect on regulating feeding behaviour.
The important point from the current work on the role of the hindbrain in energy homeostasis is that, in addition to critical circuits from the hypothalamus to the hindbrain that are important for regulating energy homeostasis, there are also ascending circuits from the hindbrain that may be just as critical. Identification of these circuits and understanding the interaction of these two CNS regions will be important in understanding the neurophysiology underlying body-weight control.
Newly identified peripheral signals
Decades of research have been spent on understanding the impact of peripherally derived signals such as GLP1, CCK, PYY and leptin on CNS regulation of short-term and long-term energy balance. Although there is no doubt that these peptides are important players in the regulation of energy homeostasis, there are more and more peripherally derived signals being discovered to play a role in communicating energy status to the CNS. One example is bile acids. Synthesized from cholesterol in the liver and secreted into the duodenum after a meal, bile acids have historically been thought to function solely as lipid emulsifiers86. However, bile acids are now recognized as critical signalling molecules. Bile acids have been found to signal through two receptors: GPCR19 (also known as TGR5 or BPBAR1) and the nuclear transcription factor farnesoid X-activated receptor (FXR; also known as NR1H4). Both receptors are expressed in several tissues, including the small intestine, liver and adipose tissue. Within the intestine, bile acid activation of TGR5 stimulates GLP1 secretion87 and increases colonic peristalsis88, whereas bile-acid-induced activation of FXR results in the secretion of fibroblast growth factor 19 (FGF19; FGF15 is the mouse orthologue)87 into the circulation. FGF15 and FGF19 signal through both FGF receptor 1 and 4 (FGFR1 and FGFR4, respectively) and its co-receptor, β-klotho, to regulate enterohepatic circulation of bile acids as well as systemic lipid and glucose metabolism89. Although liver receptors are important targets, data also suggest critical CNS action. FGF19 administration has been shown to increase energy expenditure in obese mice90. Furthermore, acute intracerebroventricular (ICV) infusion of FGF19 reduced 24 hour food intake and body weight and improved glucose tolerance in rats, whereas ICV infusion of an FGFR1 and FGFR4 combined antagonist increased food intake91. Lastly, neuronal expression of β-klotho is necessary for the long-term impact of FGF19 on body mass92.
The intestine is home to trillions of microbial species, and increasing evidence links the microbiome to regulation of feeding and energy homeostasis. Faecal transplantation is one method that researchers have used to transfer microbiota from one mouse to another. When faeces from ob/ob mice are transferred to lean, germ-free mice, the lean mice become hyperphagic and gain fat mass93,94. A critical question remains regarding the mechanism by which the microbiome communicates with the host to regulate energy homeostasis. One possibility is that this communication occurs through the tight link between the microbiome, the innate immune system and the gut–brain axis. In support of this, lipopolysaccha-ride, a pathogenic agent that exists on the bacterial cell wall, stimulates GLP1 secretion95,96. A link to the CNS is also indicated by data in which a specific intestinal pathogen (Salmonella enterica subsp. enterica serovar Typhimurium) has been found to inhibit inflammatory-induced anorexia via the vagus nerve97. Alternatively, the microbiome is also an active participant in the processing of ingested nutrients, resulting in by-products that can then act in an autocrine, paracrine or endocrine fashion to regulate host metabolism. In fact, one critical function of the microbiome is in the processing of primary bile acids into secondary bile acids, which directly impacts FXR signalling (discussed above)98. Within the colon, the microbiota also processes complex carbohydrates into short-chain fatty acids, and one of these (acetate) has been found to increase ghrelin and glucose-stimulated insulin secretion via the parasympathetic nervous system99.
Another recently discovered peptide that links the immune system to CNS regulation of feeding is growth/differentiation factor 15 (GDF15). Exposure to toxins leads to suppression of food intake as an emergency response, and GDF15 is thought to be a player in mediating this effect100. This peptide is predominantly secreted by the liver, and it was recently discovered that it binds to an orphan receptor that is a part of the glial-cell-line-derived neurotrophic factor family (GDNF) family receptor α-like (GFRAL)100–103. Interestingly, this receptor is not found in peripheral tissues but was found specifically in the AP and NTS in mice100–103.
AAV-mediated systemic overexpression of GDF15 in mice or GDF15 administration to mice, rats and nonhuman primates drastically reduced body mass and food intake both acutely and chronically100,101,104. Notably, mice that lack whole-body expression of GFRAL are resistant to dietary-induced obesity and still respond to the anorectic effect of a GLP1R agonist or leptin101–103. These results suggest that the neurons within the AP and NTS that express GFRAL do not overlap with neurons that respond to GLP1 agonists or to leptin. Furthermore, GDF15 is an example of an ascending signal from the NTS that activates a PBN–central amygdala circuit to suppress feeding100. These data also highlight a circumstance in which the NTS, independently of the hypothalamus, regulates feeding.
