

Abstract
Circulating cell-free mitochondrial DNA (ccf-mtDNA) released upon cell injury or death stimulates diverse pattern recognition receptors to activate innate immune responses and initiate systemic inflammation. In this review, we discuss the temporal changes of ccf-mtDNA during pregnancy and its potential contribution to adverse pregnancy outcomes in pregnancy complications.
Keywords: circulating DNA, gestation, maternal, mitochondria, preeclampsia
Introduction
Mitochondria are cellular organelles well known for cellular energy production through oxidative phosphorylation. Additionally, mitochondria are involved in intracellular communication and play a role in regulation of cell death, gene expression, and epigenetic changes (1–5). Even beyond intracellular signaling, mitochondria release various signaling molecules, including their genome, into the extracellular space where they facilitate systemic adaptations to stress (6, 7). Recent studies have implicated circulating cell-free mitochondrial DNA (ccf-mtDNA) in various pathological conditions such as cardiovascular disease, atherosclerosis, hypertension, autoimmune disorders, and cancer progression (8–15). Specifically, concentrations of ccf-mtDNA or circulating mtDNA copy numbers are used as a proxy of cellular stress or impaired cellular bioenergetics and mitochondrial dysfunction (15).
Circulating cell-free mtDNA is a normal constituent of the maternal blood during healthy pregnancy (16, 17), but its concentrations substantially change in pregnancy complications such as preeclampsia (18–20). These alterations in circulating mtDNA dynamics are consistent with observations of changes in placental mitochondrial morphology, impaired bioenergetics, and oxidative stress in pregnancy complications (21–24). In this review, we discuss the unique characteristics of ccf-mtDNA that define its immunostimulatory properties, describing mtDNA-associated signaling mechanisms and focusing on its association with various pathologies. Importantly, we discuss the relevance of maternal ccf-mtDNA to gestational and perinatal outcomes and its prognostic value in pregnancy complications, such as preeclampsia. Finally, we identify methodological limitations, controversies, and perspective proposals for nonclassical functions of ccf-mtDNA in pregnancy.
Mitochondria and Mitochondrial DNA
According to the endosymbiotic theory, mitochondria are derived from aerobic bacteria that were phagocytized during the evolution of the first eukaryotic cells (25, 26). The phagocytosed aerobic bacteria developed a symbiotic relationship with the eukaryotic cell, losing DNA and structural components that were not of benefit to the cell host. Evidence supporting this theory include the size of the mitochondrion, the presence of small ribosomes, its double membrane, and the circular structure and hypomethylated state of its DNA: all characteristic of prokaryotic cells. Accordingly, the human mitochondrial genome is relatively small, consisting of only 16,569 base pairs encoding 13 proteins relative to its main cellular function of oxidative phosphorylation (27, 28). The remaining ∼1,200 mitochondrial proteins are encoded by nuclear DNA, translated in the cytosol, and imported into the mitochondrion (28).
There are 2–10 copies of mtDNA per mitochondrion, and hundreds to thousands of mitochondria per cell, depending on the metabolic demand of the tissue (29). Copy numbers of mtDNA are regulated by cell-specific mechanisms in response to internal and external stimuli. For instance, a reduction in mitochondrial biogenesis, such as has been observed in aging, reduces the overall mtDNA copy number (30). Furthermore, mtDNA copy numbers are higher in tissues with greater energy demands, such as in the heart and brain (31). Notably, mitochondrial transcription factor A (TFAM) is a mitochondrial protein responsible for packaging mtDNA into its nucleoid structure (32). TFAM is required for maintenance of mtDNA in multiple cell types, and TFAM levels have been shown to be directly proportional to mtDNA copy numbers in a variety of mammalian tissues (33). Moreover, mtDNA copy number differs between men and women, and the effect of sex has been shown to be variable among age groups and tissue types (34–39).
Mitochondrial dysfunction and/or damage has been associated with aging and numerous neurodegenerative and cardiovascular diseases (40). Cellular stress, injury, or infection can induce degradation of mtDNA and initiation of mitochondrial autophagy, termed mitophagy (41). In response to cell stressors, mitochondria-derived proteins and mtDNA are released into the cytoplasm, extracellular space, and eventually circulation where they can activate innate immune system effectors and induce systemic inflammation, a common feature of many pregnancy-related pathologies (42). Accordingly, ccf-mtDNA is a proxy of cellular stress and impaired bioenergetics; thus, ccf-mtDNA may provide insight to disease progression, etiology, or response to treatment.
Mitochondrial DNA as an Inflammatory Signaling Molecule
Mitochondrial DNA not only resembles bacterial DNA in replication, structure, and hypomethylation, but also in immunogenicity. Many human innate immune receptors, termed pattern recognition receptors (PRRs), evolved to sense “foreign” invaders such as bacterial or viral products, known as pathogen-associated molecular patterns (PAMPs), that would be encountered during an infection. Since mitochondrial products resemble bacterial products, mitochondria-derived proteins and mtDNA, termed mitochondrial damage-associated molecular patterns (mtDAMPs), can be recognized by PRRs to initiate immune and proinflammatory responses to endogenous ligands in the absence of infection (i.e., sterile inflammation) (7) (FIGURE 1).
FIGURE 1.

Endosymbiotic theory and mitochondria-induced sterile inflammation Mitochondria evolve from bacteria and synthesize proteins and DNA with similar modifications as bacterial counterparts. The evolutionary similarity between mitochondria and bacteria permits the mitochondrial danger-associated molecular patterns (mtDAMPs) to stimulate the same pattern recognition receptors of the innate immune system as bacterial pathogen-associated molecular patterns (PAMPs). Image created with BioRender.com and used with permission.
Although mitochondrial products can elicit robust immune responses, there are many quality control mechanisms to inhibit release of mtDAMPs and reduce inflammatory stimuli from entering the circulation. Damaged cells can prevent release of mtDAMPs and activation of the innate immune system through autophagy processes and selective packaging of damaged mitochondrial components (43). To prevent release of mtDAMPs following cell damage, cells use a sorting mechanism to direct damaged mitochondrial products to the lysosome for degradation. However, cellular damage in the context of a potent inflammatory stimulus will induce release of mtDAMPs into the extracellular space (43).
A revolutionary study in 2004 by Collins et al. (44) first revealed the inflammatory potential of mtDNA by demonstrating oxidized mtDNA caused local inflammation when injected into the joints of mice, and direct treatment of murine splenic cells in vitro with mtDNA resulted in release of the potent inflammatory cytokine tumor necrosis factor-alpha (TNF-α). This initial study created an avenue of future investigations into the immunogenicity of mtDNA that would reveal recognition of mtDNA by multiple DNA-sensing PRRs including Toll-like receptor 9 (TLR9), inflammasomes, and the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-stimulator of interferon genes (STING) signaling axis (FIGURE 2).
FIGURE 2.
