

Abstract
Proteinaceous aggregates containing α-synuclein protein called Lewy bodies in the substantia nigra is a hallmark of Parkinson’s disease. The molecular mechanisms of Lewy body formation and associated neuronal loss remain largely unknown. To gain insights into proteins and pathways associated with Lewy body pathology, we performed quantitative profiling of the proteome. We analyzed substantia nigra tissue from 51 subjects arranged into three groups: cases with Lewy body pathology, Lewy body-negative controls with matching neuronal loss, and controls with no neuronal loss. Using a label-free liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach, we characterized the proteome both in terms of protein abundances and peptide modifications. Statistical testing for differential abundance of the most abundant 2963 proteins, followed by pathway enrichment and Bayesian learning of the causal network structure, was performed to identify likely drivers of Lewy body formation and dopaminergic neuronal loss. The identified pathways include (1) Arp2/3 complex-mediated actin nucleation; (2) synaptic function; (3) poly(A) RNA binding; (4) basement membrane and endothelium; and (5) hydrogen peroxide metabolic process. According to the data, the endothelial/basement membrane pathway is tightly connected with both pathologies and likely to be one of the drivers of neuronal loss. The poly(A) RNA-binding proteins, including the ones relevant to other neurodegenerative disorders (e.g., TDP-43 and FUS), have a strong inverse correlation with Lewy bodies and may reflect an alternative mechanism of nigral neurodegeneration.
Keywords: Lewy bodies, Parkinson’s disease, neurodegeneration, α-synuclein, TDP-43
Graphical Abstract
INTRODUCTION
Lewy bodies (LBs) are abnormal protein aggregates predominantly comprised of the α-synuclein protein discovered by Frederic Lewy in 1912 in the nerve cells of Parkinson’s disease patients.1 LB formations along with the loss of pigmented dopaminergic (DA) neurons in the substantia nigra (SN) are the defining neuropathologic features of Parkinson’s disease (PD). Synuclein pathology has also been described in association with other neurodegenerative disorders, collectively called synucleinopathies. The well-established association of LB and α-synuclein with a wide group of disorders underlines its important role in neurodegeneration.2 Incident LB pathology is considered a presymptomatic phase of PD.3 Therefore, understanding the protein changes associated with LB may lead to the development of biomarkers for early PD detection. Notably, LB formation and α-synuclein-mediated neurotoxicity are likely to be distinct, but certainly related, phenomena. In this study, we focus on LB formation.
The findings from familial genetics point to two major contributors to LB formation: mitochondrial pathway (mutations in PINK1) and lysosomal dysfunction (mutations in GBA, PLA2G6, PANK2, and NPC1).4 The gene expression profiling studies of postmortem SN tissue have repeatedly implicated dysfunction of mitochondria and protein processing (reviewed in ref 5−7). A few studies indicated the potential involvement of other pathways in PD such as splicing (reviewed in ref 8). Proteome profiling studies of the SN tissue of PD patients and controls revealed a similar theme of alterations in oxidative stress response, redox metabolism, and mitochondrial dysfunction.9−11
This study was aimed at investigating the protein abundance changes associated specifically with LB. The subjects were assigned to case and control groups based on the presence and absence of LB, correspondingly. Note that the case/control assignment did not consider the clinical PD status as the long-term goal was to identify proteins associated with LB formation prior to the onset of overt parkinsonism.3 The study design leveraged a large amount of clinical and histopathological information on the subjects involved in the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP).12,13 Out of 573 individuals with frozen SN tissue, using a case/control matching strategy that accounted for a number of demographic and pathologic confounding factors such as age, sex, DA neuronal loss,14 density of β-amyloid load, and density of tangles in the SN, we selected subjects that were assigned into three groups. Group 1 consisted of cases with the highest density of SN LB. Group 2 were LB-free controls with the same extent of DA neuronal loss. Matching by the neuronal loss was important to make sure we did not detect protein abundance differences associated with an overall loss of neurons. Finally, subjects in group 3 were the LB-free controls with a normal density of neurons, which represents the majority of the population. This double-matching approach allowed deconvolving the two main effects: LB presence and pigmented DA neuronal loss.
Using a shotgun label-free liquid chromatography-tandem mass spectrometry (LC-MS/MS) proteomic approach, we quantified 2963 of the most abundant proteins according to the iBAQ estimate.15 The data analysis confirms involvement in LB formation and neuronal loss of some prior known processes such as reactive oxygen species and synapse as well as suggesting the roles of poly(A) RNA-binding proteins, Arp2/3 complex-mediated actin nucleation, epithelial cells, and basement membrane. Moreover, we discovered that poly(A) RNA-binding proteins and Arp2/3 complex-mediated actin nucleation distinguish between LB-dependent and LB-independent neuronal loss.
METHODS
Participants
Participants were from two prospective cohort studies of aging and dementia, the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP), which began enrollment in 1994 and 1997, respectively. Both studies were approved by the Institutional Review Board of Rush University Medical Center. Participants enrolled without known dementia and agreed to an annual detailed clinical evaluation and organ donation. They signed informed consent and Anatomical Gift Act agreements. At the time of the protein data collection for this study in February 2013, more than 2400 persons had agreed to participate. The clinical follow-up exceeded 95% and the autopsy rate exceeded 90% with 764 autopsies, at the time of which 573 had frozen SN. Details of the design and conduct of the study have been previously reported.12 Both studies were conducted by the same team of investigators with a large common core of measures designed to allow efficient merging of clinical and neuropathologic data.16,17 Postmortem examinations documented a large number of phenotypes including PD-related pathological signs (Lewy bodies and dopaminergic neuronal loss) and Alzheimer’s disease-related pathology such as neuritic and diffuse plaques, and neurofibrillary tangles.13 A board-certified neuropathologist made all diagnoses blind to all clinical data. Subjects with available frozen SN (N = 573 as of February 2013) were considered for the design of this study.
Study Design
Correlation Structure of the Clinical Variables within the Cohort and Implications for Case/Control Matching.
The study design is based on a matched case/control scheme. The key difference in the case/control pairs is the presence of LB in SN. The presence of LB in SN is strongly correlated with the presence in other various brain regions. Not unexpectedly, we found a significant correlation (Spearman ρ = 0.45, p-value 3.6 × 10−8) of SN LB with the loss of pigmented neurons (Figure 1). The other demographic and neuropathologic variables, including age, sex, amyloid-β load, and density of tangles in SN, were not correlated with LBs. The association of LBs with neuronal loss is well-established.18 Given that the goal of the study is to identify changes associated with LB, this correlation suggests considering neuronal loss as an important matching variable. If the cases are not matched by neuronal loss, then it becomes challenging to distinguish the factor associated with the protein abundance change: the presence of LB or neuronal loss. However, pigmented neuronal loss is not common in the general population with no SN LB (Figure 1). Given that it is not common, there is a possibility of a different pathology driving neuronal loss in an LB-independent manner. Thus, the differences between cases and LB-free controls with matching neuronal loss may reflect the differences between the underlying pathologies, but not just the LB presence. Therefore, to deconvolve the proteome changes associated with LB pathology from confounding neuronal loss or other pathology, we decided on two types of controls (Figure 2): matched and not matched by neuronal loss. The latter control represents the general LB-free population. Throughout the text we will be denoting cases, controls matched by neuronal loss, and controls with no neuronal loss (denoted later as NL) as LB +NL+, LB−NL+, and LB−NL−, respectively.
Figure 1.

Association of LB density in the SN with DA neuronal loss. The density of LBs in the SN has been discretized into three levels (zero, below, and above median value) for the purpose of case/control design. Based on the nonparametric Kruskal−Wallis test, the association is highly significant (p-value = 4.3 × 10−7).
Figure 2.

Design with pairing cases with two types of controls. LB and NL denote Lewy bodies and neuronal loss, respectively. Each of the 17 cases (LB+NL+) matched to controls by age, sex, β-amyloid load, and tangles density in the SN. The first type of controls (LB−NL+) matched by neuronal loss. The second type of controls (LB−NL−) reflects the general population and does not have signs of the loss of pigmented neurons.
Imputation of LB, Amyloid, and Tangle Density in the SN Region Using a Machine Learning Approach.
Cases and controls are based on subjects with a high density of LB in the SN region and a complete absence of LB across the entire brain, respectively. While the presence/absence of LB pathology is available for all subjects, quantitative LB density in the SN16,19 was scored only for 140 out of 573 (∼25%) subjects. We attempted to quantify the LB density in the SN based on the presence/absence of LBs in a number of brain regions and other neuropathologic and demographic variables. In brief, we applied a random forest machine learning technique to develop a classifier for the three LB density states (zero, low, and high). Zero means no presence of LB in SN. Low and high states denote below and above median value, correspondingly. The classification accuracy was estimated using leave one out cross-validation (LOOCV) that included independent feature selection at each round. According to the confusion matrix statistics (Figure S1, Supporting Information), the overall accuracy is 0.87 with positive predictive values for the extreme classes with zero and high densities being 0.96 and 0.50. The variables consistently selected for the predictive model over 140 rounds of cross-validation are the presence of LB in any brain location, anterior cingulate cortex, angular gyrus, entorhinal cortex, midfrontal cortex, middle temporal cortex, and LB disease type (0 —not present, 1—nigra, 2—limbic system, 3—neocortex). The overall satisfactory performance allowed us to impute the LB density state for the additional 431 subjects.
Amyloid-β load and tangle density specific to the SN region14,19,20 were considered as additional matching criteria for paired case/control matching to minimize the proteome variability extraneous to LB pathology. A similar approach applied for imputing amyloid-β load in the SN region had a classification accuracy of 0.82. The consistent predictive variables were average amyloid-β loads in calcarine cortex, cingulated region, hippocampus, inferior temporal gyrus, midfrontal gyrus, angular gyrus, and superior frontal gyrus brain regions. Notably, the distribution of SN amyloid-β load across the cohort is strongly dominated (84%) by low amyloid-β. Therefore, the amyloid-β load was further considered as an inclusion (restricting cohort to low-density class only) rather than matching criteria. The classification across four classes of SN tangle density had an accuracy of 0.43 and positive predictive values ranging from 0.25 to 0.66, depending on the class. The brain regions predictive of SN tangle density are the inferior temporal gyrus, superior frontal gyrus, cingulated region, hippocampus, and calcarine cortex region.