Interestingly, most of these ‘newly’ identified peripheral signals are interrelated. The microbiome regulates the bile acid pool, whereas bile acids are upstream of the FGF15 or FGF19 secretion. There are more questions that need to be addressed to understand the complex role of the FXR system in regulating energy homeostasis, including the key CNS neuronal populations that may be involved. Furthermore, GDF15 is a newly defined hindbrain signal that is an attractive therapeutic target for obesity. Understanding the integration of this system will likely be a critical component to advancing our knowledge of the many peripheral signals necessary for CNS-induced regulation of energy homeostasis.
Findings from the bedside
Bariatric surgery
One of the clinical findings that highlight the importance of the gut–brain axis in regulation of energy homeostasis is the overwhelming success of bariatric surgery in weight loss and improvements in obesity-associated comorbidities. Roux-en Y gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG) are two of the most frequently performed bariatric surgeries in the United States. Although RYGB results in gastric size reduction as well as intestinal rearrangement, VSG only restricts stomach size (FIG. 3). Despite the drastically different anatomical rearrangements, the results of these surgeries are remarkably similar. In fact, both RYGB and VSG result in ~80% reduction in excess body mass and an ~38% remission of T2DM105. Despite this success, surgery cannot possibly be widely applied to solve the obesity epidemic. However, it is an interesting model to explore the regulation of energy homeostasis because the degree of success of surgery suggests that we can change systemic regulation of body weight by changing GI anatomy.
Figure 3 |. Roux-en Y gastric bypass and vertical sleeve gastrectomy.
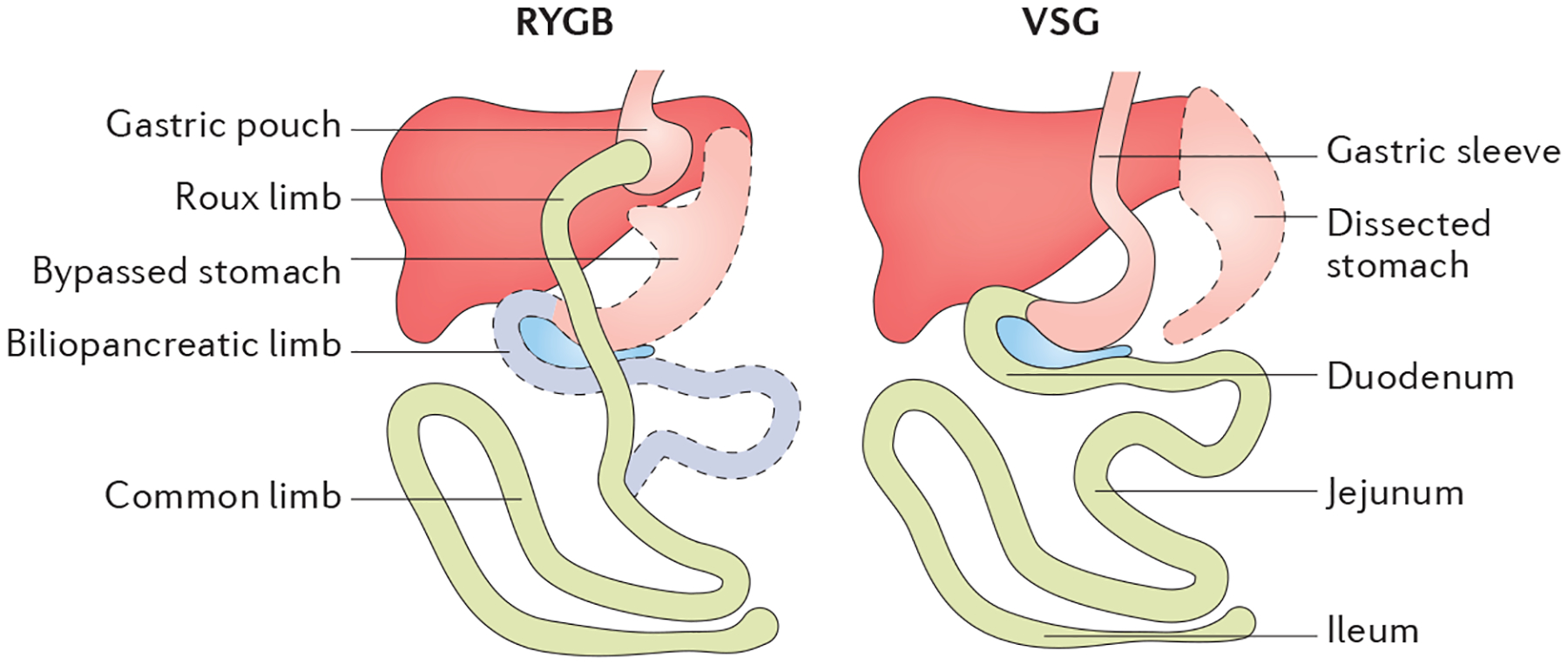
In Roux-en Y gastric bypass (RYGB), there is surgical reduction of the stomach to form a small gastric pouch. The remaining 95% of the stomach remains in the peritoneal cavity. The intestinal tract is rearranged such that the mid-jejunum is anastomosed to the gastric pouch, bypassing 95% of the stomach and the whole upper gastrointestinal tract. The distal end of the duodenum is then anastomosed to the jejunum to provide biliopancreatic digestive enzymes to ingested nutrients. By contrast, vertical sleeve gastrectomy (VSG) surgically removes about 80% of the stomach along the greater curvature with no intestinal rearrangement. Adapted with permission from Kim, K. S. & Sandoval, D. A., Endocrine function after bariatric surgery, Comprehensive Physiology, John Wiley & Sons (REF. 139). Copyright © 2013 American Physiological Society.