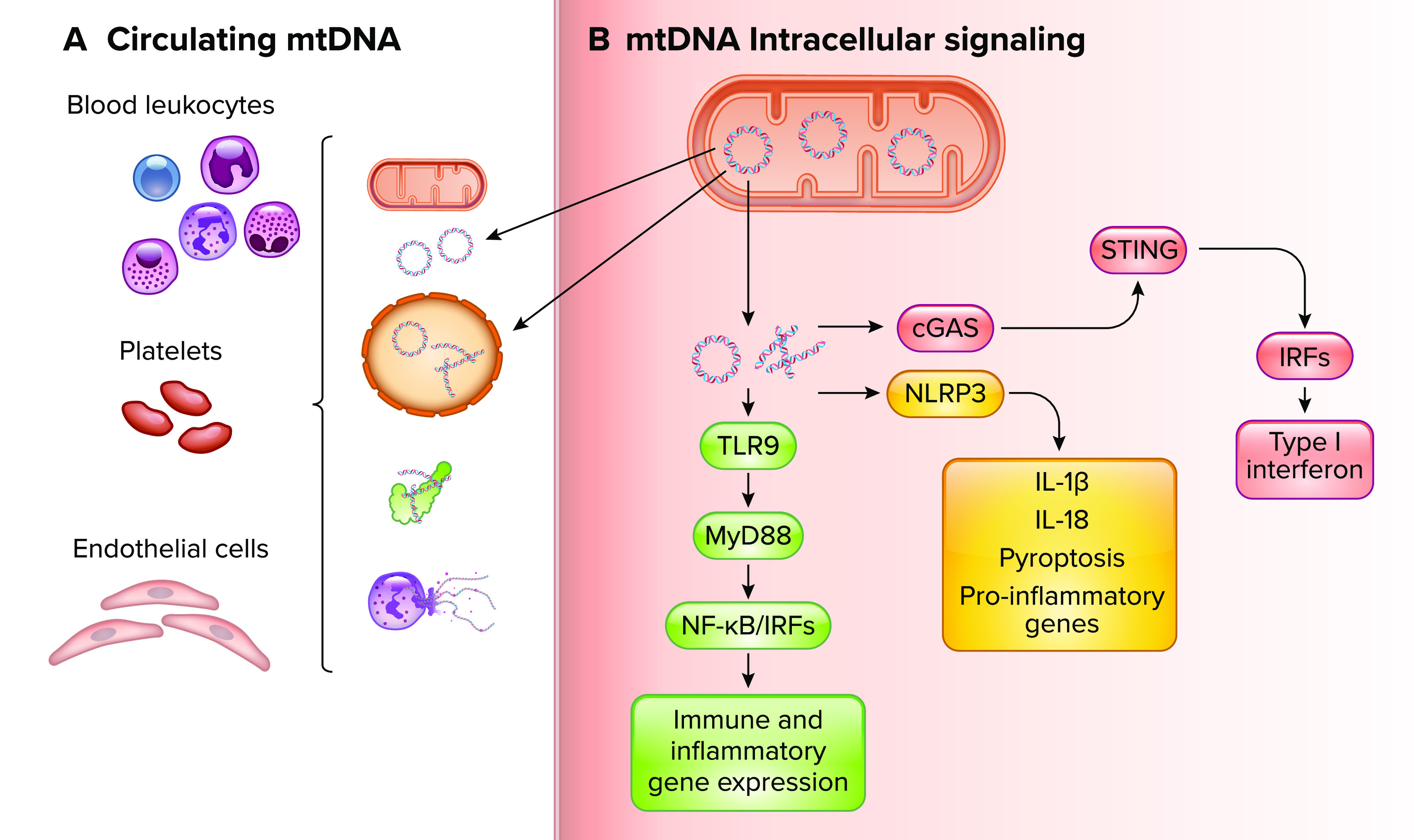
Recognition of various biological forms of circulating cell-free mitochondrial DNA in maternal circulation and mtDNA-induced intracellular proinflammatory signaling by diverse innate immune system receptors A: biological forms of ccf-mtDNA include mtDNA confined to whole mitochondria and extracellular vesicles, extruded from neutrophils in neutrophil extracellular traps (NETs), and in circular or fragmented biological forms complexed to other mitochondrial-associated proteins such as mitochondrial transcription factor A. These various biological forms can be recognized by leukocytes, platelets, and endothelial cells in the maternal circulation. B: intracellular mtDNA-induced proinflammatory signaling mechanisms contributing to innate immune system activation and release of mtDNA into the maternal circulation. TLR9, Toll-like receptor 9; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRFs, interferon regulatory factors; NLRP3, nucleotide-binding oligomerization domain, leucine-rich repeats, and pyrin domain-containing protein 3; IL, interleukin; cGAS, cyclic guanosine monophosphate-adenosine monophosphate synthase; STING, stimulator of interferon genes. Image created with BioRender.com and used with permission.
TLR9 Signaling
TLR9 is localized in the endoplasmic reticulum of resting cells and recruited to lysosomal compartments following cellular uptake of unmethylated CpG dinucleotides found in bacteria and mtDNA (45–48). Although TLR9 has been shown to have similar binding affinities to methylated and unmethylated CpG motifs, unmethylated CpG DNA distinctively stimulates TLR9 signaling by inducing the colocalization of TLR9 into endosomes containing the unmethylated CpG ligand and signaling adaptor protein myeloid differentiation primary response gene 88 (MyD88) (46, 49). Within the late endosome, TLR9 recognition of unmethylated CpG DNA results in receptor dimerization, recruitment of MyD88, and downstream signaling through a MyD88-dependent pathway resulting in activation of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) and subsequent transcription of proinflammatory cytokines, chemokines, and cell adhesion molecules (50, 51). Additionally, TLR9 stimulation induces activation of interferon regulatory factor 7 (IRF7), resulting in increased production of type I interferons that influence dendritic cell activation and migration (46).
Inflammasome Signaling
Canonical inflammasomes are multisubunit complexes of the innate immune system that are activated by PAMPs and/or DAMPs during cell stress or necrosis. Upon activation, inflammasome complexes can serve as a platform for caspase I dimerization, cleavage, and activation. Activated caspase 1 can then cleave immature forms of the proinflammatory cytokines interleukin (IL)-1β and IL-18 and stimulate formation of gasdermin D pores, resulting in an inflammatory cell death known as pyroptosis (52–56). In 2010, Nakahira et al. (57) were the first to link mtDNA with nucleotide-binding oligomerization domain, leucine-rich repeats, and pyrin domain-containing protein 3 (NLRP3) inflammasome activation by demonstrating the increased production of inflammasome-associated cytokines following mitochondrial reactive oxygen species production and release of mtDNA into the cytosol. A follow-up study by Shimada et al. (58) would reveal mtDNA directly binds to NLRP3 in the cytosol and that NLRP3 has a preference for oxidized mtDNA.
cGAS-STING Signaling
cGAS directly binds to cytosolic, double-stranded DNA to trigger strong type I interferon responses. Normally, cellular DNA is sequestered in the nucleus and mitochondria. Cell stressors and excessive DNA damage that induce DNA leakage into the cytosol can lead to constitutive and systemic activation of cGAS-STING signaling (59). mtDNA has been shown to stimulate cGAS-STING after initiation of cell death or as a response to mitochondrial stress and mis-packaged mtDNA that is transported to the cytosol (60–62).
mtDNA-Induced Inflammation and Disease
In 2010, Zhang et al. (7) first demonstrated circulating mtDAMPs induced sterile inflammation via stimulating bacteria-associated PRRs in circulating neutrophils, which resulted in a systemic inflammatory response syndrome. Since this discovery, a growing body of evidence has supported the role of ccf-mtDNA in inflammation-induced pathology underlying a wide variety of diseases including autoimmunity and cardiovascular diseases (42). Specifically, mtDNA-mediated inflammatory pathology has been characterized in human disease and models of atherosclerosis, systemic lupus erythematosus, hypertension, and cardiomyopathy (8, 10–12, 63–66). Although activation of TLR9, cGAS-STING, and inflammasome pathways has been implicated in mtDNA-induced pathologies, the mechanism by which ccf-mtDNA stimulates the innate immune system remains unclear. For instance, it is not clear whether all these pathways are stimulated by mtDNA during a certain disease state or if a specific pathway predominates. The ambiguity underlying these mechanisms lies in the various forms of ccf-mtDNA that alter ccf-mtDNA transport and interactions with cognate receptors.
