

Abstract
Background:
Hemizygous mutations in GRIA3 encoding the GluA3 subunit of the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor are known to be associated with neurodevelopmental disorders, including intellectual disability, hypotonia, an autism spectrum disorder, sleep disturbances, and epilepsy in males.
Objective:
To describe a new and consistent phenotype in 4 affected male patients associated with an undescribed deleterious variant in GRIA3.
Methods:
We evaluated a large French family in which segregate a singular phenotype according to an apparent X-linked mode of inheritance. Molecular analyses using next generation sequencing and in vitro functional studies using 2-electrode voltage clamp recordings on Xenopus laevis oocytes and a β-lactamase reporter assay in transfected human embryonic kidney (HEK293) cells were performed.
Results:
In addition to mild intellectual disability and dysarthria, affected patients presented a tightly consistent early-onset movement disorder combining an exaggerated startle reflex with generalized chorea and multifocal myoclonus. The unreported GRIA3 missense variant c.2477G > A; p.(Gly826Asp) affecting the fourth transmembrane domain of the protein was identified in index patients and their unaffected mothers. Functional studies revealed that variant receptors show decreased current response evoked by agonist (ie, kainic acid and glutamate) and reduced expression on the cell surface in favor of pathogenicity by a loss-of-function mechanism.
Conclusions:
Taken together, our results suggest that apart from known GRIA3-related disorders, an undescribed mutation-specific singular movement disorder does exist. We thus advocate considering GRIA3 mutations in the differential diagnosis of hyperekplexia and generalized chorea with myoclonus.
Keywords: AMPA receptor, chorea, glutamate receptor, GRIA3, myoclonus
Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), that are ligand-gated ionotropic glutamate receptors, mediate the fast component of excitatory synaptic transmission in the central nervous system. They are essential actors during brain development and subsequently play a major role in activity dependent synaptic plasticity.1 They result from tetrameric assemblies of GluA1-GluA4 subunits encoded by GRIA1 to GRIA4 genes.2,3 AMPAR interact with scaffolding and multiple other proteins and are localized to the postsynaptic density. Each subunit is composed of an extracellular amino-terminal domain, an agonist binding domain with 2 segments, 4 transmembrane domains, and a cytoplasmic C-terminal tail.1
A large number of genetic variations, including missense, frameshift, fusion transcript, partial tandem duplication, and interstitial deletion of chromosome scattered across all AMPAR subunits, have been identified in patients with autism spectrum disorders, epilepsy, developmental delay, and intellectual disability (ID).4–9 GRIA3 is located on the X chromosome and encodes the GluA3 subunit of AMPAR. It is highly expressed in the central nervous system. GRIA3 hemizygous mutations, either genomic rearrangements or deleterious missense point mutations, have been associated in male patients to a phenotype that includes moderate to severe ID, hypotonia, poor muscle bulk, an autism spectrum disorder, sleep disturbances, and epilepsy of various degree.10–18
In this article, we describe a singular and atypical phenotype in 4 related male patients segregating the unreported GRIA3 missense variant NM_000828.4:c.2477G > A; p.(Gly826Asp) and study its functional influence on AMPAR.
Materials and Methods
Participants
Written informed consent was obtained from all participants. All in vitro studies were conducted according to the guidelines of Emory University.
Methods
ID Gene Panel (ID286 Panel)
Targeted exons and 6 base pairs (bp) of flanking intronic sequence of 286 ID genes were analyzed by next-generation sequencing on a Miseq Platform (Illumina, San Diego, CA) and analyzed with the use of the Sophia DDM (SOPHiA GENETICS, Saint Sulpice, Switzerland) analytical platform.
Whole-Exome Sequencing
An exome capture kit (SeqCap EZ Exome probes; Roche-NimbleGen, Madison, WI) was used to target all exons. Exons capture was followed by massive parallel 150 bp paired-end sequencing (Illumina). Read mapping and variant calling were performed following standard bioinformatics procedures.19 Filtering and prioritization of the variants were conducted using an in-house interactive Paris-Descartes bioinformatics platform pipeline based on the Ensembl database (release 67).20 Variants were filtered according to their frequency (1%) against the single nucleotide polymorphism database (dbSNP) (https://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genome Project (http://www.internationalgenome.org/), ExAC (http://exac.broadinstitute.org/), and GnomAD (http://gnomad.broadinstitute.org/about) databases. In silico prediction of variant pathogenicity was performed using SIFT, Poly-Phen, and Mutation Taster. Cadd score was also integrated to the prioritization criteria, and a cutoff of 20 was used to determine whether a variant was likely deleterious.
Sanger Sequencing
Specific primers were used for polymerase chain reaction amplification of exon 15 of GRIA3. Polymerase chain reaction products were purified with NucleoFast 96 (Macherey-Nagel, Düren, Germany) polymerase chain reaction and sequenced on an automated capillary sequencer (ABI 3130xl; Applied Biosystems, Foster City, CA) with the dye terminator method.