Bariatric surgery.
A surgical dissection and/or reorganization of the gastrointestinal tract that is used to induce weight loss.
That surgery has a system effect is not just indicated by the weight loss but by the many physiological changes that also occur. For example, both RYGB and VSG drive widespread postprandial increases in gut peptides106, some of which are summarized in FIG. 4. As an example, postprandial GLP1 and PYY are almost tenfold higher after RYGB and VSG107,108.
Figure 4 |. The impact of bariatric surgery on intestinal signalling peptides.
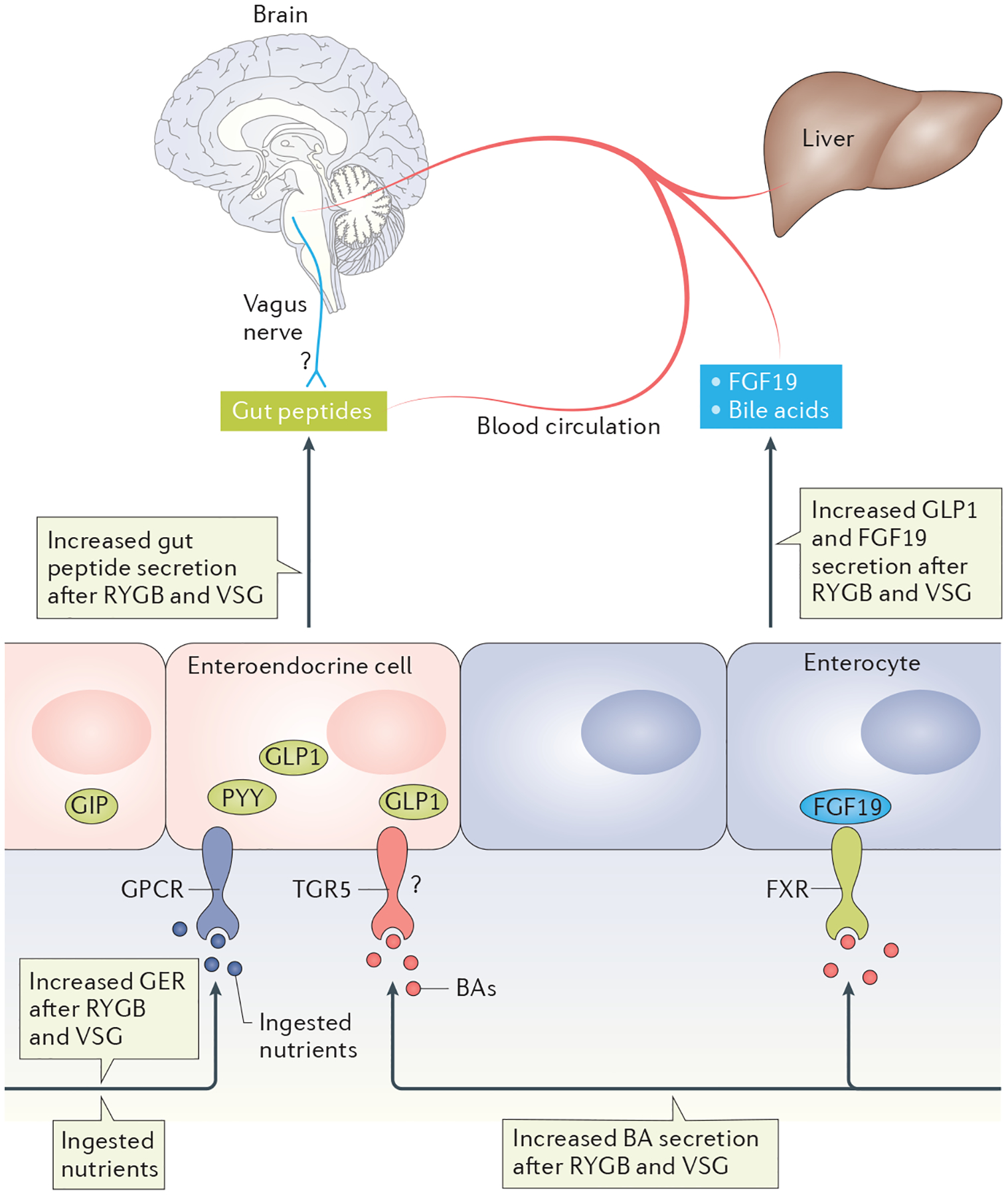
Both Roux-en Y gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG) increase the gastric-emptying rate (GER) and thus cause rapid nutrient access to the intestine, likely contributing to the large increase in postprandial secretions of several gut peptides, including glucagon-like peptide 1 (GLP1), peptide YY (PYY) and gastric inhibitory polypeptide (GIP). Both surgeries also increase circulating bile acids (BAs). BAs signal via a cell-surface G-protein-coupled receptor (GPCR) called TGR5 or via a nuclear transcription factor, farnesoid X-activated receptor (FXR). Whereas TGR5 signalling is known to regulate GLP1 secretion, its role after bariatric surgery is less clear (represented by a ?). Activation of FXR leads to secretion of fibroblast growth factor 19 (FGF19; FGF15 is the mouse orthologue) from the enterocytes. After bariatric surgery, FGF15 (or FGF19) is increased, and FXR has been demonstrated to be necessary for the metabolic success of surgery in mice.
