

Abstract
Cytotoxic effects of chemotherapy and radiation therapy (RT) used for the treatment of brain metastases results from DNA damage within cancer cells. Cells rely on highly evolved DNA damage response (DDR) pathways to repair the damage caused by these treatments. Inhibiting these repair pathways can further sensitize cancer cells to chemotherapy and RT. The catalytic subunit of DNA-dependent protein kinase, in a complex with Ku80 and Ku70, is a pivotal regulator of the DDR, and peposertib is a potent inhibitor of this catalytic subunit. The characterization of central nervous system (CNS) distributional kinetics of peposertib is critical in establishing a therapeutic index in the setting of brain metastases. Our studies demonstrate that the delivery of peposertib is severely restricted into the CNS as opposed to peripheral organs, by active efflux at the blood-brain barrier (BBB). Peposertib has a low free fraction in the brain and spinal cord, further reducing the active concentration, and distributes to the same degree within different anatomic regions of the brain. However, peposertib is heterogeneously distributed within the metastatic tumor, where its concentration is highest within the tumor core (with disrupted BBB) and substantially lower within the invasive tumor rim (with a relatively intact BBB) and surrounding normal brain. These findings are critical in guiding the potential clinical deployment of peposertib as a radiosensitizing agent for the safe and effective treatment of brain metastases.
SIGNIFICANCE STATEMENT
Effective radiosensitization of brain metastases while avoiding toxicity to the surrounding brain is critical in the development of novel radiosensitizers. The central nervous system distribution of peposertib, a potent catalytic subunit of DNA-dependent protein kinase inhibitor, is restricted by active efflux in the normal blood-brain barrier (BBB) but can reach significant concentrations in the tumor core. This finding suggests that peposertib may be an effective radiosensitizer for intracranial tumors with an open BBB, while limited distribution into normal brain will decrease the risk of enhanced radiation injury.
Introduction
Treatment of brain tumors uses a combination of approaches including surgical resection, radiotherapy (RT), and chemotherapy. Central nervous system (CNS) metastases from peripheral malignancies are the most common brain tumors, with 20%–40% of cancer patients ultimately developing brain metastases (Palmer et al., 2020). Radiation therapy is a mainstay of treatment of brain metastases, with treatment delivered by whole brain radiation therapy (WBRT) or stereotactic radiosurgery (SRS), depending on the clinical situation (Proescholdt et al., 2021). Although conformal RT and SRS strategies are designed to limit radiation dose delivery to especially sensitive regions of the brain, either technique can result in radiation-induced toxicities (Smart, 2017). Therefore, development of novel radiosensitizing agents for treatment of brain tumors should consider both their potential to augment cytotoxicity of RT in tumor cells and the possibility to enhance radiation-induced brain injury (Greene-Schloesser et al., 2012; Dragojevic et al., 2021).
Cells have evolved a robust DNA damage response (DDR) machinery to recover from genotoxic stress induced by intrinsic factors like replication stress or extrinsic factors like environmental toxins and RT. DDR is comprised of a variety of complex signaling networks that coordinate the repair of DNA lesions to allow cell cycle progression or induce apoptosis or senescence if unrepaired DNA accumulates (Huang and Zhou, 2020) (Fig. 1). In the context of endogenous genotoxic stress, DDR is essential for cell growth and survival. Cancer cells can have a heightened level of DDR signaling in association with higher levels of reactive oxygen species, elevated replication stress, and dysregulation of one or more DDR pathways (O’Connor, 2015). One of the most lethal types of DNA damage is DNA double-strand breaks that, if left unrepaired, can cause severe genomic instability and cell death. RT is a potent inducer of DNA double-strand breaks, and even a single unrepaired break can result in RT-induced cell death. There are two major pathways that mediate DNA double-strand break repair [homologous recombination and nonhomologous end-joining (NHEJ)], and the cytotoxic effects of RT can be further enhanced by inhibiting repair of either of these pathways. The catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) is a key component of NHEJ and very important for recovery from RT (Davidson et al., 2013) (Fig. 1).
Fig. 1.

DNA-PKcs inhibiton by peposertib can lead to chemo and radiosensitization of tumor cells. DNA-PK is a key regulator of DNA damage response signaling via nonhomologous end joining. DNA-PKcs inhibition using peposertib will prevent DNA repair, and the resulting unrepaired DNA damage will lead to tumor cell death.
Melanoma, non–small cell lung cancer, and renal cell cancer commonly metastasize to the brain and are often highly resistant to radiation (Goyal et al., 2015). Therefore, there is a strong rationale to develop novel radiosensitizing strategies for brain metastases. Moreover, given the critical role of NHEJ in repair of otherwise lethal DNA double-strand breaks, the development of DNA-PKcs inhibitors in a radiosensitizing strategy would be logical for brain metastases (Fig. 1). Peposertib is a potent and selective inhibitor of DNA-PKcs. In combination with RT or chemotherapy, peposertib potently represses DNA double-strand break repair and increases therapeutic efficacy in a variety of human xenograft models (Wise et al., 2019; Zenke et al., 2020; Haines et al., 2021). Peposertib is in clinical trials as a single agent and in combination with RT and chemotherapy for advanced solid tumors (Mau-Sorensen et al., 2018; van Bussel et al., 2021).
The blood-brain barrier (BBB) protects the CNS from circulating toxins and can also act as a formidable barrier for treatments intended for brain tumors by limiting the CNS distribution of small molecules due to various barrier mechanisms, including active efflux transport. Therefore, this study evaluated the CNS distribution of peposertib and examined the role of active efflux by P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) in limiting its CNS distribution. Inhibition of efflux by the dual inhibitor elacridar was employed to examine the change in delivery of peposertib to the CNS and peripheral organs. Additionally, a critical understanding of the distribution of a radiosensitizer to the intracranial tumor versus the surrounding normal brain to assess the possibility of normal tissue toxicity is important for its safe and effective utilization (Brown, 2019). To examine this spatial heterogeneity in distribution, accumulation of peposertib within an orthotopic melanoma patient-derived xenograft (PDX) tumor (M12) was conducted.
These studies elucidate the underlying mechanisms limiting the CNS distribution of peposertib and heterogeneity of drug distribution of peposertib in tumor relative to surrounding normal brain. These findings outline critical considerations in the clinical development of peposertib and other potent DNA-PKcs inhibitors as radiosensitizing agents in brain tumors.
Materials and Methods
Chemicals and Reagents
Peposertib [(S)-(2-chloro-4-fluoro-5-(7-morpholinoquinazolin-4-yl)phenyl)(6-methoxypyridazin-3-yl)methanol] and LMP-400 (2,3-dimethoxy-6-(3-morpholinopropyl)-5H-[1,3]dioxolo[4',5′:5,6]indeno[1,2-c]isoquinoline-5,12(6H)-dione) were sourced from the Cancer Therapy Evaluation Program through the National Cancer Institute (Bethesda, MD). Elacridar (N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). High-performance liquid chromatography-grade chemicals and reagents were obtained from Thermo Fisher Scientific (Waltham, MA) and Sigma Aldrich (St. Louis, MO). A rapid equilibrium dialysis (RED) device, composed of a polytetrafluoroethylene reusable base plate and cellulose membrane inserts (molecular weight cutoff, 8 kDa), was purchased from Thermo Fisher Scientific (Waltham, MA).
In Vitro Binding Studies to Determine the Unbound Fractions of Peposertib in Tissues
Binding studies for peposertib were conducted in plasma, brain homogenate, and spinal cord homogenate using RED, as per the following modifications to the manufacturer’s protocol. Briefly, brain and spinal cords from mice were homogenized in three volumes of PBS (pH 7.4). The pH of each matrix – plasma, brain homogenate, and spinal cord homogenate – was adjusted to 7.4 and spiked with peposertib in DMSO to a final concentration of 5 µM containing 0.475% DMSO. RED base plate was heated to 37°C on an orbital shaker, and RED membrane inserts were added to the base plate. Three-hundred microliters of each matrix, spiked with 5 µM peposertib, was loaded in the donor chamber, and five-hundred microliters of PBS (pH 7.4) spiked with 0.475% DMSO was loaded in the corresponding receiver chamber. The RED base plate was sealed with a self-adhesive lid and incubated at 37°C for 24 hours on an orbital shaker at 600 rpm (ShelLab, Cornelius, OR). Donor and receiver matrix samples were collected and stored at −80°C until liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to measure the free (unbound) fractions (fu). fu in the plasma was calculated using the ratio of buffer (receiver) concentration to the plasma (donor) concentration at equilibrium. For brain and spinal cord homogenates, fu was calculated using the dilution factor from the homogenate preparation (dilution factor, D = 4), as shown below (Kalvass and Maurer, 2002):
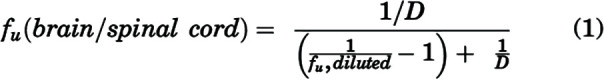 |
In Vivo Pharmacokinetic Studies to Determine the CNS Distribution of Peposertib
Animals
Friend leukemia virus strain B (FVB) wild-type, Mdr1a/b−/− (P-gp-knockout), Bcrp1−/− (Bcrp-knockout), and Mdr1a/b−/−Bcrp1−/− (triple-knockout) mice were used to conduct in vivo pharmacokinetic studies. Breeder pairs were purchased from Taconic Biosciences, and animal colonies were maintained in a standard 12-hour dark/light cycle following an established breeding protocol. Animals were housed in the Research Animal Resources animal facilities at the University of Minnesota with an unlimited supply of food and water. Animal genotypes were routinely verified using tail snip analysis for gene expression (TransnetYX, Cordova, TN). All in vivo experiments were conducted with equal number of male and female mice, in accordance with the Guide for the Care and Use of Laboratory animals established by the US National Institutes of Health and approved by the University of Minnesota Institutional Animal Care and Use Committee.
