

Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia despite substantial efforts to understand the pathophysiology of the condition and develop improved treatments. Identifying the underlying causative mechanisms of AF in individual patients is difficult and the efficacy of current therapies is suboptimal. Consequently, the incidence of AF is steadily rising and there is a pressing need for novel therapies. Research has revealed that defects in specific molecular pathways underlie AF pathogenesis, resulting in electrical conduction disorders that drive AF. The severity of this so-called electropathology correlates with the stage of AF disease progression and determines the response to AF treatment. Therefore, unravelling the molecular mechanisms underlying electropathology is expected to fuel the development of innovative personalized diagnostic tools and mechanism-based therapies. Moreover, the co-creation of AF studies with patients to implement novel diagnostic tools and therapies is a prerequisite for successful personalized AF management. Currently, various treatment modalities targeting AF-related electropathology, including lifestyle changes, pharmaceutical and nutraceutical therapy, substrate-based ablative therapy, and neuromodulation, are available to maintain sinus rhythm and might offer a novel holistic strategy to treat AF.
Atrial fibrillation (AF) is the most common serious cardiac arrhythmia in Western countries1,2. AF occurs when abnormal electrical impulses suddenly start firing in the atria and override the heart’s natural pacemaker, which can no longer control the heart’s rhythm. AF causes irregular and often abnormally fast contractions of the atrial cardiomyocytes, resulting in various symptoms, including an irregular heart rate, palpitations, dizziness, shortness of breath and tiredness. AF can be classified based on disease persistence or aetiology. When classification is based on persistence, AF is categorized into four classes. When AF episodes terminate spontaneously or with intervention within 7 days of onset, AF is defined as paroxysmal AF. Continuous AF sustained for at least 7 days or terminated by cardioversion (pharmaceutical or electrical cardioversion) after the seventh day is defined as persistent AF. Continuous AF sustained for at least 12 months and in combination with a rhythm control strategy is defined as long-standing persistent AF. In cases in which the patient and doctor decide to stop attempts to restore or maintain sinus rhythm, AF is termed permanent AF. AF is mainly found in older individuals (>70 years of age) and those with lifestyle-related conditions such as high blood pressure, diabetes mellitus and obesity1. In addition, AF might also be triggered in specific situations, for example, binge drinking (colloquially referred to as ‘holiday heart syndrome’) and stress. Based on well-described risk factors for AF, AF can be classified as ‘wear-and-tear’ AF (that is, induced by environmental factors), congenital AF or genetic AF. Wear-and-tear AF is associated with changes related to ageing and with Western dietary and lifestyle risk factors such as hypertension, diabetes mellitus, obesity, coronary artery diseases and various other conditions, including chronic kidney disease and inflammatory diseases1. AF also occurs in some patients with a congenital heart disease (estimated 4.7%), which is termed congenital AF and results from a combination of embryogenesis defects (which themselves might be partially due to genetic mutations) and peri-operative and post-operative factors related to surgical treatment for their heart disease3. Congenital AF onset occurs at a younger age than for other forms of AF and often progresses rapidly from persistent to permanent AF4,5. For ~15% of patients with AF, the condition also occurs in family members, suggesting a genetic predisposition6–9; multiple studies have revealed a prominent role for genetic variants in driving AF. Currently, the pathophysiological mechanisms underlying wear-and-tear, congenital and genetic AF have only been partly elucidated.
To date, the selection of an optimal strategy for effective management of AF is challenging owing mainly to an incomplete understanding of the aetiology of AF, which has resulted in a lack of effective AF diagnostic instruments and therapies. As life expectancy is increasing worldwide, the steep rise in the prevalence of AF in the general population is becoming an urgent public health issue, especially in the Western world and parts of the Eastern world. Furthermore, AF might severely affect a patient’s quality of life as it is associated with serious complications, such as stroke, heart failure, cognitive impairment and sudden cardiac arrest, which result in increased morbidity and mortality10 and increasing health-care costs11.
Current treatment of AF is in large part based on catheter ablation, which aims to eliminate either the trigger initiating AF or the underlying arrhythmogenic substrate (a pre-existing condition necessary for arrhythmia induction) using either heat (radiofrequency ablation) or freezing (cryoablation). As AF episodes might be triggered by foci located predominantly within the pulmonary veins, pulmonary vein isolation — electrically isolating the pulmonary veins by creating a circumferential lesion around the ostia of the right and left pulmonary veins — was introduced in the 1990s. Over time, additional ablation strategies have been proposed, including the creation of linear lesions or targeted ablation of complex fractionated atrial electrograms, low-voltage areas, or rotational activity, amongst others. Catheter ablation is more effective than anti-arrhythmic drugs in maintaining sinus rhythm and is therefore currently the cornerstone of AF therapies1,12–14. Although invasive isolation therapy of the pulmonary veins is promising in early stages of AF, AF recurrences occur in up to 70% of patients with persistent AF within 1 year of the first pulmonary vein isolation and therefore require multiple procedures1,15. Despite these recurrences, catheter ablation has proved beneficial in reducing the AF burden and improving patient quality of life16,17.
Currently available pharmacotherapies for AF, which originate from the 1960s, target ion channels and do not prevent AF onset or progression in 85% of patients1. Lack of effect is likely because these drugs are not directed at the molecular causes of AF. Moreover, anti-AF drug usage is limited by their potential adverse effects, which can be severe or even life-threatening1. The response of an individual patient to AF therapy often cannot be predicted. Despite the identification of novel druggable targets that are involved in the pathogenesis of AF18–21, translation of these findings to clinical drug studies is limited. The absence of curative AF therapies runs in parallel with the absence of knowledge of AF pathogenesis in the individual patient.
To ensure the success of mechanism-based personalized AF therapies, a diagnostic tool to stage the severity of AF pathology is indispensable. Despite the rapid development of mobile health technologies for detecting AF, AF can currently only be accurately diagnosed with a surface electrocardiogram (ECG) when a patient already has symptomatic AF. As surface ECG recordings only provide a far-field view of the electrical patterns of activation, they do not provide information on the severity of AF and therefore cannot be used to assess the stage of AF22–24. Consequently, the selection and development of mechanism-based, personalized AF treatment modalities are severely hampered, thus prolonging the tremendous physical and psychological impact on patients and family members as well as burdening health-care systems and, ultimately, the entire society.
To improve AF therapy and diagnostics, research has increasingly focused on dissection of the molecular and electrical mechanisms underlying AF pathogenesis. Evidence from experimental and clinical studies of AF indicates a key role for so-called electropathology as a driver of AF. Electropathology is defined as electrical conduction disorders, and consequently contractile dysfunction, that are caused by molecular changes in atrial tissue that drive structural changes (including myolysis, dilation and fibrosis) and AF initiation and perpetuation. Emerging key pathways in which molecular changes might occur include protein homeostasis, stress signalling and inflammasome activation, which result in impairment of cardiomyocyte calcium handling, complex patterns of electrical activation and, thus, in contractile dysfunction. Importantly, key modulators within these pathways also represent potential druggable and diagnostic targets and therefore might aid in achieving mechanism-based and personalized AF management25–27.