Whether these changes in gut peptides are necessary for changes in feeding and body weight after surgery is less clear. In rodents, the surgery-induced reduction in food intake is transiently reduced within the first 2 weeks after surgery and is similar to that of sham-surgery animals thereafter109. However, the surgically induced increase in postprandial gut peptides seems to be a fairly permanent response dissociating the reduction in food intake from the postprandial rise in gut peptides. Furthermore, whole-body GLP1R-KO mice lose comparable amounts of weight to their wild-type littermates after both VSG110,111 and RYGB112, suggesting that GLP1 signalling is not necessary for surgery-induced weight loss. However, what drives the increase in GLP1 might still be important for understanding the physiological impact of surgery. Gastric-emptying rate is drastically increased after both RYGB and VSG113, and this increase may directly or indirectly drive the increase in postprandial gut peptides; that is, the high gastric-emptying rate pushes nutrients further into the distal small intestine where they can access more enteroendocrine cells. Indeed, this seems to be the case with RYGB, as GLP1 levels are lower in response to nutrient infusion into the bypassed limb than when the same calories are orally ingested114. Interestingly, the same study after VSG in rodents resulted in similarly elevated GLP1 responses to a duodenal versus oral glucose load113, suggesting that VSG drives either an increase in number or in the sensitivity of enteroendocrine cells to nutrients. Thus, the two surgeries may lead to increases in GLP1 via different mechanisms.
Responses to bariatric surgery have also contributed to the hypothesis that bile acids are critical regulators of body mass and glucose homeostasis as they are found to be increased by bariatric surgery in humans and rodents115,116. Using whole-body genetic KO models, TGR5 has been found to be necessary for at least some of the impact of VSG on glucose homeostasis117, although its role in VSG-induced weight loss and GLP1 secretion is disputed (see REF. 117 compared with REF. 118). By contrast, ablation of the nuclear bile acid receptor FXR blocked the effect of VSG on both body mass and glucose homeostasis119. Linked to these data, the microbiome is also altered by bariatric surgery, and at least some of those changes in the microbiome were prevented in the FXR-KO mouse119.
Given the success in weight loss by manipulating the GI tract through surgery, one would predict that the peripheral nervous system would be critical in mediating this success. However, although vagal innervation of the portal vein and liver does not seem to be necessary for the surgical success of RYGB in rats120, there are clear changes in the innervation of the gut after RYGB121. In addition, performing a coeliac branch vagotomy to target intestinal innervation blunted the impact of surgery on food intake in rats122. Interestingly, a recent study demonstrated that although the vagus nerve was not necessary for body mass changes, it was necessary for surgery-induced changes in food choice123. As we discussed above, one possibility is that the large increases in either postprandial nutrients (due to the increased gastric-emptying rate) or gut peptides could activate vagal afferent neurons. However, this has yet to be determined.
It is important to recognize that not all patients have the same degree of success in response to bariatric surgery, with some patients even gaining weight over time124. In addition, some patients, particularly after RYGB, are susceptible to dumping syndrome, which consists of a constellation of symptoms including sweating, palpitations and hypoglycaemia. These factors, in addition to the infrastructure that would be necessary, demonstrate that bariatric surgery is not the cure for obesity but instead may be better used as a tool to understand how the gut–brain axis regulates body mass. This knowledge could then be used to develop less-invasive strategies for weight loss.
From the gut to the drug pipeline
Importantly, the knowledge gained from bariatric surgery is already leading to alternative pharmacotherapies for obesity. The impact of surgery on increasing the wide array of gut peptides is discussed above. However, whether the increases in postprandial gut peptides are necessary for the impact of surgery is not clear. Multiple studies have shown that removing only one of these gut peptide signals, in and of itself, is not necessary for the weight loss or improvements in glucose homeostasis seen with surgery (see REF. 106 for review). One possibility is that it is not a single signal but rather a combination of signals that is necessary for the success of surgery. This hypothesis has led to the development of polytherapies that mimic these surgery-induced endocrine changes. Preclinical data demonstrate much greater efficacy of dual or even triple agonists for inducing weight loss than when these agonists are administered alone. A low-dose glucagon administration to the CNS reduced food intake and increased energy expenditure but also increased glucose levels125. However, combining a glucagon receptor (GLR) agonist with a GLP1R agonist leads to synergistic effects on weight loss without increasing glucose levels126. Similarly, impressive results have been seen by combining GLP1R and gastric inhibitory polypeptide receptor (GIPR) agonists, GLP1R and oestrogen receptor agonists127 and even a triple combination of GLP1R, GLR and GIPR agonists128,129. These data highlight one potential strategy that was conceptually influenced by the responses to bariatric surgery.