Inclusion Criteria.
To reduce the variability of the cohort, we applied a number of additional inclusion criteria. We retained only those subjects self-identified as white non-Hispanic (555 or 97% of the cohort with available SN). The postmortem interval was constrained to 10 h, further reducing the cohort to 86% or 478 subjects. The age at death distribution was narrowed from 66−106 to 80−95 years to reduce variance in age while retaining most of the subjects (373 or 78%). SN amyloid-β load was limited to “low”, further constraining the cohort to 269 subjects.
Case/Control Definition.
Cases were defined as subjects having both the high density of LBs in the SN and LBs spread throughout the brain, including the cortex region. The spread of LBs was measured for all subjects. Although the high density of SN LB and spread of LBs are highly correlated, double criteria ensured that subjects from the case group had a high abundance of LBs. Conversely, controls were required to be free of LB in the SN and display no evidence of LB anywhere else in the brain. The number of subjects meeting the criteria for cases and controls were 29 and 211, respectively.
Paired Case/Control Matching Procedure.
The study design (Figure 2) involved two rounds of matching. In the first round, cases were matched by sex, neuronal loss, and amyloid-β and tangle density. The case/control pairing was represented with a bipartite graph in which vertices corresponding to cases connected to vertices representing controls if there was a match. The edge weight is equal to the inverse positive difference in age (less age difference means higher weight). We minimized the overall difference in age across all pairs using a maximum weighted bipartite matching algorithm (Figure S2, Supporting Information).21,22 Out of 29 cases, 18 passed the first matching round. In the situation where multiple cases matched the same control, we retained only one case. The second round of matching followed the same algorithm, except neuronal loss was left out as a matching criterion. The summary of the final 51 subjects corresponding to the matching triples of the 17 left cases is shown in Table 1. Subject to group assignment, loss of pigmented neurons, nigral density of LB, and randomization are reported in Table S1, Supporting Information.
Table 1.
Descriptive Characteristic of the Participants Included in the Study
| LB+NL+ (N = 17) | LB−NL+ (N = 17) |
LB−NL− (N = 17) | |
|---|---|---|---|
|
| |||
| demographic | |||
| age at death, (years) | 87.7 ± 3.0 | 88.5 ± 4.1 | 87.8 ± 3.1 |
| female sex (n, %) | 10 (59%) | 10 (59%) | 10 (59%) |
| education, (years) | 16.1 ± 3.3 | 15.1 ± 3.3 | 15.2 ± 3.2 |
| postmortem interval (h) | 7.3 ± 1.6 | 6.2 ± 1.8 | 5.7 ± 1.3 |
| APOE e4 allele (n, %) | 6 (35%) | 5 (29%) | 3 (18%) |
| MMSE | 18.8 ± 9.0 | 20.1 ± 10.1 | 24.1 ± 9.4 |
| global parkinsonism, max 100 | |||
| Parkinsonian gait | 46.2 ± 19.3 | 42.9 ± 19.3 | 30.2 ± 19.2 |
| rigidity | 9.1 ± 12.3 | 13.8 ± 21.4 | 7.9 ± 11.7 |
| tremor | 4.1 ± 5.5 | 5.5 ± 13.7 | 3.2 ± 8.7 |
| bradykinesia | 18.2 ± 14.1 | 25.5 ± 18.7 | 12.4 ± 17.3 |
| pathological (n, %) | |||
| PD pathological diagnosis | 6 (35%) | 0 (0%) | 0 (0%) |
| AD pathological diagnosis | 14 (82%) | 11 (65%) | 9 (53%) |
| macroscopic infarcts | 5 (29%) | 6 (35%) | 6 (35%) |
| microinfarcts | 4 (24%) | 6 (35%) | 5 (29%) |
| Lewy bodies positive | 17 (100%) | 0 (0%) | 0 (0%) |
| hippocampal sclerosis |
4 (24%) | 1 (6%) | 2 (12%) |
| TDP-43 inclusion positive | 11 (65%) | 9 (56%) | 9 (60%) |
| cerebral amyloid angiopathy positive | 15 (88%) | 16 (94%) | 14 (82%) |
| atherosclerosis | 13 (76%) | 15 (88%) | 14 (82%) |
| arteriolar sclerosis | 16 (94%) | 15 (88%) | 13 (76%) |
| loss of pigmented neurons in SN | 16 (94%) | 16 (94%) | 1 (6%) |
| β-amyloid load (score) | 6.0 ± 3.7 | 3.5 ± 2.6 | 2.8 ± 2.7 |
| PHFtau tangle density (score) | 9.3 ± 7.7 | 9.8 ± 18.7 | 8.1 ± 10.5 |
Further details on study design can be found in the Document S1 (Supporting Information), which is also available as “LB_SN_study_design” vignette in the companion R package https://github.com/vladpetyuk/LewyBodies.SN.Proteomics.BottomUp.Pub.
LC-MS/MS Proteomic Analysis
Sample Preparation.
The extraction and digestion of the proteins were performed using a conventional protocol based on denaturation of protein in 8 M urea followed by digestion with trypsin.23 Briefly, approximately 8 mg (5−10 mg range) of human SN tissue punch biopsy was resuspended in 100 μL of denaturation buffer (8 M urea, 50 mM Tris−HCl pH 8.0, 10 mM dithiothreitol (DTT), and 1 mM ethylenediaminetetraacetic acid (EDTA)) and homogenized with a motorized pestle. The amount of protein material according to the coomassie assay was 463 μg on average (133−762 μg range). Aliquots with equal amounts of 133 μg protein material were supplemented with denaturation buffer to obtain equal volumes of 100 μL. Further steps up to solid-phase extraction were performed in a 96-well plate format using an Epmotion 5075 TMX (Eppendorf) liquid handling robot. The samples were incubated for 30 min at 37 °C with gentle shaking (600 rpm) to allow for protein denaturation and reduction. Cysteine residues were alkylated by adding 10 μL of iodoacetamide 400 mM stock solution to 36 mM concentration and incubating for 1 h at 37 °C, with shaking, in the dark. Prior to the next step, the samples were stored at −80 °C overnight. After thawing, the samples were diluted by adding 690 μL of 50 mM NH4HCO3 at pH 7.8 and supplemented with 8 μL of CaCl2 100 mM stock solution. Sequencing grade modified trypsin (Promega, Cat No V5111) was added in a 1:50 ratio (w/w trypsin/protein) after preactivation and incubated for 6 h at 37°C with shaking. The sample digests were purified with solid-phase extraction using C18 columns (Discovery DSC-18, SUPELCO, 52601-U) followed by drying down to ∼50 μL in SpeedVac (ThermoFisher). The average recovered peptide amount according to the bicinchoninic acid (BCA) assay was 73 μg (41−91 μg range). Aliquots of the tryptic peptides were diluted down to 0.2 μg/μL prior to the LC-MS/MS analysis. The 7 μg aliquots were analyzed using an LC-MS/MS platform as described below.
LC-MS/MS Platform.
A Waters nanoACQUITY dual pumping ultra-performance liquid chromatography (UPLC) system (Milford, MA) was custom configured for on-line trapping of a 5 μL injection at 3 μL/min with reverse direction elution onto the analytical column at 300 nL/min. Columns were packed in-house using 360 μm o.d. fused silica (Polymicro Technologies Inc., Phoenix, AZ) with 1 cm sol−gel frits for media retention24 and contained Jupiter C18 media (Phenomenex, Torrance, CA) in 5 μm particle size for the trapping column (150 μm i.d. × 4 cm long) and 3 μm particle size for the analytical column (75 μm i.d. × 70 cm long). Mobile phases consisted of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile with the following gradient profile (min, %B): 0, 1; 2, 8; 20, 12; 75, 30; 97, 45; 100, 95; 110, 95; 115, 1; and 150, 1.
MS analysis was performed using a Q Exactive mass spectrometer (Thermo Scientific, San Jose, CA) outfitted with a custom nano-electrospray ionization interface. Electrospray emitters were homemade using 150 μm o.d. × 20 μm i.d. chemically etched fused silica.25 The heated capillary temperature and spray voltage were 250 °C and 3.0 kV, respectively. Data were collected for 100 min following a 15 min delay from sample injection. Survey MS spectra were acquired from 400 to 2000 m/z at a resolution of 70k (100 ms maximum accumulation time with 5 × 106 automatic gain control (AGC) setting), while the top higher-energy collisional dissociation (HCD)-MS/MS spectra were acquired in datadependent mode with an isolation window of 2.0 m/z and at a resolution of 17.5k (100 ms maximum accumulation time with 1 × 105 AGC setting) using a normalized collision energy of 32% and a 60 s exclusion time. The collected mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium26 via the PRIDE partner repository with the data set identifier PXD019279.
Protein Identification and Quantification.
For protein identification and quantification, the mass spectra data sets were analyzed using MaxQuant (v1.5.3.8) software. The mass spectra were searched against the UniProt protein sequence database (release 2015_04), allowing only fully tryptic peptides with static carbamidomethyl modification on the cysteine residues. The parent and fragment ion mass measurement tolerances were set to 4.5 and 8 ppm, respectively. The peptide to spectrum matching and protein identification false discovery rate thresholds were set at 1%. Proteins were quantified using the label-free quantification (LFQ) approach.27 A complete description of MaxQuant settings is provided as the mqpar.xml file in the companion R package. The protein LFQ intensity data were extracted from the proteinGroups.txt file using a custom R script also provided in the companion R package.