In this Primer, we use the 2020 European Society of Cardiology (ESC) guidelines for the diagnosis and treatment of AF as a starting point to indicate directions for improvement in AF management. First, we describe the worldwide epidemiology of AF and elaborate on associations with modifiable and non-modifiable risk factors of AF. Second, technologies for accurate diagnosis of the severity of AF electropathology (‘staging’) and identification of patients at risk of developing AF are discussed. Third, we discuss the molecular and electrical mechanisms involved in AF pathophysiology that might be useful for the development of novel and effective therapies of AF. Finally, we highlight patient participation (via co-creation of studies) and data and biomaterial sharing as prerequisites to facilitate the design of patient-tailored diagnostic tools and the implementation of high-quality studies and innovative therapies.
Epidemiology
AF shows an increasing prevalence and incidence with advancing age24, which is the strongest risk factor amongst sex, smoking, alcohol consumption, body mass index, hypertension, left ventricular hypertrophy, significant heart murmur, heart failure and myocardial infarction28,29. The global epidemiology of AF with respect to socioeconomic and geographical risk factors, age, sex and genetics has recently been comprehensively reviewed28,29. The prevalence of AF varies between geographical regions (FIG. 1) and is also related to age, sex, ethnicity and other variables, including socioeconomic status. AF prevalence and incidence are higher in men than in women, irrespective of socioeconomic status or ethnicity. A familial predisposition or white ethnicity increase the risk for incident AF. Indeed, a family history of AF (‘familial AF’) is associated with a 40% increased risk for new-onset AF30. Globally, AF is associated with an increased risk of mortality and morbidity, with a loss of 6.0 million disability-adjusted life-years worldwide in 2017, conferring 0.24% of total disability-adjusted life-years globally31.
Fig. 1 |. Global prevalence of AF.
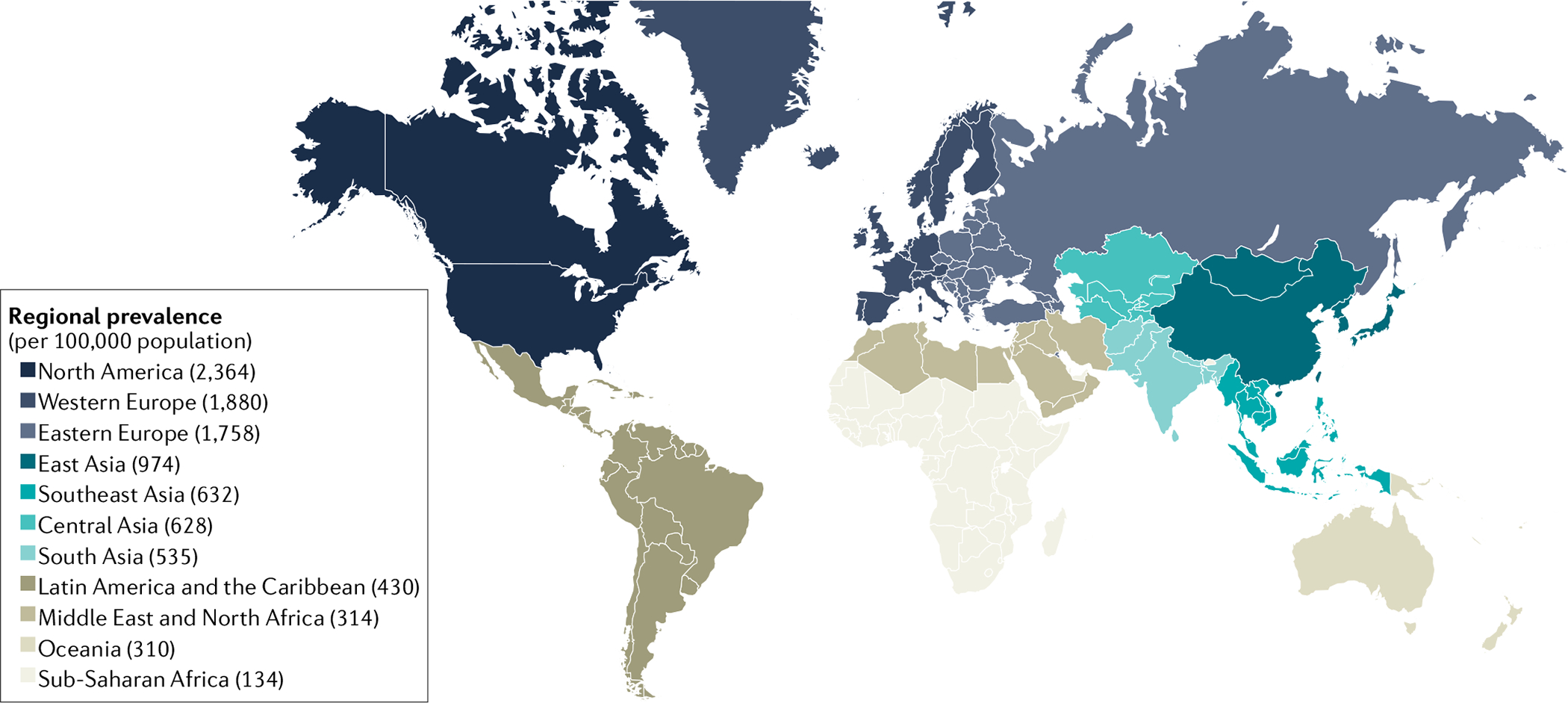
Regional prevalence (cases per 100,000 individuals) of atrial fibrillation (AF). The map shows the regions of high prevalence (Western Europe and North America) and the generally lower prevalence in South Asia, Oceania and the Middle East. Data are from the Global Burden of Disease (GBD) Collaborative Network’s GBD 2019 results (obtained using the GBD 2019 results tool).
AF prevalence is associated with high sociodemographic index regions based on average income per person, educational level and total fertility rate of each region as well as common lifestyle-related cardiovascular risk factors and comorbidities28,32. Lifestyle factors associated with increased AF risk include excessive consumption of alcohol, low-carbohydrate and high-fat intake, sleep apnoea, and sedentary behaviour or excessive physical exercise as a lifestyle33–37. Furthermore, Framingham data revealed that the risk factor burden, categorized as optimal (that is, well-controlled risk factors), borderline or elevated (that is, suboptimally managed risk factors), is associated with increased lifetime risk for AF. These risk factors include smoking, alcohol consumption, body mass index, blood pressure, diabetes, and history of heart failure or myocardial infarction38. An optimal risk factor profile was associated with a lifetime risk of AF of about 1 in 5 in adults; however, the risk of AF rose to 1 in 3 where there was at least one elevated risk factor38. Similar observations were reported for a Korean nationwide cohort, for which a combination of unhealthy lifestyle factors, including current smoking, excessive alcohol consumption (>30 g daily) and lack of regular exercise, were associated with the highest risk of incident AF39 (FiG. 2).
Fig. 2 |. Combinatorial effects of lifestyle factors on risk of AF.