Conclusion
There are a host of peripheral signals that inform the brain of acute and chronic energy status. These signals arise from circulating hormones, nutrients and nutrient by-products acting directly on the hypothalamus, the hindbrain or peripheral neurons that terminate within the NTS. New genetic technologies have revealed more details on the descending and ascending circuits that integrate these two brain regions. In addition, the success of bariatric surgery has not only led to the discovery of new peripheral signals important for metabolic regulation but has also driven the use of polyagonist therapies in the pipeline for the treatment of obesity. Taken together, this work highlights how discoveries from both clinical and preclinical studies have been, and will continue to be, instrumental in advancing our understanding of CNS regulation of energy homeostasis.
Acknowledgements
The authors’ work is supported in part by US National Institutes of Health awards DK082480 (D.A.S.) and DK093848 (R.J.S.).
Competing interests
K.-S.K. has no competing interest. R.J.S. has received research support from Ethicon Endo-Surgery, Novo Nordisk, Sanofi and Janssen. R.J.S. has served on scientific advisory boards for Ethicon Endo-Surgery, Daiichi Sankyo, Janssen, Novartis, Nestle, Takeda, Boehringer Ingelheim, Sanofi and Novo Nordisk. R.J.S. is also a paid speaker for Ethicon Endo-Surgery. D.A.S. has received research support from Ethicon Endo-Surgery, Novo Nordisk and Boehringer Ingelheim.
References
- 1.Svendsen B et al. An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156, 847–857 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Psichas A et al. Gut chemosensing mechanisms. J. Clin. Invest 125, 908–917 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinert RE et al. Ghrelin, CCK, GLP-1, and PYY (3–36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol. Rev 97, 411–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper is the pinnacle of the reviews on gut peptides that regulate feeding.
- 4.Batterham RL et al. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab 4, 223–233 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Scrocchi LA, Hill ME, Saleh J, Perkins B & Drucker DJ Elimination of glucagon-like peptide 1R signaling does not modify weight gain and islet adaptation in mice with combined disruption of leptin and GLP-1 action. Diabetes 49, 1552–1560 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Chambers AP et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25, 927–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandoval DA & D’Alessio DA Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol. Rev 95, 513–548 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Holst JJ The physiology of glucagon-like peptide 1. Physiol Rev 87, 1409–1439 (2007). [DOI] [PubMed] [Google Scholar]
- 9.West DB, Fey D & Woods SC Cholecystokinin persistently suppresses meal size but not food intake in free-feeding rats. Am. J. Physiol 246, R776–R787 (1984). [DOI] [PubMed] [Google Scholar]
- 10.Miller LJ, Holicky EL, Ulrich CD & Wieben ED Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 109, 1375–1380 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Inoue H et al. Human cholecystokinin type A receptor gene: cytogenetic localization, physical mapping, and identification of two missense variants in patients with obesity and non-insulin-dependent diabetes mellitus (NIDDM). Genomics 42, 331–335 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Bagdade JD, Bierman EL & Porte D The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J. Clin. Invest 46, 1549–1557 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Considine RV et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med 334, 292–295 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Halaas JL et al. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl Acad. Sci. USA 94, 8878–8883 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods SC, Lotter EC, McKay LD & Porte D Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505 (1979). [DOI] [PubMed] [Google Scholar]; This study is one of the first to demonstrate the role of insulin with the CNS to regulate feeding.
- 16.Sipols AJ, Baskin DG & Schwartz MW Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes 44, 147–151 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Tschöp M, Smiley DL & Heiman ML Ghrelin induces adiposity in rodents. Nature 407, 908–913 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Cummings DE et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat. Med 8, 643–644 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Zigman JM et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J. Clin. Invest 115, 3564–3572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz MW, Woods SC, Porte D, Seeley RJ & Baskin DG Central nervous system control of food intake. Nature 404, 661–671 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Leinninger GM et al. Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab 14, 313–323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhillon H et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203 (2006). [DOI] [PubMed] [Google Scholar]
- 23.van de Wall E et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 149, 1773–1785 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myers MG & Olson DP SnapShot: neural pathways that control feeding. Cell Metab 19, 732–732.e1 (2014). [DOI] [PubMed] [Google Scholar]; This paper contains many perceptive schematic figures of the neural pathways of feeding control.