Statistical Analysis
The LFQ protein abundance data were converted to the relative form by the log 2-transform and zero-centering protein arbitrary abundances prior to statistical analysis. The potential presence of unanticipated batch effects or outlying samples was assessed using principal component analysis (PCA) analysis and revealed a noticeable deviation from the group of eight samples (Figure S3, Supporting Information). Coincidently, those eight samples were allocated on the same column of the 96-well plate during the sample preparation procedure. Analysis of variance (ANOVA) analysis using 96-well plate column position as a factor confirmed its significance. Therefore, plate column grouping was further considered as a confounding factor. The differential abundance testing was performed using a mixed effects linear model with subject type as a fixed effect and batch and matching groups as random effects. The significance of over-representation of Gene Ontology (GO) terms of proteins increased or decreased in abundance was performed using the clusterProfiler R package.28 Inference of cell-type-specific changes was based on the RNA-Seq database of FACS-isolated individual cell types from the mouse brain.29 Reconstruction of the interaction network among pathways, cell types, and pathology was performed using incremental association based on the Markov blanket detection (iamb) algorithm30 implemented in the bnlearn R package.31 The exact details of statistical analysis are provided in Document S2 (Supporting Information), which is the “LB_SN_LCMS_data_analysis” vignette of the companion R package. Moreover, available R code and specification of the R environment on the docker image allow for the reproducible recomputation of all of the data analysis.
Open Post-Translational Modification (PTM) Search and Spectral Counting Analysis
Open PTM search was performed using MSFragger (v3.2 rc6) software32,33 within FragPipe (v14.0) framework. The search was performed in two steps. The first step was default open search. Then, the identified PTM masses were extracted and used for the second step, common offset search (with corresponding default settings except that mass shifts were reported as variable modifications), which is essentially a conventional dynamic PTM search, although without specification of amino acid specificities. The constrained space of the common offset search significantly helps with the peptide identification confidence.
For the differential abundance test, we selected only peptides carrying discovered modifications. The test was performed using a quasi-likelihood Poisson model34 implemented in the msmsTest R package. p-Values were adjusted for multiplicity of hypothesis testing using the Benjamini−Hochberg method.35 The exact details of MSFragger postprocessing and statistical analysis are provided in Document S3 (Supporting Information), which is the “LB_SN_LCMS_MSFragger” vignette of the companion R package.
RESULTS
Differentially Abundant Proteins
The direct, with no additional prefractionation, label-free LC-MS/MS analysis of SN tissue digest on average identified and quantified 2963 proteins (Table S2, Supporting Information). The analyses were restricted to proteins found in at least half of the analyzed samples. Relative protein abundances were modeled with a linear mixed effect approach. The fixed effect is the subject type (LB+NL+, LB−NL+, or LB−NL−). Random effects are the matching group (17 groups of 3) and the batch effect corresponds to the plate column (7 columns). The p-value histogram shows a much higher frequency of low p-values than one would expect at random (Figure 3). Despite the limited statistical power (N = 17 per group), the p-value distribution clearly indicated a significant effect of subject type on protein abundance. The p-values were adjusted for multiplicity of hypothesis testing using a q-value approach.36 Due to limited power, the q-value significance threshold was set at 0.2, resulting in 663 out of 2963 proteins called out as changing. The top 10 differentially abundant proteins are shown in Figure 4. The full results of mixed effect ANOVA analysis are reported in Table S3, Supporting Information.
Figure 3.

p-Value histogram of the mixed effect ANOVA test. Histogram bin width is 0.05. Higher than the random number of proteins with low p-values (first bin) indicates the significance of the subject type effect.
Figure 4.

Relative abundances of the top 10 most varying proteins across the groups. Proteins are ordered according to significance in the ANOVA test. The relative abundances are adjusted for matching group effect and batch effect.
In our study design, we tried to dissect the complicated nature of PD-related SN pathology, rather than dichotomizing subjects into disease vs control groups as is commonly done. Thus, to untangle what exactly is causing the protein abundance differences (LB presence or NL), we applied a post hoc contrast analysis. The differential abundance was tested across all contrasts including three pairwise comparisons and three comparisons of a particular subject type vs the other two. Adjustment for the multiplicity of comparisons was performed using z-statistic.37 The top three differentially abundant proteins for each of the pairwise comparisons are shown in Table 2. The full results of post hoc contrast analysis are reported in Table S4, Supporting Information.
Table 2.
Top Three Most Significant Proteins for Each Pairwise Comparison
| contrast | protein | estimate | p-valuea |
|---|---|---|---|
|
| |||
| LB+NL+ vs LB−NL+ | RGS6 | −0.25 | 4.94 × 10−6 |
| GANAB | −0.15 | 3.41 × 10−4 | |
| CD59 | 0.15 | 4.07 × 10−4 | |
| LB+NL+ vs LB−NL− | CD59 | 0.15 | 5.01 × 10−4 |
| SRPK2 | −0.27 | 7.10 × 10−4 | |
| COL4A1 | 0.56 | 8.94 × 10−4 | |
| LB−NL+ vs LB−NL− | ITGB8 | 0.47 | 3.02 × 10−5 |
| EIF3E | 0.28 | 1.71 × 10−4 | |
| IGHV3-23 | 0.77 | 2.22 × 10−4 | |
Adjusted for the multiplicity of comparisons.
While some insight can be gathered by following up on individual proteins, a more comprehensive picture can be inferred by putting the data into a functional context. To shed light on relationships among LB, NL, α-synuclein, and potentially other involved biological processes in SN degeneration, we interrogated the data using three complementary strategies. First, we determined if there were any functional pathways or GO ontologies that significantly enriched among the concordantly changing proteins. Second, we analyzed whether the observed protein abundance differences might be explained by cell-type population differences. Third, we examined the major factors potentially responsible for α-synuclein protein abundance, the key component of LB. Finally, we integrated these results in a Bayesian model to pinpoint causal relationships between proteins, pathways, cell-type differences, and LBs.
Differentially Abundant Modifications
To get a more detailed look at the SN proteome and potentially discover PTMs associated with LB and NL pathologies, we searched the data using MSFragger.32,33 The default setting open search resulted in the discovery of 268 mass shifts or tentative PTMs (Table S5, Supporting Information). The annotation of the modifications is based on mass alone. Essentially, it is the closest mass within 10 ppm in UniMod database.38 Thus, the mass shift annotations are qualified as tentative only. The full list of peptides with discovered modifications and corresponding spectral counts is provided in Table S6, Supporting Information.
It is also important to keep in mind the distinction between modifications introduced during sample preparation and instrumental analysis and those present because of biology-relevant reasons. The top five mass shifts were attributed to the following UniMod entries: pyro-glu from Q/Loss of ammonia (−17.026549 Da), amidination of lysines or N-terminal amines with methyl acetimidate (41.026549 Da), deamidation (0.984016 Da), iodoacetamide derivative (57.021464 Da), replacement of three protons by iron (52.911464 Da), and carbamylation (43.005814 Da). Oxidation follows as the sixth most frequent modification. Since N-terminal acetylation and cysteine iodoacetamide modification were part of the MS/MS search settings, they are excluded from the summary of the discovered mass shifts. Nonetheless, the footprint of these modifications is still present. The second modification, amidination of lysines or N-terminal amines with methyl acetimidate, is likely to be disguised as ubiquitous N-terminal acetylation (42.010565 Da) under the circumstances when the monoisotopic peak is confused with the +1 13C isotope. The reported iodoacetamide derivative modification corresponds to alkylation of free N-terminal residues, a known but commonly overlooked artifact.39
Modifications that are clearly related to physiology, such as methylation, phosphorylation, and non-N-terminal acetylation, were reported as rank 17, 19, and 27, correspondingly. Proteins with the most discovered phosphopeptides: microtubule-associated protein 1B (MAP1B), MBP, NEFM, microtubule-associated protein tau (MAPT), and ADD1, contain 99, 29, 22, 43, and 29 phosphorylation sites according to the UniProt database. For example, the discovered phosphorylation of MAPT (also known as tau) includes the well-characterized AT270, PHF1/AD2, AT8, and AT180/TG3 sites.40 Another example of a modification that cannot be attributed to a processing/analysis artifact is O-GlcNAc. A couple of examples include NEFM and SPTBN1 proteins with peptides covering prior sites reported in UniProt O-GlcNAc at 431 and 2324 residues.
The objective of the study was to find associations of the LB and NL pathology with the proteomic footprint. Thus, we were mostly interested in modifications that are differentially abundant between the three groups. In particular, modifications preferentially present in the LB+NL+ group represent the top interest. Differential abundance analysis was performed using QuasiTel approach34 using the data presented in Table S6, Supporting Information. The results of the spectral count differential abundance test are shown in Figure 5. Overall, there are 177 modified peptides that pass the threshold of Benjamini−Hochberg adjusted p-value <0.05. Interestingly, the majority of the peptides are exclusively present in a certain group, but not the other two. The peptides from the three clusters that are associated with each of the three groups of subjects are reported in Table S7, Supporting Information.
Figure 5.

Heatmap of spectral counts of the statistically significantly differentially abundant modified peptides. Spectral counts for each peptide are scaled by the highest value across the 51 samples. Cluster analysis partitioned the peptides into cluster characteristic for three groups of subjects. The majority of the peptides are exclusive to each of the three study design groups. The characteristics of the peptides for the LB+NL+ cluster are shown in the expansion on the right.