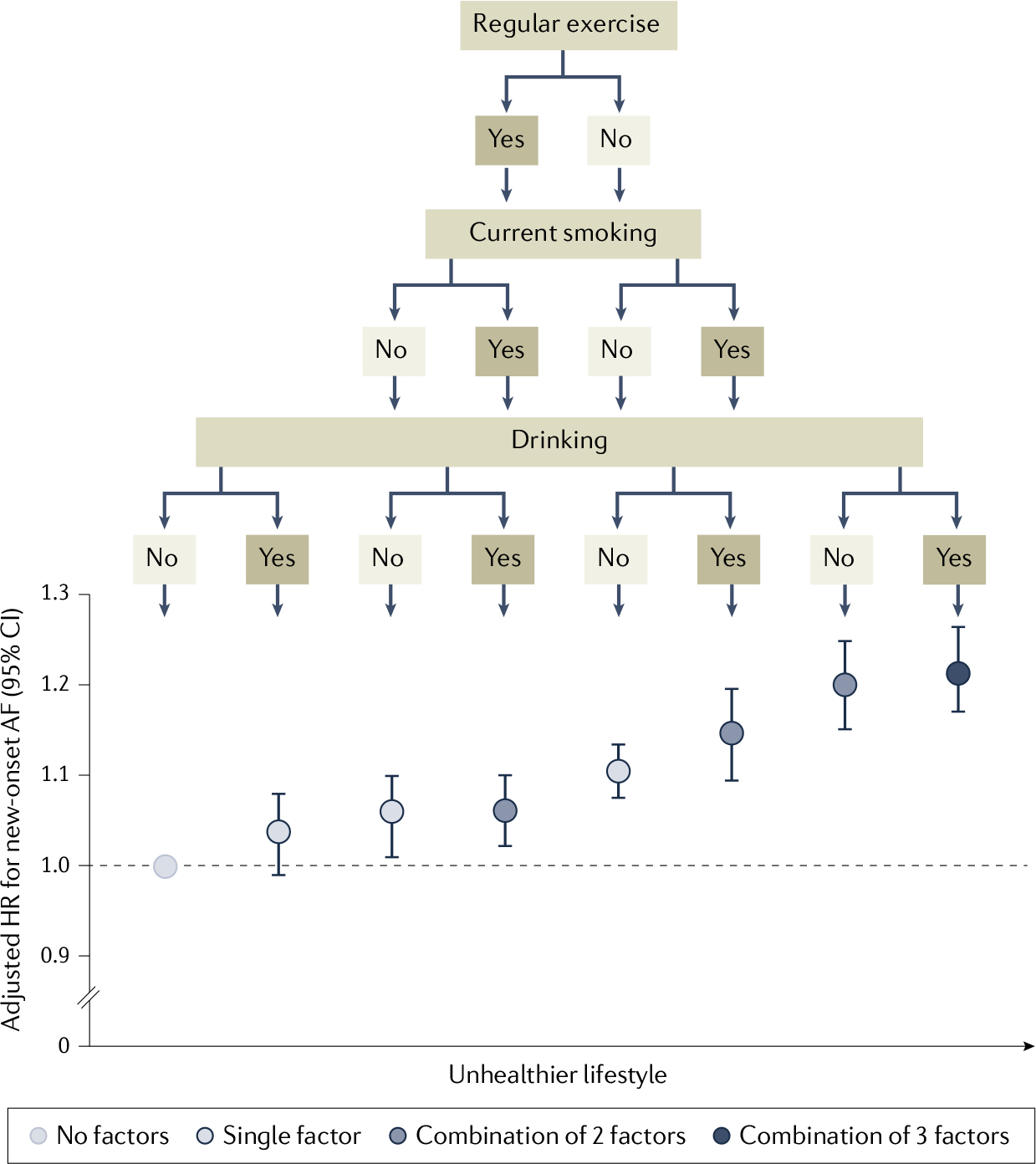
Unhealthy Lifestyle factors, such as Lack of regular exercise, smoking and excessive alcohol consumption, individually show no to moderate association with risk of new-onset atrial fibrillation (AF), with a lack of regular exercise having the strongest association with increased risk of AF (adjusted hazard ratio (HR) 1.11). However, the combined presence of smoking and lack of regular exercise markedly increases AF risk (HR 1.21), which is similar to the increase in risk of AF in individuals with all three risk factors (HR 1.22). Data are from a nationwide population-based study of 1,719,401 Korean individuals of 66 years of age39. Adapted from REF.39, CC BY 4.0.
The prevalence of AF is lower in Asian individuals (~1%) than in white individuals (~2%), notwithstanding the fact that Asian individuals experience a much higher overall disease burden given the proportionally larger aged population28,40. It is estimated that ~5.2 million men and ~3.1 million women over the age of 60 in China will suffer from AF by the year 2050, which is ~2.3-fold higher than the equivalent predicted prevalence for the United States28,40. This difference is likely due to the rising incidence of chronic diseases, including hypertension, metabolic syndrome and diabetes mellitus, related to urban lifestyle and dietary changes in the Chinese population40.
In approximately 15% of patients with AF, AF occurs in the absence of common wear-and-tear risk factors and onset is at a younger age6,7. AF might be familial in these patients, suggesting a heritable genetic predisposition. Familial AF is associated with variants in genes encoding ion channels, transcription factors, and cell coupling, cytoskeletal and intermediate filament proteins (TABLE 1). In addition, large population-based genome-wide association studies identified AF-associated single-nucleotide polymorphisms, which include those in genes encoding transcription factors involved in cardiac gene expression41. Genome-wide association studies have identified ~260 single-nucleotide polymorphisms in 166 loci, as associated with AF pathogenesis in multiple ethnicities, indicating a complex gene expression matrix regulated by a group of transcription factors, including PITX2, TBX5, GATA4, NKX2.5, SHOX2, ZFHX3, ETV1, PRRX1 and JUN7,42–48. The genes at the AF-associated loci broadly implicate pathways involved in cardiac development, calcium handling, contractile function and ion channel function.
Table 1 |.