Molecular Biology and Voltage-Clamp Current Recordings From Xenopus Oocytes
Variants were introduced into human cDNAs encoding human GluA3 receptors (NM_007325) in pCI Neo vector by the QuikChange protocol.21 cRNA were synthesized from cDNA and injected into Xenopus laevis oocytes (defolliculated stages V–VI),21 prepared from commercially available ovaries (Xenopus One Inc, Dexter, MI).22 AMPAR-mediated currents were recorded under two-electrode voltage-clamp (TEVC) from Xenopus laevis oocytes.21 The recording solution contained 90 mM NaCl, 1 mM KCl, 10 mM HEPES, 0.5 mM BaCl2, 0.01 mM EDTA (pH 7.4). Unless stated otherwise, the membrane potential was held at −40 mV and temperature was 23°C. Glutamate concentration-response curves were recorded by varying the agonist concentrations. The current response amplitudes at each concentration were fitted by
where EC50 is the concentration that produces a half maximal response, [agonist] is the concentration of agonist, and N is the Hill slope.
Assessment of Surface Expression in Mammalian Cells
Cell surface and total protein levels were measured using a β-lactamase reporter assay in transfected HEK293 (cells in 96-well plates expressing AMPAR tagged extracellularly with β-lactamase (β-lactamase–GluA3, hereafter b-lac-GluA3) using Fugene6 (Promega, Madison, WI) to evaluate the influence of the variant on cell surface expression.23 AMPAR competitive antagonists (100 μM 7-CKA, 7-Chlorokynurenic acid) were included in Dulbecco’s modified Eagle’s medium to block cell death mediated by AMPAR over activation. The background absorbance was determined in wells that only received Fugene6 treatment for surface β-lactamase activity. Eight wells were transfected for each test condition, and the surface and total β-lactamase activity were measured. Surface and total activities were measured in 4 wells each at 24 hours after transfection. To determine total activity, the cells were lysed in 50 μL dH2O for 30 minutes prior to the addition of 200 μM nitrocefin (50 μL). The absorbance was read at 468 nm every minute for 30 minutes at 30°C using a microplate reader. The rate of increase in absorbance was determined from the slope of a linear fit of the data.
Results
Clinical Manifestations
Clinical data are summarized in Table 1.
TABLE 1.
Clinical data
| Clinical Data | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Pregnancy and delivery | Normal | Monochorial monoamniotic twin pregnancy of late discovery, C-section at 36 GW | Normal | |
| Ancestry | France | France | France | |
| Age at walking | 15 m | 13 m | 13 m | 18 m |
| Language delay and dysarthria | + | + | + | + |
| Behavioral disturbances | + Oppositional disorder Psychomotor instability, attention deficit, anxiety |
− | − | − |
| Learning difficulties | + Special education from 8 y | + Special education from 6 y | + Special education from 6 y | + Special education from 6 y works now in a sheltered employment |
| Age at last examination | 14 y | 15 y | 15 y | 24 y |
| Growth parameters | H: 155.5 cm (M) W: 47 kg (M) OFC: 54 cm (−0.5 SD) |
H: 176 cm (+1.2 SD) W: 74 kg (+2 SD ) OFC: 56.8 cm (+1.5 SD) |
H: 176 cm (+1.2 SD) W: 77 kg (+2.5 SD) OFC: 57 cm (+1.5 SD) |
H: 176.5 cm (+0.2 SD) W: 67 kg (+0.2 SD) OFC: 56.5 cm (−0.5 SD) |
| Facial dysmorphism | − | − | − | − |
| Intellectual disability | +, Mild, cannot read or write | +, Mild, cannot read or write | +, Mild, cannot read or write | +, Mild, can read and write |
| Exaggerated startle reflex (AO) |
+ (Birth) |
+ (Birth) |
+ (Birth) |
+ (Birth) |
| Generalized chorea (AO) |
+ (<3 y) |
+ (<3 y) |
+ (<3 y) |
+ (Birth) |
| Multifocal myoclonus (AO) | + (<3 y) |
+ (<3 y) |
+ (<3 y) |
+ (Birth) |
| Eye movements | Smooth pursuit and normal saccade tests | Smooth pursuit and normal saccade tests | Smooth pursuit and normal saccade tests | Smooth pursuit and normal saccade tests |
| Seizures | No | No | No | No |
| Gait disorder | No | No | No | No |
| Postural instability | No | No | No | No |
| Deep tendon reflexes | Brisk | Brisk | Brisk | Brisk |
| Hypotonia | No | No | No | No |
| Spasticity | No | No | No | No |
| Muscle weakness | No | No | No | No |
| Peripheral neuropathy | No | No | No | No |
| Hearing impairment | No | No | No | No |
| Visual impairment | No | Mild hypermetropia | Mild hypermetropia | Squint (operated at 8 y) |
| Other | Bilateral hydrocele (operated at 2 y) | - | - | - |
| CT brain scan | NA | NA | NA | Normal |
| Brain MRI | Normal | Normal | Normal | NA |
| EEG | Normal | Normal | Normal | Normal |
| Eye fundus | Normal | NA | NA | Normal |
| Metabolic screening | Normal | Normal | Normal | Normal |
| CSF concentration of neurotransmitters | Normal | Normal | Normal | NA |
| Ongoing treatment | No | Levetiracetam, clonazepam, tetrabenazine (mild improvement) | Levetiracetam, clonazepam, tetrabenazine (mild improvement) | No (sodium valproate treatment was tried at birth for several years without success) |
AO, age of onset; CT, computed tomography; MRI, magnetic resonance imaging; EEG, electroencephalography; CSF, cerebrospinal fluid; GW, gestation weeks; m, months; y, years; NA, not available; H, height; W, weight; OFC, occipito-frontal circumference; M, mean; SD, standard deviation.
