

Abstract
Objective
Systemic juvenile idiopathic arthritis (sJIA) is characterized by fever, arthritis, rash, hepatosplenomegaly, and macrophage activation syndrome; however, its pathogenesis is still unclear. Elevated serum interleukin (IL)‐18 concentrations and decreased natural killer (NK) cell activity are characteristic of active disease; thus, we examined IL‐18 signaling in NK cells from sJIA.
Methods
We analyzed mitogen‐activated protein kinase (MAPK) p38 and nuclear factor κ light chain enhancer of activated B cells (NFκB) p65 phosphorylation in NK cells after in vitro recombinant IL‐18 (rIL‐18) stimulation in 31 patients with sJIA. Associations between clinical features, serum IL‐18, and phosphorylation intensity were analyzed. Furthermore, we investigated the effects of high IL‐18 concentrations on phosphorylation in NK cells.
Results
Patients were divided according to their disease activity: systemic features (n = 8), chronic arthritis (n = 7), remission on medication (n = 10), and remission off medication (n = 6). MAPK p38 and NFκB p65 phosphorylation intensity were the highest in healthy controls, followed by remission off medication, remission on medication (vs. control; MAPK p38, P < 0.01; NFκB p65, P < 0.05), chronic arthritis (P < 0.001, P < 0.001), and systemic features (P < 0.001, P < 0.001). The systemic features group showed a complete defect in phosphorylation. Serum IL‐18 was the highest in the systemic features group followed by chronic arthritis, remission on medication (P < 0.01), remission off medication (P < 0.01), and healthy controls (P < 0.01). Phosphorylation intensity was negatively correlated with serum IL‐18 (MAPK p38, r 2 = 0.42; NFκB p65, r 2 = 0.54). Furthermore, healthy control NK cells were cultured with rIL‐18; impaired phosphorylation was reproduced in vitro.
Conclusion
Impaired IL‐18 signaling in NK cells correlated with disease activity in sJIA. High serum IL‐18 exposure induces impaired MAPK and NFκB phosphorylation in NK cells.
INTRODUCTION
Systemic juvenile idiopathic arthritis (sJIA) is characterized by fever, arthritis, skin rash, and hepatosplenomegaly and is often complicated by macrophage activation syndrome (MAS). MAS is a life‐threatening complication of sJIA with massive inflammatory activation. Despite improved outcomes owing to the development of biologics targeting interleukin (IL)‐1β and IL‐6, the pathogenesis of sJIA is still unclear (1, 2, 3, 4).
Previous reports showed that significant elevation of serum IL‐18 was characteristic of active sJIA and MAS and was correlated with disease activity (5). IL‐18 is a proinflammatory cytokine, a member of the IL‐1 family, which is initially produced as an inactive pro‐form and then secreted following maturation by caspase‐1 (6, 7). After IL‐18 binds to the IL‐18 receptor α (IL‐18Rα) expressed on the surface of various cells, such as Th1 T cells and natural killer (NK) cells (8), IL‐18Rβ is recruited and forms a heterotrimeric complex with IL‐18 and IL‐18Rα. The complex induces the phosphorylation of intracellular signaling molecules, myeloid differentiation factor 88 (MyD88), human interleukin 1 receptor associated kinase (IRAK), and tumor necrosis factor receptor associated factor (TRAF6), followed by nuclear factor κ light chain enhancer of activated B cells (NFκB) phosphorylation and activation. Furthermore, the complex induces the phosphorylation of mitogen‐activated protein kinase (MAPK) p38 (9, 10, 11). In NK cells, IL‐18 activates intracellular MAPK p38 signal, leading to interferon (IFN)‐γ secretion and cytotoxic activity (12, 13, 14). Meanwhile, it has been reported that despite high serum IL‐18 concentrations in patients with active sJIA, their NK cells remain dysfunctional. Put et al demonstrated that NK cells from patients with sJIA with active disease showed decreased granzyme K expression, normal granzyme B expression, and impaired IL‐18‐driven IFNγ production. Impaired extracellular signal‐regulated kinase (ERK) 1/2 and MAPK p38 phosphorylation after recombinant IL‐18 (rIL‐18) stimulation of NK cells was also demonstrated (14). Jager et al demonstrated that NK cells from patients with sJIA with active disease failed to upregulate intracellular perforin and IFNγ production after IL‐18 stimulation. They conclude that the impaired NK cell response to IL‐18 stimulation could result from defective IL‐18Rβ phosphorylation (9).
We hypothesize that impaired IL‐18 signaling in NK cells could be associated with sJIA activity and clinical course. In this study, we examined the relationship between IL‐18/IL‐18R signaling in NK cells from patients with sJIA and clinical features of disease. To evaluate IL‐18 signaling in NK cells and its potential use as a biomarker, we analyzed the intensity of MAPKp38 and NFκB phosphorylation after rIL‐18 stimulation using flow cytometry. Additionally, we analyzed IL‐18/IL‐18R signaling in healthy NK cells cultured with rIL‐18 to assess the effects of high IL‐18 concentrations on NK cell signaling.