Gene variants identified by whole-genome sequencing that are associated with familial AF
| Protein type | Gene variant | Pathogenetic mechanism | Refs | |
|---|---|---|---|---|
| Electrical | Molecular or functional | |||
| Sodium channels | ||||
| Voltage-gated channel β-subunit | SCN1B, SCN2B, SCN3B, SCN4B | LOF INa | NA | 259–262 |
| Voltage-gated channel α-subunit | SCN5A | GOF INa, spontaneous AP firing | NA | 263–265 |
| SCN10A | LOF and GOF | NA | 266 | |
| Potassium channels | ||||
| Pacemaker current in sinoatrial node | HCN4 | LOF and GOF If | Defective trafficking to cell membrane | 267,268 |
| Voltage-gated channels | ABCC9 | LOF IKATP, defective ADP-dependent channel opening | NA | 269 |
| KCNA5 | LOF and GOF IKur | Altered CaT, contraction | 270–273 | |
| KCND2 | GOF Ito | Nocturnal expression | 274 | |
| KCND3 | GOF Ito | Increased expression | 275,276 | |
| KCNE1 | GOF IKs | NA | 277,278 | |
| KCNE2 | GOF IKr | NA | 279,280 | |
| KCNE3 | GOF multiple currents | NA | 281 | |
| KCNE4 | GOF IKs | NA | 282 | |
| KCNE5 | GOF IKs | NA | 283 | |
| KCNH2 | GOF IKr | NA | 284,285 | |
| KCNQ1 | LOF and GOF IKs | GOF blocked by HMR-1556 | 286–289 | |
| Inward rectifier channels | KCNJ2 | GOF IK1 | NA | 290,291 |
| KCNJ3 | GOF IKACh | NIP151 blocks AF (zebrafish) | 292 | |
| KCNJ8 | NA | NA | 293 | |
| Calcium channels | ||||
| Voltage-gated α-subunit and β-subunit | CACNB2, CACNA2D4 | NA | NA | 294 |
| Cytoskeletal or cytoskeleton- associated proteins | ||||
| Desmin | DES | No effect | Increase in protein aggregates, PQC and autophagy | 295,296 |
| Lamin A/C | LMNA | ↓INa | Increase in PQC, HSP, myolysis, nuclear blebbing | 297–301 |
| Titin | TTN | Abnormal ECG | Disruption of sarcomeres, fibrosis (zebrafish) | 302–304 |
| Myosin heavy chain | MYH6, MYH7 | NA | Hypertrophy | 304–306 |
| Connexin 40, 43 | GJA5, GJA1 | LOF electrical conduction | LOF gap junction coupling | 307–309 |
| Junctophilin 2 | JPH2 | NA | LOF impaired RyR2 stabilization, spontaneous Ca2+ release | 310 |
| Nucleoporin 155 | NUP155 | ECG abnormalities, ↓APD | LOF nuclear localization, loss of nuclear permeability for HSP70 | 311 |
| Nesprin 2 | SYNE2 | NA | NA | 312 |
| Transcription factors | ||||
| Cardiac development | GATA4 | AP abnormalities | LOF gene expression, interaction TBX5, Ca2+ | 313–315 |
| GATA5 | NA | LOF gene expression | 316 | |
| GATA6 | NA | GOF gene expression | 317 | |
| NKX2–5 | NA | LOF gene expression | 318,319 | |
| NKX2–6 | NA | LOF gene expression | 320 | |
| TBX5 | ↑APD | GOF gene expression | 44,321,322 | |
| Asymmetrical development of organs | PITX2 | NA | GOF gene expression | 319,323,324 |
| Zinc finger homeobox protein | ZFHX3 | NA | LOF gene expression, damage of the ZFHX3 protein structure | 312 |
| Others | ||||
| Atrial differentiation | GREM2 | NA | NA | 325 |
| Hormone, extracellular fluid and electrolyte homeostasis | NPPA | GOF IKs, ↓APD and ECG abnormalities | ↑Inflammation, fibrosis | 288,326,327 |
AF, atrial fibrillation; AP, action potential; APD, action potential duration; CaT, calcium transient; ECG, electrocardiogram; GOF, gain of function; HSP, heat shock protein; If, pacemaker current; IKACh, acetylcholine-s ensitive potassium current; IKATP, ATP-s ensitive potassium current; IK1, inward rectifying potassium current; IKr, rapid delayed rectifier potassium current; IKs, outward potassium current; IKur, ultra-r apid delayed rectifier potassium current; INa, inward sodium current; Ito, transient outward potassium current; LOF, loss of function; NA, not available; PQC, protein quality control; RyR2, ryanodine receptor 2.
In addition to wear-and-tear AF and genetic AF, the incidence of congenital AF is increasing due to improved survival of patients with congenital heart disease owing to a refinement of surgical techniques and a higher quality of post-operative clinical care49,50. Consequently, the prevalence and incidence of AF are steeply increasing, especially in high sociodemographic index regions. This steep increase results in a major public health burden. The prevalence of risk factors related to wear-and-tear AF are steadily rising owing to lifestyle changes associated with urban life and dietary habits. Improved genetic AF testing will shed light on ethnicity-dependent genetic variants for AF and aid in the development of genetic risk scoring in routine clinical care. Knowledge of the underlying causes of risk factors that drive AF might stimulate novel approaches for improved AF management.
Mechanisms/pathophysiology
Electropathology as a root cause of AF
The general concept of AF is that paroxysms of AF are caused by ectopic activities. Ectopic activities represent spontaneous depolarizations of atrial tissue outside the sinoatrial node at rates faster than the sinus rhythm. Ectopic activities commonly originate from the pulmonary veins (95% versus 5% from the inferior and superior caval veins)51. Therefore, in theory, isolation of ectopic activity by circular lesions at the ostium of the pulmonary veins would eliminate AF. However, AF recurrences occur frequently after a successful pulmonary vein isolation52. These AF recurrences might be due to re-conduction across the scarred circular lesion53, although they might also be caused by the presence of an extensive arrhythmogenic substrate at various regions in the atria. It is generally assumed that progression from paroxysmal to (long-standing) persistent AF reflects progression from an arrhythmogenic trigger-mediated initiation of AF to an electropathology-mediated arrhythmia. As such, AF recurrence is caused by structural impairment of atrial tissue.
Evidence for electropathology as the root cause of clinical AF originates from intra-operative high-resolution electrical mapping studies of the epicardial surface of the entire right and left atrium, including the Bachmann bundle. The Bachmann bundle is a muscular bundle on the atrial septal roof connecting the right and left atrial appendages. Prior mapping studies in humans demonstrated that the Bachmann bundle is mainly activated by a wavefront propagating from the right to the left side but also by wavefronts emerging in the central part of the bundle. These variable patterns of activation are most likely caused by variations in the atrial architecture. Consequently, the Bachmann bundle is a preferred site for conduction disorders (Supplementary Table 1). In addition, the presence of AF episodes is associated with more severe conduction disorders at the Bachmann bundle54. Intra-operative mapping studies have provided important insights into the extent of electropathology in patients with a variety of underlying cardiovascular diseases and risk factors and who underwent cardiac surgery (summarized in Supplementary Table 1). Patients with non-dilated atria and normal left ventricular ejection fraction in whom AF was acutely induced differed significantly from patients with long-standing persistent AF with respect to longitudinal dissociation in conduction and prevalence of focal fibrillation waves. Focal fibrillation waves appear in the middle of the mapping area and expand towards the surrounding atrial tissue as a result of enhanced electrical asynchrony between the endocardial and epicardial layers of the atria55–57(Supplementary Box 1).