Main clinical features consistently found in affected patients are indicated in bold.
Patients 1, 2, 3, and 4 were, respectively, evaluated at ages 14, 15, 15, and 24 years. They had normal growth parameters including occipito-frontal circumference. Language delay, dysarthria, learning difficulties requiring a specialized education, and early-onset movement disorders were observed in all. Neurological assessment at the time of diagnosis was similar in the 4 affected males. An exaggerated startle reflex (ie, flexor spasms of the trunk) to unexpected auditory stimuli, which started from birth on was a paramount feature. The startle response did not habituate. During the first years of life, they developed multifocal myoclonus of the face, trunk, and the 4 limbs combined with generalized chorea. Myoclonus were present at rest, on keeping posture, and to a lesser extent during action. They were also elicited by auditory stimuli. All patients had generalized chorea with motor impersistence. None of them had neither ataxia nor dystonia. Deep tendon reflexes were brisk without Babinski sign. An inexhaustible eye blinking was also observed. Muscular tone was normal without spasticity. Walking was unaltered. Patients 2 and 3 are mountain bikers (Videos 1, 2, 3, 4). Fine motor skills were impaired resulting in clumsiness and dysgraphia. During their medical work-ups, the diagnosis of hyperekplexia was suggested in patients 1 and 4, whereas hereditary benign chorea has been evoked in patients 2 and 3. According to their relatives and the old traced-back medical records, exaggerated startle reflex, chorea, and myoclonus have had a stable course during the years, and their topographical distribution did not change. None of the patients experienced any epileptic manifestation.
Patient 5 was not examined. He was described as having dysarthria, ID, and movement disorders similar to those of his maternal cousins. He is now 38 years old and works in a sheltered employment.
Patients 2 and 3 were placed under levetiracetam, clonazepam, and tetrabenazine therapy with mild improvement of myoclonic jerks and chorea. Patient 4 was on sodium valproate for several years without any improvement.
Genetic Studies
Array-comparative genomic hybridization did not show any pathogenic copy number variant (in patients 1, 2, 3). Sequencing and multiplex ligation-dependent probe amplification analysis of GLRA1, GLRB, and SLC6A5 did not identify any deleterious variant in patient 1. Sequencing of NKX2 did not show any abnormality in patients 2 and 3. Targeted sequencing for 127 genes associated with movement disorders did not identify any pathogenic variations in patients 2 and 3.
Using 2 different screening molecular approaches (ID gene panel in patient 1 and whole-exome sequencing in patients 2 and 3), the missense GRIA3 variant chrX: 122616687 G > A, ENST00000371256_X; c.2477G > A; p.Gly826Asp was identified in all affected male patients. Sanger sequencing in the 4 probands and their 3 unaffected mothers confirmed the presence of the variant in all (Fig. 1). No other pathogenic or likely pathogenic variation was identified by whole-exome sequencing or with the ID286 panel. The nucleotide substitution affects a conserved amino-acid position of the protein. It is predicted pathogenic by all in silico tools tested. It is not found in gnomAD (https://gnomad.broadinstitute.org/) with a good coverage of the nucleotide position.
FIG. 1.
