

Abstract
Mitochondrial remodeling through fusion and fission is crucial for progenitor cell differentiation but its role in myogenesis is poorly understood. Here, we characterized the function of mitofusin 2 (Mfn2), a mitochondrial outer membrane protein critical for mitochondrial fusion, in muscle progenitor cells (myoblasts). Mfn2 expression is upregulated during myoblast differentiation in vitro and muscle regeneration in vivo. Targeted deletion of Mfn2 gene in myoblasts (Mfn2MKO) increases oxygen-consumption rates (OCR) associated with the maximal respiration and spare respiratory capacity, and increased levels of reactive oxygen species (ROS). Skeletal muscles of Mfn2MKO mice exhibit robust mitochondrial swelling with normal mitochondrial DNA content. Additionally, mitochondria isolated from Mfn2MKO muscles have reduced OCR at basal state and for complex I respiration, associated with decreased levels of complex I proteins NDUFB8 (NADH ubiquinone oxidoreductase subunit B8) and NDUFS3 (NADH ubiquinone oxidoreductase subunit S3). However, Mfn2MKO has no obvious effects on myoblast differentiation, muscle development and function, and muscle regeneration. These results demonstrate a novel role of Mfn2 in regulating mitochondrial complex I protein abundance and respiratory functions in myogenic progenitors and myofibers.
Keywords: Mfn2, mitochondrion, myogenesis, myogenic progenitor cells, oxidative respiration
1 |. INTRODUCTION
Skeletal muscle represents the most abundant tissue in human body, and enables the locomotor activities of individuals as well as serves metabolic and endocrine roles to maintain the whole-body homeostasis.1,2 Skeletal muscle is composed of long, thin, and multinucleated fibers (myofibers) that are generated during embryonic and fetal myogenesis through differentiation and fusion of MyoD+ myogenic progenitors (myoblasts) arisen from embryonic Pax3/7-expressing somitic cells.3,4 A portion of the myogenic progenitors downregulate the expression of Pax3 and MyoD, and become Pax7+ muscle resident stem cells, known as muscle satellite cells (MuSCs). MuSCs in homeostatic adult muscles retain in a quiescent state,5,6 but can be rapidly activated by acute muscle injury, re-expressing MyoD and entering the cell cycle.7,8 Subsequently, the proliferating myoblasts differentiate and fuse into a damaged muscle or return to the quiescent state (self-renew) to maintain the stem cell pool.9,10 Thus, proliferation and differentiation of MyoD-expressing myoblasts are extremely important in muscle development and regeneration.
Mitochondrion is the main organelle for the production of bioenergy, adenosine triphosphate (ATP), through oxidative phosphorylation mediated by enzymes in the electron transport chain (ETC) complexes.11 Differentiation of myoblasts is associated with alterations in cellular metabolism and a tremendous higher energy demand.12–14 As myoblasts transit from proliferation to differentiation, the main energy source switches from glycolysis to oxidative phosphorylation.15,16 As such, the abundance and function of mitochondria, reflected by mitochondrial DNA (mtDNA) copy number, mitochondrial volume and structure, and respiratory capacity, increase greatly during differentiation.16 In mature multinucleated myofibers, mitochondria are concentrated in the intermyofibrillar and subsarcolemmal spaces, where they produce ATP for cellular metabolism, biochemical reactions, and sarcomere contraction.17 Mitochondria in myofibers are highly dynamic and able to change their shape, size, and energy efficiency in response to various physiological conditions, such as growth, fiber-type switching, exercise, and aging.16,18,19 These morphological and functional transitions of mitochondria are known to be facilitated by fission and fusion, which cause increase or decrease in mitochondria number, respectively.12,20 Specifically, mitochondrial fusion is mediated by the mitofusin proteins 1 and 2 located on the outer mitochondrial membrane.21–24
Mitofusin 2 (Mfn2) is a member of the transmembrane GTPase family. Mfn2 is originally identified from the mitochondrial outer membrane and shown to participate in mitochondrial fusion that requires its GTPase activity.25,26 Mfn2-mediated mitochondrial fusion is crucial for content mixing, mtDNA integrity, energy production, and cellular respiration.27 It has been reported that Mfn2 knockout or mutation impairs thermogenesis of brown adipocytes, promotes adiposity, and leads to type 2A Charcot-Marie-Tooth disease.28–30 In cultured muscle cells, Mfn2 has been reported to modulate cell respiration, substrate oxidation, and expression of oxidative phosphorylation subunit proteins.24 In C2C12 and L6E9 myoblast cell lines, Mfn2 levels increase after differentiation, to promote mitochondrial activity and facilitate myocyte fusion.25,31 Repression of Mfn2 causes fragmentation of the mitochondrial network in myotubes.25,32 However, the in vivo physiological function of Mfn2 in myogenic progenitors and skeletal muscle development has not been explored. In the present study, we addressed this question using MyodCre-driven conditional Mfn2 knockout (Mfn2MKO) mice that deletes Mfn2 gene in all embryonic myogenic progenitors and resulting muscles and MuSCs. We found that Mfn2MKO enhanced spare respiratory capacity and increased the level of reactive oxygen species (ROS) in myoblasts. Mfn2Mko muscles exhibited mitochondrial swelling and had reduced levels of complex I proteins in adult mice. However, muscle development and regeneration were normal in Mfn2MKO mice. These results demonstrate that Mfn2 is required for normal mitochondrial size maintenance and respiratory function but dispensable for embryonic and postnatal myogenic function of progenitor cells.
2 |. MATERIALS AND METHODS
2.1 |. Mice care and handing
All experiments involving mice were performed according to procedures approved by Purdue University Animal Care and Use Committee. All mice used in our experiments were C57BL/6J strain and obtained from Jackson Laboratory (Bar Harbor, ME) under stock numbers 014140 (MyoDCre ) and 026525 (Mfn2loxP/loxP ). Both male and female Mfn2MKO (MyoDCre/Mfn2loxP/loxP) and littermate WT (Mfn2loxP/loxP) mice were used for experiments at 6–8 weeks of age. Mice were maintained on a 12:12 light and dark cycle, had ad libitum access to water and standard rodent chow, and were housed in ventilated cages. All knockout (KO) and littermate WT mice were sacrificed at the same time of day in each experiment, consisting of cervical dislocation after anesthetization with carbon dioxide.
2.2 |. Muscle injury and regeneration
Muscle injury and regeneration were induced by CTX (Cardiotoxin, Sigma) injecting in tibialis anterior (TA) muscle of left hind leg. Adult mice (8 weeks old) were anesthetized by intraperitoneally injecting ketamine-xylazine cocktail, and 50 μL CTX (10 μM) was injected into the central belly of TA muscles. TA muscles were sampled at 5.5 days after CTX injection.
2.3 |. Grip strength
Muscle strength of Mfn2MKO and control mice (8 weeks) was evaluated using a DFE II Series Digital Force Gauge (Ametek DFE II 2-LBF 10-N) with an attached metal grid (10 × 10 cm2). Mice were allowed to grasp the metal grid with four limbs and gently pulled along the axis of the grid by the tip of the tail. Maximal strength (Newtons) with which mice pulled the grid was measured in triplicate trials over 3 consecutive days.
2.4 |. Treadmill measurement
Mfn2MKO and WT mice (8 weeks old) were tested on a Treadmill (Eco3/6 treadmill; Columbus Instruments, Columbus, OH). Before the exhaustion test, mice were subjected to an acclimation training for 3 consecutive days with constant 10 m/min speed for 5 minutes. Electric stimulus of 1 Hz was employed to force mice to run. Formal test was started at 10 m/min for 5 minutes and the speed was increased by 2 m/min every 2 minutes until mice were exhausted or reached to maximal speed (46 m/min). The exhaustion was defined as the inability of the animal to run on the treadmill for 10 seconds despite electrical stimulation. Maximum exercise capacity was estimated from measured Running time (min), running distance (m), and maximum speed (m/min).
2.5 |. Isolation, culture, and differentiation of primary myoblast
Primary myoblasts were isolated from hind limb skeletal muscles of Mfn2MKO and control mice at 6-week old.33 Muscles were minced and then digested in 0.1% type I collagenase and 0.24% Dispase B mixture (Roche Applied Science). The digestions were stopped by adding F-10 Ham's medium (Thermo Fisher Scientific) containing 20% fetal bovine serum (FBS, Atlanta). Cells were then filtered through 70-μm filter to remove debris, centrifuged at 1700 rpm (Thermo Fisher CL2) for 5 minutes, and cultured in F-10 Ham's medium supplemented with 20% FBS, 4 ng/mL basic fibroblast growth factor (bFGF, Thermo Fisher Scientific), and 1% penicillin–streptomycin (Thermo Fisher Scientific) on collagen-coated cell culture plates at 37°C, 5% CO2. For differentiation, primary myoblasts were plated on Matrigel (Corning No. 354234) coated cell culture plates at 70% confluence. Myoblasts were induced to differentiate in differentiation media: DMEM (Thermo Fisher Scientific) supplemented with 2% horse serum and 1% penicillin–streptomycin.
2.6 |. Hematoxylin-eosin and immunofluorescence staining
Whole muscle tissues were dissected and frozen immediately in OCT compound. Frozen muscles were cross-sectioned (10 μm) using a Leica CM1850 cryostat. The sections were subjected to histological hematoxylin-eosin (HE) staining or immunofluorescence staining. For HE staining, the slides were soaked in hematoxylin for 15 minutes, rinsed with tap water, and then stained with eosin for 2 minutes. And the slides were dehydrated in 70%, 95%, and 100% ethanol each for 20 seconds and then in xylene for 3 minutes, mounted with Permount Mounting Medium. Stained images were captured with a digital camera attached to a Leica DM6000 (Leica) inverted microscope.
For immunofluorescence staining, cross-sections or cultured cells were fixed in 4% PFA (paraformaldehyde) for 15 minutes, quenched with 100 mM glycine for 10 minutes, and incubated in blocking buffer (5% goat serum, 2% bovine serum albumin, 0.1% Triton X-100, and 0.1% sodium azide in PBS) for 1 hour. Then, samples were incubated with primary antibodies for 12 hours at 4°C. After PBS washing for 5 minutes over three times, the samples were incubated with secondary antibodies and DAPI (see Table S1 for a list of antibodies) for 1 hour at room temperature. All images were captured with a Coolsnap HQ CCD camera attached to a Leica DM 6000B microscope and identical settings were used for WT and knockout samples. All images shown are representative results of at least three biological repeats.