Within the circulation, ccf-mtDNA can be trafficked inside circulating whole mitochondria or vesicular structures, such as extracellular vesicles (5). In addition, ccf-mtDNA can be transported in its circular form, as fragments, or complexed to proteins (FIGURE 2). There is growing evidence that the majority of ccf-mtDNA is encapsulated and trafficked in extracellular vesicles that protect ccf-mtDNA from circulating nucleases but also prevent receptor recognition (67, 68). Overall, a major knowledge gap regarding biological forms of ccf-mtDNA is the relative function associated with the various biological forms. It is currently unknown if the different biological forms of ccf-mtDNA alter transport to specific recipient cell types. Furthermore, the fate of ccf-mtDNA after it has been taken up by a recipient cell is not well understood. Studies examining the source, abundance, and fate of the different biological forms of ccf-mtDNA will help elucidate their physiological role in health and disease.
Pregnancy and ccf-mtDNA
Healthy pregnancy is often described as an inflammatory state, during which the immune system continuously adapts to facilitate placental and fetal growth and provide fetal and maternal protection. In the establishment, maintenance, and conclusion of pregnancy, there are inflammatory and tolerogenic immunological phases (69). During the first trimester of pregnancy, implantation and placentation induce a proinflammatory state marked by cellular damage, repair, and removal of cellular debris. Following the initial stage of pregnancy establishment is a stage of rapid fetal growth and development that requires a second immunological phase characterized by a controlled systemic inflammatory response, which protects the mother and growing fetus from pathogens and disease while also avoiding detrimental maternal allogeneic immune responses targeting the growing fetus and placenta. During this immunological phase, the maternal innate immune system is biased toward a T-helper 2 (Th2) cytokine profile that induces an anti-inflammatory state (70). The last immunological phase occurs at parturition when a renewed proinflammatory environment promotes contractions and delivery of the fetus and placenta (71).
Placental apoptosis occurs as a result of normal cellular turnover and repair and increases throughout healthy gestation, with the highest levels of apoptosis occurring in the third trimester (72). Notably, placental apoptosis contributes to the release of cell-free fetal DNA (73). In humans, higher primates, and rodents, the hemochorial arrangement of the placenta allows the maternal blood to have direct access to the fetal chorion, and therefore, placental cell turnover can result in the release of cellular constituents to the maternal circulation (74). Accordingly, it has been hypothesized that increases in placental apoptosis may also mediate increases in maternal ccf-mtDNA (17). Indeed, previous studies have revealed concentrations of maternal ccf-mtDNA increases with advancing gestational age in healthy pregnancies and returns to nonpregnant state values within 6–8 wk after delivery (17, 75). Whether an increase in ccf-mtDNA during healthy pregnancy reflects the transition from a proinflammatory stage at the first trimester to a second immunological phase of controlled inflammation later in pregnancy is currently unknown.
Pregnancy complications can occur when the innate immune system becomes dysregulated or in response to increased exposure to DAMPs or PAMPs, resulting in an exaggerated and pathogenic systemic inflammation that can be detrimental to both mother and fetus (FIGURE 3). Although the innate immune system is activated during pregnancy, and even more so during complicated pregnancies, the underlying mechanisms contributing to the regulation of this phenomenon remain unclear and could include differences in activation thresholds of immune cells, relative expression of PRRs, and relative concentration of PAMPs and DAMPs that could be contributing to an overactive immune response.
FIGURE 3.
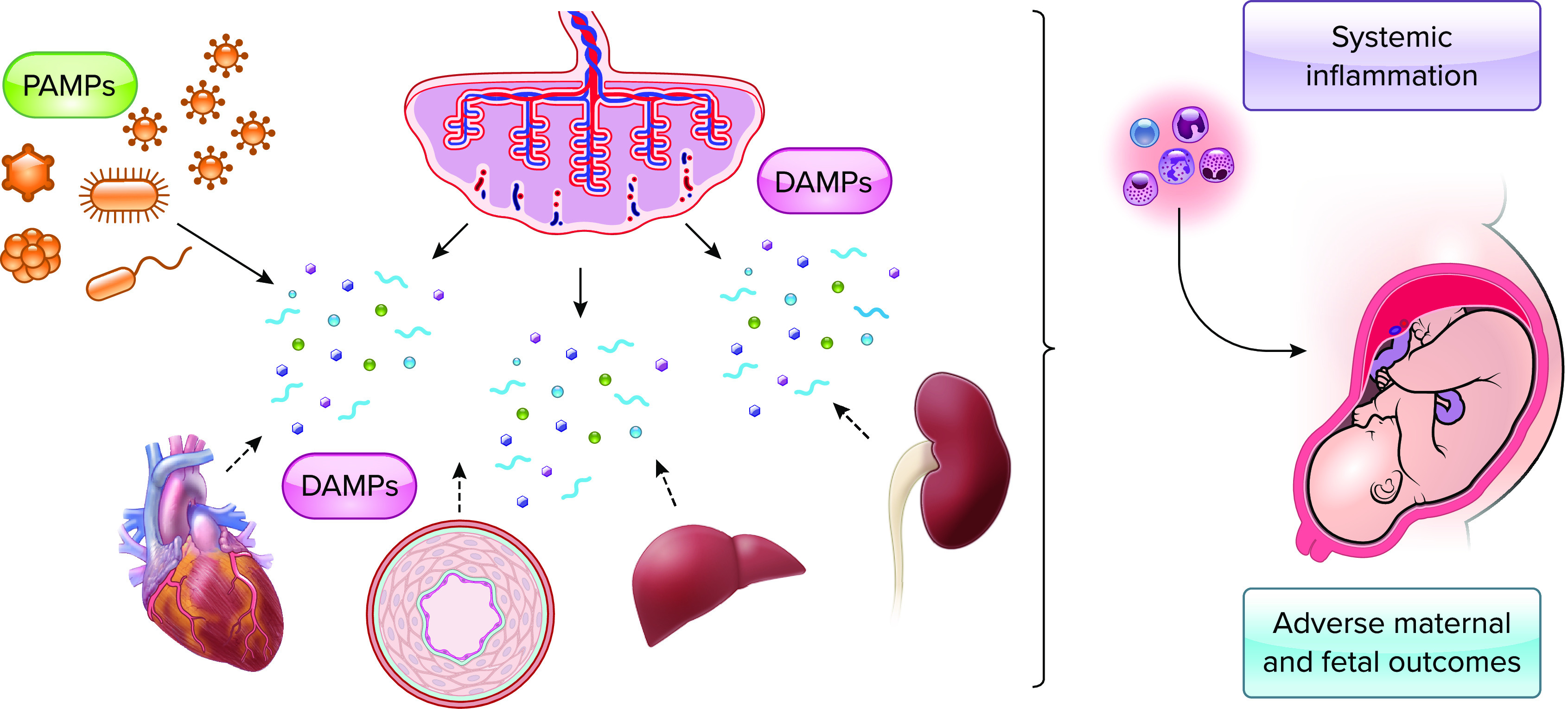
Hyperactivation of innate immune responses during pregnancies with complications During pregnancy, increased circulating pathogen-associated molecular patterns (PAMPs) due to bacterial infections or danger-associated molecular patterns (DAMPs; e.g., mitochondrial DNA) due to placental or maternal organ damage and cell death can cause hyperactivation of innate immune mechanisms resulting in pathogenic systemic inflammation that can be detrimental to both mother and fetus, inducing adverse pregnancy outcomes (e.g., preeclampsia). Image created with BioRender.com and used with permission.