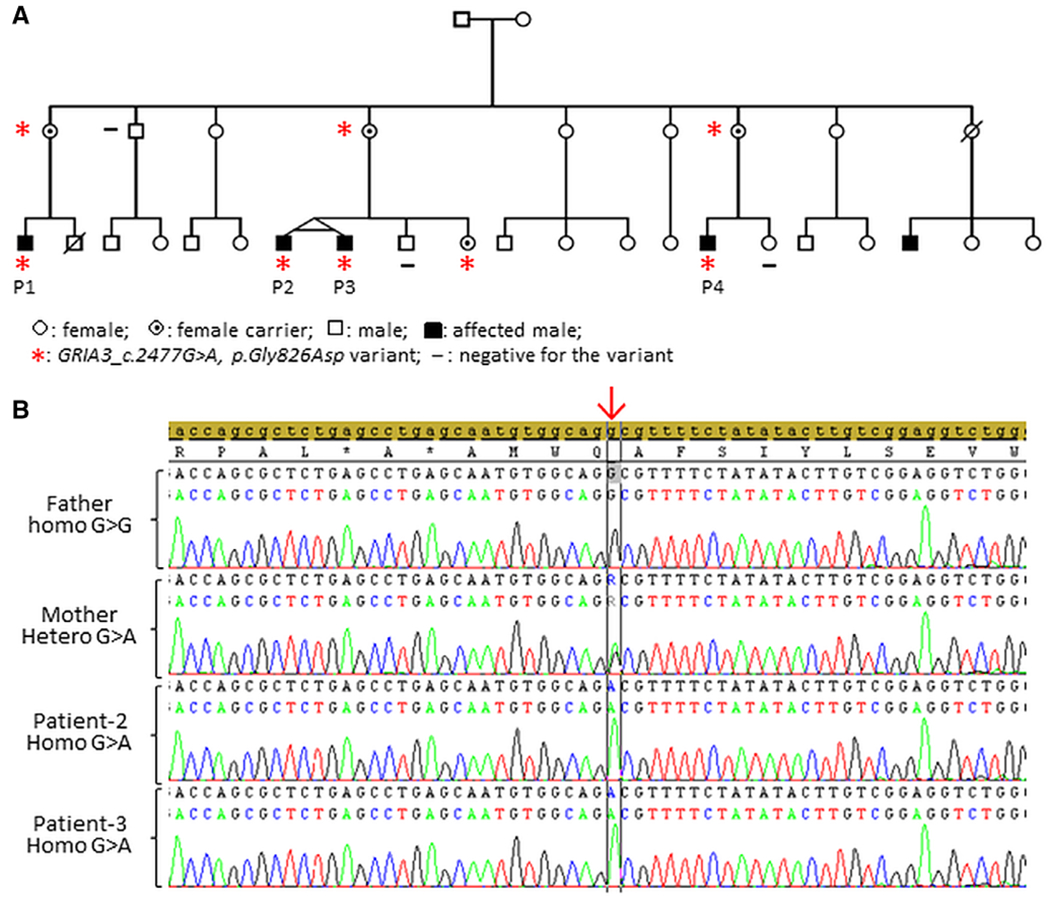
Identification of a GRIA3 missense variant in 4 affected male patients. A: Family pedigree showing the inheritance of the GRIA3 variant. B: GRIA3 electropherograms confirming the c.2477G > A hemizygous variant in index patients, absent in the father and present at the heterozygous state in the carrier mother. P1, patient 1; P2, patient 2; P3, patient 3; P4, patient 4. [Color figure can be viewed at wileyonlinelibrary.com]
Electrophysiological Study
Combined electroencephalography/electromyography recording in patient 1 showed a myoclonic pattern with repetitive spontaneous bursts of muscular activity and isolated synchronous myoclonic burst of 55 milliseconds elicited by auditory stimuli. Moreover, electroencephalography recording showed a physiological posterior alpha-band activity without any epileptiform discharges notably during intermittent photic stimulation (supplemental online material). It should be noted that we were unable to perform jerk locked back averaging and are accordingly unable to define the origin (cortical vs. noncortical) of the myoclonus.
Neuropsychological Assessment
A detailed assessment of patients 1, 2, and 3 was performed. Cognitive abilities were evaluated according to the Wechsler Intelligence Scale for Children (5th edition). Scores indicated a heterogeneous profile. The discrepancy renders calculation and interpretation of full-scale IQ score inappropriate.
The results for the indexes for patient 1 were Verbal Comprehension Index (VCI) = 68, Visual Spatial Index (VSI) = 84, Fluid Reasoning Index (FRI) = 85, Working Memory Index (WMI) = 62, and Processing Speed Index (PSI) = 66. Difficulties in language affecting both receptive and expressive language were particularly striking with a low vocabulary level. Working memory and fine motor skills were also impaired. Additional subtests from Children Memory Scale and NEPSY-II indicated performances in the range of the general population of his age for memory, visuo-spatial, and visuoconstructive abilities. However, attentional processes and executive functions were delayed. Adaptive skills (VINELAND-II) fell in the average to very low range for 3 domains (communication, day living skills, socialization).
The assessments of patients 2 and 3 revealed mild ID. The results for patient 2 were VCI = 59, VSI = 75, FRI= 69, WMI = 65, and PSI = 66. The results of patient 3 showed the same intellectual profile: VCI = 50, VSI = 78, FRI = 61, WMI = 62, and PSI = 75. A language disorder mainly affecting word production and orofacial praxis was prominent in both patients. The assessment of attentional processes and executive functions revealed differences concerning inhibition process and sustained attention. Scores of adaptive skills were very low for the 3 main domains.
In Vitro Functional Evaluation
The variant is located in transmembrane domain M4 (Fig. 2A,B), a critical region that controls channel gating.1,21,24,25 The glycine residue in position 826 is conserved not only across the vertebral species but also through other GRIA/GluA genes/subunits (Fig. 2A), suggesting a potential important role in channel function. To investigate the influence of the variant on AMPAR function, we first assessed the effects of the variant on agonist potency by TEVC current recordings from Xenopus oocytes expressing wild-type (WT) GluA3 or the GluA3-G826D variant. The EC50 of kainic acid for the variant cannot be determined because of its small current response to a maximal concentration (3000 μM) of kainic acid (11 ± 2.0 nA compared with 126 ± 18 nA of WT at a holding potential of −40 mV, P < 0.001, unpaired Student t test compared to WT; Fig. 3A,B; Table 2). We subsequently evaluated the glutamate concentration-effect relationship on human WT or GluA3 variant non-desensitizing L513Y constructs (hereafter GluA3-LY and GluA3-LY-G826D). The receptors hosting the G826D variant showed a comparable glutamate EC50 values (32 μM for GluA3-LY-G826D vs. 30 μM for GluA3-LY, P = 0.45; Table 2). However, the amplitude of the current responses to 3000 μM glutamate for the variant was significantly smaller than the control (574 ± 94 nA for GluA3-LY-G826D vs. 3020 ± 379 nA for GluA3-LY, P < 0.01; Fig. 3B; Table 2).