For mito-tracker staining, live cells were incubated with a growth medium containing 0.2% mito-tracker for 1 hour. Then cells were fixed in ice methanol for 30 minutes, and the subsequent procedure starting with incubate blocking buffer was consistent with immunofluorescence staining.
For ROS staining, live cells were incubated with 5 μM ROX and Hoechst 33342 in growth media for 45 minutes. Then cells were washed with PBS three times and imaged with a Leica DM 6000B microscope. ROS intensity was quantified by staining the cell-permeant with 2′,7′-dichlorod ihydrofluorescein diacetate (H2DCFDA) and detected with a microplate reader at 485 nm excitation and 535 nm emission wavelengths.
2.7 |. DNA extraction and mtDNA quantitation
The total DNA containing mitochondria DNA (mtDNA) and nuclear DNA were isolated from primary myoblasts or differentiated myotubes using the high salt method. The DNA from WT and KO cells were diluted to identical concentrations and for quantifying the relative amounts of mtDNA and nuclear DNA using qRT-PCR. The primers of quantifying mtDNA and nuclear DNA are listed in Table S2, as previously described.34
2.8 |. RNA extraction and qRT-PCR
Total RNA was extracted from primary myoblasts or muscle tissues using TRIzol (Thermo Fisher Scientific) reagent according to the manufacturer's instruction. The reverse-transcribed total RNA was 2 μg. The quantitative real-time PCR was carried out in a Roche Lightcycler 96 PCR system with SYBR Green Master Mix and gene-specific primers were listed in Table S2. The relative mRNA level of all genes was normalized to β-actin gene and calculated by the 2−ΔΔT method.
2.9 |. Protein extraction and western blot
Total protein was isolated from cultured cells or homogenized muscle tissues, using radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS), phenylmethylsulfonyl fluoride (PMSF), and phosphatase inhibitors NaF and Na3 VO4. Protein concentrations were measured using Pierce BCA Protein Assay Reagent (Pierce Biotechnology) and NanoDrop 1000 (Thermo Fisher). Proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (PVDF, Millipore Corporation), blocked in 5% fat-free milk for 1 hour at room temperature, and then incubated with primary antibodies in 5% milk overnight at 4°C. After three washes in PBS for 10 minutes each, the membrane was then incubated with secondary antibody for 1 hour at room temperature. Signals were detected using enhanced chemiluminescence western blotting substrate (Santa Cruz Biotechnology) on a Fluor Chem R System (ProteinSimple).
2.10 |. Co-Immunoprecipitation (Co-IP) assay
Mitochondrial protein was extracted from 293T cells after transfected with CMV promoter-Myc-MCS-Mfn2 pEGFP-C1 plasmid, using 1 mL lysate buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 2 mM MgCl2, 0.5 mM EDTA and 0.5% NP-40). The lysate was precleared with 30 μL protein A/G agarose (#F2419, Santa Cruz Biotechnology) at 4°C for 3 hours. At the same reaction condition, 4 μg of primary antibody (cMyc antibody, #9E10, Santa Cruz Biotechnology) or homologous IgG (#2729, Cell Signaling) was incubated with mitochondria lysates containing 400 μg total protein, rotating at 4°C overnight. The samples were washed at least three times and subjected to western blot analysis.33
2.11 |. Isolation of mitochondria from muscle
Mitochondria were isolated from whole hind limb skeletal muscles of Mfn2MKO and control mice at 8 weeks of age, as previously reported.35 Muscles were washed three times in PBS and cut into pieces and homogenized with a Glass/Teflon Potter Elvehjem homogenizer (5 mL) grinder in 1 mL IBm1 buffer (0.067 M sucrose, 0.05 M Tris/HCl, 0.05 M KCl, 0.01 M EDTA, and 0.2% BSA in distilled water, pH 7.4) for more than 20 times. The homogenate was centrifuged at 800 RCF for 10 minutes. The supernatant containing mitochondria were centrifuged again at 8000 RCF for 10 minutes and the sediment was collected. The sediment mitochondria were resuspended in 50 μL IBm1 buffer and the concentration was determined by using Pierce BCA Protein Assay Reagent (Pierce Biotechnology) and NanoDrop 1000.
2.12 |. Mitochondrial respiratory capacity assessment
The Seahorse assay was used to measure oxygen consumption rate, OCR (pmol/min) of primary myoblasts, and isolated mitochondria, as detailed in Materials and methods 2.5 and 2.11. For myoblasts, 3 × 104 cells/well were plated into Seahorse XF24 plates and cultured overnight. The XF assay medium (Seahorse Bioscience) was prepared by mixing 48.5 mL Seahorse XF RPMI Medium, 0.5 mL Seahorse XF Glucose (1.0 M solution), 0.5 mL Seahorse XF Pyruvate (100 mM solution), and 0.5 mL Seahorse XF L-glutamine (200 mM solution), pH 7.4. After washing for three times with XF medium, the myoblasts were incubated in XF medium for 1 hour at 37°C in a non-CO2 incubator. Mitochondrial respiration was monitored at basal state and after sequential injection of the mitochondrial modulators oligomycin (3 μM), FCCP (6 μM) and antimycin (2.5 μM) that induce mitochondrial stress.36
For a Seahorse assay of mitochondria, 3 μg mitochondria were placed into each well of Seahorse XF24 plates and centrifuged at 8000 RCF for 15 minutes to attach the mitochondria onto the bottom.37 To measure mitochondrial OCR in coupling assay, the mitochondria were initially incubated in 500 μL of mitochondrial assay solution (MAS) containing 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA, and 0.2% (w/v) fatty acid-free BSA, pH 7.2 in each well, together with succinate (11 mM) and rotenone (2.2 mM). Mitochondrial respiration was stimulated with the sequential injection of ADP (20 mM), oligomycin (20 μM), FCCP (40 μM), and antimycin A (40 μM). In the mitochondria electron transport chain assay, mitochondria was incubated in 500 μL MAS buffer with pyruvate (11 mM), malate (2.2 mM), and FCCP (4.4 mM) in each well, and mitochondrial respiration was stimulated with the sequential injection of rotenone (20 μM), succinate (100 mM), antimycin A (40 μM), ascorbate (100 mM), and TMPD (1 mM). All chemicals were purchased from Sigma.
2.13 |. Transmission electron microscopy
The Sol muscle was isolated and immediately fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, post-fixed in buffered 1% osmium tetroxide containing 0.8% potassium ferricyanide, and enbloc stained in 1% aqueous uranyl acetate. They were then dehydrated with a graded series of acetonitrile and embedded in EMbed-812 resin. Thin sections (80 nm) were cut on a Leica EM UC6 ultramicrotome and stained with 2% uranyl acetate and lead citrate. Images were acquired using a Gatan US1000 2K CCD camera on FEI Tecnai G220 electron microscope equipped with a LaB6 source and operating at 100 kV or 200 kV.
2.14 |. Statistical analysis
The relative gray value of western blot bands and the average cross-sectional area (CSA) of regeneration myofiber were measured and calculated using ImageJ software (NIH). All experimental data were presented as mean ± standard error mean (SEM) and all statistical differences were compared by Studenťs t-test with a two-tail distribution. A P value of < .05 was considered statistically significant. All data were additionally fitted with a biphasic function using GraphPad Prism, version 6 (GraphPad Software, La Jolla, CA).
3 |. RESULTS
3.1 |. Mfn2 expression is upregulated during muscle regeneration and myogenic differentiation
To establish the relevance of Mfn2 in myogenesis, we first examined the expression of Mfn2 during the regeneration of tibialis anterior (TA) muscle from 8-week-old WT mice after injured by CTX. In uninjured TA muscle, a strong Mfn2 immunofluorescence signal was only detected around the membrane (sarcolemma) of myofiber (Figure 1A). At 3 days postinjury (3 dpi), Mfn2 signal became stronger and spread throughout newly regenerated myofibers (indicated by central-nucleation) (Figure 1A). Mfn2 signal maintained strong but became more diffused and more restricted to the periphery of myofibers as they gradually enlarge in size at 5 and 7 dpi (Figure 1A). At 21 dpi when muscle regeneration was completed, Mfn2 expression was weak and mainly found in perinuclear and subsarcolemmal regions (Figure 1A). These results indicate that Mfn2 expression is especially upregulated in newly formed myofibers during regeneration.
FIGURE 1.