CNS Distribution of Peposertib Following Intravenous and Oral Administration
Peposertib was administered as an intravenous bolus dose (tail vein injection) of 10 mg/kg [vehicle, 30% (1:1 – EtOh, Cremophor EL) and 70% saline] or a single oral dose (using oral gavage) of 20 mg/kg (vehicle, 0.25% Methocel K4M Premium) + 0.25% Tween 20 in sodium citrate buffer (500 mM, pH 2.5) to FVB wild-type, P-gp-knockout, Bcrp-knockout, and triple-knockout mice (n = 4 per time point). Mice were euthanized using CO2, and blood, brain, and spinal cord samples were collected from 0.167 to 10 hours following intravenous dosing and 0.167 to 16 hours following oral dosing. Blood was collected and stored on ice in preheparinized tubes using cardiac puncture and immediately centrifuged at 3500 rpm at 4°C for 15 minutes to separate plasma. Brain and spinal cord were collected and dipped in ice-cold saline to remove excess blood by blotting with tissues and stored on ice for the duration of sample collection. Collected samples were stored at −80°C, followed by drug concentration determination by LC-MS/MS. Concentration in the brain was corrected for the residual blood in the brain using the brain vascular space, which was determined earlier as 1.4% of the whole brain volume (Dai et al., 2003), and the blood-to-plasma ratio, which was calculated to be unity for peposertib.
Regional Distribution within the CNS and Peripheral Organ Distribution of Peposertib
Peposertib was administered as a single oral dose (oral gavage) of 20 mg/kg to FVB wild-type and triple-knockout mice (n = 4 per time point). In a separate cohort of mice, elacridar, a pharmacological inhibitor of P-gp and Bcrp, was administered as a single intraperitoneal dose of 10 mg/kg in a microemulsion formulation (Sane et al., 2013), simultaneously with a single oral dose of peposertib at 20 mg/kg to FVB wild-type mice (n = 4 per time point). Blood, brain, spinal cord, heart, kidney, lungs, liver, and intestine were collected 2 and 4 hours after drug dose. Blood was collected and stored on ice in preheparinized tubes using cardiac puncture and immediately centrifuged at 3500 rpm at 4°C for 15 minutes to separate plasma. The brain was carefully dissected into the following regions: cortex, cerebellum, midbrain, pons, medulla, and hypothalamus and thalamus. Plasma, brain regions, spinal cord, and the peripheral organs were stored at −80°C, followed by drug concentration determination by LC-MS/MS.
In Vivo Tumor Spatial Distribution Studies of Peposertib
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee, Mayo Clinic, Rochester. Studies involving tumor implantation used female athymic nude mice (Hsd:athymic Nude-Foxn1nu; Envigo, Indianapolis, IN) at the age of 4 to 5 weeks, housed in a standard 12-hour dark/light cycle with unlimited access to food and water. Mice were implanted intracranially with 100 000 cells of a short-term explant culture of the melanoma PDX M12 line transduced with a lentiviral vector for expression of enhanced green fluorescent protein and firefly luciferase 2 (M12-eGFP-FLUC2) (Sarkaria et al., 2014). Briefly, after the exposure of the calvarium on anesthetized mice, the bregma of the mouse cranium was used as a landmark for tumor implantation: 1 mm lateral (right) and 2 mm anterior, introducing the injection syringe to a depth of 3 mm below the cranium. Tumors were allowed to grow for 12 days, and mice were dosed with 50 mg/kg peposertib on day 12, postinjection.
Spatial Distribution of Peposertib in an Intracranial Melanoma Brain Metastasis (M12) Model
Mice were dosed orally with 50 mg/kg peposertib on day 14 postintracranial injection of M12 cells. Blood and tumor-bearing brains were collected at 2 and 6 hours postdose (n = 5 per time point), followed by flash freezing of whole brains. Plasma was separated from the blood by centrifugation at 3500 rpm at 4°C for 15 minutes. A fluorescence-guided punch biopsy technique was used to isolate tumor core, tumor rim (region adjacent to tumor core), and normal brain (without the tumor) from the M12-eGFP-FLUC2–labeled tumor-bearing brains as described in earlier reports (Gampa et al., 2020; Talele et al., 2021). Biopsy punches were used to separate tumor core (5-fold or higher fluorescence signal relative to background) and tumor rim (3- to 5-fold higher fluorescence signal relative to background). All the samples were stored at −80°C, followed by drug concentration determination by LC-MS/MS.
LC-MS/MS Analysis
The brain, tumor regions, and organs were homogenized with three volumes of 5% BSA). Plasma (25 μL) and brain homogenate (100 μL) samples were prepared for analysis using liquid-liquid extraction with one volume of ice-cold pH 11 buffer and five volumes of ice-cold ethyl acetate as the extracting solvent. Each sample and standard tube was spiked with 50 ng of the internal standard (LMP-400). The microcentrifuge tubes were vigorously shaken for 5 minutes and centrifuged at 7500 rpm and 4°C for 10 minutes. The organic supernatant layer separated after centrifugation and was dried under nitrogen, followed by reconstitution in 100 µL mobile phase composed of 58% HPLC-grade water with 0.1% formic acid and 42% acetonitrile with 0.1% formic acid. Peposertib concentrations in these samples was determined using a LC-MS/MS assay. An ACQUITY ultra performance liquid chromatography system (Waters Corporation, Milford, MA) with a Synergy 4 μm Polar-RP 80Å column (75 × 2 mm; Phenomenex, Torrance, CA) was used for the chromatographic analysis. An isocratic method with 4-minute run and a flow rate of 0.5 mL/min was used. The retention time was 1.14 minutes for peposertib and 0.98 minutes for LMP-400. The column output from liquid chromatography was analyzed by a Micromass Quattro Ultima mass spectrometer (Waters, Milford, MA) in positive-ionization mode. The mass-to-charge (m/z) transitions were 482.17 > 446.25 for peposertib and 479.4 > 392.3 for LMP-400 (internal standard). Sensitivity and linearity of the calibration curve was observed over the range of 0.1–2000 ng/mL (weighting factor of 1/Y2), with a coefficient of variation at all concentrations of less than 20%. For each sample analysis, all measured concentrations fell within the range of the calibration curve.
Pharmacokinetic Data Analysis
Noncompartmental Analysis
Phoenix WinNonlin version 8.3 (Certara USA, Inc., Princeton, NJ) was used to perform noncompartmental analysis (NCA) and to obtain pharmacokinetic parameters from the concentration-time profiles in plasma, brain, and spinal cord following intravenous bolus and single oral dosing. Areas under the concentration-time curve for plasma (AUCplasma), brain (AUCbrain), and spinal cord (AUCspinal cord) were calculated using the linear trapezoidal integration method till the last measured time point. Extrapolation of the AUC from the last measured time point (tlast) to infinity was calculated by dividing the last concentration measured by the first-order rate constant associated with the terminal (log-linear) portion of the curve, estimated by the linear regression of time versus log concentration. The percentage AUC extrapolation from tlast to infinity was <10% in all cases, indicating that our study design was able to adequately capture peposertib exposure. Pharmacokinetic parameters including half-life (t1/2), systemic clearance (CL), and volume of distribution (Vd) were calculated using NCA. The standard errors around the means of AUC0-∞ (area under the concentration-time curve from 0 to infinity) were determined as described earlier using the Bailer modification of the Yuan method used by NCA (Bailer, 1988; Yuan, 1993). A tissue partition coefficient (brain/spinal cord-to-plasma area ratio), or Kp, was quantified the ratio of AUC0-∞,brain/spinal cord to AUC0-∞,plasma for single oral and intravenous bolus doses. Kpbrain (brain partition coefficient calculated by the ratio of AUC0-∞,brain to AUC0-∞,plasma) and Kpspinal cord (spinal cord partition coefficient calculated by the ratio of AUC0-∞,spinal cord to AUC0-∞,plasma) were calculated accordingly.