Specifically, the expression of TLR9 has been shown to be elevated in the placenta and dendritic cells from women with preeclampsia compared with healthy pregnant controls (76, 77). Plasma from women with preeclampsia with elevated circulating mtDNA copy number induces greater TLR9 stimulation compared with plasma from gestational age-matched healthy pregnant women (78). Moreover, TLR9 agonism during pregnancy induces adverse pregnancy outcomes in animal models. Unmethylated CpG oligodeoxynucleotides (ODNs) are synthetic CpG dinucleotides that stimulate TLR9 and produce immunostimulatory effects through mechanisms similar to mtDNA interactions with TLR9 (79, 80). Stimulation of TLR9 with ODN 2395 (synthetic CpG ODN) in pregnant rats increased maternal blood pressure (81–83), while age-matched nonpregnant rats did not have a hypertensive response to ODN 2395, indicating that the response to TLR9 activation was pregnancy specific (82). In addition, treatment with ODN 2395 during rat pregnancy increased maternal vasoconstriction via upregulation of cyclooxygenase-dependent mechanisms and downregulation of basal levels of nitric oxide, a potent vasodilatory and anticontractile molecule (82). These first observations were confirmed in subsequent studies in pregnant mice that developed hypertension, placental inflammation, and fetal demise after treated with CpG ODN during the second trimester (84). In addition to TLR9 involvement, recent studies have revealed increased expression and activation of NLRP3 inflammasome components in the placentas of women with preeclampsia compared with healthy pregnant controls (85, 86). These data suggest the involvement of TLR9 and NLRP3 signaling pathways in pregnancies with preeclampsia but do not directly link ccf-mtDNA to these signaling pathways. In addition, it remains unknown whether these signaling pathways are contributors to the pathogenesis or maintenance of the disease.
When compared with healthy pregnancies, genetic polymorphisms in placenta-derived and circulating mtDNA, as well as alterations in the concentrations of ccf-mtDNA, have been associated with pregnancy complications (16, 87). The ccf-mtDNA variant A4917G of haplogroup T has been associated with preterm birth in a Caucasian pregnancy cohort (87). Additionally, a higher placental mtDNA mutational load has been associated with reduced gestational length and birthweight in a racially and ethnically diverse pregnancy cohort (88). In 2010, Colleoni et al. (16) were the first to describe circulating mtDNA in pregnancy, and the authors reported patients with intrauterine growth restriction (obstetric condition, in which the estimated weight of a fetus is smaller than expected for its gestational age) had greater levels of circulating mtDNA compared with gestational age-matched healthy pregnant women. In a separate study, Williams and colleagues (19) reported that women admitted to the hospital for placental abruption had greater odds of having elevated whole blood mtDNA than near term pregnant women. A limitation of this study was the experimental design did not account for gestational age, and gestational age of women with preeclampsia at sampling was significantly lower at 35.5 ± 4.3 wk as compared with their controls at 37.9 ± 3.4 (P < .001). Previous research had already demonstrated a dynamic change in circulating mtDNA throughout pregnancy, suggesting an influence of gestational age on mtDNA determination (16).
Numerous studies have associated alterations in ccf-mtDNA with the maternal cardiovascular syndrome of preeclampsia. Qiu et al. (89) found that whole blood from women with preeclampsia had greater mtDNA concentrations than whole blood from healthy pregnant women. Measuring mtDNA in blood plasma, McCarthy and Kenny (90) noted a 5.5-fold increase in plasma mtDNA from women with preeclampsia when compared with healthy controls. These authors also demonstrated that plasma from women with preeclampsia induced more TLR9 gene expression, inflammation, and adhesion molecule expression in endothelial cells. In agreement with previous studies, Ozeki et al. (18) demonstrated that women with preeclampsia in the third trimester have greater serum mtDNA than healthy controls. Furthermore, they reported that serum from women with preeclampsia induced production of inflammatory cytokines in trophoblast cells in vitro. This inflammatory response was reduced in the presence of DNase, an enzyme that degrades DNA (18). Thus this study by Ozeki et al. provided evidence linking ccf-mtDNA to inflammation in the placenta.
In addition to gestational age and preeclampsia, prepregnancy maternal obesity and gestational diabetes mellitus (GDM), defined as high blood sugar and glucose intolerance occurring during pregnancy, affect circulating levels of mtDNA. Anelli et al. (91) found that women who were admitted for c-section during the third trimester had greater blood mtDNA if they were obese before pregnancy, and that prepregnancy obesity correlated with greater mtDNA on admission. A study by McElwain et al. (92) demonstrated pregnant women diagnosed with GDM had higher plasma ccf-mtDNA levels at 20 wk of gestation compared with women who had healthy pregnancies. Obesity and diabetes, like healthy pregnancy and preeclampsia, are states of chronic inflammation (93). Indeed, nonpregnant subjects with type 2 diabetes and obesity had greater circulating plasma mtDNA than controls (94). Additionally, obesity and diabetes are risk factors for the development of preeclampsia during pregnancy (95, 96). It is possible that pregnancy, on a background of chronic, low-grade inflammation in obesity and diabetes, results in a greater risk of pregnancy complications. A summary of human studies examining ccf-mtDNA concentrations during pregnancy complications can be found in Table 1.
TABLE 1.