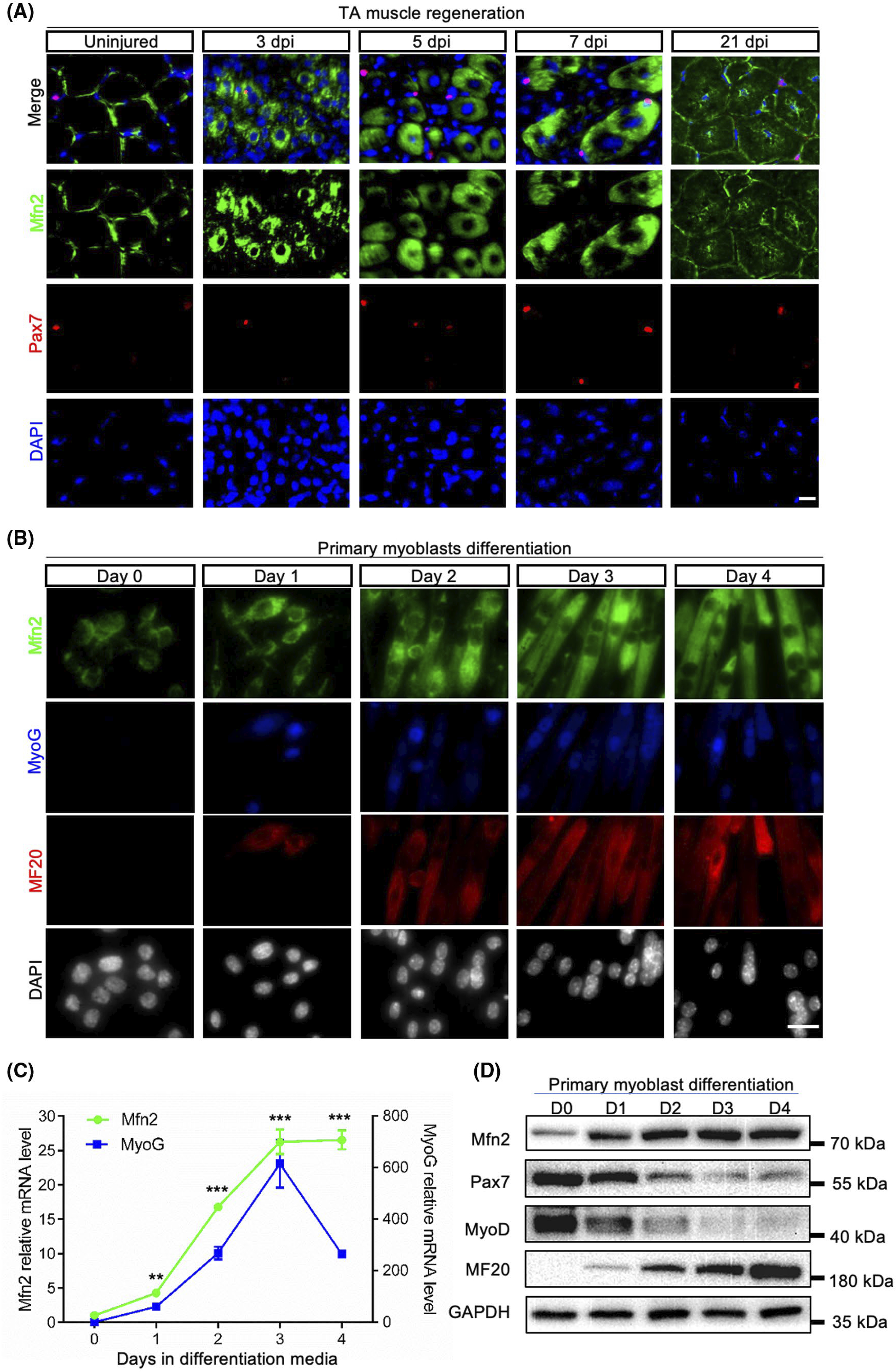
Mfn2 expression is upregulated during muscle regeneration and myoblast differentiation. A, Mfn2 immunofluorescence in of uninjured and injured TA muscle sections at 3, 5, 7, and 21 days post-injury (dpi). Scale bar: 20 μm. B, Mfn2 immunofluorescence in primary myoblasts in growth medium (Day 0) and after differentiated for 1–4 days. Scale bar: 20 μm. C, qRT-PCR analysis of relative mRNA levels of Mfn2 and Myog genes (n = 3 each group). Data are shown as mean ± SEM (t test: **P < .01, ***P < .001). D, Western blots showing relative levels of Mfn2 and myogenesis-related proteins (Pax7, MyoD, and MF20) at various stages of myoblast differentiation
We also isolated primary myoblasts from 6-week-old mice and examined the expression of Mfn2 during their differentiation. Mfn2 signal appeared weak in proliferating myoblasts cultured in growth medium (Figure 1B). After induced to differentiate, the expression of Mfn2 was significantly increased within a day and kept elevating until day 3. Notably, the upregulation of Mfn2 mirrored that of the expression patterns of myogenic differentiation markers, myogenin (MyoG), and myosin heavy chain (marked by MF20) (Figure 1B). Consistently, Mfn2 and Myog mRNA levels both peaked at day 3 after differentiation (Figure 1C). Western blot using the same samples confirmed the upregulation of Mfn2 and MF20, and downregulation of Pax7 and MyoD in protein levels during differentiation of primary myoblasts (Figure 1D). Collectively, these data demonstrated that expression of Mfn2 is elevated during myogenic differentiation, with the highest level in newly formed myotubes.
3.2 |. Mfn2-null myogenic progenitors exhibit abnormal mitochondrial respiration and elevated reactive oxygen species
To explore the function of Mfn2 in the primary myoblasts, we generated an embryonic myogenic progenitor specific Mfn2 knockout mouse model (Mfn2MKO) by crossing MyodCre mice with Mfn2loxP/loxP mice, in which two loxP sites flank the exon 6 encoding for the canonical GTPase motif (Figure 2A). Myod1-driven Cre recombinase should excise exon 6 and cause a frameshift that is predicted to generate a truncated protein with only 158 amino acids, missing all functional domains (Figure 2A). We confirmed the efficient reduction of Mfn2 in isolated myoblast from Mfn2MKO mice by immunofluorescence staining and western blot (Figure 2B). As Mfn2 is involved in mitochondria fusion, we first investigated how deletion of Mfn2 affects the mitochondria network in myoblasts. In proliferating primary myoblasts, the intensity and distribution of mito-tracker fluorescence were similar in WT and Mfn2MKO myoblasts (Figure 2C). Consistently, mtDNA content (normalized to nuclear DNA content) were identical in Mfn2MKO and WT myoblasts (Figure 2D). The mRNA levels of Mfn1 were also similar in WT and Mfn2-null myoblasts (Figure 2E), indicating that deletion of Mfn2 does not lead to compensatory increase of Mfn1 expression.
FIGURE 2.
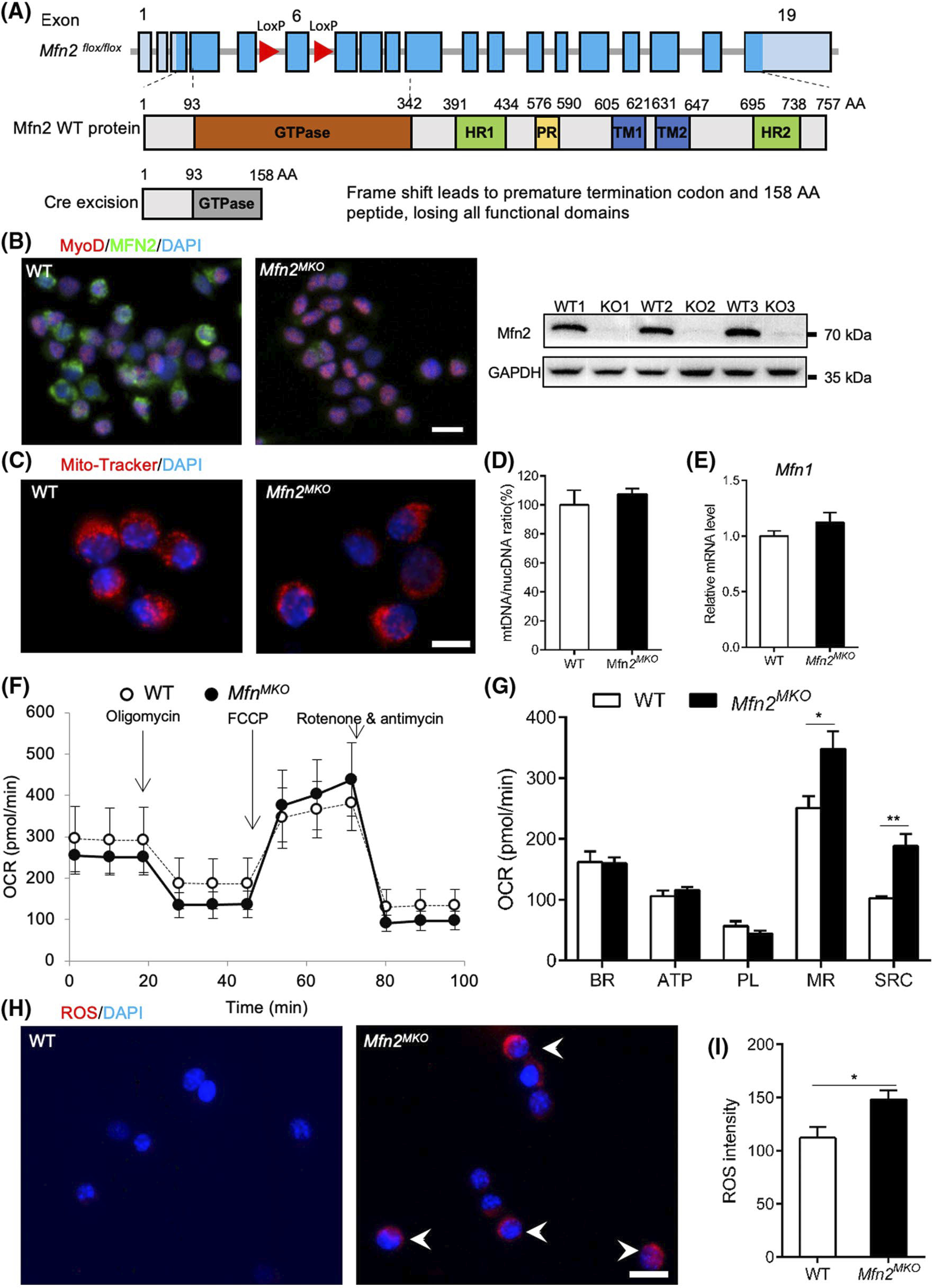
Mfn2-null myoblasts exhibit abnormal respiratory capacity and elevated reactive oxygen species. A, Strategy for myoblast-specific deletion of Mfn2 driven by Myod1Cre (Mfn2MKO). B, Immunofluorescence staining and Western blotting confirm the loss Mfn2 expression in Mfn2MKO primary myoblasts, scale bar: 20 μm. C, Mito-tracker staining of mitochondria in WT and Mfn2MKO myoblasts. Scale bar: 10 μm. D and E, qRT-PCR analysis showing the ratio of mitochondria DNA to nucleotide DNA (E) and mRNA levels of Mfn1 (F) in WT and Mfn2MKO myoblasts (n = 4 for each group). F, Seahorse cell assay showing oxygen consumption rates (OCR) between WT and Mfn2MKO primary myoblasts. G, Quantification of OCR in basal respiration (BR), ATP production (ATP), proton leak (PL), maximal respiration (MR), and spare respiratory capacity (SRC) (n = 6 each group). H, Fluorescence staining showing reactive oxygen species (ROS) in WT and Mfn2MKO myoblasts. White arrows indicate ROS positive myoblasts. Scale bar: 20 μm. I, Quantification of ROS intensity in WT and Mfn2MKO myoblasts (n = 4 each group). All data are shown as mean ± SEM (t test: *P < .05, **P < .01)
To assess the role of Mfn2 in mitochondrial respiration, we used a Seahorse Analyzer to measure the OCR of Mfn2MKO and WT primary myoblasts. OCR associated with basal respiration (BR), ATP production (ATP), and proton leak (PL) were identical between WT and Mfn2-null myoblasts (Figure 2F). However, the OCR for maximal respiration (MR) and spare respiratory capacity (SRC) were significantly higher in Mfn2-null than those in WT myoblasts (Figure 2G). The elevated MR and SRC were associated with notable higher levels of complex V protein ATP5A in the Mfn2-null myblasts when compared to WT myoblasts (Figure S1). Meanwhile, ROS staining also showed a robust increase in ROS signal in Mfn2MKO compared to WT myoblasts (Figure 2H). The average ROS intensity of Mfn2MKO myoblasts was significantly higher than that of WT myoblasts (Figure 2I). Finally, we also checked the expression of mitochondrial complex proteins (complexes I-V) in pimary myoblast but there was little difference (Figure 2J). These results suggest the loss of Mfn2 in myoblast leads to abnormal mitochondria respiration and elevated oxidative stress.