PATIENTS AND METHODS
Patients
A total of 31 patients with sJIA and six healthy controls were recruited from the Yokohama City University Hospital between 2016 and 2020. The patients were diagnosed with sJIA using the International League of Associations for Rheumatology criteria (15). They were classified into the following four groups according to their disease activity: 1) systemic features (recent onset or recurrent disease; systemic symptoms with or without arthritis), 2) chronic arthritis (active arthritis for more than 6 months without systemic symptoms), 3) remission on medication, and 4) remission off medication. Remission was defined as being asymptomatic for more than 3 months. In this study, patients with sJIA complicated with MAS were excluded because their NK cell counts were very low and it was difficult to analyze their IL‐18/IL‐18R signaling. Blood samples were collected from the patients and healthy controls after obtaining written informed consent from them or their parents or guardians. This study was performed in accordance with the Declaration of Helsinki and approved by the institutional ethics committees of Yokohama City University School of Medicine (approval date: August 2016; approval number: B160804004).
IL‐18 analysis
Serum IL‐18 concentrations were measured by a human IL‐18 enzyme linked immunosorbent assay (ELISA) kit (Medical & Biological Laboratories [MBL]) according to the manufacturer's instructions.
Phosphorylation analysis
We analyzed the phosphorylation of MAPK p38 and NFκB p65 after the in vitro rIL‐18 stimulation of NK cells by flow cytometry. Immediately after collection, whole blood was separated into peripheral blood mononuclear cells (PBMCs) by Lymphoprep (Axis‐Shield PoC AS). PBMCs (1.0 × 106 cells) were incubated with 100 ng/mL rIL‐18 (MBL) for 15 minutes at 37°C and stained with cell surface markers (APC antihuman CD56 antibody and PE/Cy7 antihuman CD3 antibody [Biolegend]). The PBMCs were fixed in a fixation buffer (BD Biosciences) and permeabilized in BD Phosflow Perm buffer (BD Biosciences). The cells were then incubated with BD Phosflow PE anti‐p38 MAPK (pT180/pY182) antibody and PE anti‐NFκB p65 (pS529) antibody (BD Biosciences) for 60 minutes at room temperature. The cells were washed and centrifuged at 1500 × g for 1 minute, twice. The pellets were then resuspended and immediately analyzed by flow cytometry (EC800, Sony Biotechnology). The data analysis was performed using FlowJo version 7.6.5 software (TreeStar Inc). NK cells were defined as the CD3−CD56+ population after lymphocyte gating, and the intensity of phosphorylation was evaluated by mean fluorescent intensity (MFI).
Analysis of IL‐18Rα expression on NK cells
The PBMCs were incubated with PE antihuman IL‐18Rα antibody (Biolegend), APC antihuman CD56 antibody (Biolegend), and PE/Cy7 antihuman CD3 antibody (Biolegend) for 30 minutes at 4°C. The cells were analyzed by flow cytometry (EC800, Sony Biotechnology). The degree of IL‐18Rα expression in NK cells was evaluated by MFI.
Culture of PBMCs with rIL‐18
We evaluated the association between clinical features of sJIA, serum IL‐18 concentrations, and the phosphorylation intensity of MAPK p38 and NFκB p65 in NK cells. We cultured six healthy control PBMCs (1.0 × 106 cells) in RPMI 1640 medium (Wako Pure Chemical industries) with and without rIL‐18 (MBL) for 12 hours. The rIL‐18 concentrations were 0.1 ng/mL, 1 ng/mL, and 10 ng/mL. After culture with rIL‐18, we analyzed the phosphorylation of MAPK and NFκB on NK cells as aforementioned.
Statistical analysis
Statistical analysis was performed using EZR (Saitama Medical Center, Jichi Medical University Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing) (16). Significance for comparison between samples was analyzed by one‐way analysis of variance with Bonferroni post hoc test. A P value of <0.05 was considered to be statistically significant. Associations between two parameters were analyzed by the Pearson correlation coefficient.
RESULTS
Patient background, serum IL‐18 concentration, and IL‐18Rα expression in NK cells
A total of 31 patients with sJIA were enrolled. The patients’ characteristics are shown in Table 1. According to their disease activity and status, they were classified into the following four groups: systemic features (n = 8 [6 recent onset and 2 recurrent]), chronic arthritis (n = 7), remission on medication (n = 10), and remission off medication (n = 6). In the systemic features group, all patients presented with fever, skin rash, and arthritis, except two patients who did not have arthritis. Conversely, none of the patients in the other groups presented with a fever or rash. The patients in the chronic arthritis group all received oral prednisolone (PSL), methotrexate (MTX), and tocilizumab (TCZ). The remission on medication group all received TCZ, with four patients receiving PSL and one patient receiving MTX, in addition. None of the patients had disease complicated by MAS. Five patients (chronic arthritis group [n = 1], remission on medication group [n = 4]) had a history of MAS.