Patients with long-standing persistent AF have extensive conduction abnormalities along the lateral boundaries of the atrial musculature, resulting in a more than sixfold higher incidence of intra-atrial conduction block than in patients with acute AF58. Multiples lines of intra-atrial conduction block were also associated with an increase in the number of fibrillation waves58 (FiG. 3). These observations indicate that quantification of electrophysiological parameters, such as the amount of conduction block and the number of focal fibrillation waves, might be utilized to stage the severity of AF, which is important as the AF stage is most likely a major determinant of anti-arrhythmic therapy effectiveness. Interestingly, (random) reentry was only observed in patients with acutely induced AF and not in any patients with (long-standing) persistent AF58. A series of mapping studies have investigated whether electropathology is already present during sinus rhythm and whether it is influenced by the presence of cardiovascular comorbidities and risk factors (Supplementary Table 1). For this purpose, patterns of activation, including conduction disorders, such as conduction delay and conduction block, have been measured in large cohorts of patients (~400 patients) undergoing cardiac surgery for coronary artery disease, congenital heart disease and valvular heart disease. Collectively, these studies reveal that a certain degree of electropathology is present during sinus rhythm in all these patients. Importantly, the type of underlying heart disease has no effect on the degree and extensiveness of conduction disorders occurring during sinus rhythm. However, patients with AF episodes show more conduction disorders throughout both atria, which are most severe at the Bachmann bundle. In addition, patients with obesity present more often with both higher prevalence and more extensive and severe conduction disorders during sinus rhythm than patients without obesity (Supplementary Table 1).
Fig. 3 |. Overview of electrical conduction abnormalities in AF.

Electrical conduction is observed by high-resolution mapping of the atria during cardiac surgery. a | Simultaneously acquired endocardial and epicardial activation maps of the right atrial free wall obtained from two different patients with coronary artery disease undergoing cardiac surgery during sinus rhythm, demonstrating synchronous and asynchronous activation. Thus, asynchronous activation of the atrial wall might already be present during normal heart rhythms. Colours indicate the timing of activation (colour bar) in different parts of the mapping area. b | Asynchronous activation of the atrial wall is a prerequisite for the occurrence of transmural propagation of fibrillation waves, giving rise to focal waves. The focal wave maps demonstrate the incidence of focal fibrillation waves (red stars) emerging at each recording site (squares) during 8 s of acute atrial fibrillation (AF) (left) and long-standing persistent AF (centre) at the right atrial wall. Each square represents a recording site. During long-standing persistent AF, focal fibrillation waves (epicardial breakthroughs (EBs)) occur not only more frequently at the same site (although not repetitively) but also occur at more recording sites. The occurrence of focal waves in the patient with long-standing persistent AF was 0.57 per median AF cycle length per squared centimetre compared with only 0.05 during acute AF. The schematic (right) shows a wave map depicting each individual fibrillation wave containing 16 fibrillation waves within an area as small as 16 cm2.
Electrical and calcium remodelling in atrial cardio-myocytes.
AF is also related to changes in electrical remodelling of cardiac ion channels (such as Ca2+ and K+ channels)59,60. In patients with AF, ectopic activity occurs most often in the pulmonary veins. Experimental studies revealed that early afterdepolarization and delayed afterdepolarization (DAD) might underlie ectopic activity and thereby trigger AF onset61. The underlying experimentally ascertained molecular mechanisms of this ectopic activity include an increase in diastolic Ca2+ release from sarcoplasmic reticulum (SR) Ca2+-stores via leaky ryanodine receptor 2 (RyR2) Ca2+-release channels and altered ion channel function, resulting in shortening of action potential duration (APD)61 (Supplementary Box 2). Ca2+ entry via L-type Ca channels (ICaL), together with a smaller Ca2+ influx via the sodium–potassium exchanger NCX, activates Ca2+ release from the SR through RyRs in myocytes (FIG. 4). During systole, this Ca2+-triggered release of Ca2+ from the SR creates a large intracellular Ca2+ ([Ca]i) transient that drives myocyte contraction62. During diastole, RyRs are usually closed and the excess cytosolic Ca2+ is cleared from the cytosol either by pumping Ca2+ back into the SR by SERCA2 or removing Ca2+ from the cell, mostly by NCX62. However, RyRs can spontaneously (albeit rarely) open during diastole, which might produce a non-spark SR Ca2+ leak when individual RyRs open or a spark-mediated SR Ca2+ leak when opening of multiple RyRs drives local inter-RyR Ca2+-induced Ca2+ release. Of note, unusually large and frequent sparks might trigger propagating diastolic Ca2+ waves. Atypically high SR Ca2+ leak reduces SR Ca2+ levels and consequently decreases systolic fractional SR Ca2+ release for a given L-type voltage-gated Ca2+ current (ICaL) trigger63,64. Propagating Ca2+ waves result in excess NCX function during diastole, which is electrogenic (3 Na+ imported for each Ca2+ exported) and might generate abnormal triggered activities (such as DADs) and initiate arrhythmias62. Compared with the ventricles, the amplitude of atrial Ca2+ transient is smaller, the rate of intracellular Ca2+ decay is higher (due to increased SERCA uptake and NCX function) and SR Ca2+ content is higher65. When RyRs channels are sensitized under certain pathological conditions, this higher level of SR Ca2+ content makes atrial cardiomyocytes prone to spontaneous diastolic SR Ca2+ release62,66–69. However, a reduced SR Ca2+ content due to impaired Ca2+ uptake (owing to reduced SERCA2 activity) but significantly increased diastolic SR Ca2+ leakage results in increased cytosolic Ca2+ levels and abnormal triggered Ca2+ activities (Ca2+ sparks and waves), which consequently lead to abnormal triggered activities (such as DADs) and initiate atrial arrhythmias62. However, atria from patients with paroxysmal AF and animal models of ageing or of holiday heart syndrome show a different pathomechanism as increased diastolic SR Ca2+ leak is accompanied by an elevated SR Ca2+ content via increased SERCA2 activity70–72. This combined diastolic SR Ca2+ leak and overload aggravates arrhythmic Ca2+ waves and DADs and, ultimately, might trigger arrhythmias71–73.
Fig. 4 |. Overview of molecular pathways driving electropathology and AF.