3.3 |. Myofibers derived from Mfn2-null myogenic cells exhibit mitochondrial swelling
As Mfn2 was specifically deleted in all embryonic myoblasts that give rise to postnatal myofibers in Mfn2MKO mice, we investigated whether loss of Mfn2 would affect the number and structure of mitochondria in adult muscle. We first used transmission electron microscopy (TEM) to analyze the ultrastructure of mitochondria in Soleus (Sol) muscles of WT and Mfn2MKO mice. Mitochondria in myofibers were mainly found along subsarcolemmal and intermyofibrillar regions in both WT and Mfn2MKO mice (Figure 3A,B). Morphologically, the Mfn2-null mitochondria were much larger and contained irregularly arranged cristae when compared to the WT mitochondria (Figure 3A,B). Specifically, the size (measured by area) of WT subsarcolemmal mitochondria ranged from 0.1 to 1.3 μm2, while that of Mfn2MKO mitochondria was 0.2–3.1 μm2, averaging a nearly threefold increase in size (Figure 3C). In consistent, a twofold expansion of intermyofibrillar mitochondrial size was observed in Mfn2MKO myofibers (WT: 0.05–0.65 μm2, Mfn2MKO: 0.05–1.45 μm2) (Figure 3D). Interestingly, the enlarged size of Mfn2-null mitochondria was not associated with changes in mtDNA contents, which were identical between Mfn2MKO and WT groups in various muscle groups, including TA, EDL, Sol, and Gas (Figure 3E). The mRNA levels of fusion and fission related genes, including Mfn1, OPA1, Drp1, and Fis1, were also identical between WT and Mfn2MKO muscles (Figure 3F). In addition, mito-tracker staining patterns, mtDNA content, and Mfn1 expression of newly differentiated myotubes were very similar in the WT and Mfn2MKO groups (Figure 3G–I). These results demonstrate that mitochondria from Mfn2 knockout mice undergo swelling.
FIGURE 3.
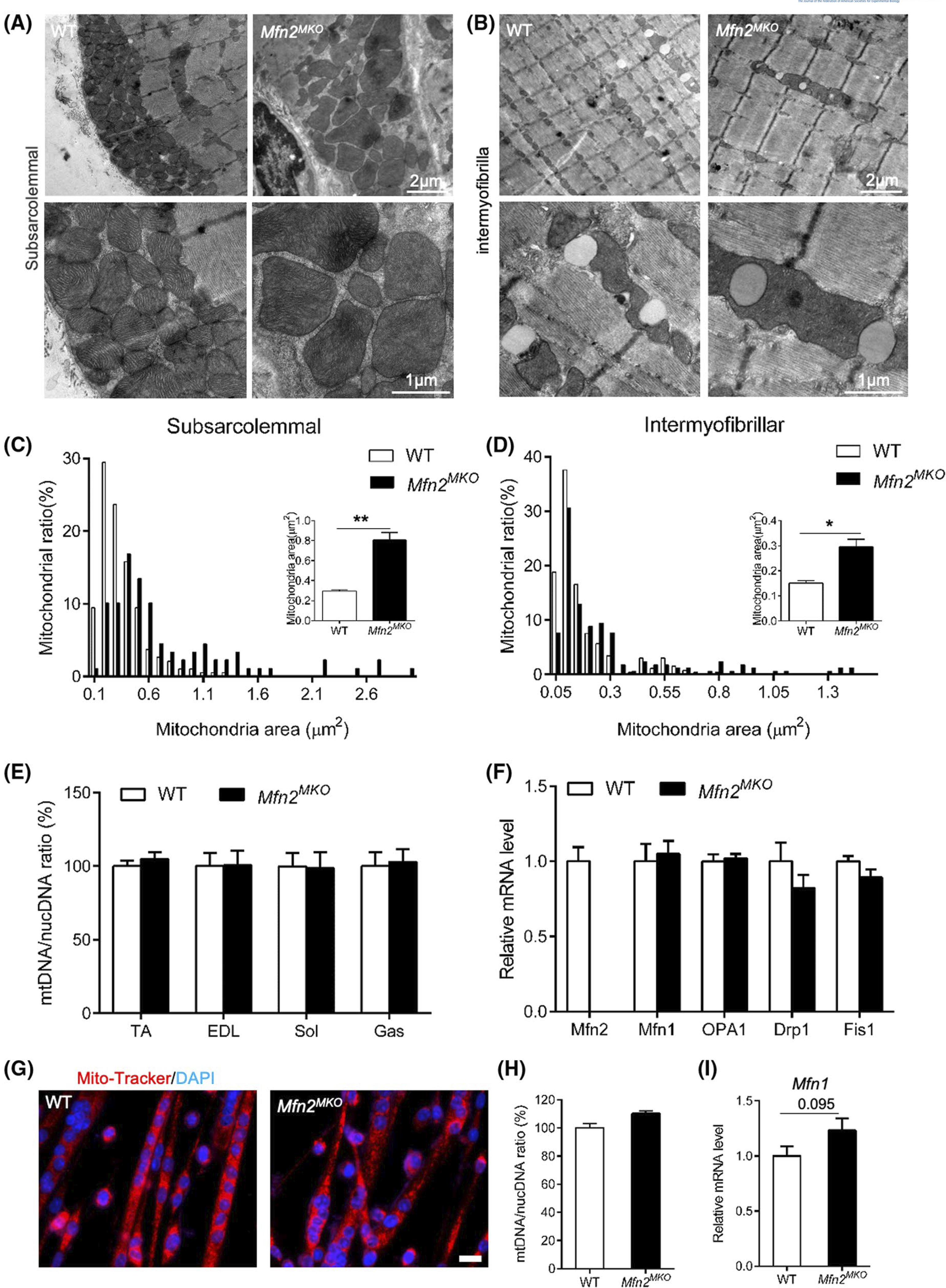
Mfn2-null myoblasts give rise to myofibers containing enlarged mitochondria. A and B, Representative transmission electron micrograph (TEM) of subsarcolemmal (A) and intermyofibrillar (B) mitochondria in WT and Mfn2MKO soleus muscles. C and D, Distribution analysis of subsarcolemmal (C) and intermyofibrillar (D) mitochondrial area in WT and Mfn2MKO muscles. Insets show the average mitochondrial areas (n = 3 pairs of mice). E, qRT-PCR analysis showing ratios of mitochondria DNA to nucleotide DNA in TA, EDL, Sol, and Gas muscles between WT and Mfn2MKO mice (n = 4 pairs of mice). F, qRT-PCR showing relative mRNA levels of fusion and fission genes in WT and Mfn2MKO Sol muscles (n = 4 pairs of mice). G, Immunofluorescence of mito-tracker in myotubes differentiated for 3 days. Scale bar: 20 μm. H and I, qRT-PCR analysis showing ratios of mitochondria DNA to nucleotide DNA (H) and mRNA levels of Mfn1 (I) in myotubes as shown in G (n = 4 pairs of mice). All data are shown as mean ± SEM (t test: *P < .05, **P < .01)
3.4 |. Loss of Mfn2 reduces the level of mitochondrial complex I proteins and affects electron transport
To examine the respiratory function of the swelled mitochondria in Mfn2MKO mice, we performed respiratory coupling and ETC assays using mitochondria isolated from WT and Mfn2MKO muscles. The Mfn2MKO mitochondria had normal state IIIADP, state IVo, and state IIIu OCR (Figure 4A,B). However, the Mfn2MKO mitochondrial had lower state IIbase OCR than WT mitochondria (176.06 ± 7.23 pmol/min in Mfn2MKO, 274.08 ± 24.92 pmol/min in WT; P < .01) (Figure 4B). The ETC assay indicated that the Mfn2MKO mitochondria exhibited a significantly lower (P < .01) uncoupled complex I respiration (66.86 ± 4.20 pmol/min), when compared to WT mitochondria (137.74 ± 13.07 pmol/min) (Figure 4C,D). However, the respiration of complex II and complex IV were similar in WT and Mfn2MKO mitochondria (Figure 4D). To further assess the structural basis of dysfunction of complex I, we checked the expression of mitochondrial complex proteins (complexes I-V) in different muscle groups, including TA, EDL, Sol, and Gas muscles. Consistently, only complex I (NDUFB8) proteins were specifically decreased (by ~20%, P < .05) in all Mfn2MKO muscles compared with WT muscles (Figure 4E,F). These results suggest that loss of Mfn2 reduces complex I protein content and respiratory function.
FIGURE 4.

Loss of Mfn2 reduces the level of mitochondrial complex I protein and affects electron transport. A, Seahorse coupling assay showing oxygen consumption rates (OCR) in mitochondria isolated from WT and Mfn2MKO Sol muscles. B, Quantification of OCR in state IIbase (Basal respiration), state IIIADP (ADP-stimulated respiration), state IVo (non-ADP-stimulated respiration, after oligomycin treatment), and state IIIu (uncoupled respiration, after FCCP treatment) (n = 3 pairs of mice). C, Seahorse electron transport chain assay showing oxygen consumption rates (OCR) in mitochondria isolated from WT and Mfn2MKO Sol muscles. D, Quantification of OCR in complex I, complex II, and complex IV (n = 3–5 pairs of mice). E, Western blots showing the protein levels of complexes I (NDUFB8), II (SDHB) III (UQCRC2), IV (MTCO1) and V (ATP5A) in TA, EDL, Sol and Gas muscles of WT and Mfn2MKO mice. F, Quantification of the relative levels of complex I (normalized to GAPDH) in TA, EDL, Sol, and Gas muscle (n = 4–6 pairs of mice). All data are shown as mean ± SEM (t test: *P < .05, **P < .01)
We also examined the protein levels of two additional complex I proteins, NDUFS3 and ECSIT. As a result, the level of NDUFS3 but not ECSIT was significantly decreased in Mfn2MKO compared with WT muscle (Figure 5A,B). Mfn2 has been reported to mediate mitochondrial complex I function through directly interacting with subunits p39 in rat heart.38 To investigate if Mfn2 interacts with complex I proteins, we constructed an Mfn2 overexpression vector, pEGFP-C1-Myc-MCS-Mfn2 plasmid where the EGFP gene in pEGFP-C1 is replaced by the Mfn2 gene (Figure 5C). Then we performed a Co-IP assay using the mitochondrial protein obtained from HEK293T cells that overexpress Mfn2. We detected a strong interaction between Mfn2 and NDUFS3, but not between Mfn2 and NDUFB8 or ECSIT (Figure 5D). These results suggest that Mfn2 may interacts with NDUFS3 to regulate the assembly and function of mitochondrial respiratory complexes.