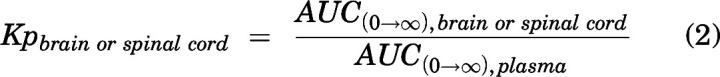 |
Oral bioavailability of peposertib was calculated using the following equation:
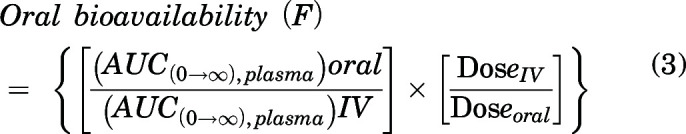 |
An instantaneous tissue partition coefficient (e.g., brain region/organ/tumor-to-plasma concentration ratio), or Kpt, was quantified as the ratio of total tissue concentration to total plasma concentration at a particular time point for the regional brain distribution, organ distribution, and tumor spatial distribution studies.
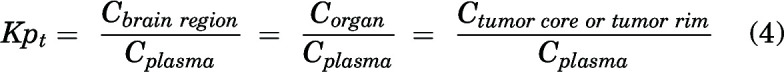 |
The free tissue partitioning ratio (Kpuu) was calculated using the following equation:
 |
Relative free drug exposure in the brain between wild-type and knockout mice was compared using the free distribution advantage (DAfree) as described:
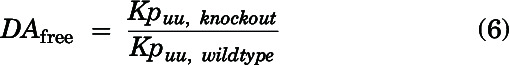 |
Statistical Analysis
For in vivo experiments, the analyst was not blinded to the treatment. The animals were randomized into groups of similar ages with equal males and females. The sample size in this study was determined from a power analysis assuming 20% variance and an α value of 0.05, where the power is about 80% (hence, β = 0.2) to detect a true difference between the anticipated means (about 50%). Data were presented using GraphPad Prism (Version 8; GraphPad Software, La Jolla, California). Statistical tests were also performed using GraphPad Prism, and comparisons between two groups were made using an unpaired t test to obtain a P value. Comparisons between multiple groups were made using one-way ANOVA, followed by multiple comparisons between groups using Bonferroni’s test to obtain adjusted P values. In all cases, P < 0.05 was considered statistically significant. All experimental data are presented as mean ± S.D.
Results
Peposertib Binding in Plasma, Brain, and Spinal Cord
The unbound fraction of peposertib in plasma, brain homogenate, and spinal cord homogenate was determined using RED following a 24-hour equilibration at a concentration of 5 µM. Table 1 summarizes the free fraction values for each matrix. Peposertib exhibits extensive binding to brain and spinal cord tissues. The unbound fraction of peposertib is similar in the brain (%fu = 4.1%) and spinal cord (%fu = 4.8%) and is 2.6-fold lower than the unbound fraction of peposertib in the plasma (%fu = 11.4%). The unbound partition coefficient (Kpuu) for peposertib was determined using fu values in the respective tissues. The differences in binding of peposertib to the brain and spinal cord versus the plasma will impact the free concentrations available to exert pharmacological activity in these tissues. The RED method uses brain and spinal cord homogenates to determine drug binding and therefore has the limitation that we cannot determine the exact nature and specific location of peposertib binding sites within the brain and spinal cord.
TABLE 1.
Unbound fraction of peposertib in plasma, brain homogenate and spinal cord homogenate
| Matrix | fu (Unbound Fraction) | % fu (% Unbound Fraction) |
|---|---|---|
| Plasma | 0.114 ± 0.09 | 11.4 ± 0.9 |
| Brain homogenate | 0.041 ± 0.06 | 4.1 ± 0.6 |
| Spinal cord homogenate | 0.048 ± 0.01 | 4.8 ± 0.1 |
Pharmacokinetics and CNS Distribution of Peposertib Following an Intravenous Bolus Dose
Concentrations in the plasma, brain, and spinal cord were determined following a single intravenous bolus dose of 10 mg/kg in FVB wild-type, P-gp-knockout, Bcrp-knockout, and triple-knockout mice (Fig. 2, A-C). Plasma concentration-time profiles were similar across all four genotypes (Fig. 2A). Brain and spinal cord concentrations were maximum in the triple-knockout mice, followed by the P-gp-knockout mice, and were the lowest in the wild-type and Bcrp-knockout mice (Fig. 2, B and C). This same pattern was seen in the Kpbrain (Fig. 2D) and Kpspinal cord (Fig. 2E) time profiles across all four genotypes. Pharmacokinetic parameters for all four genotypes are listed in Table 2. t1/2, Vd, and CL were similar across all four genotypes. AUC0→∞ plasma was not significantly different among wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice (P > 0.05). AUC0→∞ brain and spinal cord were the lowest in the wild-type mice and not significantly different in the Bcrp-knockout mice (P > 0.05) when compared with the wild-type mice. AUC0→∞ brain and spinal cord were higher in the P-gp-knockout mice (P < 0.05), followed by an even greater increase in the triple-knockout mice (P < 0.05) as compared with the wild-type and Bcrp-knockout mice. Subsequently, the Kpbrain values were 0.09, 0.09, 0.15, and 1.64 in wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice, respectively. Similar to the Kpbrain values, the Kpspinal cord values were 0.08, 0.08, 0.19, and 1.6 in wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice, respectively. These results indicate that efflux mediated by P-gp is a major factor limiting the brain and spinal cord distribution of peposertib. Although efflux mediated by Bcrp does not appear to be important when comparing wild-type with Bcrp-knockout mice, the deletion of both P-gp and Bcrp in the triple-knockout mice shows a marked increase of the Kp values in the brain and spinal cord. After incorporating the fu values in plasma, brain, and spinal cord, the unbound partition coefficient, Kpuu, indicates a limited distribution of unbound peposertib to the brain and spinal cord in wild-type and Bcrp-knockout mice. Kpuu,brain values are 0.03, 0.03, 0.05, and 0.6 in wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice, respectively. These values indicate that the free distribution advantage, DAfree, of peposertib to the brain in the absence of P-gp and both P-gp and Bcrp are 1.7- and 20-fold, respectively. Similarly, Kpuu,spinal cord values are 0.03, 0.03, 0.08, and 0.67 in wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice, respectively. These values indicate that the free distribution advantage, DAfree, of peposertib to the spinal cord in the P-gp-knockout and triple-knockout mice are 2.7 and 22-fold, respectively. High binding of peposertib to brain and spinal cord, in addition to efflux mediated by P-gp and Bcrp, limits the CNS exposure of free peposertib.
Fig. 2.
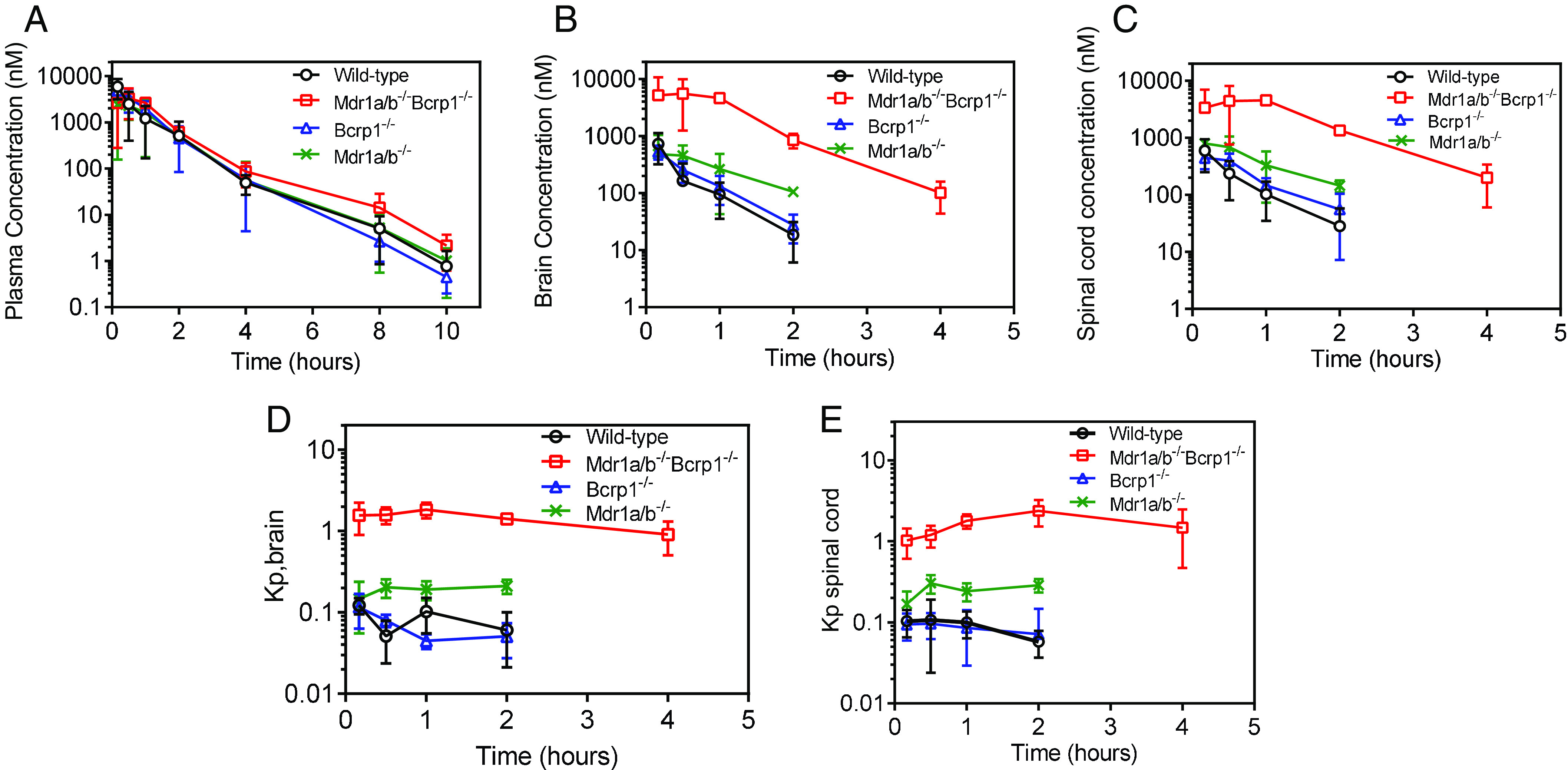
Pharmacokinetics of peposertib following intravenous administration in FVB wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice. (A) Plasma concentration-time profile, (B) brain concentration-time profile, (C) Spinal cord concentration-time profile, (D) brain-to-plasma concentration ratios, and (E) spinal cord–to-plasma concentration ratios following a single intravenous bolus dose of 10 mg/kg in FVB wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice. Data represent mean ± S.D., n = 4.