FIG. 2.
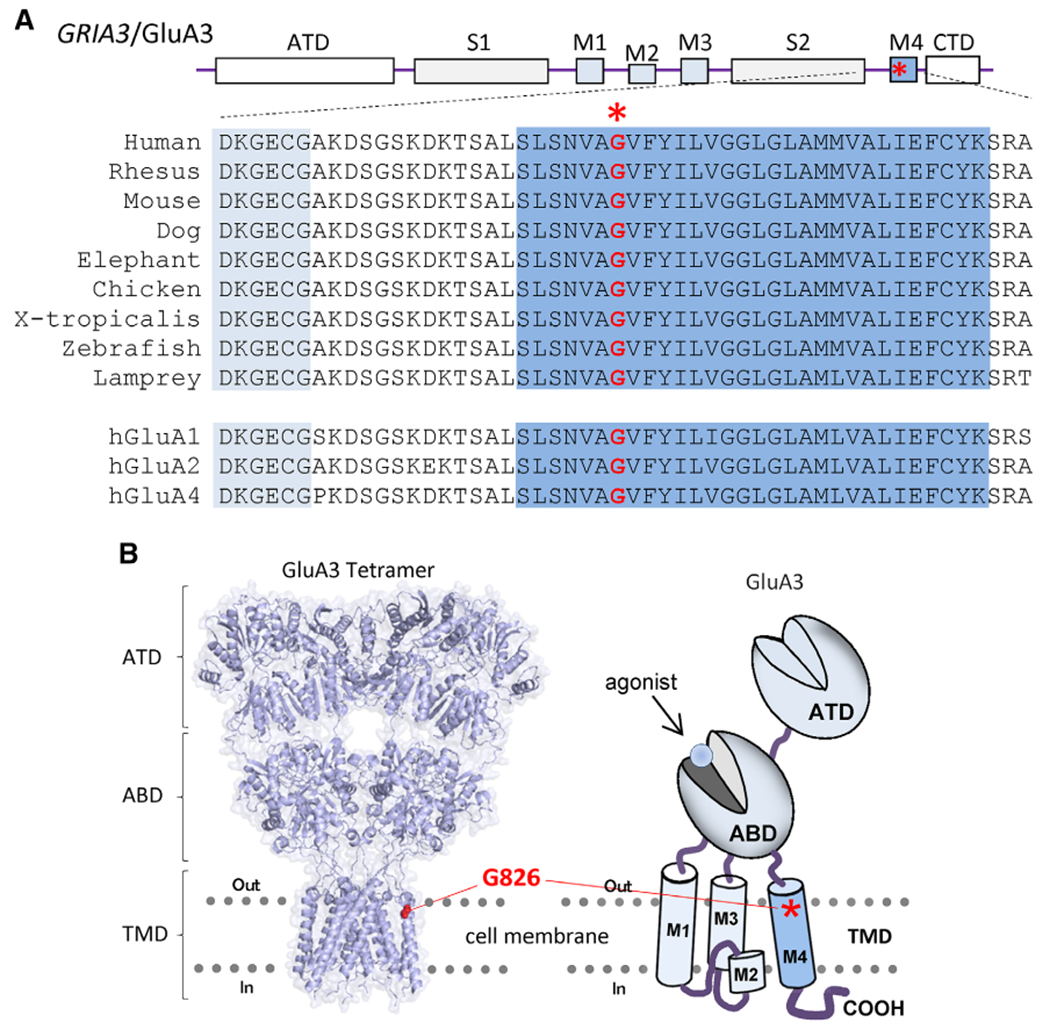
GRIA3_c.2477G > A, p.Gly826Asp variant information. A: Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors architecture and sequence alignment. B: Tetrameric AMPA receptor model built from GluA2 structure (5WEO 32 [left panel] and cartoon for a single GluA3 subunit [right panel]) indicate the location of the residue hosting the variant. ABD, agonist binding domain; ATD, amino-terminal domain; CTD, cytoplasmic C-terminal tail; TMD, 3 transmembrane domain (M1, M3, and M4) and 1 re-entrant pore-forming loop M2; S1, segment 1; S2, segment 2; TMD, 4 transmembrane domain. [Color figure can be viewed at wileyonlinelibrary.com]
FIG. 3.