Summary of evidence demonstrating changes in circulating cell-free mitochondrial DNA in human pregnancy complications
| Pregnancy Complication/ccf-mtDNA Compared with Healthy Pregnancy | Gestational Age at Sample | Blood Component | Reference |
|---|---|---|---|
| Preeclampsia | |||
| Increased | Term/delivery | Whole blood | Qiu et al. (89) |
| Decreased | First trimester | Whole blood | Busnelli et al. (97) |
| Increased | 37–40 wk | Plasma | McCarthy and Lenny (90) |
| Greater increase from 15 to 20 wk | 15–20 wk | Plasma | Williamson et al. (20) |
| Increased | 30–31 wk | Serum | Marschalek et al. (98) |
| Increased | 34–39 wk | Serum | Ozeki et al. (18) |
| Placental abruption | |||
| No significant difference | 35–38 wk | Whole blood | Williams et al. (19) |
| IUGR | |||
| Increased | Third trimester | Whole blood | Colleoni et al. (16) |
| GDM | |||
| Increased | 19–21 wk | Plasma | McElwain and McCarthy (92) |
ccf-mtDNA, circulating cell-free mitochondrial DNA; IUGR, intrauterine growth restriction; GDM, gestational diabetes mellitus.
Ccf-mtDNA as a Predictor of Pregnancy Complications
Circulating factors, such as ccf-mtDNA, can be detected during pregnancy using noninvasive techniques and are currently being studied as biomarkers used to predict pregnancy complications before symptoms are present. In a recent study, Busnelli et al. (97) demonstrated that women who had reduced whole blood mtDNA in the first trimester were more likely to develop preeclampsia when compared with women who went on to have healthy pregnancies. Moreover, mtDNA was reduced even further in women diagnosed with early onset preeclampsia (most severe form of the syndrome) compared with women who were diagnosed with preeclampsia after the 34th wk of gestation (97). These data suggest that suppression of mtDNA in the first trimester may be associated with severity of pregnancy outcomes and mechanisms of preeclampsia pathogenesis. In addition, this study provides promising preliminary data for prediction of preeclampsia using circulating mtDNA in the first trimester.
Williamson et al. (20) found no difference in plasma mtDNA concentrations at two individual timepoints of gestation (15 wk or 20 wk), when comparing women who went on to develop preeclampsia to women who had healthy pregnancies. However, the difference between mtDNA concentrations at 15 wk and 20 wk was greater in women who later developed preeclampsia compared with women who had healthy pregnancies, thus providing some evidence for a potential predictive value of serial plasma mtDNA measurements (20). Since these measurements were performed in the second trimester, the findings from this study could reflect a transitional period in preeclampsia between a suppressed mtDNA profile, as seen in Busnelli et al. (97), to the increased mtDNA profile in the third trimester as seen in the majority of publications on preeclampsia (18, 89, 90, 98). Using serum, Marschalek and McCarthy (98) demonstrated greater third trimester (31 wk) mtDNA in women who would later be diagnosed with preeclampsia. This study shows how late pregnancy measurement of mtDNA could still be used to predict near-term incidence of preeclampsia.
In addition to preeclampsia, ccf-mtDNA has also been investigated as a potential predictor of GDM. McElwain et al revealed greater ccf-mtDNA levels at 20 wk of gestation in women who were later diagnosed with GDM at 24 wk of gestation when compared with women who had healthy pregnancies (92). Further, this study demonstrated that maternal obesity affected the association of ccf-mtDNA with a GDM diagnosis, which suggests that maternal obesity modifies ccf-mtDNA levels. Although recent studies have investigated the utility of ccf-mtDNA as a predictor of pregnancy complications such as preeclampsia and GDM, additional studies are needed to increase the power of ccf-mtDNA to predict pregnancy complications in pregnant women of diverse backgrounds. Moreover, pregnancy complications are multifactorial diseases that will likely require a panel of biomarkers to estimate risk for developing a pregnancy complication, and current ongoing studies are assessing the use of combined biomarkers and clinical parameters in algorithms to predict adverse pregnancy outcomes.
Limitations of Previous Studies and Considerations for Determining the Impact of ccf-mtDNA During Pregnancy
The genetic contributions (maternal, fetal, or paternal), tissue-specific sources (placenta, endothelium, etc.), biological forms and transport, as well as optimal technical approaches for ccf-mtDNA isolation and quantification are all significant factors that affect data reliability and interpretation when evaluating the biological significance and function of ccf-mtDNA during pregnancy. Interestingly, ccf-mtDNA copy number can vary depending on maternal age, gestational age, health status, and time of blood draw (99–103); however, many of these factors affecting ccf-mtDNA concentrations were not considered or controlled in previous studies. Moreover, the sample type chosen (maternal whole blood, plasma, or serum) greatly varies among previous studies, which prevents cross-study comparisons and does not adequately represent ccf-mtDNA since whole blood and coagulated blood, such as serum, contain cellular contaminants that grossly overestimate ccf-mtDNA levels (42). Other challenges limiting valid and reliable mtDNA quantification relate to the effects of hypervariability and pseudogene contamination since a significant part of the mitochondrial genome is duplicated in the nuclear genome (17). Amplifying mitochondrial genome regions that have few known mutations and are absent from the nuclear genome is of paramount importance and could protect against these limitations. With regards to the genetic basis of pregnancy complications and adverse pregnancy outcomes, the majority of previous studies focus on the maternal nuclear genome and disregard the mitochondrial genome, as well as genetic factors of fetal or paternal origin contributing to mitochondrial dysfunction and release of ccf-mtDNA.
Overall, previous human observational studies have focused on the quantity of ccf-mtDNA with a lack of understanding of the quality of ccf-mtDNA in pregnancy and pregnancy complications. In regards to animal studies, there is a need for natural ccf-mtDNA, rather than a synthetic TLR9 agonist, to be incorporated in an animal model of pregnancy and pregnancy complications to tease out causal mechanisms underlying the correlative findings in human studies. The greatest challenges to elucidating the role of ccf-mtDNA in pregnancy is understanding the mechanisms underlying the release of mtDNA into the circulation and defining specific targets of ccf-mtDNA in the circulation. To gain a better understanding of the role of ccf-mtDNA in pregnancy and pregnancy complications, future studies will need to address these limitations using standardized methodology to isolate specific biological forms of ccf-mtDNA in plasma blood fractions. After proper isolation of ccf-mtDNA is employed, the use of absolute quantification methodology alongside novel techniques to identify mutational load and base pair modifications in ccf-mtDNA will help identify the source of ccf-mtDNA and define its functional role and immunostimulatory potential in the circulation.
Conclusions and Prospective Proposals
Circulating cell-free mtDNA increases with advancing gestational age in healthy pregnancies and returns to prepregnancy values after delivery of the placenta. This evidence suggests the feto-placental unit may be the largest contributor to ccf-mtDNA. Mechanisms underlying mtDNA release from mitochondria, extrusion from the cell, and interactions in maternal circulation remain elusive and are subject of exciting ongoing studies. Interestingly, experimental models examining responses to systemic TLR9 stimulation have revealed hemodynamic and inflammatory responses to synthetic TLR9 agonists are pregnancy specific, suggesting that pregnancy increases sensitivity to ccf-mtDNA-associated signaling. Measurements of ccf-mtDNA with higher temporal resolution in the prepregnancy, pregnancy, and postpartum periods will help elucidate the physiological and pathophysiological contributions of ccf-mtDNA during pregnancy, as well as determine its utility as a potential biomarker for maternal risk of having a complicated pregnancy. In addition, studies examining the heritability and genetic basis, such as mtDNA gene variants, of pregnancy complications will increase the power and resolution of predictive models assessing maternal risk and underlying causes of complex diseases such as the maternal syndrome of preeclampsia. Finally, this review has primarily focused on evidence related to the immunostimulatory potential of ccf-mtDNA and its potential role as a pathogenic trigger or biomarker of pregnancy complications. Recent studies, however, have proposed novel, noninflammatory properties of ccf-mtDNA. It is noteworthy that ccf-mtDNA is abundant in healthy individuals in the absence of inflammation (5, 104), suggesting possible functions of ccf-mtDNA beyond inflammation, such as antibacterial effects and cell-to-cell communication for clearance of dysfunctional mitochondria and bioenergetic rescue (5). The presence of ccf-mtDNA in maternal blood in healthy pregnancies supports the notion that mtDNA in the maternal circulation may have properties beyond inflammation such as maternal-fetal communication or priming for parturition.