TABLE 2.
Pharmacokinetic parameters in FVB wild-type, Bcrp-knockout, P-gp-knockout, and triple-knockout mice following an intravenous bolus dose of peposertib
| Parameter | Units | Wild-Type | Bcrp1−/− | Mdr1a/b−/− | Mdr1a/b−/− Bcrp1−/− |
|---|---|---|---|---|---|
| t1/2 | hour | 0.9 | 0.82 | 0.94 | 1.06 |
| Vd | L/kg | 3.1 | 3.1 | 5.5 | 4.2 |
| CL | L/hr/kg | 4.0 | 3.8 | 5.3 | 3.8 |
| AUC 0-∞,plasma | hr*ng/ml | 2452 ± 375 | 2644 ± 289 | 2317 ± 389 | 2618 ± 293 |
| AUC 0-∞,brain | hr*ng/g | 226 ± 34 | 224 ± 19 | 346 ± 87 | 4301 ± 566 |
| AUC 0-∞,spinal cord | hr*ng/g | 207 ± 31 | 219 ± 29 | 446 ± 66 | 4207 ± 447 |
| Kpbrain | 0.09 | 0.09 | 0.15 | 1.64 | |
| Kpuu,brain | 0.03 | 0.03 | 0.05 | 0.60 | |
| DAfree,brain | 1 | 1 | 1.7 | 20 | |
| Kpspinal cord | 0.08 | 0.08 | 0.19 | 1.60 | |
| Kpuu,spinal cord | 0.03 | 0.03 | 0.08 | 0.67 | |
| DAfree,spinal cord | 1 | 1 | 2.7 | 22 |
AUC0-∞, plasma/brain/spinal cord, area under the concentration-time curve from 0 to infinity; DAfree, ratio of Kpuu, knockout mice to Kpuu, wild-type mice; Kpbrain/spinal cord, brain or spinal cord partition coefficient calculated by the ratio of AUC0-∞,brain/spinal cord to AUC0-∞,plasma; Kpuu,brain/spinal cord, Kpbrain/spinal cord multiplied by the ratio of fu,brain/spinal cord and fu,plasma.
CNS Distribution of Peposertib Following a Single Oral Dose
Plasma, brain, and spinal cord concentrations were determined following a single oral dose of 20 mg/kg in FVB wild-type and triple-knockout mice (Fig. 3, A and B). In wild-type mice, concentrations in the plasma were higher than the brain, which was higher than the spinal cord at all time-points. In triple-knockout mice, concentrations in plasma, brain, and spinal cord were similar to one another. Kpbrain and Kpspinal cord values were lower in wild-type mice as compared with triple-knockout mice at every time point (Fig. 3, C and D). AUC0→∞ plasma was similar in FVB wild-type and triple-knockout mice (P > 0.05), whereas the AUC0→∞ brain and spinal cord were significantly higher in the triple-knockout mice as compared with wild-type mice (P < 0.05). Therefore, the Kpbrain was 0.16 and 1.26 in wild-type and triple-knockout mice, respectively (Table 3). The Kpspinal cord was 0.07 and 0.68 in wild-type and triple-knockout mice, respectively (Table 3). This indicates that the overall distribution of peposertib to the spinal cord is lower than that to the brain. Further, on incorporating the fu values in plasma, brain, and spinal cord, the unbound partition coefficient, Kpuu, indicates a limited distribution (circa 10-fold) of unbound peposertib to the brain and spinal cord in wild-type mice as compared with the triple-knockout mice, as was observed in the intravenous dosing study above. Kpuu,brain values are 0.05 and 0.45 in wild-type and triple-knockout mice, respectively. These values indicate that the free distribution advantage, DAfree, of peposertib to the brain in the absence of both P-gp and Bcrp is 9-fold (Table 3). Similarly, Kpuu,spinal cord values are 0.03 and 0.29 in wild-type and triple-knockout mice, respectively. These values indicate that the free distribution advantage, DAfree, of peposertib to the spinal cord in the absence of both P-gp and Bcrp is 10-fold (Table 3). Additionally, oral bioavailability was determined in both wild-type and triple-knockout mice as 32% and 45%, respectively. Similarity in oral bioavailability between the wild-type and triple-knockout mice (oral AUC0→∞ plasma are not different) indicate that the systemic bioavailability of peposertib is unaffected by P-gp and Bcrp, even though they play a significant role in limiting its CNS distribution.
Fig. 3.

Pharmacokinetics of peposertib following oral administration in FVB wild-type and triple-knockout mice. Plasma, brain, and spinal cord concentration-time profiles following a single oral dose of 20 mg/kg in (A) wild-type and (B) triple-knockout mice. (C) Brain-to-plasma concentration ratios and (D) spinal cord–to-plasma concentration ratios following a single oral dose of 20 mg/kg in FVB wild-type and triple-knockout mice. Data represent mean ± S.D., n = 4.
TABLE 3.
Pharmacokinetic parameters in FVB wild-type and triple-knockout mice following a single oral dose of peposertib
| Parameter | Units | Wild- Type | Mdr1a/b−/− Bcrp1−/− |
|---|---|---|---|
| t1/2 | hour | 2.4 | 2.0 |
| tmax | hour | 2 | 4 |
| Cmax | ng/ml | 286 | 499 |
| Vd/F | L/kg | 45 | 25 |
| CL/F | L/hr/kg | 12.7 | 8.5 |
| AUC 0-∞,plasma | hr*ng/ml | 1566 ± 382 | 2355 ± 911 |
| Bioavailability | 0.32 | 0.45 | |
| AUC 0-∞,brain | hr*ng/g | 228 ± 34 | 2954 ± 864 |
| AUC 0∞,spinal cord | hr*ng/g | 103 ± 13 | 1603 ± 142 |
| Kpbrain | 0.15 | 1.25 | |
| Kpuu,brain | 0.05 | 0.45 | |
| DAfree,brain | 1 | 9 | |
| Kpspinal cord | 0.07 | 0.68 | |
| Kpuu,spinal cord | 0.03 | 0.29 | |
| DAfree,spinal cord | 1 | 10 |
CL/F, apparent clearance; Cmax, maximum observed concentration; F, oral bioavailability; sKpuu,brain/spinal cord, Kpbrain/spinal cord multiplied by the ratio of fu,brain/spinal cord to fu,plasma; tmax, time at which maximum concentration is observed; Vd/F, apparent volume of distribution.