Table 1.
Characteristics of patients with sJIA
| Systemic features | Chronic arthritis | Remission on medication | Remission off medication | ||
|---|---|---|---|---|---|
| Recent onset | Recurrence | ||||
| Number | 6 | 2 | 7 | 10 | 6 |
| Male/female | 3/3 | 2/0 | 2/5 | 5/5 | 2/4 |
| Age, y, median (range) | 4.0 (0.5‐15) | 14.0 (13‐18) | 15.0 (10‐35) | 13.5 (3‐24) | 9.5 (3‐13) |
| Symptoms, n | |||||
| Fever | 6 | 2 | 0 | 0 | 0 |
| Rash | 6 | 2 | 0 | 0 | 0 |
| Arthritis | 6 | 0 | 7 | 0 | 0 |
| History of MAS, n | 0 | 0 | 1 | 4 | 0 |
| Medications, n | |||||
| NSAIDs | 0 | 0 | 0 | 0 | 0 |
| Prednisolone | 1 | 1 | 7 | 4 | 0 |
| MTX | 0 | 0 | 7 | 1 | 0 |
| Tocilizmab | 0 | 1 | 7 | 10 | 0 |
| Laboratory data | |||||
| WBC, median, × 103/μL | 12.9 | 21.3 | 8.8 | 5.3 | 6.1 |
| CRP, median, mg/L | 51.7 | 77.3 | 0.1 | 0.2 | 0.3 |
| Serum IL‐18, pg/mL, median (range) | 84,355 (11,155‐148,646) | 5430.9 (1023‐9839) | 3210.6 (1709‐14,056) | 646.0 (246‐4274) | 195.3 (157‐450) |
Abbreviations: CRP, C‐reactive protein; IL‐18, interleukin‐18; MAS, macrophage activation syndrome; MTX, methotrexate; NSAID, non‐steroidal antiinflammatory drug; sJIA, systemic juvenile idiopathic arthritis; WBC, white blood cell.
Serum IL‐18 was the highest in the systemic features group followed by chronic arthritis, remission on medication (vs. systemic features group; P < 0.01), remission off medication (vs. systemic features group; P < 0.01), and healthy controls (vs. systemic features group; P < 0.01) (Figure 1). IL‐18Rα expression on NK cells from patients with sJIA and healthy controls are shown in Supplementary Figure S1. The expression is slightly lower in the systemic features group; however, the difference between each group is not statistically significant.
Figure 1.
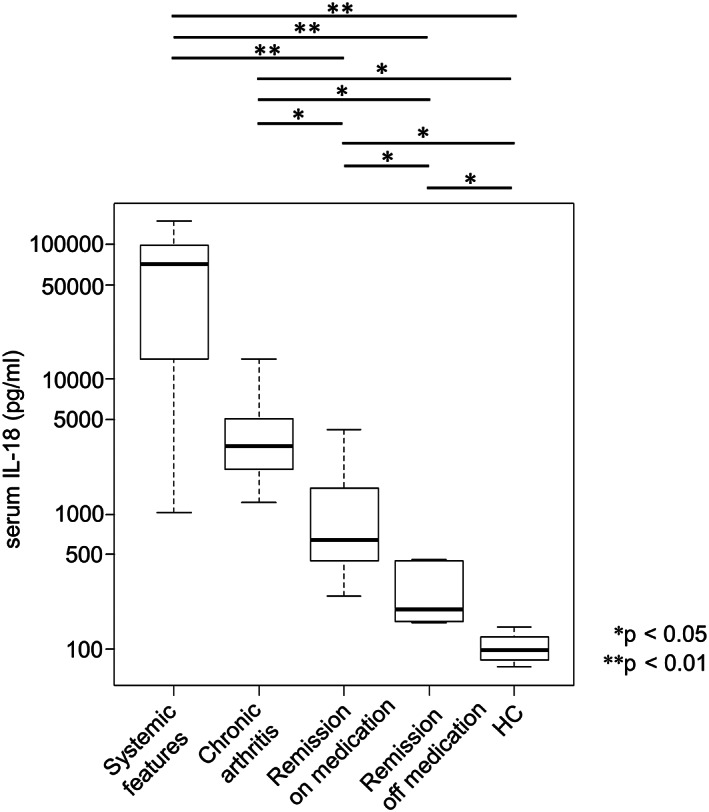
Serum IL‐18 concentration in each group of patients with sJIA and healthy controls. The systemic features group showed the highest serum IL‐18 concentration, followed (in order) by chronic arthritis, remission on medication, and remission off medication. The difference between each group is statistically significant. Data are shown as box plots, in which the boxes represent the first to third quartiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values. Statistical significance is indicated by P values. HC, healthy control; IL, interleukin; sJIA, systemic juvenile idiopathic arthritis.