Acknowledgments
Research in the authors’ laboratories is supported by National Institutes of Health Grants R01 HL-0146562, HL-0146562-02S1, and T32 AG 020494; American Heart Association Grants 19TPA34850131 and AHA903250; and the Texas Alzheimer’s Research and Care Consortium.
No conflicts of interest, financial or otherwise, are declared by the authors.
J.L.B., S.C.C., and S.G. conceived and designed research; S.G. prepared figures; J.L.B., S.C.C., and S.G. drafted manuscript; J.L.B., S.C.C., N.R.P., and S.G. edited and revised manuscript; J.L.B., S.C.C., N.R.P., and S.G. approved final version of manuscript.
References
- 1.Friedman JR, Nunnari J. Mitochondrial form and function. Nature 505: 335–343, 2014. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28: 272–275, 2001. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 3.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson H, Trounce IA, Procaccio V, Wallace DC. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A 111: E4033–4042, 2014. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165: 1197–1208, 2016. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, Lindqvist D, Fernstrom J, Moser D, Kaufman BA, Picard M. Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion 59: 225–245, 2021. doi: 10.1016/j.mito.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci U S A 112: E6614–6623, 2015. doi: 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, Wright T, Punaro M, Palucka K, Guiducci C, Banchereau J, Pascual V. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213: 697–713, 2016. doi: 10.1084/jem.20151876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, Webb RC. Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. Am J Hypertens 30: 173–181, 2017. doi: 10.1093/ajh/hpw113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107: 119–130, 2015. doi: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial dna and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol 67: 3190–3200, 2015. doi: 10.1002/art.39296. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Meng P, Han Y, Shen C, Li B, Hakim MA, Zhang X, Lu Q, Rong M, Lai R. Mitochondrial DNA-LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43: 1137–1147, 2015. doi: 10.1016/j.immuni.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Silzer T, Barber R, Sun J, Pathak G, Johnson L, O’Bryant S, Phillips N. Circulating mitochondrial DNA: New indices of type 2 diabetes-related cognitive impairment in Mexican Americans. PLoS One 14: e0213527, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohd Khair SZ, Abd Radzak SM, Mohamed Yusoff AA. The uprising of mitochondrial dna biomarker in cancer. Dis Markers 2021: 7675269, 2021. doi: 10.1155/2021/7675269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu M. Circulating cell-free mitochondrial DNA as a novel cancer biomarker: opportunities and challenges. Mitochondrial DNA 23: 329–332, 2012. doi: 10.3109/19401736.2012.696625. [DOI] [PubMed] [Google Scholar]
- 16.Colleoni F, Lattuada D, Garretto A, Massari M, Mando C, Somigliana E, Cetin I. Maternal blood mitochondrial DNA content during normal and intrauterine growth restricted (IUGR) pregnancy. Am J Obstet Gynecol 203: 365, 2010. doi: 10.1016/j.ajog.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 17.Cushen SC, Sprouse ML, Blessing A, Sun J, Jarvis SS, Okada Y, Fu Q, Romero SA, Phillips NR, Goulopoulou S. Cell-free mitochondrial DNA increases in maternal circulation during healthy pregnancy: a prospective, longitudinal study. Am J Physiol Regul Integr Comp Physiol 318: R445–R452, 2020. doi: 10.1152/ajpregu.00324.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozeki A, Tani K, Takahashi H, Suzuki H, Nagayama S, Hirashima C, Iwata H, Kuwayama T, Ohkuchi A, Shirasuna K. Preeclamptic patient-derived circulating cell-free DNA activates the production of inflammatory cytokines via toll-like receptor 9 signalling in the human placenta. J Hypertens 37: 2452–2460, 2019. doi: 10.1097/HJH.0000000000002208. [DOI] [PubMed] [Google Scholar]
- 19.Williams MA, Sanchez SE, Ananth CV, Hevner K, Qiu C, Enquobahrie DA. Maternal blood mitochondrial DNA copy number and placental abruption risk: results from a preliminary study. Int J Mol Epidemiol Genet 4: 120–127, 2013. [PMC free article] [PubMed] [Google Scholar]
- 20.Williamson RD, McCarthy FP, Khashan AS, Totorika A, Kenny LC, McCarthy C. Exploring the role of mitochondrial dysfunction in the pathophysiology of pre-eclampsia. Pregnancy Hypertens 13: 248–253, 2018. doi: 10.1016/j.preghy.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Beyramzadeh M, Dikmen ZG, Erturk NK, Tuncer ZS, Akbiyik F. Placental respiratory chain complex activities in high risk pregnancies. J Matern Fetal Neonatal Med 30: 2911–2917, 2017. doi: 10.1080/14767058.2016.1268594. [DOI] [PubMed] [Google Scholar]
- 22.Mando C, Marino MA, Miriam F, Palma CD, Borelli M, Trabattoni D, Stampalija T, Ferrazzi E, Clementi E, Cetin I. Mitochondrial content and function in placental cells and tissues of preeclampsia and IUGR. Pregnancy Hypertens 2: 203, 2012. doi: 10.1016/j.preghy.2012.04.049. [DOI] [PubMed] [Google Scholar]
- 23.Zsengellér ZK, Rajakumar A, Hunter JT, Salahuddin S, Rana S, Stillman IE, Ananth Karumanchi S. Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens 6: 313–319, 2016. doi: 10.1016/j.preghy.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Illsinger S, Janzen N, Sander S, Schmidt KH, Bednarczyk J, Mallunat L, Bode J, Hagebölling F, Hoy L, Lücke T, Hass R, Das AM. Preeclampsia and HELLP syndrome: Impaired mitochondrial function in umbilical endothelial cells. Reprod Sci 17: 219–226, 2010. doi: 10.1177/1933719109351597. [DOI] [PubMed] [Google Scholar]
- 25.Archibald JM. Endosymbiosis and eukaryotic cell evolution. Curr Biol 25: R911–921, 2015. doi: 10.1016/j.cub.2015.07.055. [DOI] [PubMed] [Google Scholar]
- 26.Roger AJ, Munoz-Gomez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol 27: R1177–R1192, 2017. doi: 10.1016/j.cub.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Stewart JB, Chinnery PF. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet 22: 106–118, 2021. doi: 10.1038/s41576-020-00284-x. [DOI] [PubMed] [Google Scholar]