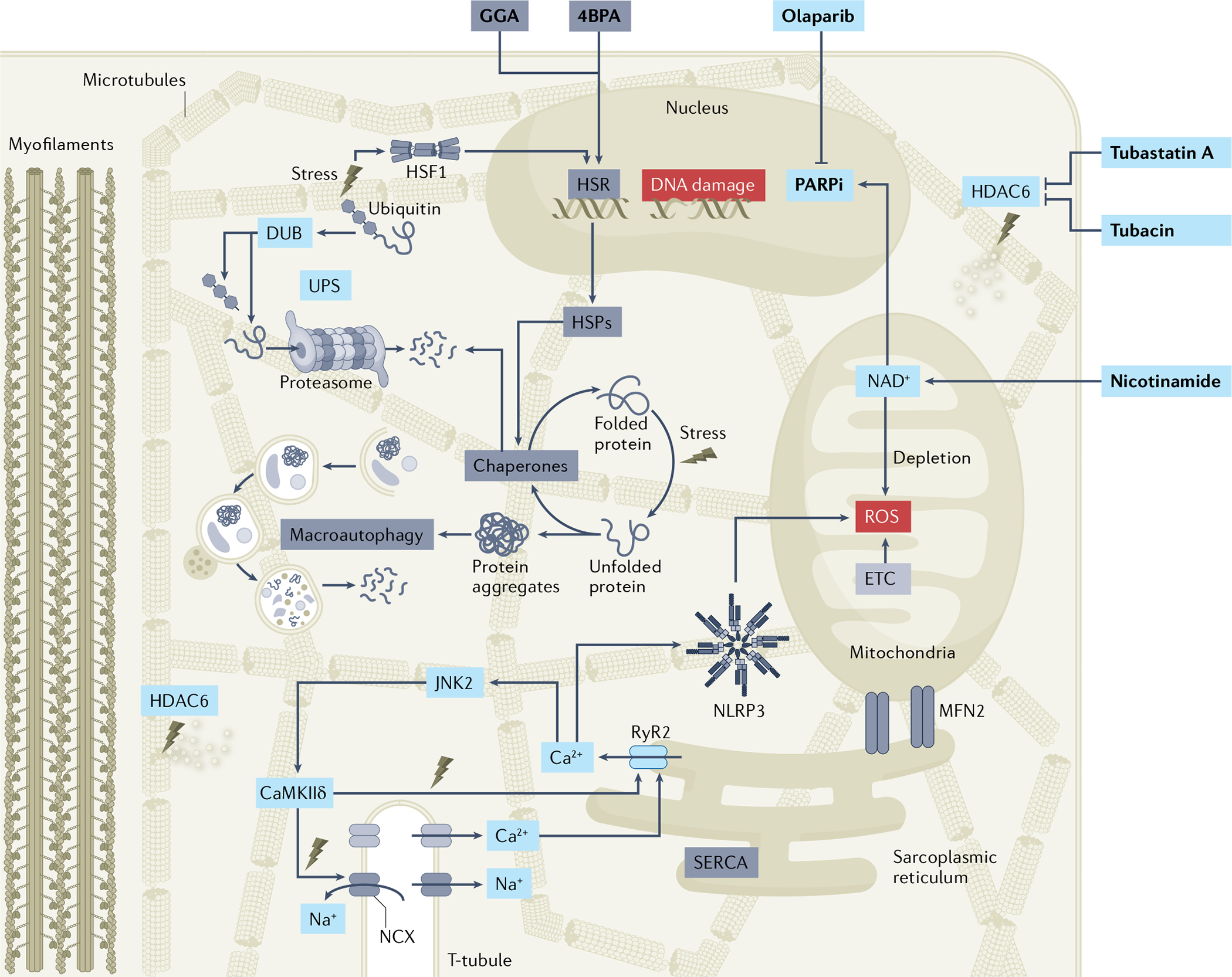
Atrial fibrillation (AF) causes loss of protein quality control via downregulation of the heat shock protein (HSP)-mediated heat shock response (HSR) and subsequent reduction in HSP expression levels and loss of chaperone activity. As HSPs represent the cell’s first line of defence against stress, the loss of stress response induces further endoplasmic reticulum stress and downstream excessive activation of the macroautophagy protein degradation pathway. AF also increases the activity of the histone deacetylase HDAC6, resulting in deacetylation of microtubules and destabilization of the microtubule network, Ca2+ handling alterations, and loss of cardiomyocyte contractile function (BOX 1). AF triggers DNA damage, poly(ADP-ribose) polymerase 1 (PARP1) activation and depletion of mitochondrial NAD+ levels, thereby causing electrophysiological and contractile impairment. Pharmacological treatments (in bold) that boost the HSR (for example, geranylgeranylacetone (GGA)), prevent endoplasmic reticulum stress (for example, 4-phenylbutyrate (4PBA)), inhibit HDAC6 (for example, tubastatin A and tubacin) or PARP1 (for example, olaparib) activity, or supplement NAD+ (for example, nicotinamide) protect against proteostasis dysfunction and AF progression in experimental model systems. As several of the proteoceutical compounds are marketed and off-patent, drug repurposing approaches might be within reach. The increased Ca2+ release from the sarcoplasmic reticulum (sarcoplasmic reticulum Ca2+ leak) in cardiomyocytes might directly activate the NLRP3 inflammasome by facilitating interactions between inflammasome components or might have indirect effects by promoting mitochondrial reactive oxygen species (ROS) production. In addition, increased levels of ROS can activate JUN N-terminal kinase 2 (JNK2) and Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ), resulting in excessive abnormal Ca2+ signalling. DUB, deubiquitylase; ETC, electron transport chain; MFN2, mitofusin 2; PARPi, PARP inhibitor; RyR2, ryanodine receptor 2; UPS, ubiquitin–proteasome system.
Molecular defects in electropathology
Complementary to the emerging findings on electrophysiological abnormalities as a readout for personalized AF diagnostics and therapies, identification of the underlying molecular causes of these abnormalities might greatly enhance AF management. Understanding the molecular defects might aid in the development of mechanism-based, potentially more effective, AF therapies. Analysis of human atrial tissue samples has revealed that AF results in sustained structural damage in atrial cardiomyocytes, including breakdown of the mitochondrial, cytoskeletal and sarcomeric networks (BOX 1), which is accompanied by autophagosome formation, suggestive for activation of autophagic protein degradation and changes in chromatin composition21,25. Molecular pathways involved in these structural changes include defective proteostasis, activation of stress signalling, genome instability and inflammatory signalling (FIG. 4). Elucidating the role of key modulators within these molecular pathways is the first step in developing novel diagnostic tools and therapies that specifically address electrical and contractile impairment and might aid in the treatment of AF at various stages.
Box 1 |. Sarcomeric cytoskeleton: backbone of the cardiomyocyte structure and function.
For balanced communication between components of the proteostasis machinery and correct cardiomyocyte function, an intact and functional sarcomeric cytoskeleton is crucial93,346. The cytoskeleton, which consists of actin filaments, intermediate filament proteins and microtubules, provides a network for communication between various organelles and contractile proteins. The cytoskeleton interacts with membrane-associated proteins (including connexins and desmosomes), sarcomeric proteins, various organelles and the nuclear envelope93. Together, the sarcomeric cytoskeleton supports mechanical contractions, signal transduction between organelles (such as the sarcoplasmic reticulum (SR), endoplasmic reticulum (ER), mitochondria and the nucleus) and transport of ubiquitylated proteins within the proteostasis network, and provides the shape and architecture of the cardiomyocytes. As such, the cytoskeleton represents the backbone of cardiomyocyte structure and function. In atrial fibrillation (AF), loss of the sarcomeric cytoskeleton network is precipitated by activation of HDAC6, which deacetylates and thereby depolymerizes α-tubulin; in this state, the microtubule network is susceptible to degradation by the protease calpain, which is activated by excess cytoplasmic Ca2+ accumulation19. Interestingly, a dominant-negative mutation in the α-tubulin catalytic domain of HDAC6 preserved the microtubule network and, as such, maintained electrical and contractile function19. Furthermore, AF-induced loss of contacts between the microtubule network and the SR, ER and mitochondria, via mitofusin 2 (MFN2), results in Ca2+ overload in organelles, upregulation of the unfolded protein response in the ER, and mitochondrial dysfunction86,124,223 and might potentially underlie spontaneous SR Ca2+ releases that lead to ectopic activity, triggering AF72,347,348. Moreover, excess Ca2+ accumulation in the cytosol activates calpain, resulting in sarcomeric, ion channel and cytoskeletal protein cleavage and, consequently, structural damage in atrial cardiomyocytes84,85. These findings underscore the central role of HDAC6 activation in AF-related structural damage and offer a novel therapeutic target in AF.