Inference of Pathways and Common Ontologies Explaining the Proteome Variance
To test for the over-represented GO terms among the differentially abundant proteins, we applied the Fisher’s exact test. The top three enriched terms are reported in Table 3 (full list reported as Table S8, Supporting Information). Given the substantial overlap of genes across the related GO terms and a possibility that some proteins may be significantly different in multiple pairwise comparisons, the interpretation of over-represented terms and corresponding pathways is not straightforward. To eliminate this redundancy, we inferred a parsimonious set of GO terms using an iterative procedure. For each iteration, we selected the most statistically significant over-represented GO term regardless of the contrast and directionality of change. Then, proteins corresponding to this GO term were removed from further consideration and the GO enrichment statistics were recomputed for the remaining data set. The iterations continued until no GO term was significant. The procedure resulted in the following five GO terms: poly(A) RNA binding (GO:0044822), synapse (GO:0045202), basement membrane (GO:0005604), Arp2/3 complex-mediated actin nucleation (GO:0034314), and hydrogen peroxide metabolic process (GO:0042743). GO term relative abundances were computed as centroids of the corresponding proteins (Figure 6). Notably, we detected two major patterns of change in LB+NL+ or LB−NL+ subjects compared to LB−NL− controls. Several pathways (hydrogen peroxide metabolic process, basement membrane, and synapse) show consistent change regardless of the presence of LB pathology. Interestingly, the Arp2/3 complex and poly(A) RNA-binding proteins show a divergent pattern, with directionality depending on the presence of LB. The individual protein relative abundances mapped to the GO terms are shown in Figure 7.
Table 3.
Top GO Ontology Terms for Each of the Pairwise Contrasts and Directions of Change
| comparison | dir | ont | GO ID | description | gene ratioa | bg. ratiob | p-adjust |
|---|---|---|---|---|---|---|---|
|
| |||||||
| LB+NL+ vs LB− NL+ | up | BP | 0034314 | Arp2/3 complex-mediated actin nucleation | 8/96 | 19/2798 | 4.2 × 10−5 |
| CC | 0015629 | actin cytoskeleton | 16/97 | 166/2872 | 8.2 × 10−3 | ||
| MF | 0008092 | cytoskeletal protein binding | 26/94 | 313/2745 | 4.9 × 10−4 | ||
| down | BP | 0006397 | mRNA processing | 22/82 | 67/2798 | 2.4 × 10−16 | |
| CC | 0071013 | catalytic step 2 spliceosome | 12/84 | 25/2872 | 3.8 × 10−11 | ||
| MF | 0044822 | poly(A) RNA binding | 44/80 | 370/2745 | 1.7 × 10−17 | ||
| LB+NL+ vs LB− NL− | up | BP | 0030198 | extracellular matrix organization | 11/51 | 82/2798 | 1.4 × 10−5 |
| CC | 0005604 | basement membrane | 8/51 | 25/2872 | 1.3 × 10−7 | ||
| MF | 0016684 | oxidoreductase activity, acting on peroxide as acceptor | 5/50 | 20/2745 | 4.5 × 10−4 | ||
| down | BP | 1903829 | positive regulation of cellular protein localization | 5/35 | 92/2798 | 2.7 × 10−1 | |
| CC | 0044432 | endoplasmic reticulum part | 7/37 | 250/2872 | 3.3 × 10−1 | ||
| MF | 0044877 | macromolecular complex binding | 7/36 | 293/2745 | 4.3 × 10−1 | ||
| LB−NL+ vs LB− NL− | up | BP | 0000377 | RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 13/101 | 53/2798 | 3.9 × 10−6 |
| CC | 0071013 | catalytic step 2 spliceosome | 8/102 | 25/2872 | 9.0 × 10−5 | ||
| MF | 0003723 | RNA binding | 32/102 | 443/2745 | 4.4 × 10−3 | ||
| down | BP | 0023061 | signal release | 20/125 | 133/2798 | 5.0 × 10−4 | |
| CC | 0045202 | synapse | 32/127 | 262/2872 | 4.0 × 10−6 | ||
| MF | 0030695 | GTPase regulator activity | 11/119 | 82/2745 | 2.6 × 10−2 | ||
Gene ratio: number of genes mapped to the ontology over the total number of considered genes.
bg. ratio: number of genes mapped to the ontology from the background (not changing genes).
Figure 6.

Nonredundant GO terms. X-axis corresponds to a median log2 fold change of the corresponding proteins. The Control group representing the general population (LB−NL−) serves as a reference. The rectangle height (Y-axis direction) is proportional to the number of proteins mapped to the term.
Figure 7.

Relative abundances of the individual proteins mapping to the nonredundant significant GO terms. (A) mRNA processing GO:0006397, (B) post-translational protein modification GO:0043687, (C) basement membrane GO:0005604, (D) hydrogen peroxide metabolic process GO:0042743, (E) synapse GO:0045202, and (F) Arp2/3 complex-mediated actin nucleation GO:0034314.
Inference of Cell-Type Level Changes
The five major cell types in the brain tissue are neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells. To derive cell-type-specific gene signatures, we leveraged a database with estimates of mRNA abundances across individually isolated cell types from the mouse cortex.29 First, we constrained the genes only to those that were present in our proteomics data set. Second, we required that the gene must be within the top 10% most expressed in any of the cell types. Third, for a gene to be a part of cell-type-specific signature, we specified that 80% of mRNA abundance should originate from a given cell type. That is, at most 20% of expression was allowed to be derived from other cell types. The final set of cell-type markers included 37, 40, 27, 16, and 15 proteins for astrocyte, neuron, oligodendrocyte, microglia, and endothelial cells, respectively. Based on the protein centroid, two out of five cell types showed significant changes across the subject types (Figure 8). Specifically, the endothelial cells increased in abundance in the case of neuronal loss regardless of LB pathology (LB+NL+ or LB−NL−). The profiles of individual marker proteins for the endothelial cell are shown in Figure 9.
Figure 8.

Estimates of cell-type level changes based on the sets of marker proteins. Gross cell-level changes were estimated based on the median relative abundance value of the marker protein sets. The p-values corresponding to the nonparametric Kruskal−Wallis test are shown in the boxes below.
Figure 9.

Relative abundances of the endothelial marker proteins. The color key of relative abundances is the same as in Figure 7.
Association of α-Synuclein Abundance with LB Presence
The α-synuclein protein deserves particular attention because it is the main component of LB, and increased α-synuclein expression has been genetically linked to both familial and sporadic PD.41−43 Given the iBAQ estimates of absolute abundance,15 α-synuclein protein was ranked as the 93rd most abundant protein in the SN. Importantly, α-synuclein is expressed specifically in neurons, and our data suggest that neuronal loss indeed reduces the amount of α-synuclein at the whole tissue level (LB−NL+ vs LB−NL−, p-value: 0.016). When matching by pigmented neuronal loss pathology (i.e., LB−NL+ vs LB+NL+ comparison), α-synuclein protein levels were 35% greater in the presence of LB (p-value = 0.021) (Table S4, Supporting Information). Interestingly, when comparing LB+NL+ cases to the LB−NL− controls without neuronal loss, α-synuclein protein levels were not differentially abundant (p-value: 0.99). This is likely due to the opposing effects of LB pathology and neuronal loss. This highlights the importance of our experimental design strategy of matching samples based on SN neuronal loss to detect changes in the proteome specifically associated with LB pathology.
Inference of the Causal Model Explaining LB and NL
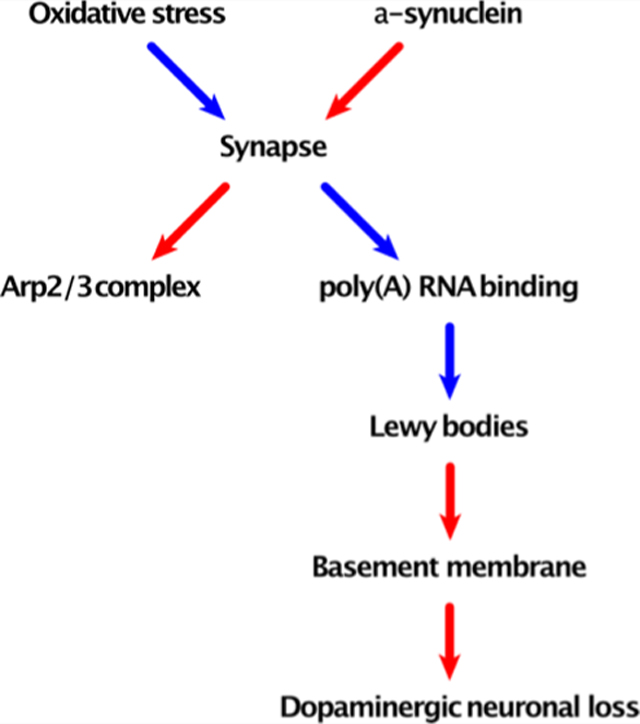
While identifying top associated pathways (Figure 6) and cell types is informative for understanding SN pathology, it is of particular interest to understand their causal relationships. The causal model (Figure 10) was inferred using the incremental association Markov blanket algorithm.30 Probabilities of the edges and their causal directionality were computed based on 1000 bootstrap resamplings. The edge probability threshold was set to the level where all of the nodes were connected into a single graph. In most of the cases, the directionality of the correlation must be interpreted as merely suggestive. Also, the edges represent interactions based on statistics of correlations. Thus, the physical meaning of the node interaction must be interpreted with caution.
Figure 10.

Drivers of LB formation and DA neuronal loss. The representation of the causal model and associated uncertainties inferred by bootstrap analysis. Edge color represents the sign of correlation (red—positive, blue—negative). Uncertainties in the causal graph inference are shown as the probability of the edge strength (s value) and its directionality (d value).
Nonetheless, the data suggest that α-synuclein is not linked directly to LB. Rather, it is connected to LB through synapse and poly(A) RNA-binding proteins. Reactive oxygen species constitute another top causal node that acts through synapse and propagates its effect down to LB and neuronal loss. Interestingly, the graph suggests that LB-cased NL is mediated by the basement membrane or endothelial cells. The Arp2/3 complex-mediated actin nucleation (GO:0034314) is certainly related to synapse. The graph suggested it as a consequence rather than a cause, although the directionality of its relationship with synapse is rather marginal. The causal chain of synapse, poly(A) RNA-binding proteins, and finally LBs is very prominent. The strong negative correlation between poly(A) RNA-binding proteins and LBs may represent either some sort of protective mechanism or perhaps alternative pathology.