CNS Regional Distribution of Peposertib with Elacridar
Distribution of peposertib to different anatomic regions of the brain and to the spinal cord was examined following oral administration to wild-type and triple-knockout mice at 2 and 4 hours postdose. In addition, the change in peposertib distribution to different anatomic regions of the brain and to the spinal cord was examined following coadministration of peposertib and elacridar, a dual inhibitor of P-gp and Bcrp, in wild-type mice. Concentrations at 2 hours in the cortex, cerebellum, hypothalamus and thalamus, midbrain, medulla, pons, and spinal cord are depicted in Fig. 4A. The subsequent brain region-to-plasma concentration ratios are depicted in Fig. 4B. Distribution of peposertib in the wild-type mice, as measured by this concentration ratio, was similar within the different anatomic regions of the brain, with no significant differences in the concentrations within cortex, cerebellum, hypothalamus and thalamus, midbrain, medulla, and pons (P > 0.05) (Fig. 4A). This indicates that for peposertib, the basal functional efflux activity of P-gp and Bcrp is similar across these different brain regions. However, concentration in the spinal cord was lower and significantly different than each of the brain regions (P < 0.05) (Fig. 4A). On coadministration of elacridar with peposertib, concentrations to each of the brain regions and spinal cord increased significantly (P < 0.05), indicating that inhibition of P-gp and Bcrp can increase CNS distribution of peposertib (Fig. 4A). However, even with the coadministration of elacridar, concentration of peposertib was similar within the different anatomic regions of the brain (P > 0.05), indicating that inhibition of P-gp and Bcrp-mediated efflux by elacridar is functionally similar across these different brain regions. Concentration of peposertib in the spinal cord was lower and significantly different than each of the brain regions (P < 0.05) with coadministration of elacridar, even in light of the increase in concentration resulting from inhibition of P-gp and Bcrp. In case of triple-knockout mice, peposertib concentration within each of the brain regions and spinal cord increases significantly as compared with wild-type mice and the coadministration of elacridar (P < 0.05) (Fig. 4A). This indicates that knockdown of P-gp and Bcrp significantly increases peposertib distribution to the CNS to a greater degree than using elacridar at the current dose. In triple-knockout mice, the concentration of peposertib was similar within the different anatomic regions of the brain (P > 0.05) and significantly lower in the spinal cord as compared with each of these regions (P < 0.05). Brain region-to-plasma concentration ratios show a similar trend to the concentrations (Fig. 4B) in each group. Brain region-to-plasma concentration ratios of the different brain regions are lowest in the wild-type mice, followed by an increase with the coadministration of elacridar, and are the maximum in the triple-knockout mice (Fig. 4B). Within each group, although the brain region-to-plasma concentration ratios are not significantly different within different regions (P > 0.05), the spinal cord-to-plasma concentration ratio was significantly lower than the other regions (P < 0.05) (Fig. 4B).
Fig. 4.
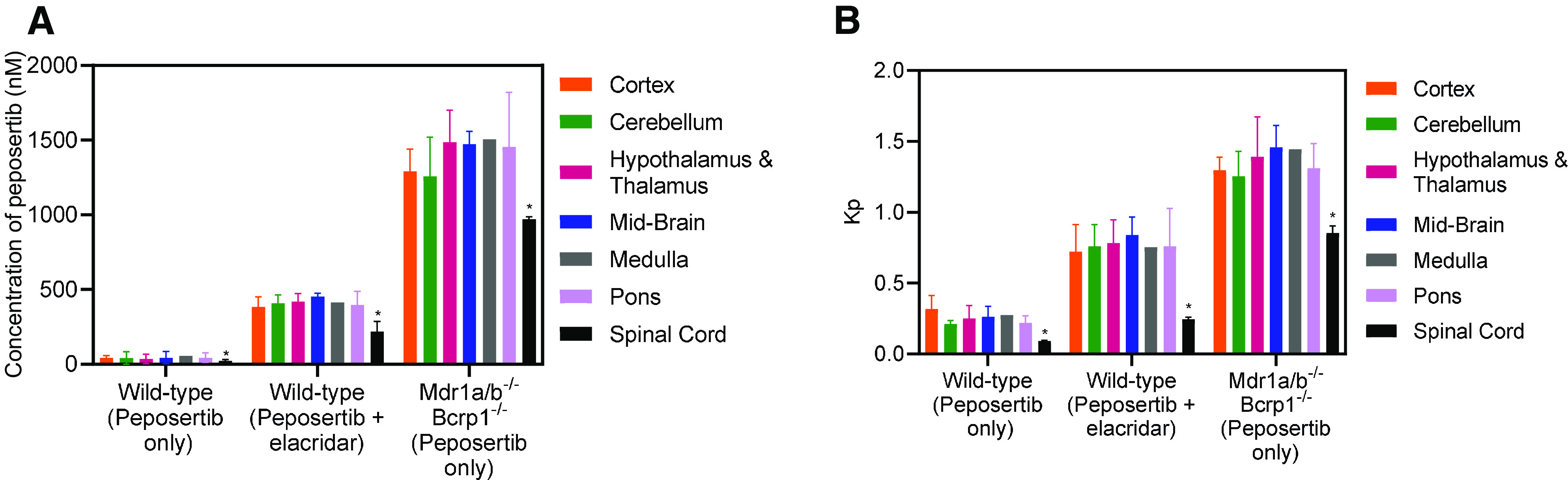
Distribution of peposertib within different anatomic regions of the CNS in wild-type and triple-knockout mice and the effect of pharmacological inhibition of efflux transport using elacridar in wild-type mice. (A) Concentrations and (B) CNS region-to-plasma concentration ratios of peposertib within cortex, cerebellum, hypothalamus and thalamus, midbrain, medulla, pons, and spinal cord in wild-type mice with and without efflux inhibition using coadministration of elacridar and in triple-knockout mice at 2 hours postdose. Data represent mean ± S.D., n = 4. * P < 0.05
The regional concentrations and brain region-to-plasma concentration ratios at 4 hours are depicted in Supplemental Materials (Supplemental Fig. 1, A and B) and follow the same trends as the 2-hour data described above.
Organ Distribution of Peposertib
Distribution of peposertib to peripheral organs other than the CNS also was determined. After the oral administration of 20 mg/kg peposertib in wild-type mice, the concentration at 2 hours postdose in the heart, kidney, liver, lungs, and plasma is not affected by the coadministration of elacridar or when given to the triple-knockout mice (P > 0.05) (Fig. 5A). This indicates that neither the inhibition of efflux nor the knockdown of P-gp and Bcrp impacts the distribution of peposertib to these tissues. Concentrations in the intestine have a significant variation between these three groups (P < 0.05). Importantly, concentrations in the cortex, as described earlier, increase significantly (P < 0.05) on the inhibition of efflux and the knockdown of P-gp and Bcrp (Fig. 5A). This indicates that efflux mediated by P-gp and Bcrp greatly influences the distribution of peposertib to the CNS as opposed to no significant effect that was seen in the major peripheral organs. More importantly, partition coefficients of the organs depicted in Fig. 5B indicate that distribution to all the peripheral organs is high (>1), irrespective of the inhibition or knockdown of efflux. However, Kpcortex is significantly lower than 1 in the wild-type mice, increases upon the inhibition by elacridar, and further increases in the triple-knockout mice. These findings are critical in the toxicological considerations of using peposertib as a radiosensitizer. In the peripheral organs, where peposertib distribution is not as limited as in the CNS, its potent activity to inhibit DNA damage repair mediated by DNA-PK could lead to severe tissue toxicities.
Fig. 5.

Organ distribution of peposertib in wild-type and triple-knockout mice and the effect of pharmacological inhibition of efflux transport using elacridar in wild-type mice. (A) Concentrations and (B) organ-to-plasma concentration ratios of peposertib within heart, kidney, liver, intestine, lungs, cortex, and plasma in wild-type mice with and without efflux inhibition using coadministration of elacridar and in triple-knockout mice at 2 hours postdose. Data represent mean ± S.D., n = 4. * P < 0.05
The organ concentrations and organ-to-plasma concentration ratios at 4 hours are depicted in Supplemental Materials (Supplemental Fig. 2, A and B) and follow the same trends as the 2-hour data described above.
Spatial Distribution of Peposertib in an Intracranial PDX Model (M12)
Distribution of peposertib within the brain in an intracranial PDX model of metastatic melanoma tumor (M12) is critical to study for its safe application as a radiosensitizer. We used M12 tumors stably transduced with a lentiviral construct for expression of fLuc2-eGFP. M12-eGFPfLuc2 tumors were implanted intracranially, and, after 14 days of tumor growth, tumor-bearing brains as well as plasma were harvested at 2 and 6 hours after an oral administration of peposertib (50 mg/kg). Fluorescence-guided punch biopsy was used to separate the brain regions into core, rim, and normal brain, followed by LC-MS/MS analysis of peposertib concentrations in these regions (Fig. 6, A and B). Peposertib distribution is heterogenous within different regions of tumor-bearing brains, with significantly higher concentration within the tumor core as compared with the normal brain (P < 0.05) (Fig. 6C). Peposertib concentration within the tumor core is also significantly higher than the tumor rim (P < 0.05) (Fig. 6C), whereas concentrations within the normal brain and tumor rim are not significantly different (P > 0.05). Similarly, the partition coefficients of tumor core, rim, and normal brain also show a similar trend, with Kptumor core significantly higher than the Kptumor rim and Kpnormal brain (P < 0.05) (Fig. 6D). This heterogenous distribution in tumor-bearing brains, with maximum concentration within the tumor core and severely restricted distribution to the surrounding normal brain tissue, has important implications in the use of peposertib as a safe yet effective radiosensitizer for metastatic brain tumors.
Fig. 6.