Defective proteostasis: the role of the protein quality control system.
Proteins are versatile, complex macromolecules that are involved in the correct functioning of all cells, including atrial cardiomyocytes. Human cardiomyocytes synthesize ~14,000 different proteins on ribosomes, representing 73% of the proteins encoded in the human genome74. To ensure maintenance of the proteome integrity, protein synthesis, maturation, function, transport and breakdown (that is, proteostasis) are closely monitored by a complex system called the protein quality control (PQC) system75,76. The main players in the PQC system include chaperone proteins, especially the stress response-related heat shock proteins (HSPs) that assist in the folding and refolding of proteins77–79, and the ubiquitin–proteasome and macroautophagy protein degradation pathways80,81. Protein degradation pathways clear expired, irreversibly misfolded or damaged proteins from the cell to prevent toxic protein aggregation and malfunction of the cardiomyocyte. The rapid activation rate of the atria during AF is a trigger for mechanical stress and cytoskeletal protein damage and, together with concomitant failure of the PQC system, results in activation of the stress response, increased formation of reactive oxygen species (ROS), and consequent oxidative damage to proteins and DNA21 (FIG. 4). In physiological conditions, cardiomyocytes express high levels of specific small HSPs, including the small HSP27, which localize to contractile proteins and the microtubule network, thereby stabilizing the structure and conserving contractile and electrophysiological functions of cardiomyocytes18,82,83. At the severe stage of persistent AF, human HSP27 levels become depleted in atrial tissue samples83. Consistent with this observation, boosting HSP expression by genetic and pharmacological means attenuates electropathology (including Ca2+ handling, ICaL, APD and structural abnormalities) and AF promotion in various experimental models of AF18. Thus, depletion of HSP levels is a prominent contributor to electropathology and thereby drives AF.
As mentioned previously, the rapid activation rate during AF causes marked structural stress to the cytoskeleton, resulting in degradation of cytoskeletal and sarcomeric proteins with concomitant excessive activation of macroautophagy and calcium overload-induced calpain protein degradation pathways, whereas the ubiquitin–proteasome system only has a minor role84–87. Macroautophagy is an evolutionarily conserved protein degradation pathway that clears misfolded and damaged proteins and organelles by sequestering them into autophagosomes, which fuse with lysosomes, resulting in degradation of their contents. The degradation end-products comprise amino and fatty acids that are recycled to generate ATP88. Although macroautophagy was originally described as a vital cellular process, excessive activation of this pathway, as observed in AF, triggers degradation of the cytoskeleton, impairment of calcium handling, and atrial electrical (ICaL and APD abnormalities) and contractile dysfunction86,89. The endoplasmic reticulum (ER)/SR stress response is an upstream trigger for autophagic protein degradation via the so-called unfolded protein response (UPR). The UPR induces the phosphorylation of the ER/SR stress sensor eIF2α at Ser31, resulting in protein translation inhibition and concomitant selective expression of stress-response transcripts, including activating transcription factor 4 (ATF4) and ATF6, respectively90. In turn, ATF4 and ATF6 signalling increases the expression and activation of CCAAT/enhancer-binding protein (C/EBP) homology protein (CHOP) and various autophagy proteins, including ATG12, MAP1LC3B (also known as LC3) and BiP (also known as HSPA5), which together stimulate elongation of autophagosomes and activate autophagic protein degradation91–94. In AF, ER stress-induced (excessive) activation of autophagy constitutes an important mechanism of electropathology as blocking ER stress by administration of the chemical chaperone 4-phenylbutyrate (4PBA), overexpression of the ER chaperone BiP or mutation of the gene encoding eIF2α inhibits excessive autophagy activation and thereby prevents electrical and contractile dysfunction86. Of note, although autophagy is needed for normal physiological function, excessive autophagy is toxic.
AF risk factor-evoked stress signalling pathways underlying electropathology.
A number of AF risk factors, including advanced age, heart failure, diabetes mellitus, alcohol abuse, intra-operative or post-operative atrial injury, and myocardial ischaemia, result in increased cellular stress1. Emerging evidence suggests that stress signalling pathways have an important role in AF pathogenesis. In addition to the role of the ER/SR stress and UPR pathways, mitogen-activated protein kinase (MAPK) stress signalling pathways are involved in the pathogenesis of AF. Key members of the MAPK family, JUN N-terminal kinases (JNKs), are activated in response to various stresses and are considered markers of ER stress95. JNKs orchestrate cellular stress responses and regulate cell differentiation, survival and migration, which, when aberrant, contribute to the development of cardiovascular diseases, including AF46,96. In particular, JNK2 but not JNK1 has a crucial role in the development of AF in humans and animal models of advanced age or exposed to alcohol binge drinking71–73,96. In these contexts, JNK2 has dual roles as it activates diastolic Ca2+ leak from the SR, which is known to have a proarrhythmogenic effect, while JNK2 simultaneously increases SR Ca2+ content by accelerating Ca2+ uptake into the SR through stimulation of SERCA2 activity71–73. While this accelerated Ca2+ uptake in the SR partially compensates for the toxic SR Ca2+ leak, elevated SR Ca2+ content further enhances the diastolic SR Ca2+ leakage, which together aggravates arrhythmic susceptibility. Moreover, JNK2-induced diastolic Ca2+ leak is a crucial activator of Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ, the predominant cardiac isoform), which is a validated pro-arrhythmic signaling molecule71,73. The pro-arrhythmic effects of CaMKIIδ are related to its promotion of RyR2 dysfunction and, consequently, Ca2+ handling abnormalities in human atrial samples and animal models67,68,71–73,97. Furthermore, activation of CamKIIδ results in dysregulation of the ‘late’ sodium current (INa,late), causing pro-arrhythmic activity in atrial cardiomyocytes from patients with AF and sleep-disordered breathing98,99. Extensive studies have also shown an important role of CamKIIδ in AF pathogenesis, while elevated ROS promote CaMKIIδ activation by oxidation of Met280 and Met281 in the protein, representing an alternative, dynamic, calcium-independent mechanism of CaMKIIδ activation99,100. In addition, ROS can also alter the function of gap junctions by activating CaMKIIδ or JNK2, whereas JNK2-specific regulation of CaMKIIδ activation and SR Ca2+ mishandling is independent of both the intracellular Ca2+ concentration and oxidative stress101,102. However, in non-cardiac myocytes, ROS are known to activate NLRP3 inflammasomes, which can lead to increased atrial fibrosis20,103–105. In post-operative patients with AF (with no history of AF prior to open-heart surgery), activation of NLRP3 inflammasome signalling augments CaMKIIδ-dependent hyperphosphorylation of RyR2 and arrhythmic Ca2+ activities68,69,97,106–109. This augmented CaMKIIδ-dependent SR Ca2+ mishandling was further found to underlie AF pathological remodelling in an atrial tachypaced canine AF model and in a CREM-transgene mouse model of spontaneous AF110–112. Thus, current studies have demonstrated that dysregulation of a number of different stress-evoked signalling pathways underlies atrial molecular and electrophysiological remodelling and drives AF promotion. Although electrical remodelling of cardiac membrane ion channels leads to altered action potentials and/or triggered activities, these alterations alone might not be sufficient to provide an arrhythmogenic substrate62.