FIGURE 5.
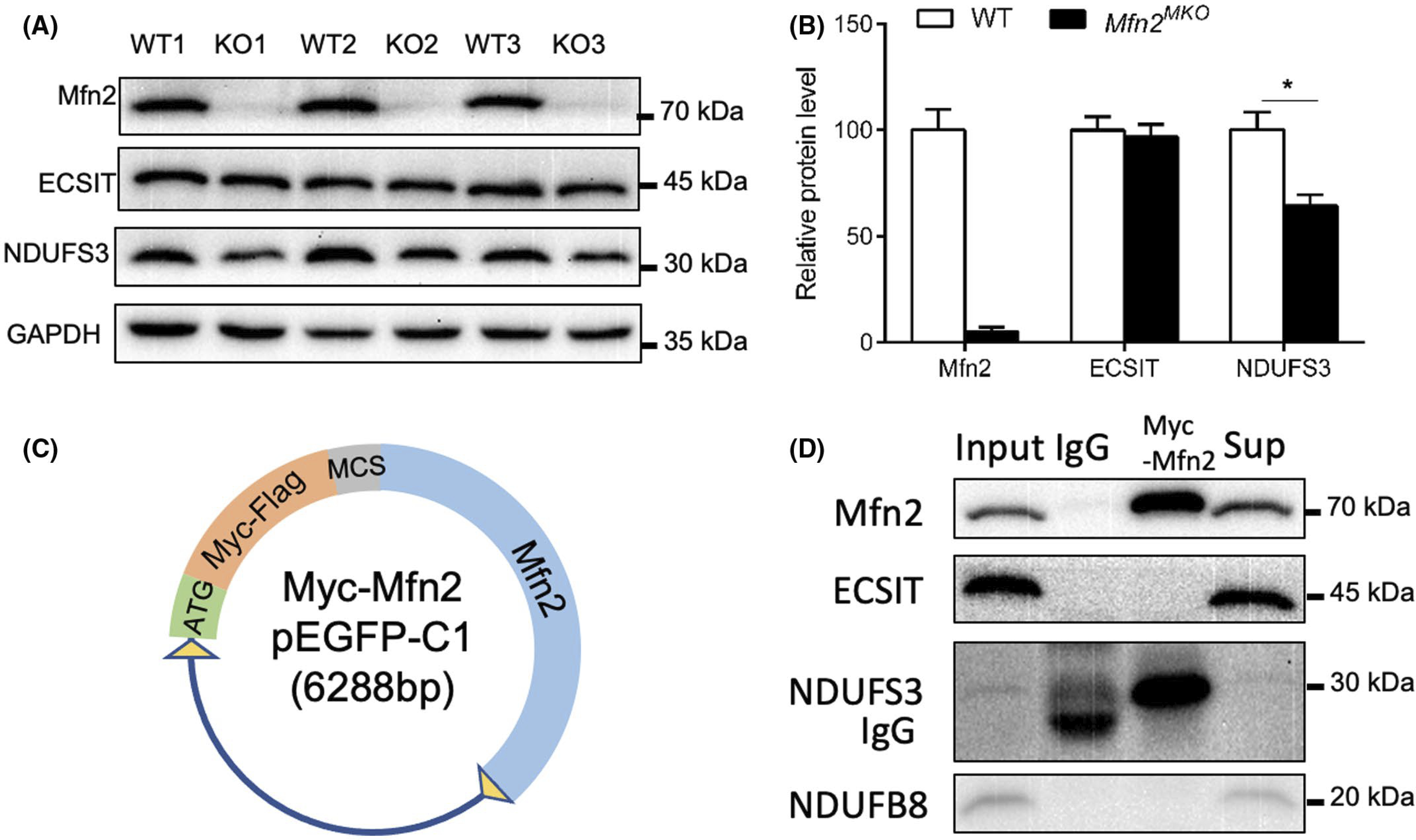
Mfn2 interacts with the complex I protein NDUFS3. A and B, Western blot images (A) and quantification (B) of the relative levels of Mfn2 and complex I subunits (ECSIT, NDUFS3) in Sol muscles of WT and Mfn2MKO mice. Data are shown as mean ± SEM (t test: *P < .05, n = 4–6 pairs of mice). C, pEGFP-C1-Myc-MCS-Mfn2 vector used to overexpress in 293T cells. D, Co-immunoprecipitation of Mfn2 and ECSIT, NDUFS3 or NDUFB8 using cell lysates from 293T cells transfected with the vector shown in C
3.5 |. Mfn2 is dispensable for embryonic and postnatal myogenesis
Given the requirement of Mfn2 in the mitochondrial function of myogenic progenitors, we evaluated how the loss of Mfn2 affects muscle development and regeneration. WT and Mfn2MKO mice were born normally, shared similar body size, weight, and lean and fat masses at 8 weeks old (Figure 6A–C). Various organs including brown and white adipose tissues, heart, kidney, liver, and spleen were all indistinguishable among WT, heterozygous, and Mfn2MKO (Figure S2), indicating the specificity of the KO. Morphology and weight of various muscles including TA, EDL, Sol, and Gas were also identical between WT and Mfn2MKO mice (Figure 6D,E). Immunofluorescence of dystrophin, the average myofiber size, and total myofiber number per microscopic area were indistinguishable between WT and Mfn2MKO mice (Figure 6F–H). These observations suggest that the loss of Mfn2 in embryonic myogenic progenitors has no obvious effect on muscle development and growth.
FIGURE 6.
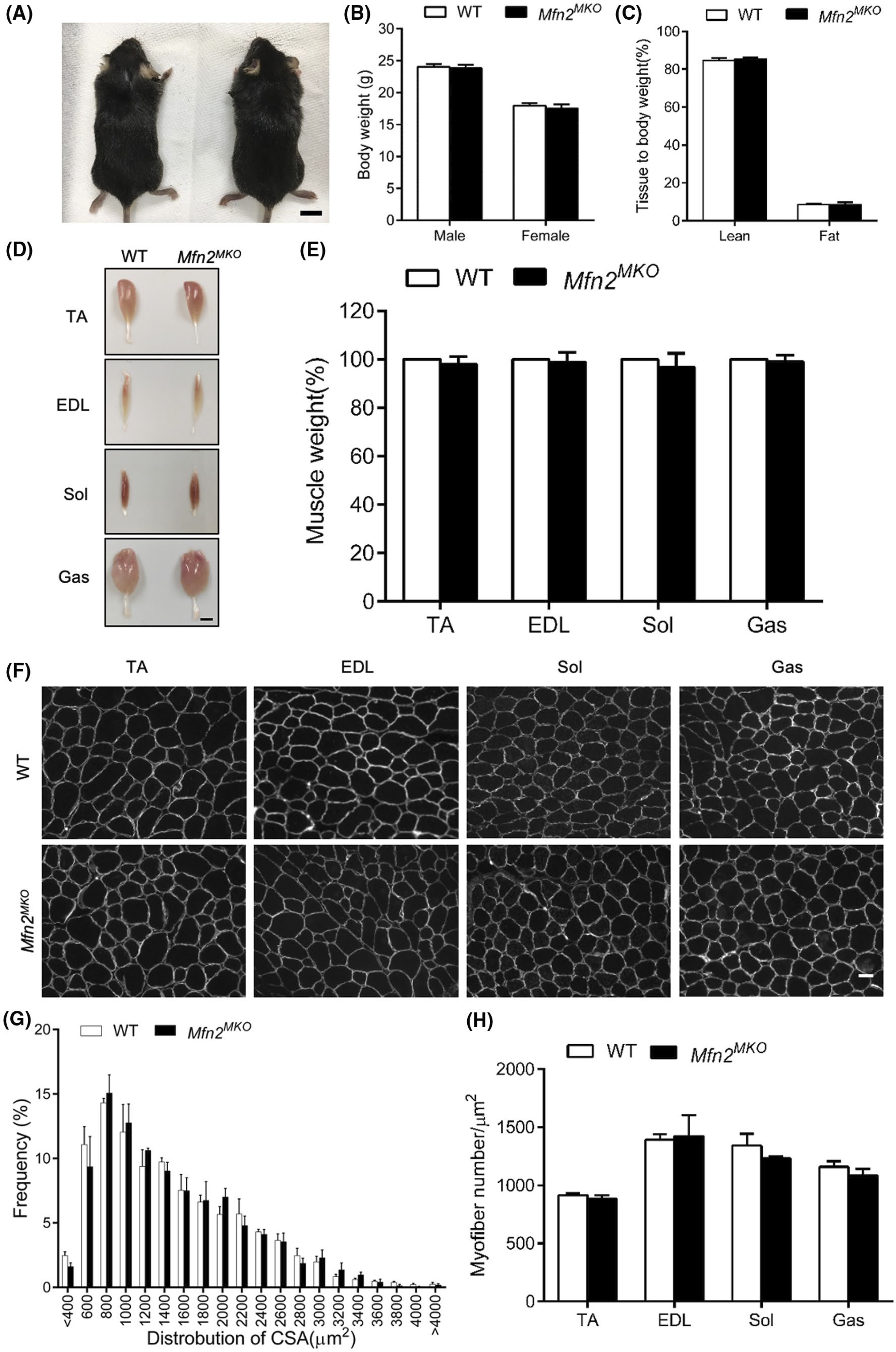
Conditional knockout of Mfn2 in myogenic progenitors does not affect muscle development. A, Representative images of WT and Mfn2MKO mice, scale bar: 1 cm. B, Body weights of male and female WT and Mfn2MKO mice at 2 months old (n = 10 pairs of mice). C, EchoMRI analysis showing ratios of lean and fat mass relative to body weight (n = 3 pairs of mice). D and E, Representative images (D) of TA, EDL Sol, and Gas muscles isolated from adult WT and Mfn2MKO mice (scale bar: 2 mm) and their relative weights (WT weights are normalized to 100, n = 6–8 pairs of mice). F, Dystrophin immunofluorescence outlining myofiber membrane to reveal myofiber size in WT and Mfn2MKO muscles. Scale bar: 50 μm. G, Distribution of myofiber cross-sectional areas (CSA, μm2) of Sol muscles (n = 4 each group). H, Average numbers of myofiber per μm2 in various muscles (n = 5–6 pairs of mice). All data represent mean ± SEM
Human Charcot-Marie-Tooth (CMT) patients harboring a frameshift mutation in Mfn2 exhibit inherited neuromuscular dysfunctions30,39; we therefore evaluated the motor performance of WT and Mfn2MKO mice using a treadmill and the grip force. The grip strengths of WT and gender-matched Mfn2MKO mice both ranged from 1.0 to 1.2 newtons (N) with no significant differences in the average strengths (Figure 7A). There were also no differences in maximum speed, running distance, and total running time between WT and KO mice (Figure 7B–D). In consistent, the composition of fiber type in both slow-twitch and fast-twitch muscles (Sol and EDL) from WT and Mfn2MKO mice were identical (Figure 7E–H). These observations indicate that loss of Mfn2 in myogenic progenitors do not affect the contractile function of the resulting myofibers.