Phosphorylation analysis in NK cells from patients with sJIA and healthy controls
Figure 2a demonstrates the representative pattern of phosphorylation of MAPK p38 and NFκB p65 in NK cells after rIL‐18 stimulation. The phosphorylation intensity was similar in the healthy controls and remission off medication patients; however, phosphorylation was completely absent in patients with systemic features. Figure 2b shows a comparison of the phosphorylation intensity in each group. Phosphorylation intensity was the highest in healthy controls, followed by remission off medication, remission on medication (vs. control; MAPK p38, P < 0.01; NFκB p65, P < 0.05), and chronic arthritis (P < 0.001, P < 0.001) and absent from the systemic features group (P < 0.001, P < 0.001). Both MAPK p38 and NFκB phosphorylation showed no significant difference between the systemic features group and the chronic arthritis group, or between the healthy control and remission off medication groups.
Figure 2.
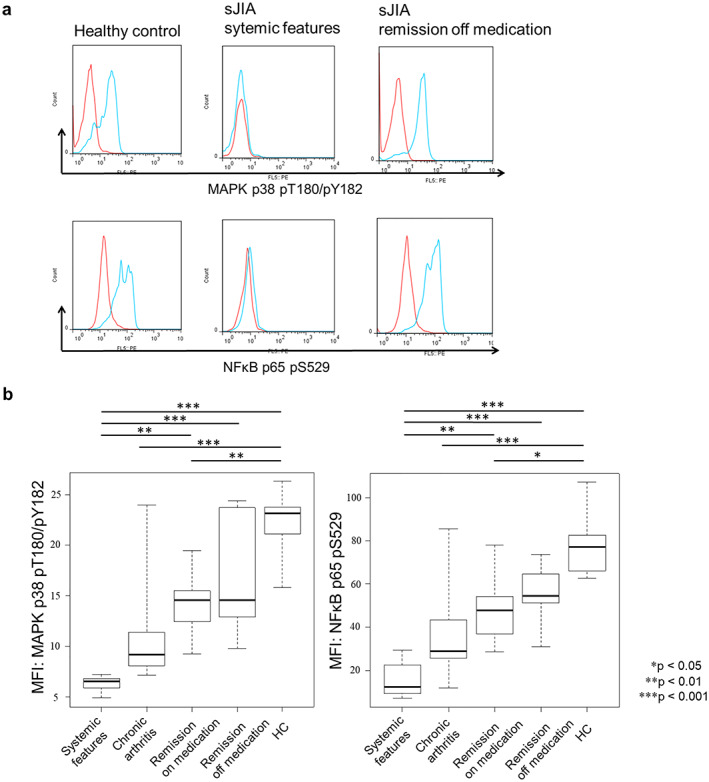
Phosphorylation of MAPK p38 and NFκB p65 in NK cells from patients with sJIA and healthy controls after rIL‐18 stimulation. (a) The phosphorylation of MAPK p38 and NFκB p65 after rIL‐18 stimulation of NK cells of healthy controls, systemic features group, and remission off medication group patients. The NK cells from healthy controls and remission off medication group patients showed similar levels of MAPK p38 and NFκB p65 phosphorylation. In the systemic features group patients, there was no phosphorylation. Red peak: no stimulation. Blue peak: rIL‐18 stimulation. (b) A comparison of each group and the healthy controls. In the systemic features group, the phosphorylation of MAPK p38 and NFκB p65 was the lowest, followed by the chronic arthritis group and remission on medication group. The phosphorylation was statistically different between the systemic features group and remission on medication group (MAPK p38, P < 0.01; NFκB p65, P < 0.01). No statistically significant difference was seen between the remission off medication group and healthy controls. Statistical significance is indicated by P values. HC, healthy control; IL, interleukin; MAPK, mitogen‐activated protein kinase; MFI, mean fluorescent intensity; NFκB, nuclear factor κ light chain enhancer of activated B cells; NK, natural killer; sJIA, systemic juvenile idiopathic arthritis.
Correlation between the phosphorylation of MAPK p38 and NFκB p65 in NK cells and serum IL‐18 concentration
We analyzed the correlation between the phosphorylation of MAPK p38 and NFκB p65 in NK cells after rIL‐18 stimulation and serum IL‐18 concentration in patients with sJIA. The phosphorylation of MAPK p38 and NFκB was impaired when serum IL‐18 was high and negatively correlated with serum IL‐18 concentration (MAPK p38, r 2 = 0.4237; NFκB p65, r 2 = 0.5417) (Figure 3).
Figure 3.