- 25.Marston OJ, Garfield AS & Heisler LK Role of central serotonin and melanocortin systems in the control of energy balance. Eur. J. Pharmacol 660, 70–79 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Burmeister MA et al. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes 66, 372–384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batterham RL et al. Gut hormone PYY3–36 physiologically inhibits food intake. Nature 418, 650–654 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Wren AM et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes 50, 2540–2547 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Sandoval DA, Bagnol D, Woods SC, D’Alessio DA & Seeley RJ Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 57, 2046–2054 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinzig KP, D’Alessio DA & Seeley RJ The diverse roles of CNS GLP-1 in the control of food intake and the mediation of visceral illness. J. Neurosci 22, 10470–10476 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods SC, Begg DP & Woods SC The endocrinology of food intake. Nat. Rev. Endocrinol 9, 584–597 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Aponte Y, Atasoy D & Sternson SM AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci 14, 351–355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses optogenetics to demonstrate the melanocortin-independent effects of AGRP neurons on regulation of feeding behaviour.
- 33.Krashes MJ et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest 121, 1424–1428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses DREADD technology to demonstrate the role of AGRP neurons on regulating feeding behaviour.
- 34.Zhan C et al. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J. Neurosci 33, 3624–3632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steculorum SM et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 165, 125–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berthoud HR, Blackshaw LA, Brookes SJH & Grundy D Neuroanatomy of extrinsic afferents supplying the gastrointestinal tract. Neurogastroenterol. Motil 16, 28–33 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Berthoud H-R Anatomy and function of sensory hepatic nerves. Anat. Rec 280A, 827–835 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Woods SC Metabolic signals and food intake. Forty years of progress. Appetite 71, 440–444 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Wang PY et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 452, 1012–1016 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Grabauskas G, Song I, Zhou S & Owyang C Electrophysiological identification of glucose-sensing neurons in rat nodose ganglia. J. Physiol 588, 617–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lal S, Kirkup AJ, Brunsden AM, Thompson DG & Grundy D Vagal afferent responses to fatty acids of different chain length in the rat. Am. J. Physiol 281, G907–G915 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Randich A et al. Responses of celiac and cervical vagal afferents to infusions of lipids in the jejunum or ileum of the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol 278, R34–R43 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Liu C et al. PPARγ in vagal neurons regulates high-fat diet induced thermogenesis. Cell Metab 19, 722–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansuy-Aubert V et al. Loss of the liver X receptor LXRα/β in peripheral sensory neurons modifies energy expenditure. eLife 4, e06667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ritter S & Taylor JS Vagal sensory neurons are required for lipoprivic but not glucoprivic feeding in rats. Am. J. Physiol 258, R1395–R1401 (1990). [DOI] [PubMed] [Google Scholar]
- 46.Darling RA et al. Mercaptoacetate and fatty acids exert direct and antagonistic effects on nodose neurons via GPR40 fatty acid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol 307, R35–R43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li A-J, Wang Q, Dinh TT, Simasko SM & Ritter S Mercaptoacetate blocks fatty acid-induced GLP-1 secretion in male rats by directly antagonizing GPR40 fatty acid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol 310, R724–R732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams EK et al. Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166, 209–221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study finds that there are distinct projections of vagal neurons to the GI tract that provide nutritive and mechanical information from the gut to the CNS.
- 49.Moran TH, Baldessarini AR, Salorio CF, Lowery T & Schwartz GJ Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am. J. Physiol 272, R1245–R1251 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Raybould HE et al. Expression of 5-HT3 receptors by extrinsic duodenal afferents contribute to intestinal inhibition of gastric emptying. Am. J. Physiol. Gastrointest. Liver Physiol 284, G367–G372 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Babic T, Troy AE, Fortna SR & Browning KN Glucose-dependent trafficking of 5-HT3 receptors in rat gastrointestinal vagal afferent neurons. Neurogastroenterol. Motil 24, e476–e488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgartner I et al. Hepatic-portal vein infusions of glucagon-like peptide-1 reduce meal size and increase c-Fos expression in the nucleus tractus solitarii, area postrema and central nucleus of the amygdala in rats. J. Neuroendocrinol 22, 557–563 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Kim D-H, D’Alessio DA, Woods SC & Seeley RJ The effects of GLP-1 infusion in the hepatic portal region on food intake. Regul. Pept 155, 110–114 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayes MR et al. The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. Am. J. Physiol. Regul. Integr. Comp. Physiol 301, R1479–1485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abbott CR et al. The inhibitory effects of peripheral administration of peptide YY3–36 and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal–brainstem–hypothalamic pathway. Brain Res 1044, 127–131 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Plamboeck A et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am. J. Physiol. Gastrointest. Liver Physiol 304, G1117–G127 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Kanoski SE, Fortin SM, Arnold M, Grill HJ & Hayes MR Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152, 3103–3112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baraboi E-D et al. Effects of albumin-conjugated PYY on food intake: the respective roles of the circumventricular organs and vagus nerve. Eur. J. Neurosci 32, 826–839 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Halatchev IG & Cone RD Peripheral administration of PYY3–36 produces conditioned taste aversion in mice. Cell Metab 1, 159–168 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Ripken D et al. Cholecystokinin regulates satiation independently of the abdominal vagal nerve in a pig model of total subdiaphragmatic vagotomy. Physiol. Behav 139, 167–176 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Reidelberger RD, Hernandez J, Fritzsch B & Hulce M Abdominal vagal mediation of the satiety effects of CCK in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 286, R1005–R1012 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Diepenbroek C et al. Validation and characterization of a novel method for selective vagal deafferentation of the gut. Am. J. Physiol. Gastrointest. Liver Physiol 313, G342–G352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Lartigue G, Ronveaux CC & Raybould HE Deletion of leptin signaling in vagal afferent neurons results in hyperphagia and obesity. Mol. Metab 3, 595–607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sisley S et al. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Invest 124, 2456–2463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krieger J-P et al. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 65, db150973 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Ritter RC Gastrointestinal mechanisms of satiation for food. Physiol. Behav 81, 249–273 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Hayes MR et al. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab 13, 320–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hisadome K, Reimann F, Gribble FM & Trapp S Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like peptide 1 neurons. Diabetes 59, 1890–1898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrera JG et al. Hyperphagia and increased fat accumulation in two models of chronic CNS glucagon-like peptide-1 loss of function. J. Neurosci 31, 3904–3913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaykema RP et al. Activation of murine pre-proglucagon–producing neurons reduces food intake and body weight. J. Clin. Invest 127, 1031–1045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses DREADD technology to understand the role of preproglucagon neurons in the regulation of energy homeostasis.