FIGURE 7.
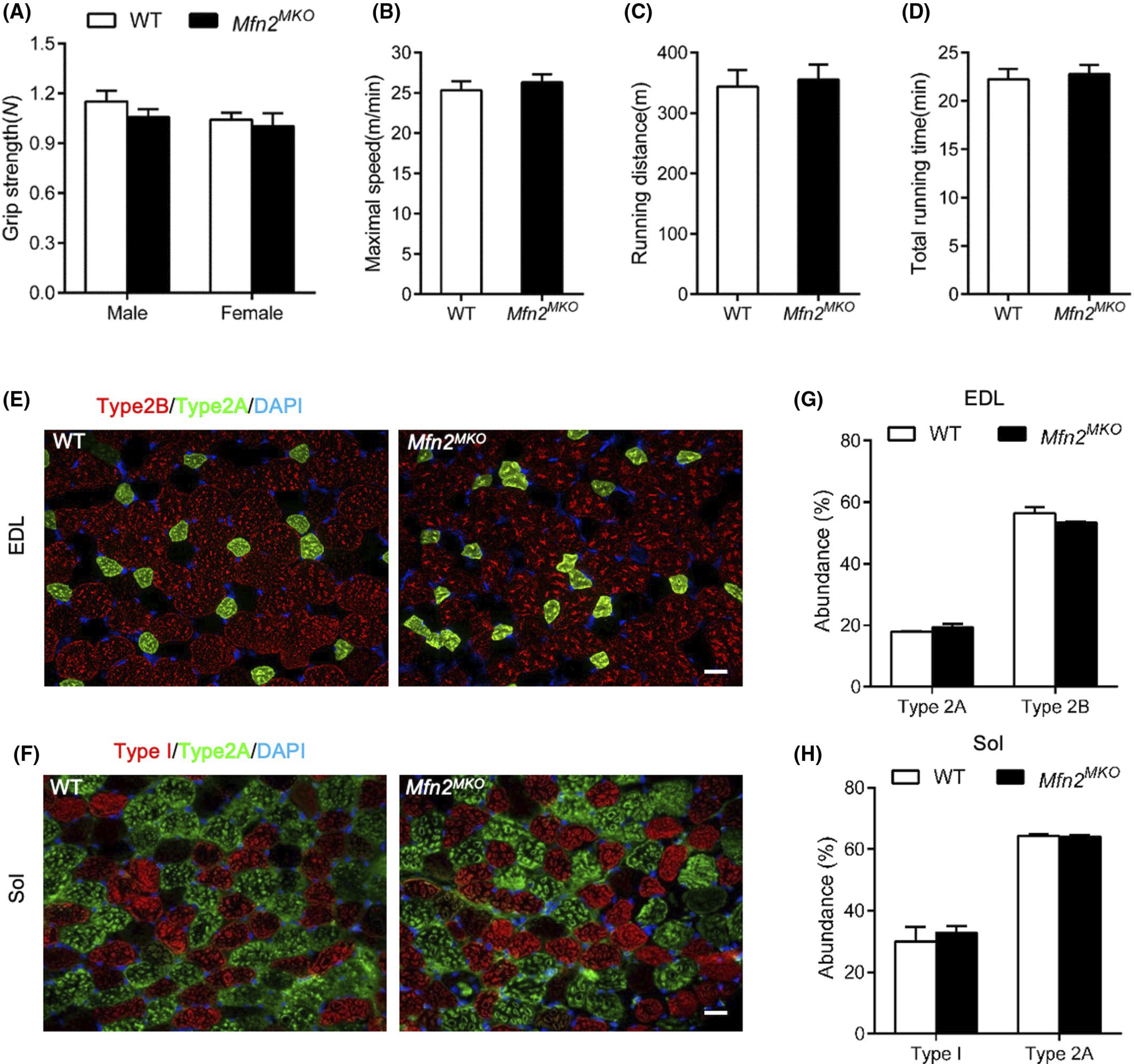
Mfn2MKO mice have normal exercise performance and myofiber type composition. A, Grip strengths of WT and Mfn2MKO mice at 8 weeks old (n = 6 and 8 pairs of mice for males and females, respectively). B–D, Treadmill exercise performance of male WT and Mfn2MKO mice (n = 6 pairs of mice), evaluated based on maximum running speed (B), running distance (C), and total running time (D). E and F, Immunofluorescence showing the distribution of type I, type 2A, and type 2B myofibers in EDL (E) and Sol (F) muscle sections. Scale bar: 20 μm. G and H, Percentage distribution of various types of myofiber in EDL (G) and Sol (H) muscles (n = 6 pairs of mice). All data represented mean ± SEM
We further examined MuSCs in WT and Mfn2MKO mice. Immunofluorescence staining of Pax7 and dystrophin showed no significant differences in the number of Pax7+ MuSCs that were attached to dystrophin-expressing sarcolemma per microscopic area between TA muscles of WT and Mfn2MKO mice (Figure 8A,B). Primary myoblasts derived from Mfn2-null MuSCs also had normal proliferation and differentiation capacity (data not shown). We further induced TA muscle injury by CTX injection and analyzed regeneration of the injured muscles (Figure 8C). At 5.5 dpi, TA muscles from WT and Mfn2MKO mice had similar masses and recovery rate (to ~60% of contralateral non-injured muscle weights) (Figure 8D,E). Histological sections showed that Mfn2MKO mice displayed similar morphological features to WT in numbers and sizes of the regenerated central-nucleated myofibers (Figure 8F–I). Similarly, immunofluorescence staining of dystrophin and Pax7 revealed no differences in myofiber size and MuSC numbers between WT and Mfn2MKO mice (Figure 8J,K). Thus, Mfn2 null MuSCs can regenerate injured muscles normally.
FIGURE 8.
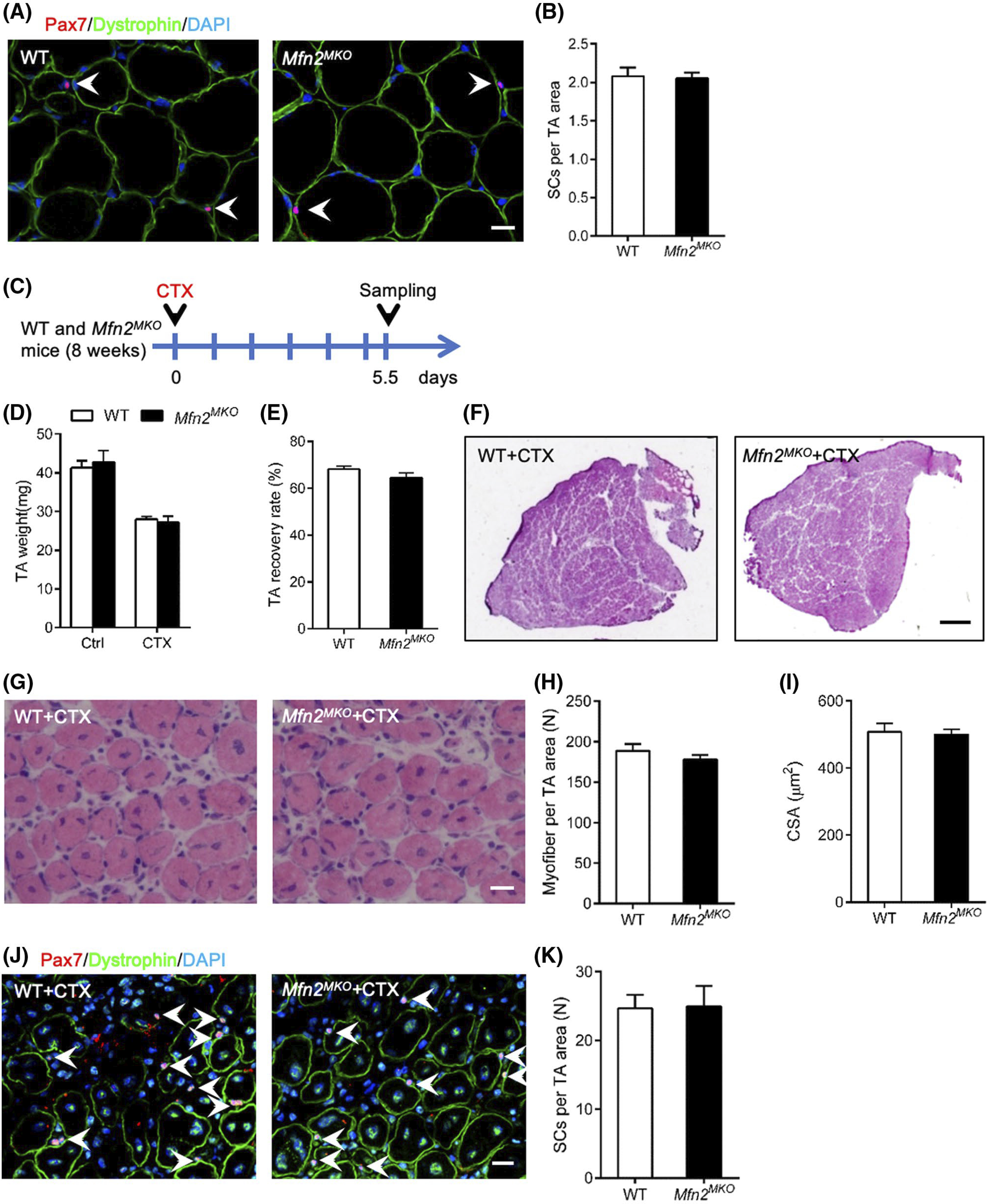
Mfn2MKO mice have normal regenerative capacity upon muscle injury. A, Immunofluorescence of Pax7 and dystrophin in TA muscle cross-sections at 8-week age. White arrows point to Pax7+ satellite cells (SCs). Scale bar: 20 μm. B, Average number of MuSCs per microscopic area (n = 6 pairs of mice). C, Experimental design for CTX injection and sample collecting. D and E, TA weight (D) and recovery rate (E, ratio of injured to uninjured muscle weights) (n = 6 pairs of mice). F and G, H&E staining of TA muscle cross-sections showing whole muscle view (F) and area view (G) of regenerated muscle at 5.5 days after CTX injury. Scale bar: 500 μm (F), 20 μm (G). H and I, Average numbers of regenerated myofibers per microscopic area (H) and cross-sectional area (CSA) of regenerated myofibers (I) (n = 6 pairs of mice, four to five microscopic areas per mouse). J, Immunofluorescence of Pax7 and Dystrophin on TA muscle cross-sections of WT and Mfn2MKO mice at 5.5 days after CTX injection. Scale bar: 20 μm. K, Quantification of the number of Pax7+ SCs (n = 5 pairs of mice, five microscope areas per mouse). All data represented mean ± SEM
4 |. DISCUSSION
Mfn2 is a key regulator of mitochondrial fusion that is essential for the remodeling of mitochondrial networks during cell differentiation, but the function of Mfn2 in myogenic cells and myogenesis remains unclear. In the present study, we discover a previously unrecognized role of Mfn2 in maintaining mitochondria size and complex I protein levels and function. Myogenic progenitor-specific knockout of Mfn2 increases OCR associated with maximal respiration and elevates ROS production and leads to mitochondrial swelling and reduction of complex I protein levels in adult skeletal muscle. Interestingly, Mfn2 directly interacts with a mitochondrial complex I subunits protein, NDUFS3. However, these structural and functional alterations of mitochondria in Mfn2MKO myoblasts do not affect their proliferation and differentiation, nor do they affect embryonic muscle development and postnatal muscle regeneration. These results demonstrate that myogenic progenitor cells lacking Mfn2 may have developed compensatory metabolic pathways to maintain normal myogenic function.