- 28.Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11: 25–44, 2010. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Hara R, Tedone E, Ludlow A, Huang E, Arosio B, Mari D, Shay JW. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res 29: 1878–1888, 2019. doi: 10.1101/gr.250480.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pyle A, Anugrha H, Kurzawa-Akanbi M, Yarnall A, Burn D, Hudson G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging 38: 216, 2016. doi: 10.1016/j.neurobiolaging.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 53: 214–223, 2020. doi: 10.1016/j.mito.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filograna R, Mennuni M, Alsina D, Larsson NG. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett 595: 976–1002, 2021. doi: 10.1002/1873-3468.14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13: 935–944, 2004. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 34.Ding J, Sidore C, Butler TJ, Wing MK, Qian Y, Meirelles O, Busonero F, Tsoi LC, Maschio A, Angius A, Kang HM, Nagaraja R, Cucca F, Abecasis GR, Schlessinger D. Assessing mitochondrial dna variation and copy number in lymphocytes of ∼2,000 sardinians using tailored sequencing analysis tools. PLoS Genet 11: e1005306, 2015. doi: 10.1371/journal.pgen.1005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Leeuwen N, Beekman M, Deelen J, van den Akker EB, de Craen AJ, Slagboom PE, T Hart LM. Low mitochondrial DNA content associates with familial longevity: The Leiden Longevity Study. Age (Dordr) 36: 9629, 2014. doi: 10.1007/s11357-014-9629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Echem C, Costa TJ, Oliveira V, Giglio Colli L, Landgraf MA, Rodrigues SF, Franco M, Landgraf RG, Santos-Eichler RA, Bomfim GF, Akamine EH, de Carvalho MH. Mitochondrial DNA: a new driver for sex differences in spontaneous hypertension. Pharmacol Res 144: 142–150, 2019. doi: 10.1016/j.phrs.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Sharma J, Johnston MV, Hossain MA. Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation. BMC Neurosci 15: 2, 2014. doi: 10.1186/1471-2202-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amengual-Cladera E, Llado I, Gianotti M, Proenza AM. Sex differences in the effect of high-fat diet feeding on rat white adipose tissue mitochondrial function and insulin sensitivity. Metabolism 61: 1108–1117, 2012. doi: 10.1016/j.metabol.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 39.Guevara R, Gianotti M, Roca P, Oliver J. Age and sex-related changes in rat brain mitochondrial function. Cell Physiol Biochem 27: 201–206, 2011. doi: 10.1159/000327945. [DOI] [PubMed] [Google Scholar]
- 40.Nakahira K, Hisata S, Choi AM. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal 23: 1329–1350, 2015. doi: 10.1089/ars.2015.6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medeiros TC, Graef M. Autophagy determines mtDNA copy number dynamics during starvation. Autophagy 15: 178–179, 2019. doi: 10.1080/15548627.2018.1532263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17: 363–375, 2017. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pepin G, Germain M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun 12: 1971, 2021. doi: 10.1038/s41467-021-21984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 75: 995–1000, 2004. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 45.Nishimoto S, Fukuda D, Sata M. Emerging roles of Toll-like receptor 9 in cardiometabolic disorders. Inflamm Regen 40: 18, 2020. doi: 10.1186/s41232-020-00118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karapetyan L, Luke JJ, Davar D. Toll-like receptor 9 agonists in cancer. Onco Targets Ther 13: 10039–10060, 2020. doi: 10.2147/OTT.S247050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol 87: 209–217, 2009. doi: 10.1038/icb.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 5: 190–198, 2004. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 49.de Jong SD, Basha G, Wilson KD, Kazem M, Cullis P, Jefferies W, Tam Y. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J Immunol 184: 6092–6102, 2010. doi: 10.4049/jimmunol.0802442. [DOI] [PubMed] [Google Scholar]
- 50.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2: 17023, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 13: 460–469, 2007. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022, 2014. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 53.Suarez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Alvarez-Cordoba M, Villalon-Garcia I, Talaveron-Rey M, Suarez-Carrillo A, Munuera-Cabeza M, Sanchez-Alcazar JA. From mitochondria to atherosclerosis: the inflammation path. Biomedicines 9: 258, 2021. doi: 10.3390/biomedicines9030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, Wong W, Kamen R, Tracey D, Allen H. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386: 619–623, 1997. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 56.Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593: 607–611, 2021. doi: 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21: 501–521, 2020. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 60.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DC, Kile BT. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159: 1549–1562, 2014. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577, 2014. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557, 2015. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep 3: 1077, 2013. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, Gottlieb RA, Chen S, Arditi M. Ogg1-dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ Res 119: e76-90–e90, 2016. doi: 10.1161/CIRCRESAHA.116.308362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22: 146–153, 2016. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lazo S, Noren Hooten N, Green J, Eitan E, Mode NA, Liu QR, Zonderman AB, Ezike N, Mattson MP, Ghosh P, Evans MK. Mitochondrial DNA in extracellular vesicles declines with age. Aging Cell 20: e13283, 2021. doi: 10.1111/acel.13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L, Galkin A, Thakur BK, Soplop N, Uryu K, Hoshino A, Norton L, Bonafe M, Cricca M, Gasparre G, Lyden D, Bromberg J. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A 114: E9066–E9075, 2017. doi: 10.1073/pnas.1704862114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mor G, Cardenas I, Abrahams V, Guller S. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann N Y Acad Sci 1221: 80–87, 2011. doi: 10.1111/j.1749-6632.2010.05938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abu-Raya B, Michalski C, Sadarangani M, Lavoie PM. Maternal immunological adaptation during normal pregnancy. Front Immunol 11: 575197, 2020. doi: 10.3389/fimmu.2020.575197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel LA, Nien JK. Inflammation in preterm and term labour and delivery. Semin Fetal Neonatal Med 11: 317–326, 2006. doi: 10.1016/j.siny.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharp AN, Heazell AE, Crocker IP, Mor G. Placental apoptosis in health and disease. Am J Reprod Immunol 64: 159–169, 2010. doi: 10.1111/j.1600-0897.2010.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aucamp J, Bronkhorst AJ, Badenhorst CP, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc 93: 1649–1683, 2018. doi: 10.1111/brv.12413. [DOI] [PubMed] [Google Scholar]