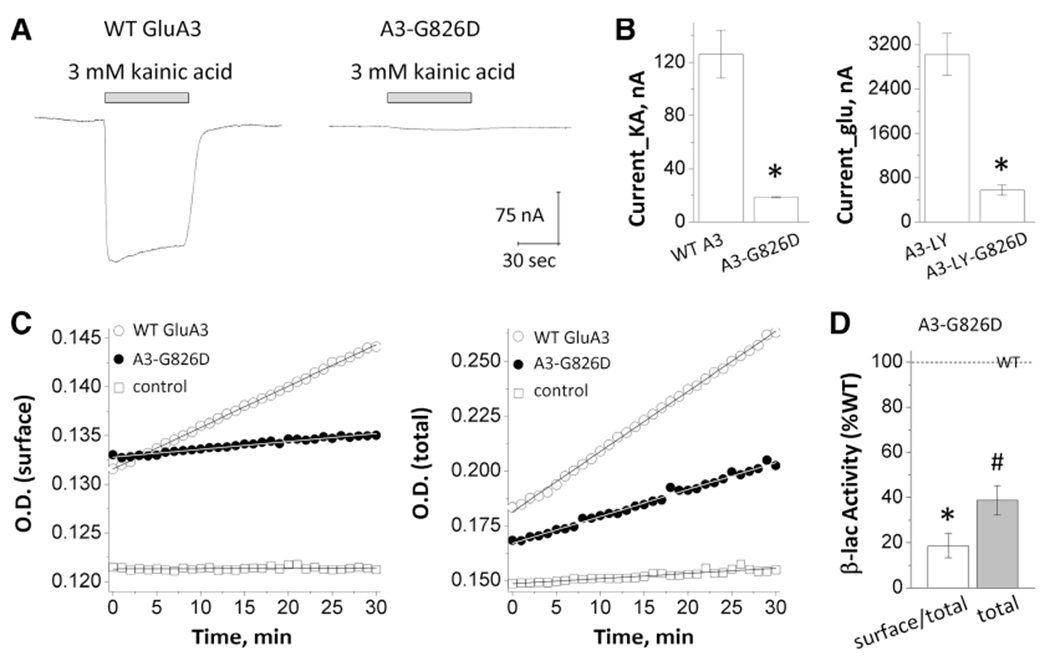
GRIA3_p.Gly826Asp variant influences current response and receptor expression on cell. A: Representative traces of current response to 3000 mM kainic acid. B: Summary of current amplitudes. C: Representative plots of nitrocefin absorbance. D: The slopes of O.D. versus time course. O.D., optical density; WT, wild type.
TABLE 2.
Summary of pharmacological data and receptor surface expression
| Parameters | WT GluA3 | GluA3-G826D |
|---|---|---|
| Current amplitude_KA, nAa b | 126 ± 18 (22) | 11 ± 2.0 (23)e |
| Current amplitude_glutamate, nAa c | 3020 ± 379 (23) | 574 ± 94 (21)e f |
| EC50glutamate, μMa c | 30 ± 2.8 (23) | 32 ± 2.4 (21)f |
| Surface/total ratio (β-lac)d | 1.0 ± 0.11 (4) | 0.19 ± 0.05 (4)e |
| Total level (β-lac)d | 1.0 (4) | 0.39 ± 0.06 (4)e |
Data were generated by 2 electrode voltage clamp recordings from Xenopus oocytes with −40 mV of holding potential and are expressed as mean ± standard error of the mean (n).
Current responses to 3000 μM KA were generated from human WT or GluA3 variant constructs.
Glutamate concentration response curves and current responses to 3000 μM glutamate were generated from human WT or GluA3 variant non-desensitizing L-Y construct.
Data were generated from transiently transfected HEK293 cells.
P < 0.01 unpaired Student t test.
Controlled family-wise error rate was adjusted.
WT, wild type; KA, kainic acid; EC50, half-maximally effective concentration.
To test if the variant influenced AMPAR surface expression, the cell surface protein level and total protein level were determined using a reporter assay in which β-lactamase was fused to the extracellular amino-terminal domain of WT or the GluA3 variant (β-lac–GluA3 or β-lac–GluA3–G826D). The surface receptor expression was measured by the β-lactamase cleavage of the cell-impermeable chromogenic substrate nitrocefin in the extracellular solution.23,26,27 AMPARs that contained the GluA3-G826D variant showed a significant reduction of surface-to-total protein level (19 ± 5% of WT, P < 0.001) as well as the total protein level (39 ± 6% of WT, P < 0.001; Fig. 3C,D; Table 2). These data suggest that the GluA3-G826D variant showed a strong reduction of receptor expression on the cell surface.
Taken together, the combined effects of decreased current responses and reduced receptor cell surface expression suggest that the GluA3-G826D variant is a loss-of-function mutation.
Discussion
The distinctive phenotype of the 4 affected males hereby described delineate a novel clinical syndrome with an X-linked pattern of inheritance. The main features include mild ID, dysarthria, and a tightly consistent movement disorder starting from birth on and combining an exaggerated startle reflex with multifocal myoclonus and generalized chorea. Although the co-occurrence of language disorders and ID is frequent, the association of ID with early-onset movement disorders is an uncommon combination, apart from brain damage resulting from fetal or neonatal suffering.