Our observation that Mfn2 is predominantly expressed in differentiated myotubes is consistent with a previous report that Mfn2 is induced during L6E9 myoblast differentiation in vitro and contributes to the maintenance and operation of the mitochondrial network.25 It was also reported that myogenic progenitor cells increase mitochondrial mass, DNA copy number and respiration during myogenesis.40,41 The upregulation of Mfn2 expression during differentiation may function to facilitate mitochondria remodeling to meet the metabolic demands of the growing skeletal muscle.42 In addition, we show that Mfn2 is mainly located around the myofiber membrane in mature myofibers but throughout the entire cytoplasm of newly differentiated myotubes. The temporal changes in Mfn2 intracellular localization may reflect dynamic remodeling of mitochondrial network and maturation of myofibrils during the late stages of myogenesis.
Previous studies have reported that deletion of Mfn2 in stem cell and thermogenic tissue increased mitochondrial respiration.28,39 Especially, disruption of Mfn2’s GTPase due to an arginine 94 to glutamine mutation causes uncoupling of respiration possibly by a less efficient mitochondrial quality control,30 augmenting cellular respiration to resist mild oxidative stress.30,39 These results indicate that loss of GTPase activity leads to respiratory dysfunction and oxidative stress in stem cells. In our model, KO of Mfn2 gene similarly led to elevated OCR tied to maximal mitochondrial respiration in primary myoblasts. The higher maximal respiratory capacity may also be linked to the increased mitochondrial size. In contrast, others have also reported that antisense oligonucleotides mediated knockdown of Mfn2 reduces mitochondrial membrane potential, cellular respiration, and proton leak in a muscle cell line.25 These contrasting results suggest that Mfn2 knockdown and knockout may have different functional consequences, or Mfn2 function may be cell type dependent.
Interestingly, this additional respiratory potency of Mfn2-null myoblasts is accompanied by elevated levels of ROS, consistent with previous reports.43,44 ROS are mainly formed from electron transfer reactions and oxygen is the major molecular source of redox equivalents at the cell and organism level.45 ROS production has been reported as an important indicator that tissues or cells are in an oxidative stress state.43,45 It has been reported that loss of Mfn2 induced an oxygen oxidative stress with elevated ROS level in rat skeletal muscle cell line,43 and a higher ROS level increases mitochondrial respiratory capacity in C2C12 cell line.46 Similar observations were reported in fibroblasts, where Mfn2 ablation caused an oxidative stress response and increased mitochondria maximal respiratory capacity through increased pyruvate shuttling into mitochondria.39 Therefore, we speculate that the increased maximal respiration and spare respiratory capacity in Mfn2-null myoblasts may be due to the elevated oxidative stress. Meanwhile, it is also possible that loss of Mfn2 in myoblasts increases the coupled respiratory capacity by improving the efficiency of fatty acids oxidation.28 Although elevated ROS production may be detrimental to myoblasts, the ROS signaling may serve as a feedback mechanism to protect the Mfn2-null myoblasts.
We did not detect any significant change either in adult satellite cell number or in myoblast proliferation in Mfn2 KO mice, suggesting that loss of Mfn2 has no effect on proliferation. The result is different from previous reports that loss of Mfn2 inhibits proliferation and cell-cycle via Ras-NF-κB signaling pathway in human cervical carcinoma cells,47 indicating a cell type-specific effect of Mfn2 on proliferation. Myogenic differentiation is accompanied by a metabolic switch from higher rates of glycolysis to primarily fatty acid oxidation.48 During muscle regeneration, activated myoblasts exhibit a high level of glycolysis after injury.49 The reliance on glycolysis spares other energy sources and provides the proliferating myogenic progenitor cells with macromolecules to meet their anabolic demands,50 also unnecessitated mitochondria dynamics and Mfn2 function. This explains the relative normal proliferation and differentiation of Mfn2 null myoblasts.
Mfn2 null embryonic myoblasts give rise to postnatal myofibers containing swelled mitochondria with cristae disorganization. These phenotypes are similar to the observation that mitochondria form spheres of widely varying sizes in other Mfn2-null cells and tissues. For instance, in Purkinje cells, deletion of Mfn2 leads to enlarged and spherical mitochondria.51 The diameters of the spherical mitochondria in Mfn2 mutant mouse embryonic fibroblasts and trophoblast stem cells are several times larger than the diameters of mitochondrial tubules in wild-type cells.22 The extra-large spherical mitochondria in Mfn2 null cells suggest that Mfn2 may be involved in maintaining the mitochondrial tubular shape in addition to its function in fusion. We found that the enlarged mitochondria in Mfn2-null myofibers were predominant in both the subsarcolemmal and the interfibrillar compartment, similar to those in cardiomyocytes of Mfn2 KO mice.52 As ROS also promotes mitochondrial expansion in the brain and liver,53–55 the elevated level of ROS in myoblasts may have contributed to mitochondrial swelling in the Mfn2-null myofibers.
We observed a specific reduction in the level of complex I subunits, NDUFB8 and NDUFS3, accompanied by reduced oxygen consumption from complex I in Mfn2 deficient muscles. Decreased mitochondrial complex I protein expression and activity have also been observed in Mfn2-deficiet heart, liver, brown, or white adipose tissue.28,38,56,57 However, adult myofiber-specific knockout of Mfn2 in homeostatic muscles did not lead to the reduction of complex I protein,34,56 suggesting perturbation of complex I proteins occurs during development and growth of muscles involving mitochondria remodeling. We provide evidence that the decrease of complex I proteins in Mfn2 defective tissues might be linked to a potential direct interaction between Mfn2 and NDUFS3. Similar interaction between Mfn2 and complex I subunit p39 via an intermembrane loop was reported in cardiomyocytes.38 Importantly, the reduction in complex I proteins is consistent with the reduced complex I OCR.
Mitochondrial fusion is affected by GTPase activity of Mfn1 and Mfn2 on mitochondrial outer membrane.22,26 It has been reported that Mfn2 regulates thermogenesis, adiposity, and Type 2A CMT disease phenotypes independent of mitochondrial fusion,29,57,58 suggesting that Mfn2 may be functionally distinct from Mfn1. However, the altered mitochondrial function in Mfn2-null myoblasts does not affect muscle development and regeneration. According to previous reports, single KO of Mfn1 or Mfn2 in muscle tissue has little impact on mitochondria shape, muscle fiber size and exercise performance, but double KO of Mfn1 and Mfn2 in muscle causes severe phenotype and leads to mitochondrial dysfunction.34,56,59 In our current myoblast-specific Mfn2 KO model, we found that Mfn1 expression was unaltered in myoblasts, myotubes, and mature myofibers (Figures 2E and 3F,I). Consistently, Mfn1 mRNA and protein levels were unchanged after KO of Mfn2 in adult skeletal muscles.56 The lack of compensatory upregulation of Mfn1 in our and other studies indicate redundant functions of Mfn1 and Mfn2 in myoblasts and myofibers. Gene disruption experiments in cultured cells also support that active GTPase domains from both Mfn1 and Mfn2 are essential for the mitochondrial fusion.22,23 Despite similar protein structures, Mfn1 exhibits eightfold higher GTPase activities than Mfn2 in an in vitro study.60 Thus, these results suggest that Mfn1 GTPase activity is sufficient to mediate normal muscle development and regeneration in Mfn2MKO mice.