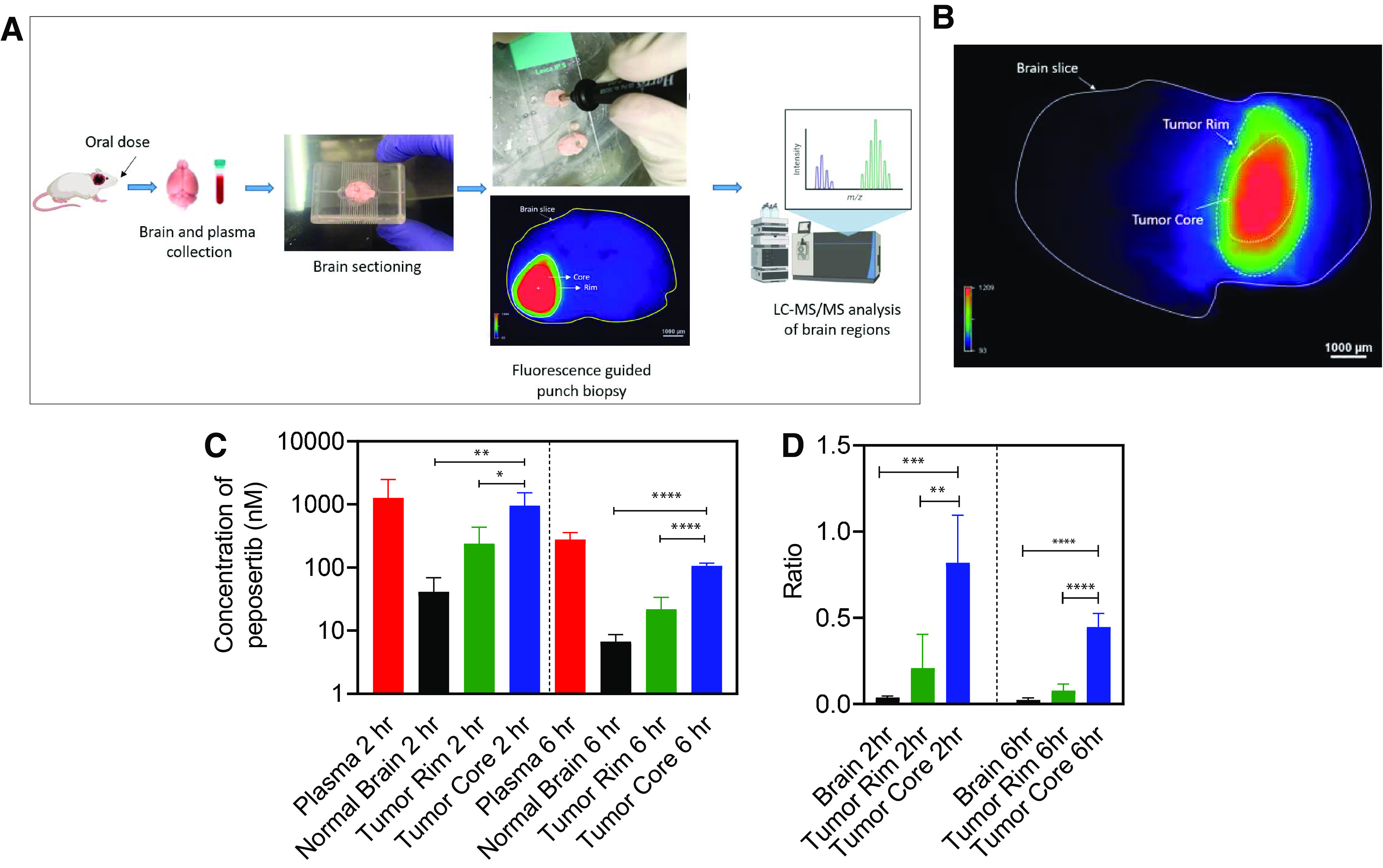
Spatial distribution of peposertib in different regions around the brain tumor following oral administration in mice with intracranial M12 tumors. (A) Schematic of the brain-slice method to study intracranial tumor spatial distribution of peposertib. (B) Representative image of mouse brain slice marked with tumor core and tumor rim regions. (C) Concentrations in plasma, normal brain, tumor rim, and tumor core, and (D) region-to-plasma concentration ratios in brain, tumor rim, and tumor core at 2 and 6 hours following a single oral dose of 50 mg/kg peposertib. Data represent mean ± S.D., n = 5. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Discussion
Brain metastases remain incurable despite multidisciplinary management using surgery, RT, and chemotherapy (Palmer et al., 2020). Of these brain metastases, melanoma has a high propensity to metastasize to the brain and is associated with a dismal prognosis (Kim et al., 2018). RT, particularly WBRT and SRS, are used to control tumor growth of melanoma brain metastases; however, radio resistance and acute and late-stage radiation-induced toxicities limit the dose of RT. Novel radiosensitizers that target DDR and can be adequately delivered to the brain metastases while sparing the normal brain tissues would be a significant breakthrough in the management of brain metastases.
Targeting DNA-PKcs, a key component of DDR, for the treatment of cancers is being actively evaluated in a variety of clinical trials. Peposertib, a potent DNA-PKcs inhibitor, is being studied as a single agent and as a potential radiosensitizer in peripheral tumors. A critical consideration for the development of a potent radiosensitizer is ensuring that it targets the tumor while sparing toxicities to nearby normal tissues, as stressed by J. Martin Brown in a recent commentary (Brown, 2019). Normal tissue toxicities, including dysphagia, mucosal inflammation, radiation-related skin injury, and mucositis have been reported in a phase 1B trial with peposertib and RT (clinicaltrials.gov identifier: NCT02516813) (Mau-Sorensen et al., 2018; Triest et al., 2018). DNA-PKcs inhibition using peposertib can play a critical role in normal tissue radiosensitivity. A classic example of how the activity of DNA-PKcs can influence the susceptibility of normal tissues for radiotoxicity is severe combined immunodeficiency (SCID). SCID is a rare disorder caused by NHEJ defects, including germline mutations in DNA-PKcs, that confer hypersensitivity to RT (Fulop and Phillips, 1990; Woodbine et al., 2013). SCID mice demonstrate significant enhancement of radiation toxicities like gastrointestinal and bone marrow toxicities, skin irritation, mucositis, and lethality (Biedermann et al., 1991). A recently published report for AZD7648, a DNA-PK inhibitor under investigation as a chemo- and radiosensitizer (Fok et al., 2019), showed potent radiosensitizing activity in head and neck squamous cell carcinoma tumors but also demonstrated normal tissue radiation toxicity to the oral mucosa and small intestine, leading to significant morbidity and body weight loss in mice (Hong et al., 2021). The inhibition of DNA-PK and the subsequent inhibition of repair by NHEJ should therefore be carefully evaluated mechanistically and clinically with respect to the implications for normal tissue toxicity. Thus, to evaluate the use of peposertib as a safe and effective radiosensitizing agent for the treatment of brain metastases, we conducted thorough CNS pharmacokinetic and regional distribution studies in normal brain, drug binding assays, and spatial distribution studies in tumor, in a relevant PDX model of melanoma brain metastasis.
Our in vivo studies demonstrate that efflux mediated by P-gp is the dominant factor limiting the CNS delivery of peposertib. Although efflux mediated by Bcrp does not seem to play a major role in the presence of P-gp in restricting the CNS delivery of peposertib, in the absence of both P-gp and Bcrp in the triple-knockout mice, the Kpbrain and Kpspinal cord both show greater-than-additive increase as compared with the individual increase observed in the Bcrp-knockout and P-gp-knockout mice, respectively. This points toward a mechanism of functional compensation of P-gp and Bcrp at the BBB in limiting the distribution of peposertib to the CNS (Polli et al., 2008; Chen et al., 2009; Kodaira et al., 2010; Laramy et al., 2018; Talele et al., 2021). It has been shown that the genetic deletion of P-gp and/or Bcrp across the knockout models used in our studies does not influence the expression of other efflux transporters or other selected BBB proteins such as influx transporters, tight junction proteins, and some receptors (Agarwal et al., 2012). Oral bioavailability of peposertib was determined in wild-type and triple-knockout mice and was found to be 32% and 45%, respectively. This indicates that even though P-gp and Bcrp efflux activity at the BBB impacts the CNS distribution of peposertib, these transporters in the intestine did not dramatically affect the oral bioavailability of peposertib. This observation may be attributed to the saturation of P-gp and Bcrp, due to high intestinal concentrations as compared with the plasma at the administered dose of peposertib (Lin and Yamazaki, 2003).
The free drug hypothesis (Gillette, 1973) states that it is the free or unbound drug that is able to exert pharmacological activity at the site of action. The free fractions in both plasma, the driving force concentration for brain distribution, and in the brain tissue are key determinants to evaluate the distributional mechanisms of a drug across the BBB into and out of the CNS (Hammarlund-Udenaes et al., 2008). Binding studies indicate that peposertib is highly bound to both brain and spinal cord. Therefore, in addition to the limited distribution of peposertib to the CNS by active efflux, its binding to brain and spinal cord further lowers the free drug partitioning. In wild-type mice, Kpuu,brain and Kpuu, spinal cord are 5% and 3%, respectively, following oral administration, indicating severely restricted CNS distribution of free peposertib.