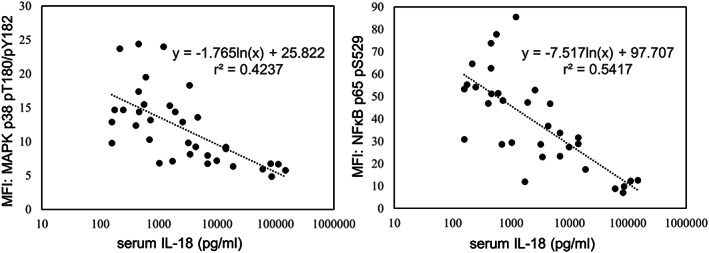
Association between the phosphorylation intensity of MAPK p38 and NFκB p65 in NK cells after rIL‐18 stimulation and serum IL‐18 concentration in patients with sJIA. The phosphorylation of MAPK p38 and NFκB p65 was impaired when serum IL‐18 was high and was negatively correlated with serum IL‐18 concentration (MAPK p38, r 2 = 0.4237; NFκB p65, r 2 = 0.5417). IL, interleukin; MAPK, mitogen‐activated protein kinase; MFI, mean fluorescent intensity; NFκB, nuclear factor κ light chain enhancer of activated B cells; NK, natural killer; rIL‐18, recombinant IL‐18; sJIA, systemic juvenile idiopathic arthritis.
Clinical course and recovery of phosphorylation of MAPK p38 and NFκB p65 in rIL‐18‐stimulated NK cells from patients with sJIA
Figure 4 shows the sequential change in MAPK p38 and NFκB p65 phosphorylation and serum IL‐18 in three patients with sJIA (Figure 4a‐c). These patients were from the systemic features group and were followed up either once or twice during their clinical course. All these patients showed a recovery in their MAPK p38 and NFκB p65 phosphorylation toward levels seen in healthy controls and a decrease in serum IL‐18 in accordance with clinical remission following successful treatment.
Figure 4.
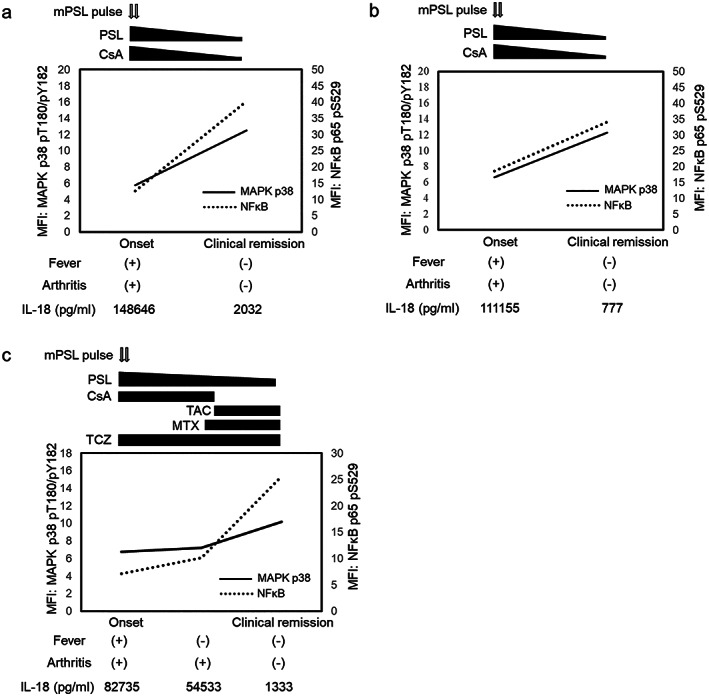
Clinical courses in three patients with sJIA and the phosphorylation of MAPK p38 and NFκB p65 in NK cells after rIL‐18 stimulation (a, b, and c). Phosphorylation was analyzed in three patients at disease onset without treatment and followed up at one or two points, subsequently. With the reduction of serum IL‐18 concentration, the phosphorylation of MAPK p38 and NFκB p65 was recovered, which was consistent with clinical improvement. CsA, cyclosporin A; IL, interleukin; MAPK, mitogen‐activated protein kinase; MFI, mean fluorescent intensity; mPSL, methyl prednisolone; MTX, methotrexate; NFκB, nuclear factor κ light chain enhancer of activated B cells; NK, natural killer; PSL, prednisolone; rIL‐18, recombinant IL‐18; sJIA, systemic juvenile idiopathic arthritis; TAC, tacrolimus; TCZ, tocilizumab.
Phosphorylation analysis in healthy NK cells after culture with rIL‐18
To investigate the mechanism of suppression of MAPK p38 and NFκB p65 phosphorylation in NK cells subjected to high serum IL‐18 concentrations, we cultured PBMCs from six healthy donors with rIL‐18 in vitro and analyzed the phosphorylation of MAPK p38 and NFκB p65. The phosphorylation of MAPK p38 and NFκB p65 was suppressed in a dose‐dependent manner (Figure 5). Impaired MAPK p38 and NFκB p65 phosphorylation in NK cells was successfully reproduced in vitro by high IL‐18 exposure.