DISCUSSION
Differentially Abundant Proteins
Some of the differentially abundant proteins have been previously associated with PD or neurodegeneration in general. Among our top results, we discovered sharply reduced levels of regulator of G-protein signaling 6 (RGS6) in the presence of LB pathology (Figure 4) when compared with matched samples either with or without SN neuronal loss. RGS6 is a GTPase-activating protein that modulates G-protein-mediated signaling, which is broadly impactful in central nervous system (CNS) development, maintenance, and function.44 Interestingly, RGS6 expression is enriched in SN DA neurons and is a target of Pitx3, a transcription factor with a role in programming differentiation of DA neurons in the ventral SN.45 Moreover, Rgs6−/− null mice develop a late-onset loss of DA neurons in the ventral SN by 1 year of age. It has been suggested that RGS6 may promote the survival of DA neurons by inhibiting DRD2 dopamine receptor signaling and preventing the accumulation of cytotoxic byproducts such as 3,4-dihydroxyphenylacetaldehyde (DOPAL) and 3,4-dihydroxyphenylacetic acid (DOPAC).44 Our results raise the intriguing possibility that synuclein-mediated DA neuronal loss may occur via a similar mechanism.
Eukaryotic translation initiation factor 3 subunit E (EIF3E) was the second most differentially abundant protein. It is one of the 13 subunits of the 800 kDa eIF-3 complex that play a role in the initiation of protein synthesis. A recent study found that components of the eIF-3 complex are increased ∼2-fold in the SN of the subjects with late-stage PD (Braak 5–6).46 Interestingly, missense mutations in another component of the eIF-3 complex, EIF4G1, have been linked to autosomal-dominant parkinsonism.47 The identified key mutations (p.Ala502Val and p.Arg1205His) disrupt the binding of EIF4G1 to EIF3E. Along with other experimental evidence,48,49 our findings suggest that LB formation in PD pathogenesis is associated with disruptions in the protein translation process or potentially other functions of the eIF-3 complex. At the same time, ribosomal proteins per se did not appear as differentially abundant at individual or pathway levels, thus indicating that distortion of protein synthesis is limited only to certain steps such as initiation and potentially mRNA to ribosome transport (poly(A) RNA-binding group).
The CD59 protein abundance pattern was exclusively associated with the presence of LBs. It is a membrane glycoprotein that prevents complement from attacking the cells.50 While CD59 has not previously been linked to PD, the innate immune system has been strongly implicated in Alzheimer’s disease.51 In one notable study,52 CD59 protein levels were decreased in the frontal cortex and hippocampus of Alzheimer’s disease subjects. Further, in a mouse model of traumatic brain injury, loss of CD59 null resulted in enhanced neuronal loss and evidence of impaired performance based on several neurobehavioral assays. There is also accumulating evidence that abnormal forms of α-synuclein trigger innate and adaptive immunity responses in PD.53,54 Given this evidence, one hypothesis may be that CD59 prevents the immune system from destroying malfunctioning LB-containing neurons, thus leading to their accumulation.
The integrin β8 protein (ITGB8) was found to be associated with pigmented neuronal loss, particularly in the absence of LB pathology. ITGB8 heterodimerizes with ITGAV to form an integrin cell surface adhesion receptor (αvβ8 type).55 Though integrins have broad roles in the CNS, ITGB8 has been specifically implicated in brain angiogenesis/vascularization56,57 as well as axonal survival.58 The finding that ITGB8 is increased in the case of neuronal loss may suggest either a causal or compensatory effect involving vascularization and/or axonal integrity in the setting of SN neurodegeneration.
Differentially Abundant Modifications
The open search capability of MSFragger is the latest development in MS/MS spectra analysis. It offers a realistic opportunity for the discovery of a broad spectrum of modifications that includes both sample processing artifacts and the ones related to physiology. Identification of the modification is based purely on mass, and thus confidence that the identification is correct requires additional validation. Nonetheless, MSFragger identified a number of interesting PTM leads that are exclusively present in one subject group but not the other two.
One example of such PTM is phosphorylation of MBP evident by peptide LGGRDSRSGSPMAR that covers known phosphosites at serines 295 and 299. The exact localization of the modification based on the MS/MS spectra is not certain. It is known that the latter site is phosphorylated by UHMK1 kinase.59 The kinase is highly expressed in the brain and has been associated with schizophrenia60 and neuron projection remodeling.61 The other example is N-acetylhexosamine (likely O-GlcNAc) modification of BSN covered by HSDSGSDSKHDATASSSSAAATVR. It has previously been detected in global profiling of the O-GlcNAc-modified proteome of mouse synapses.62 Lysine methylation of ACTC1 at position 86, covered by YPIEHGIITNWDDMEK peptide, has also been observed before.63 There are also a number of related redox PTMs exclusively associated with LB +NL+. Historically oxidation-related modifications do not receive as much attention as more complex ones (including the three aforementioned). For example, UniProt does not follow these modifications, perhaps due to concerns of their potentially artifactual nature. However, when they are statistically significantly associated with a certain pathologic condition, oxidation PTMs deserve attention.
In summary, MSFragger identified 65 modified peptides that are exclusively associated with LB+ subjects. Since it is a novel approach, without additional validation using alternative methods (e.g., targeted SRM quantification using synthetic peptides), it is hard to assess the reliability of the findings. Nonetheless, these results provided a diverse set of interesting candidate markers for further exploration.
Quantitative Relationship of α-Synuclein and LBs
α-Synuclein is the predominant component of LBs, and increased α-synuclein protein expression, due to either rare locus duplication/triplication41,42 or common polymorphisms affecting the transcriptional promoter,43 is a strong causal factor for PD. When controlling for DA neuronal loss, we detected a 35% increase in α-synuclein in the presence of LB pathology. It is possible that this is an underestimation of the actual difference for the following reason: although we used detergent (8 M urea) during the protein denaturation and solubilization step, it is possible that LBs were not completely solubilized. Thus, we may have measured only a fraction of the total α-synuclein. Nonetheless, our finding of a 20% increase in α-synuclein protein in LB presence is in agreement with the magnitude of changes discovered from studies of PD risk alleles at the SNCA locus. Specifically, SNCA gene duplications, which cause a 50% increase in expression, are sufficient to cause highly penetrant, familial early-onset PD. Common polymorphisms at the SNCA locus associated with relatively modest PD risk were recently shown to cause only a 6−18% increase in expression.43 Given that LB precedes the onset of PD, it is likely that LB incidence is even higher in the subjects with gene duplication/triplication and risk alleles. Thus, LB formation is likely to be even more sensitive to α-synuclein concentration as our data suggest.
The current proteomic study was focused on measuring bulk levels of α-synuclein regardless of structure, solubility, and localization. This limitation needs to be addressed in the future. Some studies suggest particular soluble, oligomeric forms of α-synuclein species that mediate neuronal injury in PD. It has been suggested that larger insoluble aggregates, such as LB, may have a bystander role or even be protective similar to aggresomes.64−67 Therefore, although this is the first study that established a statistically significant relationship between α-synuclein protein abundance measurements and LB presence, further granularity in the quantification of specific protein forms may be informative for filling in potentially missing links. This will help to dissect the different roles of α-synuclein species including LBs.
Drivers of LB Formation and DA Neuron Loss
This study highlights five major pathways or biological themes that are strongly implicated in LB formation and DA neuronal loss (Figures 6 and 10): (1) Arp2/3 complex-mediated actin nucleation; (2) synapse; (3) poly(A) RNA binding; (4) basement membrane and endothelium; and (5) hydrogen peroxide metabolic process. The synaptic dysfunction and the role of hydrogen peroxide metabolism, which is linked to oxidative stress, are well-researched subjects and reviewed in refs 68−76 and 77−83, respectively. Therefore, we focus our below discussion on three novel findings.
Arp2/3 Complex-Mediated Actin Nucleation.
The Arp2/3 complex plays a major role in actin cytoskeletal organization and is essential for neuronal development, function, and maintenance in aging.84,85 Of particular relevance to PD and neurodegeneration, Arp2/3 has been implicated in intraneuronal vesicular/endosomal transport, autophagy, and synaptic function.86−90 Substantial evidence from human genetics highlights the likely importance of endosomal transport in PD risk and pathogenesis.91−93 Specifically, mutations in VPS35 cause autosomal-dominant, familial PD (OMIM #614203).94 VPS35 is a part of the retromer complex, which is responsible for the retrograde transport of target proteins from the late endosome to the Golgi or plasma membrane. Engagement of Arp2/3 is essential for proper retromer function, and the PD-associated D620N mutation disrupts the recruitment of a key Arp2/3-activator. The Wiskott−Aldrich syndrome protein and scar homolog (WASH) complex,95−97 Arp2/3, and VPS35 are also implicated in autophagy.89 Autophagy is an important pathway for α-synuclein clearance and is linked to PD pathogenesis.98−101 Our results therefore support prior studies suggesting that potential derangements in the Arp2/3 complex may accompany LB formation and DA neuronal loss. However, the inferred causal graph suggests that the observed changes in Arp2/3 are reactive to the changes in the synapse. Indeed, the complex plays a supportive role in synapse maturation and function.102−104 Thus, despite the statistical significance, the exact nature of the association with LB presence is a fruitful direction for future investigations.
Poly(A) RNA Binding.
Our proteomic analyses also identified that a significant change among mRNA-binding proteins is inversely associated with LB formation. Most of the 44 proteins in the enriched gene set (GO:0044822) are nuclear proteins with established roles in RNA metabolism and processing, such as splicing.105 Many of these proteins also participate in the formation of the cytoplasmic stress granules, which are RNA and protein-containing particles that can form in response to neuronal injury.106,107 In recent years, a strong connection has also been established between these ribonucleoproteins and neurodegenerative disorders.8,108−115 In particular, three proteins in the highlighted set, TARDBP (TDP-43), FUS, and HNRPA3, are established risk factors for amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). These are fairly rare disorders and none of the 51 subjects had ALS or FTLD. However, LB−NL+ had faster cognitive decline (Wilcoxon test p-value = 0.0411) compared to the LB−NL− group. Note that the LB+NL+ group had the fastest cognitive decline of all three.