An important consideration to inform the safe, yet effective, use of a radiosensitizer is to know its distribution to different anatomic areas of the brain. Although effective delivery to the tumor-bearing brain region is essential for potent radiosensitizing activity, regions of high concentration in normal brain can lead to significant toxicity, especially when combined with WBRT. Therefore, it is important to determine regional differences in the mechanisms, such as active efflux, that influence drug distribution across the BBB.
We observed that although peposertib distribution is similar within different anatomic regions of the brain, it is more restricted in the spinal cord. This is an important finding for peposertib, since it indicates an increased protection of the spinal cord when combined with RT. The administration of elacridar, a dual inhibitor of P-gp and Bcrp, increases the concentration of peposertib in different anatomic regions, similar to the triple-knockout mice. However, this pharmacological inhibition did not reach the same magnitude of increase in the local concentration of peposertib as the genetic knockout. Even though elacridar is a potent inhibitor of both P-gp and Bcrp, the magnitude of transport inhibition is dependent on the local concentration, inhibitory potency of elacridar, and the expression of the transport systems at that location. Therefore, if the inhibitory concentration is not significantly above the Ki (inhibition constant), there will be incomplete inhibition of the transporter action when compared with the knockout. These results indicate that the chances of a drug-drug interaction at the BBB while using an inhibitor of efflux are low due to the inhibitory concentrations available locally being significantly lower than Ki (Kalvass et al., 2013).
Our results indicate that the functional activity of P-gp and Bcrp-mediated efflux and its inhibition, as well as genetic knockdown, is uniform across several anatomic regions of the CNS. The current study is the first comprehensive regional CNS distribution study for an anticancer agent in wild-type mice with P-gp and Bcrp inhibition using elacridar as well as in triple-knockout mice. Other studies that have examined the effect of P-gp inhibition in different regions of the brain for molecules like flavonoids, where certain regional differences were observed, and verapamil, where no regional differences were observed (Youdim et al., 2004; Eyal et al., 2010).
The distribution of peposertib to peripheral organs was determined to critically evaluate if this potent radiosensitizer could contribute to off-target toxicity. The current data suggest that peposertib readily distributes to, and accumulates in, peripheral organs with Kporgan values greater than 1. Neither inhibition of P-gp and Bcrp using elacridar nor the knockdown of these efflux transporters significantly affected the distribution of peposertib to peripheral organs. This is important because high concentrations of peposertib, when combined with RT in peripheral tumors, may lead to severe toxicities in surrounding normal tissues that have been noted in ongoing clinical trials of peposertib (Mau-Sorensen et al., 2018).
We then evaluated if peposertib can be effectively delivered to the brain tumor while sparing the surrounding normal brain in a melanoma brain metastatic PDX model, M12. Our studies indicate that peposertib distribution in and around the tumor core was heterogeneous, with maximum distribution to the tumor core, followed by tumor rim and normal brain. When using peposertib as a radiosensitizer for brain metastases, the results are promising, considering that peposertib distribution is high in tumor core regions where it can exhibit potent radiosensitizing activity. Fortuitously, in adjacent areas of normal brain tissues, where peposertib distribution is restricted, these areas will be protected from possible toxicities from the combined treatment of peposertib and RT.
In conclusion, key considerations for the use of peposertib as a safe yet effective radiosensitizer for brain tumors, in particular brain metastases, were evaluated. We observed that peposertib distribution into the CNS is severely restricted due to active efflux at the intact BBB and has high binding to brain tissue. Furthermore, we observed that the functional efflux activity of P-gp and Bcrp, as determined by pharmacological inhibition and genetic deletion, to limit peposertib distribution does not vary among different anatomic regions of the brain. However, it is interesting to note that the distribution to the spinal cord is lower than to the brain. This difference in the barrier in different regions of the CNS can be important when treating spinal metastases with RT. Active efflux does not limit the distribution of peposertib to peripheral organs as it is readily distributed to these organs, indicating a need for safety evaluation with RT when using peposertib for peripheral tumor treatment.
Taken as a whole, these results indicate that a potent DNA-PKcs radiosensitizer, such as peposertib, may be of great use in exerting local control over metastatic brain tumors that are otherwise radiation resistant or cannot be surgically resected while sparing the normal brain tissue around the tumor and thereby preventing the risk of radiation-associated toxicities with either WBRT or SRS (Fig. 7). As more potent DNA-PKcs radiosensitizers are being developed (Fok et al., 2019; Willoughby et al., 2020), appreciating their local distribution in the CNS for brain tumors and other organs for peripheral tumors will be important to avoid severe radiation-associated toxicities in patients (Dragojevic et al., 2021). It will be of paramount importance to assess if these agents can selectively target tumor cells and spare surrounding normal tissues for their effective and safe radiosensitization.
Fig. 7.

Importance of heterogeneous spatial tumor distribution of peposertib in brain metastases for safe and effective radiosensitization.
Acknowledgments
The authors thank Jim Fisher, Clinical Pharmacology Analytical Laboratory, University of Minnesota, for his support in the development of the LC-MS/MS assay. The healthcare business of Merck KGaA, Darmstadt, Germany reviewed the manuscript for medical accuracy only before submission. The authors are fully responsible for the content of this manuscript, and the views and opinions described in the manuscript reflect solely those of the authors.
Abbreviations
- AUC
area under the concentration-time curve
- AUC 0-∞
area under the concentration-time curve from 0 to infinity
- BBB
blood-brain barrier
- Bcrp
breast cancer resistance protein
- CL
clearance
- CNS
central nervous system
- DAfree
free distribution advantage
- DDR
DNA damage response
- DNA-PKcs
catalytic subunit of DNA-dependent protein kinase
- fu
free (unbound) fraction
- FVB
Friend leukemia virus strain B
- Kp
tissue-to-plasma ratio
- Kpbrain
brain partition coefficient calculated by the ratio of AUC0-∞, brain to AUC0-∞, plasma
- Kpspinal cord
spinal cord partition coefficient calculated by the ratio of AUC0-∞, spinal cord to AUC0-∞, plasma
- Kpuu
unbound (free) tissue-to-plasma ratio
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LMP-400
2, 3-dimethoxy-6-(3-morpholinopropyl)-5H-[1, 3]dioxolo[4', 5′:5, 6]indeno[1, 2-c]isoquinoline-5, 12(6H)-dione
- NCA
noncompartmental analysis
- NHEJ
nonhomologous end joining
- PDX
patient-derived xenograft
- P-gp
P-glycoprotein
- RED
rapid equilibrium dialysis
- RT
radiotherapy
- SCID
severe combined immunodeficiency
- SRS
stereotactic radiosurgery
- t1/2
half-life
- Vd
volume of distribution
- WBRT
whole brain radiation therapy
Authorship Contributions
Participated in research design: Talele, Elmquist.
Conducted experiments: Talele, Zhang, Oh, Burgenske, Mladek.
Performed data analysis: Talele, Dragojevic, Elmquist.
Wrote or contributed to the writing of the manuscript: Talele, Sarkaria, Elmquist.
Footnotes
This work was supported by National Institutes of Health National Cancer Institute [Grant RO1-CA138437], [Grant U54-CA210181], [Grant U01-CA227954], and [Grant P50-CA108960] and National Institute of Neurological Disorders and Stroke [Grant RO1-NS077921] (to J.N.S. and W.F.E). This research was supported by EMD Serono (CrossRef Funder ID: 10.13039/100004755), who provided peposertib through the Cancer Therapy Evaluation Program (CTEP) of the NCI. S.T. was supported by the Rory P. Remmel and Cheryl L. Zimmerman fellowship in Drug Metabolism and Pharmacokinetics, Edward G. Rippie fellowship, Bighley Graduate fellowship, Ronald J. Sawchuk fellowship in Pharmacokinetics, and Doctoral Dissertation fellowship.