Figure 5.
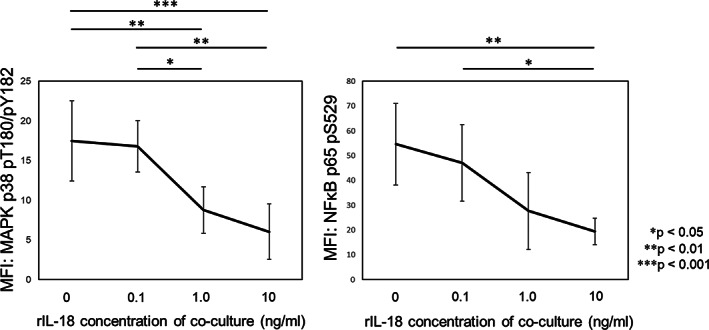
Phosphorylation analysis in NK cells from healthy controls after culture with rIL‐18 in vitro. We cultured PBMCs from healthy controls (n = 6) with rIL‐18 for 12 hours and then analyzed the phosphorylation of MAPK p38 and NFκB p65 after stimulation with 100 ng/mL rIL‐18. The phosphorylation of MAPK p38 and NFκB p65 after rIL‐18 stimulation was impaired after culture with rIL‐18, and the degree of impaired phosphorylation increased with rIL‐18 concentration. Error bars indicate SD. Statistical significance is indicated by P values. MAPK, mitogen‐activated protein kinase; MFI, mean fluorescent intensity; NFκB, nuclear factor κ light chain enhancer of activated B cells; NK, natural killer; PBMC, peripheral blood mononuclear cell; rIL‐18, recombinant interleukin‐18.
DISCUSSION
NK cell dysfunction and abnormally high serum IL‐18 concentrations are characteristic immunological abnormalities seen in sJIA. Previous studies revealed that serum IL‐18 is significantly correlated with disease activity (5, 17). Meanwhile, there have been few reports investigating a correlation between NK cell dysfunction and disease activity. In this study, we discovered that impaired IL‐18‐driven MAPK p38 and NFκB p65 phosphorylation in NK cells was significantly correlated with disease activity in patients with sJIA. Furthermore, MAPK p38 and NFκB p65 phosphorylation were markedly suppressed in acute sJIA with systemic features. Additionally, we found that NK cell dysfunction could be induced following exposure to persistently high concentrations of IL‐18.
Owing to the development of biological agents targeting IL‐6 and IL‐1β, the outcome of sJIA has dramatically improved; however, 10%‐15% of patients develop MAS, which is a life‐threatening complication (2, 18, 19, 20). Furthermore, MAS can occur despite the use of biologics. Additionally, approximately 30% of patients develop chronic arthritis even when treated with biologics (21); thus, establishing new treatments against chronic arthritis in sJIA is urgently needed. Many studies regarding NK cell dysfunction focus on the acute phase of arthritic disease, not chronic arthritis or during remission. Our study revealed suppressed MAPK p38 and NFκB p65 phosphorylation reflecting NK cell dysfunction persisting in patients with chronic arthritis and remission on medication. Furthermore, the suppressed phosphorylation observed in the chronic arthritis group was as severe as that in the systemic features group, suggesting that the underlying immunological abnormality was not altered during the switch to chronic disease.
Interestingly, suppressed phosphorylation intensity was significantly correlated with serum IL‐18, and previous studies have shown that serum IL‐18 reflects disease activity in patients with sJIA. However, serum IL‐18 measured by conventional ELISA contains inactive forms of IL‐18 and active IL‐18 and IL‐18 binding protein (IL‐18BP) complexes. Recently, it was reported that IL‐18BP deficiency in mice was associated with elevated plasma levels of free IL‐18 and an enhanced IFNγ signature (22). Therefore, the measurement of free IL‐18 would be optimal to accurately determine active IL‐18 levels; however, it is still technically difficult to do (23, 24). As shown in Figure 4, evaluation of MAPK p38 and NFκB p65 phosphorylation in NK cells may allow the direct evaluation of immunological abnormalities induced by high IL‐18, which is an interesting biomarker candidate. Further studies investigating the use of MAPK p38 and NFκB p65 phosphorylation in NK cells as a biomarker for therapy intensification and discontinuation need to be performed.