Exaggerated startle reflex is reminiscing of hyperekplexia, a rare genetic condition characterized by regressive generalized congenital stiffness, excessive startle reflex, and mostly the absence of cognitive impairment. This sequence differs significantly from our patients. Pathogenic variants in GLRA1 encoding the glycine receptor subunit α1 are known to cause hyperekplexia, which is less frequently associated with deleterious variants in SLC6A5, GLRB, GPHN, and ARHGEF9.28 Sanger sequencing of GLRA1, GLRB, and SLC6A5 was indeed first performed in patient 1 and did not disclose any mutation. Movement disorders of our patients were also reminiscent of other neurological diseases combining chorea with myoclonus, such as the so-called NKX2-1-related “benign” hereditary chorea,29 ADCY5-related dyskinesia,30 or juvenile Huntington’s disease. Importantly, exaggerated startle reflex does not belong to the spectrum of these diseases. Finally, the combination of movement disorders tightly superimposable in the 4 index cases is unique and makes the phenotype recognizable. Our clinical, molecular, and functional studies were able to correlate this new syndrome with an undescribed GRIA3 mutation allowing one to extend the clinical spectrum of GRIA3-related conditions.
According to the nomenclature recommended by the Movement Disorder Society,31 we suggest to name the genetically determined movement disorder we described MYC/CHOR-GRIA3 and to include it in the second group of the classification in addition to the 9 genes already involved in combined myoclonus syndromes.31 In our cases, accompanying signs were mild ID and dysarthria without ataxia, dystonia, epilepsy, or cognitive decline.
Our functional studies indicated almost no current response to kainic acid detected and a significant reduction of receptor expression on cell surface, suggesting that the missense GluA3-G826D variant, located in a highly conserved residue in a key functional domain of the protein (TM4), acts through a loss-of-function mechanism. Interestingly, another missense variant (G833R) located in the TM4 domain of GluA3 reported by Wu and colleagues12 in 2007 was also associated with moderate ID and a movement disorder resembling myoclonic jerks in the 2 affected male relatives described. Expression studies demonstrated that this variant results in a substantially reduced amount of iGluR3 attributed to the rapid degradation of an abnormally folded receptor protein involving proteasome. Because the only reported patient carrying a proven loss-of-function mutation in TM4 was also experiencing myoclonus, it is tempting to consider that the movement disorder could be the result of a locus-specific effect. With a very limited number of GRIA3 variants described and functional studies performed, it is yet not possible to connect the specific phenotype to the TM4 location of the variant.
Taken together, our results suggest that apart from known GRIA3-related disorders, a new syndrome including ID, dysarthria, and a specific complex movement disorder is associated with the missense loss-of-function GluA3-G826D variant. We thus advocate considering GRIA3 mutations in the differential diagnosis of hyperekplexia and generalized chorea with myoclonus, especially in cases where dysarthria and ID are also present. Although the current observation delineates convincingly a novel clinical entity and its molecular basis, further reports are warranted to confirm evidence for a GluA3-TM4–related phenotype.
Supplementary Material
Acknowledgments:
The authors thank Jing Zhang, Gil Shaulsky, Sukhan Kim, and Phoung Le for their outstanding technical assistance.
Funding agencies:
During this work, H.Y. was supported by the National Institutes of Health–the Eunice Kennedy Shriver National Institute of Child Health & Human Development (Grant R01HD082373) and by the University Research Committee (00085889) from Emory University. S.F.T. was supported by the National Institutes of Health–National Institute of Neurologic Disorders and Stroke (Grants R01NS036654, R01NS065371, and R24NS092989). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. T.W. received funding from the Revue Neurologique to perform genetic analysis. Exome sequencing analysis was financed by a grant from the France Parkinson association.
Relevant conflicts of interests/financial disclosures:
H.Y. is principal investigator on a research grant from Sage Therapeutics to Emory University School of Medicine. S.F.T. is principal investigator on a research grant from Allergan to Emory University School of Medicine, is a member of the SAB for Sage Therapeutics, is cofounder of NeurOp Inc., and receives royalties for software. S.F.T. is coinventor on Emory-owned intellectual property that includes allosteric modulators of NMDA receptor function. T.W. received research grants from the APTES Association, The Planiol’s Foundation, and the Revue Neurologique.