- 71.Lachey JL et al. The role of central glucagon-like peptide-1 in mediating the effects of visceral illness: differential effects in rats and mice. Endocrinology 146, 458–462 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Scott MM, Williams KW, Rossi J, Lee CE & Elmquist JK Leptin receptor expression in hindbrain Glp-1 neurons regulates food intake and energy balance in mice. J. Clin. Invest 121, 2413–2421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hayes MR et al. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11, 77–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grill HJ et al. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 143, 239–246 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Garfield AS et al. Neurochemical characterization of body weight-regulating leptin receptor neurons in the nucleus of the solitary tract. Endocrinology 153, 4600–4607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hisadome K, Reimann F, Gribble FM & Trapp S CCK stimulation of GLP-1 neurons involves α1-adrenoceptor-mediated increase in glutamatergic synaptic inputs. Diabetes 60, 2701–2709 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alhadeff AL, Golub D, Hayes MR & Grill HJ Peptide YY signaling in the lateral parabrachial nucleus increases food intake through the Y1 receptor. Am. J. Physiol. Endocrinol. Metab 309, E759–E766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alhadeff AL, Baird J-P, Swick JC, Hayes MR & Grill HJ Glucagon-like peptide-1 receptor signaling in the lateral parabrachial nucleus contributes to the control of food intake and motivation to feed. Neuropsychopharmacology 39, 2233–2243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alhadeff AL, Hayes MR & Grill HJ Leptin receptor signaling in the lateral parabrachial nucleus contributes to the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol 307, R1338–R1344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campos CA, Bowen AJ, Schwartz MW & Palmiter RD Parabrachial CGRP neurons control meal termination. Cell Metab 23, 811–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that specific populations of neurons within the parabrachial nucleus are important in regulated feeding.
- 81.Roman CW, Derkach VA & Palmiter RD Genetically and functionally defined NTS to PBN brain circuits mediating anorexia. Nat. Commun 7, 11905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Essner RA et al. AgRP neurons can increase food intake during conditions of appetite suppression and inhibit anorexigenic parabrachial neurons. J. Neurosci 37, 798–717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stachniak TJ, Ghosh A & Sternson SM Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus→midbrain pathway for feeding behavior. Neuron 82, 797–808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weissbourd B et al. Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron 83, 645–662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nectow AR et al. Identification of a brainstem circuit controlling feeding. Cell 170, 429–442.e11 (2017). [DOI] [PubMed] [Google Scholar]; This study identifies specific neurons in the dorsal raphe nucleus a novel feeding regulator.
- 86.Thomas C, Pellicciari R, Pruzanski M, Auwerx J & Schoonjans K Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov 7, 678–693 (2008). [DOI] [PubMed] [Google Scholar]
- 87.Katsuma S, Hirasawa A & Tsujimoto G Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun 329, 386–390 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Alemi F et al. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 144, 145–154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nies VJM et al. Fibroblast growth factor signaling in metabolic regulation. Front. Endocrinol 6, 193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fu L et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145, 2594–2603 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Ryan KK et al. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 154, 9–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lan T et al. FGF19, FGF21, and an FGFR1/β-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab 26, 709–718.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Turnbaugh PJ et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1131 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Turnbaugh PJ, Bäckhed F, Fulton L & Gordon JI Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 3, 213–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper links altered microbiome populations to dietary-induced obesity.
- 95.Kahles F et al. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes 63, 3221–3229 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Ellingsgaard H et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med 17, 1481–1489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rao S et al. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell 168, 503–516.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sayin SI et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Perry RJ et al. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature 534, 213–217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hsu J-Y et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 (2017). [DOI] [PubMed] [Google Scholar]
- 101.Mullican SE et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med 23, 1150–1157 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Emmerson PJ et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med 23, 1215–1219 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Yang L et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med 23, 1158–1166 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Xiong Y et al. Long-acting MIC-1/GDF15 molecules to treat obesity: evidence from mice to monkeys. Sci. Transl Med 9, eaan8732 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Schauer PR et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N. Engl. J. Med 366, 1567–1576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates the much greater efficacy of bariatric surgery in diabetes treatment than that of intensive medical therapy.