IL‐18 plays an important role in the activation of NK cells, enhancing IFNγ production and cytotoxicity via the IL‐18/IL‐18R pathway. In patients with acute sJIA, with or without MAS, IFNγ production and cytotoxicity are severely impaired (9, 14). In NK cells from patients with sJIA, it was reported that granzyme K expression and IL‐18‐driven IFNγ production were decreased (14), and perforin and CD107a expression were not increased after IL‐18 stimulation (9). Jager et al reported defective IL‐18Rβ phosphorylation after IL‐18 stimulation in NK cells from patients with acute sJIA. They also reported increased ERK1/2 phosphorylation without IL‐18 stimulation and defective c‐jun N‐terminal kinase phosphorylation after IL‐18 stimulation (9). As shown in Figure 5, we suppressed MAPK p38 and NFκB p65 phosphorylation using healthy NK cells exposed to high IL‐18 concentrations in vitro, in a dose‐dependent manner. We did not evaluate IFNγ and granzyme production in this study; however, the dysfunction in NK cell phosphorylation was apparent after only 12 hours of exposure to high IL‐18. The mechanism of this dysfunction remains to be further clarified. Intracellular cytokine signal regulators, such as suppressor of cytokine signal (SOCS) family and cytokine‐inducible Src homology‐2 containing protein (CIS), act as negative regulators of cytokine signaling (25, 26). SOCS2 and CIS regulate the cell differentiation, IFNγ production and cytotoxicity of NK cells (27). In patients with sJIA, increased SOCS3 messenger RNA expression in PBMCs and increased SOCS1 expression in monocytes have been reported (28, 29). It is possible that impaired IL‐18/IL‐18R signal in patients with sJIA could be associated with regulator proteins induced by high IL‐18 concentrations; however, IL‐18/IL‐18R signal regulators in NK cells have not been elucidated.
Our study has several limitations. First, the number of patients in our study is small. Additional studies with a greater number of patients are needed to confirm our results. Second, most of the patients were treated with TCZ, which may influence the immune responses in these patients. Third, the analysis of intracellular regulatory proteins in the NK cells was not conducted. Fourth, we could not confirm the effects of antibodies to IL‐18 and IL‐18R blocker on healthy control NK cells cultured with rIL‐18. We consider these to be issues for future study.
In conclusion, patients with sJIA showed an impaired phosphorylation of MAPK p38 and NFκB p65 in NK cells following rIL‐18 stimulation, particularly in the active phase of disease, and this was negatively correlated with clinical course and serum IL‐18 concentrations. High serum IL‐18 exposure could induce impaired phosphorylation in healthy NK cells. One mechanism in the pathogenesis of sJIA could be that high serum IL‐18 concentrations induce IL‐18 signal impairment in NK cells. Impaired phosphorylation was restored to normal levels with improvements in the patients’ clinical condition; therefore, we propose that phosphorylation of MAPK p38 and NFκB p65 in NK cells may be a good biomarker to improve the diagnosis and therapy of patients with sJIA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Ohya and Ito had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Ohya, Nishimura, Ito.
Acquisition of data
Ohya, Nishimura, Murase, Hattori, Ohara, Nozawa, Hara.
Analysis and interpretation of data
Ohya, Nishimura, Ito.
Supporting information
Disclosure Form:
Supplementary Fig S1 IL‐18Rα expression on NK cells from sJIA patients and healthy controls. The IL‐18Rα expression on NK cells was lower in the systemic features group; however, it was not statistically different between each group of sJIA patients and healthy controls. Data are shown as box plots, where the boxes represent the first to third quartiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values. HC: healthy control.
ACKNOWLEDGMENTS
We thank Mayuko Miyake for her technical support and helpful discussion. We thank Koji Yasutomo, MD, PhD, for his helpful discussion and review of the manuscript. We thank S. J. Win, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
This study was supported by Yokohama City University Basic Research Fund and JSPS KAKENHI JP20K16931.
No potential conflicts of interest relevant to this article were reported.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Facr2.11426&file=acr211426‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet 2007;369:767‐78. [DOI] [PubMed] [Google Scholar]
- 2. Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol 2011;7:416‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic‐onset juvenile idiopathic arthritis: a randomised, double‐blind, placebo‐controlled, withdrawal phase III trial. Lancet 2008;371:998‐1006. [DOI] [PubMed] [Google Scholar]
- 4. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double‐blind, placebo‐controlled trial with the interleukin‐1 receptor antagonist anakinra in patients with systemic‐onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic‐onset juvenile idiopathic arthritis‐associated macrophage activation syndrome with particular emphasis on the role of interleukin‐18 in its pathogenesis. Rheumatology (Oxford) 2010;49:1645‐53. [DOI] [PubMed] [Google Scholar]