- 74.Soares MJ, Varberg KM, Iqbal K. Hemochorial placentation: development, function, and adaptations. Biol Reprod 99: 196–211, 2018. doi: 10.1093/biolre/ioy049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lefebvre T, Roche O, Seegers V, Cherif M, Khiati S, Gueguen N, Desquiret-Dumas V, Geffroy G, Blanchet O, Reynier P, Legendre G, Lenaers G, Procaccio V, Gascoin G. Study of mitochondrial function in placental insufficiency. Placenta 67: 1–7, 2018. doi: 10.1016/j.placenta.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Panda B, Panda A, Ueda I, Abrahams VM, Norwitz ER, Stanic AK, Young BC, Ecker JL, Altfeld M, Shaw AC, Rueda BR. Dendritic cells in the circulation of women with preeclampsia demonstrate a pro-inflammatory bias secondary to dysregulation of TLR receptors. J Reprod Immunol 94: 210–215, 2012. doi: 10.1016/j.jri.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Pineda A, Verdin-Teran SL, Camacho A, Moreno-Fierros L. Expression of toll-like receptor TLR-2, TLR-3, TLR-4 and TLR-9 is increased in placentas from patients with preeclampsia. Arch Med Res 42: 382–391, 2011. doi: 10.1016/j.arcmed.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 78.Williamson RD, McCarthy FP, Kenny LC, McCarthy CM. Activation of a TLR9 mediated innate immune response in preeclampsia. Sci Rep 9: 5920, 2019. doi: 10.1038/s41598-019-42551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374: 546–549, 1995. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 80.Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A 98: 9237–9242, 2001. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Osikoya O, Jaini PA, Nguyen A, Valdes M, Goulopoulou S. Effects of low-dose aspirin on maternal blood pressure and vascular function in an experimental model of gestational hypertension. Pharmacol Res 120: 267–278, 2017. doi: 10.1016/j.phrs.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 82.Goulopoulou S, Wenceslau CF, McCarthy CG, Matsumoto T, Webb RC. Exposure to stimulatory CpG oligonucleotides during gestation induces maternal hypertension and excess vasoconstriction in pregnant rats. Am J Physiol Heart Circ Physiol 310: H1015–H1025, 2016. doi: 10.1152/ajpheart.00834.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goulopoulou S, Matsumoto T, Bomfim GF, Webb RC. Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin Sci (Lond) 123: 429–435, 2012. doi: 10.1042/CS20120130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He B, Yang X, Li Y, Huang D, Xu X, Yang W, Dai Y, Zhang H, Chen Z, Cheng W. TLR9 (Toll-like receptor 9) agonist suppresses angiogenesis by differentially regulating VEGFA (vascular endothelial growth factor A) and sFLT1 (soluble vascular endothelial growth factor receptor 1) in preeclampsia. Hypertension 71: 671–680, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10510. [DOI] [PubMed] [Google Scholar]
- 85.Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, Saito S, Sadovsky Y, Sharma S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis 10: 927, 2019. doi: 10.1038/s41419-019-2162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weel IC, Romao-Veiga M, Matias ML, Fioratti EG, Peracoli JC, Borges VT, Araujo JP, Jr, Peracoli MT. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol 123: 40–47, 2017. doi: 10.1016/j.jri.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Velez DR, Menon R, Simhan H, Fortunato S, Canter JA, Williams SM. Mitochondrial DNA variant A4917G, smoking and spontaneous preterm birth. Mitochondrion 8: 130–135, 2008. doi: 10.1016/j.mito.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Cowell W, Brunst K, Colicino E, Zhang L, Zhang X, Bloomquist TR, Baccarelli AA, Wright RJ. Placental mitochondrial DNA mutational load and perinatal outcomes: Findings from a multi-ethnic pregnancy cohort. Mitochondrion 59: 267–275, 2021. doi: 10.1016/j.mito.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiu C, Hevner K, Enquobahrie DA, Williams MA. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int J Mol Epidemiol Genet 3: 237–244, 2012. [PMC free article] [PubMed] [Google Scholar]
- 90.McCarthy C, Kenny LC. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep 6: 32683, 2016. doi: 10.1038/srep32683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anelli GM, Cardellicchio M, Novielli C, Antonazzo P, Mazzocco MI, Cetin I, Mando C. Mitochondrial content and hepcidin are increased in obese pregnant mothers. J Matern Fetal Neonatal Med 31: 2388–2395, 2018. doi: 10.1080/14767058.2017.1344209. [DOI] [PubMed] [Google Scholar]
- 92.McElwain C, McCarthy CM. Investigating mitochondrial dysfunction in gestational diabetes mellitus and elucidating if BMI is a causative mediator. Eur J Obstet Gynecol Reprod Biol 251: 60–65, 2020. doi: 10.1016/j.ejogrb.2020.04.037. [DOI] [PubMed] [Google Scholar]
- 93.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–445, 2011. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 94.Yuzefovych LV, Pastukh VM, Ruchko MV, Simmons JD, Richards WO, Rachek LI. Plasma mitochondrial DNA is elevated in obese type 2 diabetes mellitus patients and correlates positively with insulin resistance. PLoS One 14: e0222278, 2019. doi: 10.1371/journal.pone.0222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roberts JM, Bodnar LM, Patrick TE, Powers RW. The role of obesity in preeclampsia. Pregnancy Hypertens 1: 6–16, 2011. doi: 10.1016/j.preghy.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Montoro MN, Kjos SL, Chandler M, Peters RK, Xiang AH, Buchanan TA. Insulin resistance and preeclampsia in gestational diabetes mellitus. Diabetes Care 28: 1995–2000, 2005. doi: 10.2337/diacare.28.8.1995. [DOI] [PubMed] [Google Scholar]
- 97.Busnelli A, Lattuada D, Ferrari S, Reschini M, Colciaghi B, Somigliana E, Fedele L, Ferrazzi E. Mitochondrial DNA copy number in peripheral blood in the first trimester of pregnancy and different preeclampsia clinical phenotypes development: a pilot study. Reprod Sci 26: 1054–1061, 2019. doi: 10.1177/1933719118804410. [DOI] [PubMed] [Google Scholar]
- 98.Marschalek J, Wohlrab P, Ott J, Wojta J, Speidl W, Klein KU, Kiss H, Pateisky P, Zeisler H, Kuessel L. Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia - A matched case-control study. Pregnancy Hypertens 14: 195–199, 2018. doi: 10.1016/j.preghy.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 99.Miliotis S, Nicolalde B, Ortega M, Yepez J, Caicedo A. Forms of extracellular mitochondria and their impact in health. Mitochondrion 48: 16–30, 2019. doi: 10.1016/j.mito.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 100.Meddeb R, Dache ZA, Thezenas S, Otandault A, Tanos R, Pastor B, Sanchez C, Azzi J, Tousch G, Azan S, Mollevi C, Adenis A, El Messaoudi S, Blache P, Thierry AR. Quantifying circulating cell-free DNA in humans. Sci Rep 9: 5220, 2019. doi: 10.1038/s41598-019-41593-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee JY, Lee DC, Im JA, Lee JW. Mitochondrial DNA copy number in peripheral blood is independently associated with visceral fat accumulation in healthy young adults. Int J Endocrinol 2014: 1–7, 2014. doi: 10.1155/2014/586017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Knez J, Winckelmans E, Plusquin M, Thijs L, Cauwenberghs N, Gu Y, Staessen JA, Nawrot TS, Kuznetsova T. Correlates of peripheral blood mitochondrial DNA content in a general population. Am J Epidemiol 183: 138–146, 2016. doi: 10.1093/aje/kwv175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stawski R, Walczak K, Kosielski P, Meissner P, Budlewski T, Padula G, Nowak D. Repeated bouts of exhaustive exercise increase circulating cell free nuclear and mitochondrial DNA without development of tolerance in healthy men. PLoS One 12: e0178216, 2017. doi: 10.1371/journal.pone.0178216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, El Messaoudi S, Mazard T, Prevostel C, Thierry AR. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J 34: 3616–3630, 2020. doi: 10.1096/fj.201901917RR. [DOI] [PubMed] [Google Scholar]