- 106.Seeley RJ, Chambers AP & Sandoval DA The role of gut adaptation in the potent effects of multiple bariatric surgeries on obesity and diabetes. Cell Metab 21, 369–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.le Roux CW et al. Gut hormones as mediators of appetite and weight loss after Roux-en-Y gastric bypass. Ann. Surg 246, 780–785 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Yousseif A et al. Differential effects of laparoscopic sleeve gastrectomy and laparoscopic gastric bypass on appetite, circulating acyl-ghrelin, peptide YY3–36 and active GLP-1 levels in non-diabetic humans. Obes. Surg 24, 241–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilson-Pérez HE et al. The effect of vertical sleeve gastrectomy on food choice in rats. Int. J. Obes 37, 288–295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mokadem M, Zechner JF, Margolskee RF, Drucker DJ & Aguirre V Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol. Metab 3, 191–201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wilson-Pérez HE et al. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like peptide-1 receptor deficiency. Diabetes 62, 2380–2385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ye J et al. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am. J. Physiol. Regul. Integr. Comp. Physiol 306, R352–362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chambers AP et al. Regulation of gastric emptying rate and its role in nutrient-induced GLP-1 secretion in rats after vertical sleeve gastrectomy. Am. J. Physiol. Endocrinol. Metab 306, E424–E432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nguyen NQ et al. Rapid gastric and intestinal transit is a major determinant of changes in blood glucose, intestinal hormones, glucose absorption, and postprandial symptoms after gastric bypass. Obesity 22, 2003–2009 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Myronovych A et al. Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity 22, 390–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Patti M-E et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity 17, 1671–1677 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McGavigan AK et al. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 66, 226–234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ding L et al. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 64, 760–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ryan KK et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509, 183–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shin AC, Zheng H & Berthoud H-R Vagal innervation of the hepatic portal vein and liver is not necessary for Roux-en-Y gastric bypass surgery-induced hypophagia, weight loss, and hypermetabolism. Ann. Surg 255, 294–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gautron L, Zechner JF & Aguirre V Vagal innervation patterns following Roux-en-Y gastric bypass in the mouse. Int. J. Obes 37, 1603–1607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hao Z et al. Vagal innervation of intestine contributes to weight loss after Roux-en-Y gastric bypass surgery in rats. Obes. Surg 24, 2145–2151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hankir MK et al. Gastric bypass surgery recruits a gut PPAR-α-striatal D1R pathway to reduce fat appetite in obese rats. Cell Metab 25, 335–344 (2017). [DOI] [PubMed] [Google Scholar]; This study demonstrates that vagal innervation is necessary for surgery-induced changes in food choice.
- 124.Benoit SC, Hunter TD, Francis DM & De La Cruz-Munoz N Use of Bariatric Outcomes Longitudinal Database (BOLD) to study variability in patient success after bariatric surgery. Obes. Surg 24, 936–943 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Geary N in Satiation. From Gut to Brain (ed. Smith GP) 164–197 (Oxford Univ. Press, 1998). [Google Scholar]
- 126.Pocai A et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 58, 2258–2266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Finan B et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med 18, 1847–1856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Finan B et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl Med 5, 209ra151 (2013). [DOI] [PubMed] [Google Scholar]
- 129.Finan B et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med 21, 27–36 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Wren AM & Bloom SR Gut hormones and appetite control. Gastroenterology 132, 2116–2130 (2007). [DOI] [PubMed] [Google Scholar]
- 131.Williams DL, Grill HJ, Cummings DE & Kaplan JM Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology 144, 5184–5187 (2003). [DOI] [PubMed] [Google Scholar]
- 132.Masuda Y et al. Ghrelin stimulates gastric acid secretion and motility in rats. Biochem. Biophys. Res. Commun 276, 905–908 (2000). [DOI] [PubMed] [Google Scholar]
- 133.Sato T et al. Structure, regulation and function of ghrelin. J. Biochem 151, 119–128 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Foster-Schubert KE et al. Acyl and total ghrelin are suppressed strongly by ingested proteins, weakly by lipids, and biphasically by carbohydrates. J. Clin. Endocrinol. Metab 93, 1971–1979 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Batterham RL et al. Inhibition of food intake in obese subjects by peptide YY 3–36. N. Engl. J. Med 349, 941–948 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Mentlein R, Dahms P, Grandt D & Krüger R Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul. Pept 49, 133–144 (1993). [DOI] [PubMed] [Google Scholar]
- 137.Magnus CJ et al. Chemical and genetic engineering of selective ion channel-ligand interactions. Science 333, 1292–1296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gomez JL et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim K-S & Sandoval DA in Comprehensive Physiology (ed. Pollock DM) 783–798 (John Wiley & Sons, 2017). [Google Scholar]