- 6. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN‐gamma production by T cells. Nature 1995;378:88‐91. [DOI] [PubMed] [Google Scholar]
- 7. Mehta VB, Hart J, Wewers MD. ATP‐stimulated release of interleukin (IL)‐1beta and IL‐18 requires priming by lipopolysaccharide and is independent of caspase‐1 cleavage. J Biol Chem 2001;276:3820‐6. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura S, Otani T, Okura R, Ijiri Y, Motoda R, Kurimoto M, et al. Expression and responsiveness of human interleukin‐18 receptor (IL‐18R) on hematopoietic cell lines. Leukemia 2000;14:1052‐9. [DOI] [PubMed] [Google Scholar]
- 9. de Jager W, Vastert SJ, Beekman JM, Wulffraat NM, Kuis W, Coffer PJ, et al. Defective phosphorylation of interleukin‐18 receptor beta causes impaired natural killer cell function in systemic‐onset juvenile idiopathic arthritis. Arthritis Rheum 2009;60:2782‐93. [DOI] [PubMed] [Google Scholar]
- 10. Torigoe K, Ushio S, Okura T, Kobayashi S, Taniai M, Kunikata T, et al. Purification and characterization of the human interleukin‐18 receptor. J Biol Chem 1997;272:25737‐42. [DOI] [PubMed] [Google Scholar]
- 11. Boraschi D, Dinarello CA. IL‐18 in autoimmunity: review. Eur Cytokine Netw 2006;17:224‐52. [PubMed] [Google Scholar]
- 12. Trotta R, Fettucciari K, Azzoni L, Abebe B, Puorro KA, Eisenlohr LC, et al. Differential role of p38 and c‐Jun N‐terminal kinase 1 mitogen‐activated protein kinases in NK cell cytotoxicity. J Immunol 2000;165:1782‐9. [DOI] [PubMed] [Google Scholar]
- 13. Mavropoulos A, Sully G, Cope AP, Clark AR. Stabilization of IFN‐gamma mRNA by MAPK p38 in IL‐12‐ and IL‐18‐stimulated human NK cells. Blood 2005;105:282‐8. [DOI] [PubMed] [Google Scholar]
- 14. Put K, Vandenhaute J, Avau A, van Nieuwenhuijze A, Brisse E, Dierckx T, et al. Inflammatory gene expression profile and defective interferon‐γ and granzyme k in natural killer cells from systemic juvenile idiopathic arthritis patients. Arthritis Rheumatol 2017;69:213‐24. [DOI] [PubMed] [Google Scholar]
- 15. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390‐2. [PubMed] [Google Scholar]
- 16. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant 2013;48:452‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yasin S, Fall N, Brown RA, Henderlight M, Canna SW, Girard‐Guyonvarc'h C, et al. IL‐18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatology (Oxford) 2020;59:361‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woo P. Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome. Nat Clin Pract Rheumatol 2006;2:28‐34. [DOI] [PubMed] [Google Scholar]
- 19. Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun 2012;13:289‐98. [DOI] [PubMed] [Google Scholar]
- 20. Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol 2007;34:1133‐8. [PubMed] [Google Scholar]
- 21. Janow G, Schanberg LE, Setoguchi S, Hasselblad V, Mellins ED, Schneider R, et al. The systemic juvenile idiopathic arthritis cohort of the Childhood Arthritis and Rheumatology Research Alliance Registry: 2010‐2013. J Rheumatol 2016;43:1755‐62. [DOI] [PubMed] [Google Scholar]
- 22. Girard‐Guyonvarc'h C, Palomo J, Martin P, Rodriguez E, Troccaz S, Palmer G, et al. Unopposed IL‐18 signaling leads to severe TLR9‐induced macrophage activation syndrome in mice. Blood 2018;131:1430‐41. [DOI] [PubMed] [Google Scholar]
- 23. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin‐18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 1999;10:127‐36. [DOI] [PubMed] [Google Scholar]
- 24. Michels M, de Mast Q, Netea MG, Joosten LA, Dinarello CA, Rudiman PI, et al. Normal free interleukin‐18 (IL‐18) plasma levels in dengue virus infection and the need to measure both total IL‐18 and IL‐18 binding protein levels. Clin Vaccine Immunol 2015;22:650‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshimura A, Ito M, Chikuma S, Akanuma T, Nakatsukasa H. Negative regulation of cytokine signaling in immunity. Cold Spring Harb Perspect Biol 2018;10:a028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J 2011;25:863‐74. [DOI] [PubMed] [Google Scholar]
- 27. Keating N, Nicholson SE. SOCS‐mediated immunomodulation of natural killer cells. Cytokine 2019;118:64‐70. [DOI] [PubMed] [Google Scholar]
- 28. Li HW, Xie Y, Li F, Sun GC, Chen Z, Zeng HS. Effect of miR‐19a and miR‐21 on the JAK/STAT signaling pathway in the peripheral blood mononuclear cells of patients with systemic juvenile idiopathic arthritis. Exp Ther Med 2016;11:2531‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macaubas C, Wong E, Zhang Y, Nguyen KD, Lee J, Milojevic D, et al. Altered signaling in systemic juvenile idiopathic arthritis monocytes. Clin Immunol 2016;163:66‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form:
Supplementary Fig S1 IL‐18Rα expression on NK cells from sJIA patients and healthy controls. The IL‐18Rα expression on NK cells was lower in the systemic features group; however, it was not statistically different between each group of sJIA patients and healthy controls. Data are shown as box plots, where the boxes represent the first to third quartiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values. HC: healthy control.