In summary, our study demonstrates that deletion of Mfn2 improves spare respiratory capacity and elevates ROS production in myoblasts, leading to mitochondrial swelling and reduced level and function of complex I proteins in mature muscle. However, these changes do not affect muscle development and regeneration due to the potential compensatory function of Mfn1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the US National Institutes of Health (R01AR071649 to SK and R35GM119787 to QD), US Department of Agriculture (NC1184) and Purdue University Center for Cancer Research. NL is supported by a scholarship from the China Scholarship Council (No. 201806990052). The authors thank Jun Wu and Wenqing Zhou for technical assistance and members of the Kuang laboratory for valuable comments.
Funding information
HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: R01AR071649; U.S. Department of Agriculture (USDA), Grant/Award Number: NC1184; China Scholarship Council (CSC), Grant/Award Number: 201806990052; HHS | NIH | National Institute of General Medical Sciences (NIGMS), Grant/Award Number: R35GM119787
Abbreviations:
- ATP
adenosine triphosphate
- BR
basal respiration
- CSA
cross-sectional area
- CTX
cardiotoxin
- ETC
electron transport chain
- EDL
extensor digitorum longus
- Gas
gastrocnemius
- HE
hematoxylin-eosin
- MAS
mitochondrial assay solution
- Mfn1/2
mitofusin 1/2
- MR
maximal respiration
- mtDNA
mitochondrial DNA
- MuSCs
muscle satellite cells
- NDUFB8/S3
NADH ubiquinone oxidoreductase subunit B8/S3
- nuDNA
nuclear DNA
- OCR
oxygen-consumption rates
- PFA
paraformaldehyde
- PL
proton leak
- RIPA
radioimmunoprecipitation assay
- ROS
reactive oxygen species
- SEM
standard error mean
- Sol
soleus
- SRC
spare respiratory capacity
- TA
tibialis anterior
- TEM
transmission electron microscopy
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Westerblad H, Bruton JD, Katz A. Skeletal muscle: energy metabolism, fiber types, fatigue and adaptability. Exp Cell Res. 2010;316(18):3093–3099. 10.1016/j.yexcr.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Sheffieldmoore M, Urban R. An overview of the endocrinology of skeletal muscle. Trends Endocrinol Metab. 2004;15(3):110–115. 10.1016/j.tem.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Relaix F, Rocancourt D, Mansouri A, et al. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):948–953. 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 4.Kanisicak O, Mendez JJ, Yamamoto S, et al. Progenitors of skeletal muscle satellite cells express the muscle determination gene. MyoD. Dev Biol. 2009;332(1):131–141. 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seale P, Sabourin LA, Girgis-Gabardo A, et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102(6):777–786. [DOI] [PubMed] [Google Scholar]
- 6.Brack A, Rando T. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell. 2012;10(5):504–514. 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudnicki MA, Schnegelsberg PNJ, Stead RH, et al. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75(7):1351–1359. [DOI] [PubMed] [Google Scholar]
- 8.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med. 2008;14(2):82–91. 10.1016/j.molmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Jia Z, Nie Y, Yue F, et al. A requirement of Polo-like kinase 1 in murine embryonic myogenesis and adult muscle regeneration. Elife. 2019;8:e47097. 10.7554/eLife.47097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuang S, Kuroda K, Le Grand F, et al. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129(5):999–1010. 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26(6):711–723. 10.1016/j.beem.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duguez S, Féasson L, Denis C, et al. Mitochondrial biogenesis during skeletal muscle regeneration. Am J Physiol Endocrinol Metab. 2002;282(4):E802–E809. 10.1152/ajpendo.00343.2001. [DOI] [PubMed] [Google Scholar]
- 13.Ryall JG. Metabolic reprogramming as a novel regulator of skeletal muscle development and regeneration. FEBS J. 2013;280(17):4004–4013. 10.1111/febs.12189. [DOI] [PubMed] [Google Scholar]
- 14.Rochard P, Rodier A, Casas F, et al. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J Biolo Chem. 2000;275(4):2733–2744. [DOI] [PubMed] [Google Scholar]
- 15.Yu SB, Pekkurnaz G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J Mol Biol. 2018;430(21):3922–3941. 10.1016/j.jmb.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya D, Scimè A. Mitochondrial function in muscle stem cell fates. Front Cell Dev Biol. 2020;8:480. 10.3389/fcell.2020.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dirksen RT. Sarcoplasmic reticulum-mitochondrial through-space coupling in skeletal muscle. Appl Physiol Nutr Metab. 2009;34(3):389–395. 10.1139/H09-044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ljubicic V, Joseph A-M, Saleem A, et al. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 2010;1800(3):223–234. 10.1016/j.bbagen.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 19.Wyckelsma VL, Levinger I, McKenna MJ, et al. Preservation of skeletal muscle mitochondrial content in older adults: relationship between mitochondria, fibre type and high-intensity exercise training. J Physiol. 2017;595(11):3345–3359. 10.1113/JP273950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gouspillou G, Hepple RT. Editorial: mitochondria in skeletal muscle health, aging and diseases. Front Physiol. 2016;7:446. 10.3389/fphys.2016.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sin J, Andres AM, Taylor DJR, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 2016;12(2):369–380. 10.1080/15548627.2015.1115172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Detmer SA, Ewald AJ, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eura Y, Ishihara N, Yokota S, et al. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134(3):333–344. 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- 24.Zorzano A Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab. 2009;34(3):433–439. 10.1139/H09-049. [DOI] [PubMed] [Google Scholar]
- 25.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278(19):17190–17197. 10.1074/jbc.M212754200 [DOI] [PubMed] [Google Scholar]
- 26.Scott I, Youle RJ. Mitochondrial fission and fusion. Essays Biochem. 2010;47:85–98. 10.1042/bse0470085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ainbinder A, Boncompagni S, Protasi F, et al. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium. 2015;57(1):14–24. 10.1016/j.ceca.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahdaviani K, Benador IY, Su S, et al. Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep. 2017;18(7):1123–1138. 10.15252/embr.201643827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancini G, Pirruccio K, Yang X, et al. Mitofusin 2 in mature adipocytes controls adiposity and body weight. Cell Rep. 2019;26(11):2849–2858.e4. 10.1016/j.celrep.2019.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 31.Seo B, Yoon S, Do J. Mitochondrial dynamics in stem cells and differentiation. Int J Mol Sci. 2018;19(12):3893. 10.3390/ijms19123893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rochard P, Rodier A, Casas F, et al. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J Biol Chem. 2000;275(4):2733–2744. 10.1074/jbc.275.4.2733. [DOI] [PubMed] [Google Scholar]
- 33.Yue F, Bi P, Wang C, et al. Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun. 2017;8:14328. 10.1038/ncomms14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–289. 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–295. 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 36.Divakaruni AS, Paradyse A, Ferrick DA, et al. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014;547:309–354. 10.1016/B978-0-12-801415-8.00016-3. [DOI] [PubMed] [Google Scholar]
- 37.Rogers GW, Brand MD, Petrosyan S, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 2011;6(7):e21746. 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segalés J, Paz JC, Hernández-Alvarez MI, et al. A form of mitofusin 2 (Mfn2) lacking the transmembrane domains and the COOH-terminal end stimulates metabolism in muscle and liver cells. Am J Physiol Endocrinol Metab. 2013;305(10):E1208–E1221. 10.1152/ajpendo.00546.2012. [DOI] [PubMed] [Google Scholar]
- 39.Wolf C, Zimmermann R, Thaher O, et al. The Charcot-Marie Tooth disease mutation R94Q in MFN2 decreases ATP production but increases mitochondrial respiration under conditions of mild oxidative stress. Cells. 2019;8(10):1289. 10.3390/cells8101289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis. 2012;50(11):833–843. 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagatsuma A, Sakuma K. Mitochondria as a potential regulator of myogenesis. ScientificWorldJournal. 2013;2013:593267. 10.1155/2013/593267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liesa M, Shirihai O. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nie Q, Wang C, Song G, et al. Mitofusin 2 deficiency leads to oxidative stress that contributes to insulin resistance in rat skeletal muscle cells. Mol Biol Rep. 2014;41(10):6975–6983. 10.1007/s11033-014-3584-9. [DOI] [PubMed] [Google Scholar]
- 44.Kawalec M, Boratyńska-Jasińska A, Berȩsewicz M, et al. Mitofusin 2 deficiency affects energy metabolism and mitochondrial biogenesis in MEF cells. PLoS ONE. 2015;10(7):e0134162. 10.1371/journal.pone.0134162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7(8):504–511. 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oláh G, Szczesny B, Brunyánszki A, et al. Differentiation-associated downregulation of poly(ADP-Ribose) polymerase-1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS ONE. 2015;10(7):e0134227. 10.1371/journal.pone.0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Sun J, Yuan P, et al. Mfn2 inhibits proliferation and cell-cycle in Hela cells via Ras-NF-kappaB signal pathway. Cancer Cell Int. 2019;19:197. 10.1186/s12935-019-0916-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryall J, Dell’Orso S, Derfoul A, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16(2):171–183. 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pala F, Di Girolamo D, Mella S, et al. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J Cell Sci. 2018;131(14):jcs212977. 10.1242/jcs.212977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Folmes C, Dzeja P, Nelson T, et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 52.Papanicolaou KN, Khairallah RJ, Ngoh GA, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31(6):1309–1328. 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galindo MF, Jordan J, Gonzalez-Garcia C, et al. Reactive oxygen species induce swelling and cytochrome c release but not transmembrane depolarization in isolated rat mitochondria. Br J Pharmacol. 2003;139(4):797–804. 10.1038/sj.bjp.0705309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nabben M, Shabalina IG, Moonen-Kornips E, et al. Uncoupled respiration, ROS production, acute lipotoxicity and oxidative damage in isolated skeletal muscle mitochondria from UCP3-ablated mice. Biochim Biophys Acta. 2011;1807(9):1095–1105. 10.1016/j.bbabio.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 55.Wilson JD, Bigelow CE, Calkins DJ, et al. Light scattering from intact cells reports oxidative-stress-induced mitochondrial swelling. Biophys J. 2005;88(4):2929–2938. 10.1529/biophysj.104.054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bell MB, Bush Z, McGinnis GR, et al. Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J Appl Physiol. 2019;126(2):341–353. 10.1152/japplphysiol.00719.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boutant M, Kulkarni SS, Joffraud M, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017;36(11):1543–1558. 10.15252/embj.201694914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pich S, Bach D, Briones P, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14(11):1405–1415. 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 59.Mishra P, Varuzhanyan G, Pham A, et al. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 2015;22(6):1033–1044. 10.1016/j.cmet.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117(Pt 26):6535–6546. 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.