No author has an actual or perceived conflict of interest with the contents of this article.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF (2012) Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos 40:1164–1169 10.1124/dmd.112.044719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer AJ (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16:303–309 10.1007/BF01062139. [DOI] [PubMed] [Google Scholar]
- Biedermann KA, Sun JR, Giaccia AJ, Tosto LM, Brown JM (1991) scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc Natl Acad Sci USA 88:1394–1397 10.1073/PNAS.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, JM. (2019) Beware of Clinical Trials of DNA Repair Inhibitors. Int J Radiat Oncol Biol Phys. 103:1182–1183. 10.1016/J.IJROBP.2018.11.063. [DOI] [PubMed] [Google Scholar]
- van Bussel MTJ, Awada A, de Jonge MJA, Mau-Sørensen M, Nielsen D, Schöffski P, Verheul HMW, Sarholz B, Berghoff K, El Bawab S, et al. (2021). A First-in-Man Phase 1 Study of the DNA-Dependent Protein Kinase Inhibitor Peposertib (Formerly M3814) in Patients with Advanced Solid Tumours. Br J Cancer 124:728–735 10.1038/s41416-020-01151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF (2009) P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 330:956–963 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- O’Connor MJ (2015) Review Targeting the DNA Damage Response in Cancer. Mol Cell 60:547–560 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Dai H, Marbach P, Lemaire M, Hayes M, Elmquist WF (2003) Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Ther 304:1085–1092 10.1124/jpet.102.045260. [DOI] [PubMed] [Google Scholar]
- Davidson D, Amrein L, Panasci L, Aloyz R (2013) Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front Pharmacol 4:5 10.3389/FPHAR.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragojevic S, Ji J, Singh PK, Connors MA, Mutter RW, Lester SC, Talele SM, Zhang W, Carlson BL, Remmes NB, et al. (2021) Preclinical Risk Evaluation of Normal Tissue Injury With Novel Radiosensitizers. Int J Radiat Oncol Biol Phys 2021;111:E54–E62. 10.1016/J.IJROBP.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal S, Ke B, Muzi M, Link JM, Mankoff DA, Collier AC, Unadkat JD (2010) Regional P-glycoprotein activity and inhibition at the human blood-brain barrier as imaged by positron emission tomography. Clin Pharmacol Ther 87:579–585 10.1038/CLPT.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok JH, Ramos-Montoya A, Vazquez-Chantada M, Wijnhoven PW, Follia V, James N, Farrington PM, Karmokar A, Willis SE, Cairns J, et al. (2019) AZD7648 Is a Potent and Selective DNA-PK Inhibitor That Enhances Radiation, Chemotherapy and Olaparib Activity. Nature Commun 10:5065. 10.1038/s41467-019-12836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop GM, Phillips RA (1990) The Scid Mutation in Mice Causes a General Defect in DNA Repair. Nature 347:479–482. 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- Gampa G, Kenchappa RS, Mohammad AS, Parrish KE, Kim M, Crish JF, Luu A, West R, Hinojosa AQ, Sarkaria JN, et al. (2020) Enhancing Brain Retention of a KIF11 Inhibitor Significantly Improves its Efficacy in a Mouse Model of Glioblastoma. Sci Rep 10:6524 10.1038/s41598-020-63494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette JR (1973) Overview of drug-protein binding. Ann N Y Acad Sci 226:6–17 10.1111/J.1749-6632.1973.TB20464.X. [DOI] [PubMed] [Google Scholar]
- Goyal S, Silk AW, Tian S, Mehnert J, Danish S, Ranjan S, Kaufman HL (2015) Clinical Management of Multiple Melanoma Brain Metastases: A Systematic Review. JAMA Oncol 1:668–676 10.1001/JAMAONCOL.2015.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene-Schloesser D, Robbins ME, Peiffer AM, Shaw EG, Wheeler KT, Chan MD (2012) Radiation-induced brain injury: A review. Front Oncol 2:73 10.3389/fonc.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines E, Nishida Y, Carr MI, Montoya RH, Ostermann LB, Zhang W, Zenke FT, Blaukat A, Andreeff M, Vassilev LT (2021) DNA-PK inhibitor peposertib enhances p53-dependent cytotoxicity of DNA double-strand break inducing therapy in acute leukemia. Sci Rep 11:12148 10.1038/S41598-021-90500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25:1737–1750 10.1007/s11095-007-9502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CR, Buckley CD, Wong WW, Anekal PV, Dickson BD, Bogle G, Hicks KO, Hay MP, Wilson WR (2022) Radiosensitisation of SCCVII tumours and normal tissues in mice by the DNA-dependent protein kinase inhibitor AZD7648. Radiother Oncol 166:162–170 10.1016/J.RADONC.2021.11.027. [DOI] [PubMed] [Google Scholar]
- Huang R-X, Zhou P-K (2020) DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct Target Ther 5:60. 10.1038/s41392-020-0150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvass JC, Polli JW, Bourdet DL, Feng B, Huang SM, Liu X, Smith QR, Zhang LK, Zamek-Gliszczynski MJ; International Transporter Consortium (2013) Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: the ITC evidence-based position. Clin Pharmacol Ther 94:80–94 10.1038/CLPT.2013.34. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- Kim M, Kizilbash SH, Laramy JK, Gampa G, Parrish KE, Sarkaria JN, Elmquist WF (2018) Barriers to Effective Drug Treatment for Brain Metastases: A Multifactorial Problem in the Delivery of Precision Medicine. Pharm Res 35:177 10.1007/s11095-018-2455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- Laramy JK, Kim M, Parrish KE, Sarkaria JN, Elmquist WF (2018) Pharmacokinetic Assessment of Cooperative Efflux of the Multitargeted Kinase Inhibitor Ponatinib Across the Blood-Brain Barrier. J Pharmacol Exp Ther 365:249–261 10.1124/jpet.117.246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- Mau-Sorensen M, van Bussel M, Kuipers M, Nielsen DL, Verheul HM, Aftimos P, de Jonge MJA, van Triest B, Falkenius J, Debus J, et al. (2018) Safety, Clinical Activity and Pharmacological Biomarker Evaluation of the DNA-Dependent Protein Kinase (DNA-PK) Inhibitor M3814: Results from Two Phase I Trials. Ann of Oncol 29:viii654. 10.1093/ANNONC/MDY303.015. [DOI] [Google Scholar]
- Palmer JD, Trifiletti DM, Gondi V, Chan M, Minniti G, Rusthoven CG, Schild SE, Mishra MV, Bovi J, Williams N, et al. (2020) Multidisciplinary patient-centered management of brain metastases and future directions. Neurooncol Adv 2:vdaa034 10.1093/NOAJNL/VDAA034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, St. John-Williams L, Koch KM, Serabjit-Singh CJ (2008) The Role of Efflux and Uptake Transporters in N-{3-Chloro-4-[(3- Fluorobenzyl)Oxy]Phenyl}-6-[5-({[2-(Methylsulfonyl)Ethyl]Amino}methyl)-2-Furyl] -4-Quinazolinamine (GW572016, Lapatinib) Disposition and Drug Interactions. Drug Metab Dispos 36:695–701 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- Proescholdt MA, Schödel P, Doenitz C, Pukrop T, Höhne J, Schmidt NO, Schebesch K-M (2021) The Management of Brain Metastases-Systematic Review of Neurosurgical Aspects. Cancers (Basel) 13:1616 10.3390/CANCERS13071616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane R, Agarwal S, Mittapalli RK, Elmquist WF (2013) Saturable active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther 345:111–124 10.1124/jpet.112.199786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria J, Ma D, Schroeder M, Carlson B, Giannini C, Parney I (2014) PM-19DEVELOPMENT OF A PANEL OF PATIENT-DERIVED XENOGRAFT (PDX) MODELS FROM BRAIN METASTASES. Neuro Oncol 16 (Suppl 5):v173 10.1093/NEUONC/NOU268.19. [DOI] [Google Scholar]
- Smart D (2017) Radiation Toxicity in the Central Nervous System: Mechanisms and Strategies for Injury Reduction. Semin Radiat Oncol 27:332–339 10.1016/J.SEMRADONC.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele S, Zhang W, Burgenske DM, Kim M, Mohammad AS, Dragojevic S, Gupta SK, Bindra RS, Sarkaria JN, Elmquist WF (2021) Brain Distribution of Berzosertib: An ATR Inhibitor for the Treatment of Glioblastoma. J Pharmacol Exp Ther 379:343–357 10.1124/JPET.121.000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triest BV, Damstrup L, Falkenius J, Budach V, Troost E, Samuels M, Debus J, et al. (2018) A Phase Ia/Ib Trial of the DNA-PK Inhibitor M3814 in Combination with Radiotherapy (RT) in Patients (Pts) with Advanced Solid Tumors: Dose-Escalation Results (Abstract). J Clin Oncol 36:2518–2518 10.1200/JCO.2018.36.15_SUPPL.2518. [DOI] [Google Scholar]
- Willoughby CE, Jiang Y, Thomas HD, Willmore E, Kyle S, Wittner A, Phillips N, Zhao Y, Tudhope SJ, Prendergast L, et al. (2020) Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J Clin Invest 130:258–271 10.1172/JCI127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise HC, Iyer GV, Moore K, Temkin SM, Gordon S, Aghajanian C, Grisham RN (2019) Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci Rep 9:18882 10.1038/S41598-019-54796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, Dobyns WB, Ogi T, Meek K, Davies EG, et al. (2013) PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest 123:2969–2980 10.1172/JCI67349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim KA, Qaiser MZ, Begley DJ, Rice-Evans CA, Abbott NJ (2004) Flavonoid permeability across an in situ model of the blood-brain barrier. Free Radic Biol Med 36:592–604 10.1016/J.FREERADBIOMED.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Yuan J (1993) Estimation of variance for AUC in animal studies. J Pharm Sci 82:761–763 10.1002/jps.2600820718. [DOI] [PubMed] [Google Scholar]
- Zenke FT, Zimmermann A, Sirrenberg C, Dahmen H, Kirkin V, Pehl U, Grombacher T, Wilm C, Fuchss T, Amendt C, et al. (2020) Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol Cancer Ther 19:1091–1101 10.1158/1535-7163.MCT-19-0734. [DOI] [PubMed] [Google Scholar]