Footnotes
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010;62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hollmann M, O’shea-Greenfield A, Rogers SW, Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature 1989;342:643–648. [DOI] [PubMed] [Google Scholar]
- 3.Keinänen K, Wisden W, Sommer B, et al. A family of AMPA-selective glutamate receptors. Science 1990;249:556–560. [DOI] [PubMed] [Google Scholar]
- 4.Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hackmann K, Matko S, Gerlach E-M, et al. Partial deletion of GLRB and GRIA2 in a patient with intellectual disability. Eur J Hum Genet 2013;21:112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poot M Towards identification of individual etiologies by resolving genomic and biological conundrums in patients with autism spectrum disorders. Mol Syndromol 2013;4:213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Z, Yu L, Geng J, Wang J, Jin X, Huang H. A novel 47.2 Mb duplication on chromosomal bands Xq21.1-25 associated with mental retardation. Gene 2015;567:98–102. [DOI] [PubMed] [Google Scholar]
- 8.Martin S, Chamberlin A, Shinde DN, et al. De novo variants in GRIA4 lead to intellectual disability with or without seizures and gait abnormalities. Am J Hum Genet 2017;101:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strehlow V, Heyne HO, Vlaskamp DRM, et al. GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain J Neurol 2019;142:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gécz J, Barnett S, Liu J, et al. Characterization of the human glutamate receptor subunit 3 gene (GRIA3), a candidate for bipolar disorder and nonspecific X-linked mental retardation. Genomics 1999;62:356–368. [DOI] [PubMed] [Google Scholar]
- 11.Chiyonobu T, Hayashi S, Kobayashi K, et al. Partial tandem duplication of GRIA3 in a male with mental retardation. Am J Med Genet A 2007;143A:1448–1455. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Arai AC, Rumbaugh G, et al. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci U S A 2007;104:18163–18168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnet C, Leheup B, Béri M, Philippe C, Grégoire M-J, Jonveaux P. Aberrant GRIA3 transcripts with multi-exon duplications in a family with X-linked mental retardation. Am J Med Genet A 2009;149A:1280–1289. [DOI] [PubMed] [Google Scholar]
- 14.Bonnet C, Masurel-Paulet A, Khan AA, et al. Exploring the potential role of disease-causing mutation in a gene desert: duplication of noncoding elements 5’ of GRIA3 is associated with GRIA3 silencing and X-linked intellectual disability. Hum Mutat 2012;33:355–358. [DOI] [PubMed] [Google Scholar]
- 15.Philippe A, Malan V, Jacquemont M-L, et al. Xq25 duplications encompassing GRIA3 and STAG2 genes in two families convey recognizable X-linked intellectual disability with distinctive facial appearance. Am J Med Genet A 2013;161A:1370–1375. [DOI] [PubMed] [Google Scholar]
- 16.Philips AK, Sirén A, Avela K, et al. X-exome sequencing in Finnish families with intellectual disability—four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis 2014;9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies B, Brown LA, Cais O, et al. A point mutation in the ion conduction pore of AMPA receptor GRIA3 causes dramatically perturbed sleep patterns as well as intellectual disability. Hum Mol Genet 2017;26:3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyu Y, Yang Y, Liu Y, Gai Z. Analysis of a patient with X-linked mental retardation by next generation sequencing. Zhonghua Yi Xue Yi Chuan Xue Za Zhi Zhonghua Yixue Yichuanxue Zazhi Chin J Med Genet 2018;35:257–260. [DOI] [PubMed] [Google Scholar]
- 19.Broix L, Jagline H, Ivanova E, et al. Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat Genet 2016;48:1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flicek P, Amode MR, Barrell D, et al. Ensembl 2012. Nucleic Acids Res 2012;40:D84–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W, Tankovic A, Burger PB, Kusumoto H, Traynelis SF, Yuan H. Functional evaluation of a de novo GRIN2A mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol Pharmacol 2017;91:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang Wei W, Kannan V, Xu Y, et al. Heterogeneous clinical and functional features of GRIN2D-related developmental and epileptic encephalopathy. Brain 2019;142(10):3009–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swanger SA, Chen W, Wells G, et al. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet 2016;99:1261–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogden KK, Chen W, Swanger SA, et al. Molecular mechanism of disease-associated mutations in the pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genet 2017;13:e1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amin JB, Leng X, Gochman A, Zhou H-X, Wollmuth LP. A conserved glycine harboring disease-associated mutations permits NMDA receptor slow deactivation and high Ca2+ permeability. Nat Commun 2018;9:3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam VM, Beerepoot P, Angers S, Salahpour A. A novel assay for measurement of membrane-protein surface expression using a β-lactamase. Traffic Cph Den 2013;14:778–784. [DOI] [PubMed] [Google Scholar]
- 27.Li D, Yuan H, Ortiz-Gonzalez XR, et al. GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet 2016;99:802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masri A, Chung S-K, Rees MI. Hyperekplexia: Report on phenotype and genotype of 16 Jordanian patients. Brain Dev 2017;39:306–311. [DOI] [PubMed] [Google Scholar]
- 29.Parnes M, Bashir H, Jankovic J. Is benign hereditary chorea really benign? Brain-lung-thyroid syndrome caused by NKX2-1 mutations. Mov Disord Clin Pract 2019;6:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohlega SA, Abou-Al-Shaar H, AlDakheel A, et al. Autosomal recessive ADCY5-related dystonia and myoclonus: expanding the genetic spectrum of ADCY5-Related movement disorders. Parkinsonism Relat Disord 2019;64:145–149. [DOI] [PubMed] [Google Scholar]
- 31.van der Veen S, Zutt R, Klein C, Marras C, Berkovic SF, Caviness JN, Shibasaki H, de Koning TJ, Tijssen MAJ. Nomenclature of genetically determined myoclonus syndromes: recommendations of the International Parkinson and Movement Disorder Society Task Force. Mov Disord Off J Mov Disord Soc 2019;34:1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor.Nature 2009;462:745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.