While still an active area of investigation, many RNA-binding proteins have been found to have intrinsically disordered protein domains that may promote coaggregation with other disease-linked protein species (e.g., C9ORF72 in ALS or MAPT in AD).116,117 Recent evidence suggests that α-synuclein may also complex with selected ribonucleoproteins.118 One of the novelties of our study is that we uncovered a much larger set of pathology-associated ribonucleoproteins (Figure 7A). Based on strong anticorrelation, one may conjecture some type of mechanism preventing α-synuclein accumulation. However, given that there are other known neurodegenerative disorders associated with these proteins, it is more likely that this is an alternative mechanism behind the DA neuronal loss. The slightly faster cognitive decline of the LB−NL+ group compared to full control provides an additional evidence of neurodegeneration. The exact role of those poly(A) RNA binding or ribonucleoproteins in neurodegeneration is yet to be revealed.
Basement Membrane and Endothelium.
The endothelial cells in the CNS form the lining of the cerebrovascular tree. The basement membrane (GO:0005604), consisting of extracellular matrix proteins, forms an architectural substrate for endothelial cells and further mediates interactions with the surrounding neurons and glia. Thus, their common function overlaps in corresponding proteins and the strong correlation of protein abundances in our analyses suggests that changes in the basement membrane and endothelial cells reflect the same underlying biological process. Though these processes have not previously been a major focus in PD research, the neurovascular unit has received considerable recent attention in related neurodegenerative disorders, such as AD.119−123 Basement membrane thickening has been described in histopathologic studies of both the PD and AD postmortem brain.123,124 In addition, circulating endothelial progenitor cells were discovered to be increased in PD.125 It is possible that our observations reflect reactive and/or compensatory changes at the blood−brain interface, potentially in relation to neuro-inflammatory responses.126−128 Indeed, the causal graph model suggests that the basement membrane changes are downstream of LB and mediating NL.
CONCLUSIONS
Beyond α-synuclein, the proteomic data revealed a number of individual proteins (RGS6, EIF3E, CD59, ITGB8), PTMs, and pathways associated and potentially driving the LB formation and NL. While some of the biological processes such as reactive oxygen species and synapse have been previously described to play a role in LB, the novel findings include Arp2/3 complex-mediated actin nucleation, poly(A) RNA-binding proteins, and basement membrane. According to our causal model, Arp2/3 complex-mediated actin nucleation is reactive to pathologic changes in the synapse. Poly(A) RNA-binding proteins, such as TDP-43, FUS, and HNRPA3, are mutually exclusive with LB presence, thus representing either protective mechanism or alternative neuronal pathology. Based on the inferred causal model, the basement membrane and changes in epithelial cell content likely mediated between LB presence and NL. In summary, analysis of protein abundances by leveraging a rigorous study design provided novel molecular insights into pathological mechanisms associated with LB incidental disease and loss of dopaminergic neurons in substantia nigra.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to all of the ROS and MAP participants, and we also thank the staff and investigators at the Rush Alzheimer’s disease Center. The proteomic work was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Department of Energy and located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the Department of Energy under Contract DE-AC05–76RL0 1830. We are thankful to Alexey Nesvizhskii and his lab members for helping with the MSFragger tool.
Funding
This study was supported by the National Institute of Neurological Disorders and Stroke grant U18 NS082140 (V.A.P.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00747.
Study design (Document S1) (PDF)
LC-MS/MS data analysis (Document S2) (PDF) Open modification search analysis (Document S3) (PDF)
Evaluation of the random forest-based algorithm for predicting the density of LB in SN (Figure S1); the first round of matching considering neuronal loss (Figure S2); and PCA plot of relative protein abundances (Figure S3) (PDF)
Description of the samples (Table S1); protein abundance data (Table S2); results of the mixed effect ANOVA test (Table S3); results of the post hoc contrast test (Table S4); summary of modifications discovered through open MS/MS search (Table S5); spectral counts of the modified peptides (Table S6); clusters of the modified peptides associated with the study design groups (Table S7); and GO enrichment analysis (Table S8) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00747
Notes
The authors declare no competing financial interest.
The collected mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium26 via the PRIDE partner repository with the data set identifier PXD019279. A detailed description of study design steps and computational pipeline for LCMS data analysis is available at a companion R package https://github.com/vladpetyuk/LewyBodies.SN.Proteomics.BottomUp.Pub.
Contributor Information
Vladislav A. Petyuk, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Lei Yu, Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, Illinois 60612, United States; Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois 60612, United States.
Heather M. Olson, Environmental and Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States
Fengchao Yu, Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Geremy Clair, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Wei-Jun Qian, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Joshua M. Shulman, Departments of Neurology, Molecular & Human Genetics, and Neuroscience, Baylor College of Medicine, Houston, Texas 77030, United States; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, Texas 77030, United States
David A. Bennett, Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, Illinois 60612, United States; Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois 60612, United States
REFERENCES
- (1).Lewy FH Paralysis agitans. 1. Pathologische Anatomie. In Handbuch der Neurologie, Dritter Band, Spezielle Neurologie I; Lewandowsky M, Ed.; Julius Springe; r: Berlin, 1912; pp 920–933. [Google Scholar]
- (2).Wong YC; Krainc D alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1−13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).DelleDonne A; Klos KJ; Fujishiro H; Ahmed Z; Parisi JE; Josephs KA; Frigerio R; Burnett M; Wszolek ZK; Uitti RJ; Ahlskog JE; Dickson DW Incidental Lewy body disease and preclinical Parkinson disease. Arch. Neurol. 2008, 65, 1074–1080. [DOI] [PubMed] [Google Scholar]
- (4).Hardy J; Lewis P; Revesz T; Lees A; Paisan-Ruiz C The genetics of Parkinson’s syndromes: a critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [DOI] [PubMed] [Google Scholar]
- (5).Cooper-Knock J; Kirby J; Ferraiuolo L; Heath PR; Rattray M; Shaw PJ Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 518–530. [DOI] [PubMed] [Google Scholar]
- (6).Borrageiro G; Haylett W; Seedat S; Kuivaniemi H; Bardien S A review of genome-wide transcriptomics studies in Parkinson’s disease. Eur. J. Neurosci. 2018, 47, 1–16. [DOI] [PubMed] [Google Scholar]
- (7).Courtney E; Kornfeld S; Janitz K; Janitz M Transcriptome profiling in neurodegenerative disease. J. Neurosci. Methods 2010, 193, 189–202. [DOI] [PubMed] [Google Scholar]
- (8).La Cognata V; D’Agata V; Cavalcanti F; Cavallaro S Splicing: is there an alternative contribution to Parkinson’s disease? Neurogenetics 2015, 16, 245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Werner CJ; Heyny-von Haussen R; Mall G; Wolf S Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008, 6, No. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Licker V; Cote M; Lobrinus JA; Rodrigo N; Kovari E; Hochstrasser DF; Turck N; Sanchez JC; Burkhard PR Proteomic profiling of the substantia nigra demonstrates CNDP2 overexpression in Parkinson’s disease. J. Proteomics 2012, 75, 4656–4667. [DOI] [PubMed] [Google Scholar]
- (11).Jin J; Hulette C; Wang Y; Zhang T; Pan C; Wadhwa R; Zhang J Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics 2006, 5, 1193–1204. [DOI] [PubMed] [Google Scholar]
- (12).Bennett DA; Schneider JA; Arvanitakis Z; Wilson RS Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bennett DA; Schneider JA; Buchman AS; Barnes LL; Boyle PA; Wilson RS Overview and Findings From the Rush Memory and Aging Project. Curr. Alzheimer Res. 2012, 9, 646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schneider JA; Li JL; Li Y; Wilson RS; Kordower JH; Bennett DA Substantia nigra tangles are related to gait impairment in older persons. Ann. Neurol. 2006, 59, 166–173. [DOI] [PubMed] [Google Scholar]
- (15).Schwanhäusser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [DOI] [PubMed] [Google Scholar]
- (16).Buchman AS; Shulman JM; Nag S; Leurgans SE; Arnold SE; Morris MC; Schneider JA; Bennett DA Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shulman JM; Yu L; Buchman AS; Evans DA; Schneider JA; Bennett DA; De Jager PL Association of Parkinson disease risk loci with mild parkinsonian signs in older persons. JAMA Neurol. 2014, 71, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Parkkinen L; O’Sullivan SS; Collins C; Petrie A; Holton JL; Revesz T; Lees AJ Disentangling the relationship between lewy bodies and nigral neuronal loss in Parkinson’s disease. J. Parkinson’s Dis. 2011, 1, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Buchman AS; Nag S; Shulman JM; Lim AS; VanderHorst VG; Leurgans SE; Schneider JA; Bennett DA Locus coeruleus neuron density and parkinsonism in older adults without Parkinson’s disease. Mov. Disord. 2012, 27, 1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schneider JA; Bienias JL; Gilley DW; Kvarnberg DE; Mufson EJ; Bennett DA Improved detection of substantia nigra pathology in Alzheimer’s disease. J. Histochem. Cytochem. 2002, 50, 99–106. [DOI] [PubMed] [Google Scholar]
- (21).Bergstralh EJ; Kosanke JL; Jacobsen SJ Software for optimal matching in observational studies. Epidemiology 1996, 7, 331–332. [PubMed] [Google Scholar]
- (22).Hansen BB; Klopfer SO Optimal full matching and related designs via network flows. J. Comput. Graphical Stat. 2006, 15, 609–627. [Google Scholar]
- (23).Andreev VP; Petyuk VA; Brewer HM; Karpievitch YV; Xie F; Clarke J; Camp D; Smith RD; Lieberman AP; Albin RL; Nawaz Z; El Hokayem J; Myers AJ Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Maiolica A; Borsotti D; Rappsilber J Self-made frits for nanoscale columns in proteomics. Proteomics 2005, 5, 3847–3850. [DOI] [PubMed] [Google Scholar]
- (25).Kelly RT; Page JS; Luo Q; Moore RJ; Orton DJ; Tang K; Smith RD Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal. Chem. 2006, 78, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vizcaíno JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz PA; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus HJ; Albar JP; Martinez-Bartolome S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu G; Wang LG; Han Y; He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang Y; Chen K; Sloan SA; Bennett ML; Scholze AR; O’Keeffe S; Phatnani HP; Guarnieri P; Caneda C; Ruderisch N; Deng S; Liddelow SA; Zhang C; Daneman R; Maniatis T; Barres BA; Wu JQ An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tsamardinos I; Aliferis CF; Statnikov AR In Algorithms for Large Scale Markov Blanket Discovery, FLAIRS Conference, 2003. [PMC free article] [PubMed] [Google Scholar]
- (31).Scutari M Learning Bayesian Networks with the bnlearn R Package. J. Stat. Software 2010, 35, 1–22. [Google Scholar]
- (32).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yu F; Teo GC; Kong AT; Haynes SE; Avtonomov DM; Geiszler DJ; Nesvizhskii AI Identification of modified peptides using localization-aware open search. Nat. Commun. 2020, 11, No. 4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li M; Gray W; Zhang H; Chung CH; Billheimer D; Yarbrough WG; Liebler DC; Shyr Y; Slebos RJ Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J. Proteome Res. 2010, 9, 4295–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Benjamini Y; Hochberg Y Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- (36).Storey JD; Tibshirani R Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hothorn T; Bretz F; Westfall P Simultaneous inference in general parametric models. Biom J. 2008, 50, 346–363. [DOI] [PubMed] [Google Scholar]
- (38).Creasy DM; Cottrell JS Unimod: Protein modifications for mass spectrometry. Proteomics 2004, 4, 1534–1536. [DOI] [PubMed] [Google Scholar]
- (39).Suttapitugsakul S; Xiao H; Smeekens J; Wu R Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol. BioSyst. 2017, 13, 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Šimić G; Babic Leko M; Wray S; Harrington C; Delalle I; Jovanov-Milosevic N; Bazadona D; Buee L; de Silva R; Di Giovanni G; Wischik C; Hof PR Tau Protein Hyper-phosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, No. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Toft M; Ross OA Copy number variation in Parkinson’s disease. Genome Med. 2010, 2, No. 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).La Cognata V; Morello G; D’Agata V; Cavallaro S Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum. Genet. 2017, 136, 13–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Soldner F; Stelzer Y; Shivalila CS; Abraham BJ; Latourelle JC; Barrasa MI; Goldmann J; Myers RH; Young RA; Jaenisch R Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ahlers KE; Chakravarti B; Fisher RA RGS6 as a Novel Therapeutic Target in CNS Diseases and Cancer. AAPS J. 2016, 18, 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bifsha P; Yang J; Fisher RA; Drouin J Rgs6 is required for adult maintenance of dopaminergic neurons in the ventral substantia nigra. PLoS Genet. 2014, 10, No. e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Garcia-Esparcia P; Hernandez-Ortega K; Koneti A; Gil L; Delgado-Morales R; Castano E; Carmona M; Ferrer I Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, No. 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chartier-Harlin MC; Dachsel JC; Vilarino-Guell C; Lincoln SJ; Lepretre F; Hulihan MM; Kachergus J; Milnerwood AJ; Tapia L; Song MS; Le Rhun E; Mutez E; Larvor L; Duflot A; Vanbesien-Mailliot C; Kreisler A; Ross OA; Nishioka K; Soto-Ortolaza AI; Cobb SA; Melrose HL; Behrouz B; Keeling BH; Bacon JA; Hentati E; Williams L; Yanagiya A; Sonenberg N; Lockhart PJ; Zubair AC; Uitti RJ; Aasly JO; Krygowska-Wajs A; Opala G; Wszolek ZK; Frigerio R; Maraganore DM; Gosal D; Lynch T; Hutchinson M; Bentivoglio AR; Valente EM; Nichols WC; Pankratz N; Foroud T; Gibson RA; Hentati F; Dickson DW; Destee A; Farrer MJ Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Martin I Decoding Parkinson’s Disease Pathogenesis: The Role of Deregulated mRNA Translation. J Parkinsons Dis. 2016, 6, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Martin I; Kim JW; Lee BD; Kang HC; Xu JC; Jia H; Stankowski J; Kim MS; Zhong J; Kumar M; Andrabi SA; Xiong Y; Dickson DW; Wszolek ZK; Pandey A; Dawson TM; Dawson VL Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Huang Y; Qiao F; Abagyan R; Hazard S; Tomlinson S Defining the CD59-C9 binding interaction. J. Biol. Chem. 2006, 281, 27398–27404. [DOI] [PubMed] [Google Scholar]
- (51).Wyss-Coray T; Rogers J Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor Perspect. Med. 2012, 2, No. a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Stahel PF; Flierl MA; Morgan BP; Persigehl I; Stoll C; Conrad C; Touban BM; Smith WR; Beauchamp K; Schmidt OI; Ertel W; Leinhase I Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J. Neuroinflammation 2009, 6, No. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Sulzer D; Alcalay RN; Garretti F; Cote L; Kanter E; Agin-Liebes J; Liong C; McMurtrey C; Hildebrand WH; Mao X; Dawson VL; Dawson TM; Oseroff C; Pham J; Sidney J; Dillon MB; Carpenter C; Weiskopf D; Phillips E; Mallal S; Peters B; Frazier A; Lindestam Arlehamn CS; Sette A T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Allen Reish HE; Standaert DG Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinson’s Dis. 2015, 5, 1−19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Moyle M; Napier MA; McLean JW Cloning and expression of a divergent integrin subunit beta 8. J. Biol. Chem. 1991, 266, 19650–19658. [PubMed] [Google Scholar]
- (56).Vallon M; Chang J; Zhang H; Kuo CJ Developmental and pathological angiogenesis in the central nervous system. Cell. Mol. Life Sci. 2014, 71, 3489–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhu J; Motejlek K; Wang D; Zang K; Schmidt A; Reichardt LF beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 2002, 129, 2891–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).McCarty JH; Lacy-Hulbert A; Charest A; Bronson RT; Crowley D; Housman D; Savill J; Roes J; Hynes RO Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 2005, 132, 165–176. [DOI] [PubMed] [Google Scholar]
- (59).Maucuer A; Le Caer JP; Manceau V; Sobel A Specific Ser-Pro phosphorylation by the RNA-recognition motif containing kinase KIS. Eur. J. Biochem. 2000, 267, 4456–4464. [DOI] [PubMed] [Google Scholar]
- (60).Dumaine A; Maucuer A; Barbet A; Manceau V; Deshommes J; Meary A; Szoke A; Schurhoff F; Llorca PM; Lancon C; Leboyer M; Jamain S Genetic and molecular exploration of UHMK1 in schizophrenic patients. Psychiatr. Genet. 2011, 21, 315–318. [DOI] [PubMed] [Google Scholar]
- (61).Pedraza N; Ortiz R; Cornado A; Llobet A; Aldea M; Gallego C KIS, a kinase associated with microtubule regulators, enhances translation of AMPA receptors and stimulates dendritic spine remodeling. J. Neurosci. 2014, 34, 13988–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Trinidad JC; Barkan DT; Gulledge BF; Thalhammer A; Sali A; Schoepfer R; Burlingame AL Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 2012, 11, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li MM; Nilsen A; Shi Y; Fusser M; Ding YH; Fu Y; Liu B; Niu Y; Wu YS; Huang CM; Olofsson M; Jin KX; Lv Y; Xu XZ; He C; Dong MQ; Rendtlew Danielsen JM; Klungland A; Yang YG ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nat. Commun. 2013, 4, No. 1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Shults CW Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Tompkins MM; Hill WD Contribution of somal Lewy bodies to neuronal death. Brain Res. 1997, 775, 24–29. [DOI] [PubMed] [Google Scholar]
- (66).Tanaka M; Kim YM; Lee G; Junn E; Iwatsubo T; Mouradian MM Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [DOI] [PubMed] [Google Scholar]
- (67).Olanow CW; Perl DP; DeMartino GN; McNaught KS Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 2004, 3, 496–503. [DOI] [PubMed] [Google Scholar]
- (68).Picconi B; Piccoli G; Calabresi P Synaptic Dysfunction in Parkinson’s Disease. In Synaptic Plasticity; Kreutz M; Sala C, Eds.; Advances in Experimental Medicine and Biology; Springer: Vienna, 2012; Vol. 970, pp 553–572. [DOI] [PubMed] [Google Scholar]
- (69).Selnes P; Stav AL; Johansen KK; Bjornerud A; Coello C; Auning E; Kalheim L; Almdahl IS; Hessen E; Zetterberg H; Blennow K; Aarsland D; Fladby T Impaired synaptic function is linked to cognition in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 700–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Phan JA; Stokholm K; Zareba-Paslawska J; Jakobsen S; Vang K; Gjedde A; Landau AM; Romero-Ramos M Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 2017, 7, No. 6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hasegawa T; Sugeno N; Kikuchi A; Baba T; Aoki M Membrane Trafficking Illuminates a Path to Parkinson’s Disease. Tohoku J. Exp. Med. 2017, 242, 63–76. [DOI] [PubMed] [Google Scholar]
- (72).Zhai S; Tanimura A; Graves SM; Shen W; Surmeier DJ Striatal synapses, circuits, and Parkinson’s disease. Curr. Opin. Neurobiol. 2018, 48, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Saez-Atienzar S; Singleton AB Parkinson disease and clathrin coat dynamics at synapses, why not? Mov. Disord. 2017, 32, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Villalba RM; Smith Y Loss and remodeling of striatal dendritic spines in Parkinson’s disease: from homeostasis to maladaptive plasticity? J. Neural Transm. 2018, 125, 431–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bae JR; Kim SH Synapses in neurodegenerative diseases. BMB Rep. 2017, 50, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tsui A; Isacson O Functions of the nigrostriatal dopaminergic synapse and the use of neurotransplantation in Parkinson’s disease. J. Neurol. 2011, 258, 1393–1405. [DOI] [PubMed] [Google Scholar]
- (77).Owen AD; Schapira AH; Jenner P; Marsden CD Oxidative stress and Parkinson’s disease. Ann. N. Y. Acad. Sci. 1996, 786, 217–223. [DOI] [PubMed] [Google Scholar]
- (78).Al Shahrani M; Heales S; Hargreaves I; Orford M Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, No. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Angeloni C; Malaguti M; Barbalace MC; Hrelia S Bioactivity of Olive Oil Phenols in Neuroprotection. Int. J. Mol. Sci. 2017, 18, No. 2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Liu Z; Zhou T; Ziegler AC; Dimitrion P; Zuo L Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longevity 2017, 2017, No. 2525967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Bose A; Beal MF Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [DOI] [PubMed] [Google Scholar]
- (82).Ciccone CD Free-radical toxicity and antioxidant medications in Parkinson’s disease. Phys. Ther. 1998, 78, 313–319. [DOI] [PubMed] [Google Scholar]
- (83).Friedman A; Galazka-Friedman J The current state of free radicals in Parkinson’s disease. Nigral iron as a trigger of oxidative stress. Adv. Neurol. 2001, 86, 137−142. [PubMed] [Google Scholar]
- (84).Roy S; Zhang B; Lee VM; Trojanowski JQ Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [DOI] [PubMed] [Google Scholar]
- (85).Perlson E; Maday S; Fu MM; Moughamian AJ; Holzbaur EL Retrograde axonal transport: pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Schlau M; Terheyden-Keighley D; Theis V; Mannherz HG; Theiss C VEGF Triggers the Activation of Cofilin and the Arp2/3 Complex within the Growth Cone. Int. J. Mol. Sci. 2018, 19, No. 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Yang Q; Zhang XF; Pollard TD; Forscher P Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J. Cell Biol. 2012, 197, 939–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Spence EF; Soderling SH Actin Out: Regulation of the Synaptic Cytoskeleton. J. Biol. Chem. 2015, 290, 28613–28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Kast DJ; Zajac AL; Holzbaur EL; Ostap EM; Dominguez R WHAMM Directs the Arp2/3 Complex to the ER for Autophagosome Biogenesis through an Actin Comet Tail Mechanism. Curr. Biol. 2015, 25, 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Kast DJ; Dominguez R The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Staff NP; Benarroch EE; Klein CJ Neuronal intracellular transport and neurodegenerative disease. Neurology 2011, 76, 1015–1020. [DOI] [PubMed] [Google Scholar]
- (92).Millecamps S; Julien JP Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [DOI] [PubMed] [Google Scholar]
- (93).Wang S; Bellen HJ The retromer complex in development and disease. Development 2015, 142, 2392–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Vilariño-Güell C; Wider C; Ross OA; Dachsel JC; Kachergus JM; Lincoln SJ; Soto-Ortolaza AI; Cobb SA; Wilhoite GJ; Bacon JA; Behrouz B; Melrose HL; Hentati E; Puschmann A; Evans DM; Conibear E; Wasserman WW; Aasly JO; Burkhard PR; Djaldetti R; Ghika J; Hentati F; Krygowska-Wajs A; Lynch T; Melamed E; Rajput A; Rajput AH; Solida A; Wu RM; Uitti RJ; Wszolek ZK; Vingerhoets F; Farrer MJ VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Zavodszky E; Seaman MN; Moreau K; Jimenez-Sanchez M; Breusegem SY; Harbour ME; Rubinsztein DC Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, No. 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Campellone KG; Welch MD A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Rottner K; Hanisch J; Campellone KG WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol. 2010, 20, 650–661. [DOI] [PubMed] [Google Scholar]
- (98).Chu CT A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum. Mol. Genet. 2010, 19, R28–R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Rott R; Szargel R; Haskin J; Bandopadhyay R; Lees AJ; Shani V; Engelender S alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18666–18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Alegre-Abarrategui J; Christian H; Lufino MM; Mutihac R; Venda LL; Ansorge O; Wade-Martins R LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Fujikake N; Shin M; Shimizu S Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, No. 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Spence EF; Kanak DJ; Carlson BR; Soderling SH The Arp2/3 Complex Is Essential for Distinct Stages of Spine Synapse Maturation, Including Synapse Unsilencing. J. Neurosci. 2016, 36, 9696–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Racz B; Weinberg RJ Organization of the Arp2/3 complex in hippocampal spines. J. Neurosci. 2008, 28, 5654–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Wegner AM; Nebhan CA; Hu L; Majumdar D; Meier KM; Weaver AM; Webb DJ N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J. Biol. Chem. 2008, 283, 15912–15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Binder JX; Pletscher-Frankild S; Tsafou K; Stolte C; O’Donoghue SI; Schneider R; Jensen LJ COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database 2014, 2014, No. bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Mahboubi H; Stochaj U Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 884–895. [DOI] [PubMed] [Google Scholar]
- (107).Ramaswami M; Taylor JP; Parker R Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727− 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Bai B; Hales CM; Chen PC; Gozal Y; Dammer EB; Fritz JJ; Wang X; Xia Q; Duong DM; Street C; Cantero G; Cheng D; Jones DR; Wu Z; Li Y; Diner I; Heilman CJ; Rees HD; Wu H; Lin L; Szulwach KE; Gearing M; Mufson EJ; Bennett DA; Montine TJ; Seyfried NT; Wingo TS; Sun YE; Jin P; Hanfelt J; Willcock DM; Levey A; Lah JJ; Peng J U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Kim HJ; Kim NC; Wang YD; Scarborough EA; Moore J; Diaz Z; MacLea KS; Freibaum B; Li S; Molliex A; Kanagaraj AP; Carter R; Boylan KB; Wojtas AM; Rademakers R; Pinkus JL; Greenberg SA; Trojanowski JQ; Traynor BJ; Smith BN; Topp S; Gkazi AS; Miller J; Shaw CE; Kottlors M; Kirschner J; Pestronk A; Li YR; Ford AF; Gitler AD; Benatar M; King OD; Kimonis VE; Ross ED; Weihl CC; Shorter J; Taylor JP Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Love JE; Hayden EJ; Rohn TT Alternative Splicing in Alzheimer’s Disease. J. Parkinson’s Dis. Alzheimer’s Dis. 2015, 2, No. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Sreedharan J; Blair IP; Tripathi VB; Hu X; Vance C; Rogelj B; Ackerley S; Durnall JC; Williams KL; Buratti E; Baralle F; de Belleroche J; Mitchell JD; Leigh PN; Al-Chalabi A; Miller CC; Nicholson G; Shaw CE TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Berson A; Barbash S; Shaltiel G; Goll Y; Hanin G; Greenberg DS; Ketzef M; Becker AJ; Friedman A; Soreq H Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EMBO Mol. Med. 2012, 4, 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Barmada SJ Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340−351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Xiong Y; Coradetti ST; Li X; Gritsenko MA; Clauss T; Petyuk V; Camp D; Smith R; Cate JHD; Yang F; Glass NL The proteome and phosphoproteome of Neurospora crassa in response to cellulose, sucrose and carbon starvation. Fungal Genet. Biol. 2014, 72, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Belzil VV; Gendron TF; Petrucelli L RNA-mediated toxicity in neurodegenerative disease. Mol. Cell. Neurosci. 2013, 56, 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).King OD; Gitler AD; Shorter J The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Maziuk B; Ballance HI; Wolozin B Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, No. 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Chung CY; Khurana V; Yi S; Sahni N; Loh KH; Auluck PK; Baru V; Udeshi ND; Freyzon Y; Carr SA; Hill DE; Vidal M; Ting AY; Lindquist S In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst. 2017, 4, 242.e4−250.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Lewandowski SA; Nilsson I; Fredriksson L; Lonnerberg P; Muhl L; Zeitelhofer M; Adzemovic MZ; Nichterwitz S; Lawrence DA; Hedlund E; Eriksson U Presymptomatic activation of the PDGF-CC pathway accelerates onset of ALS neurodegeneration. Acta Neuropathol. 2016, 131, 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Luissint AC; Artus C; Glacial F; Ganeshamoorthy K; Couraud PO Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, No. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Miyazaki K; Ohta Y; Nagai M; Morimoto N; Kurata T; Takehisa Y; Ikeda Y; Matsuura T; Abe K Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [DOI] [PubMed] [Google Scholar]
- (122).Lepelletier F-X; Mann DM; Robinson AC; Pinteaux E; Boutin H Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2015, 167. [DOI] [PubMed] [Google Scholar]
- (123).Farkas E; De Jong GI; de Vos RA; Jansen Steur EN; Luiten PG Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000, 100, 395–402. [DOI] [PubMed] [Google Scholar]
- (124).Mancardi GL; Perdelli F; Rivano C; Leonardi A; Bugiani O Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. 1980, 49, 79–83. [DOI] [PubMed] [Google Scholar]
- (125).Cavanna F; Barichella M; Cassani E; Pezzoli G Increased levels of endothelial progenitor cells in Parkinson’s disease. Parkinsonism Relat. Disord. 2011, 17, 651–652. [DOI] [PubMed] [Google Scholar]
- (126).Andjelkovic AV; Pachter JS Central nervous system endothelium in neuroinflammatory, neuroinfectious, and neurodegenerative disease. J. Neurosci. Res. 1998, 51, 423–430. [DOI] [PubMed] [Google Scholar]
- (127).Grammas P Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, No. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Jansson D; Rustenhoven J; Feng S; Hurley D; Oldfield RL; Bergin PS; Mee EW; Faull RL; Dragunow M A role for human brain pericytes in neuroinflammation. J. Neuroinflammation 2014, 11, No. 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.