AF has been considered as a reentrant arrhythmia; both a shortened atrial effective refractory period and reduced conduction velocity of action potentials have been linked to AF113,114. Sodium channels are responsible for generating action potential in each cell through the fast sodium current (INa), while gap junctional channels directly connect adjacent cells by providing chemical and electrical communication between adjacent myocytes. Accumulating evidence from humans and animal models suggests that stressed hearts, owing to ischaemia, obesity, heart failure or ageing, have delayed atrial conduction due to altered Na+ or gap junction channels46,56,96,99,115. Recent experimental findings support the functional contribution of gap junctional channels in the reduced conductivity and reentry that might underlie AF in the setting of heart failure, myocardial ischaemia and ageing46. The pivotal role of the JNK stress kinases in suppressing the expression of CX43 (encoding the gap junction protein connexin 43), and consequently hampering cell-to-cell communication, has been recognized as an arrhythmogenic substrate for AF46,72. Additionally, overexpression of connexin 43 using atrial gene transfer corrected slow conduction caused by connexin 43 downregulation and reduced AF propensity116, implying the critical role of connexin 43 in enhanced AF risk. Of note, JNK2 is implicated in both impaired cell-to-cell communication and slowed atrial conduction velocity via reduced expression of CX43 and enhanced triggering of Ca2+ activities via JNK2-driven SR Ca2+ mishandling71–73,96,117 to generate DADs and promote AF. However, this pathway is independent of atrial fibrosis formation in the setting of ageing in both humans and animals118,119. Together, pathological molecular and structural remodelling forms a reentry substrate that facilitates the maintenance of AF, and the stress kinase JNK2 is an important molecule acting as an upstream stress integrator that drives AF pathogenesis. Future studies are needed to better understand the role of the stress signalling cascades and the crosstalk between pathways in proteostasis disruption that drives AF development.
Genome instability, DNA repair and mitochondrial dysfunction.
Maintaining the integrity of the proteostasis network is an energy-consuming process in cardiomyocytes79 and AF puts further pressure on the energy-producing capacity of the mitochondria. Flaws in mitochondrial energy production, via depletion of NAD+/NADH levels and therefore reduced electron transport chain activity, result in an increase in ROS production and oxidative protein and DNA damage120–122, and emerging evidence indicates a role for this sequela in AF123,124. AF-related electropathology is precipitated by excessive stimulation of the DNA repair machinery via poly(ADP-ribose) polymerase 1 (PARP1) activation in response to oxidative DNA damage21. In turn, PARP1-mediated synthesis of ADP-ribose chains depletes mitochondrial NAD+ levels and results in energy loss and ROS production, further driving oxidative DNA damage and impairment of electrical conduction, calcium handling, and ion channel and contractile function21. Interestingly, replenishment of NAD+ or pharmacological inhibition or genetic depletion of PARP1 prevent electrical and contractile loss of function21,125. Furthermore, inhibition of PARP1 protects against NAD+ depletion, oxidative stress, DNA damage and contractile dysfunction in experimental model systems of AF. Consistent with these findings, cardiomyocytes of patients with persistent AF also show substantial DNA damage, involving both single-strand and double-strand breaks, which correlates with PARP1 activity21,125. These findings not only indicate this DNA damage pathway as a novel mechanism driving electropathology and AF promotion but also suggest PARP1 inhibition and/or NAD+ supplementation as a possible therapeutic intervention that might preserve the proteostasis network and cardiomyocyte function in clinical AF.
Inflammasome activation.
The NLRP3 inflammasome is activated in response to danger signals, including viral or bacterial pathogens or DNA, RNA, ATP, and nuclear and cytosolic molecules released from damaged cells20. On activation, NLRP3 triggers the release of the cytokines IL-1β and IL-18, which mediate a proinflammatory response. In experimental AF, the NLRP3 inflammasome is activated in atrial cardiomyocytes, enhancing RyR2-mediated SR Ca2+ release and ectopic firing, which results in impaired electrical conduction that promotes AF onset20,25 (FiG. 4). Genetic suppression of NLRP3 levels prevents the development of AF in transgenic mouse models, supporting a causative role for NLRP3 in AF onset20,25.
In conclusion, several experimental studies have identified important molecular mechanisms underlying AF and novel druggable targets to attenuate electropathology and AF. Future research should be directed towards translating these findings into diagnostic tools for (patient-tailored) screening and prevention of AF.
Diagnosis, screening and prevention
Innovative solutions for diagnostic AF tools
Patients with AF might be asymptomatic or they might present with a variety of symptoms, including palpitations, (near) syncope, chest pain or fatigue. AF is currently staged according to the clinical presentation and duration of AF episodes as paroxysmal, (long-standing) persistent or permanent AF. Diagnosis of AF requires documentation of an AF episode lasting >30 s and recorded by either a 12-lead ECG or a single lead. In case of infrequent episodes, long-term recordings using, for example, Holter monitors are mandatory. In the past decade, several mobile health devices have been introduced that facilitate early diagnosis of AF, including hand-held devices, wearable patches, bio-textiles, smartphones and smartwatches1. Nevertheless, owing to false-positive notifications, each potential AF episode documented by mobile devices needs to be reviewed by a physician experienced in the analysis of rhythm registrations. Unfortunately, one or more surface electrocardiogram lead recordings do not provide any information on the degree of AF persistence and severity of AF-related electropathology. Therefore, a diagnostic tool capable of measuring the severity of AF-related electropathology in the atria could facilitate personalized AF diagnosis and consequently also AF therapy.
As described above, the stage of AF is correlated with the complexity of the patterns of activation in the atrial tissue as a result of conduction disorders. The complexity of patterns of activation is in turn reflected in the morphology of unipolar electrograms126. During smooth propagation, unipolar potentials consist of one single deflection, which is preceded and followed by, respectively, a positive and negative wave of variable magnitudes. In patients with paroxysmal AF, the reduction of conduction velocity is associated with a decrease in amplitudes of single potentials caused by a loss of the S-wave amplitude, which occurs particularly at Bachmanns bundle127,128. During inhomogeneous patterns of activation, which are related to areas of conduction delay or block, unipolar electrograms consist of multiple, low-amplitude, fractionated potentials instead of potentials with a single deflection (FIG. 3).
Therefore, an electrical signal fingerprint, consisting of quantified features of potential morphology, might reflect the degree of inhomogeneity in conduction and could serve as a diagnostic tool to identify electropathology and thus stage AF. Recently, 1,763,593 potentials were intra-operatively collected from the epicardial surface of 189 patients with coronary artery disease. The potentials demonstrated that quantified features of potential morphology, summarized in an electrical signal fingerprint, corresponded to the severity and extensiveness of conduction inhomogeneity129. Further studies are required to determine whether the electrical signal fingerprint, deduced by measuring the severity and extensiveness of conduction inhomogeneity, can be used to identify patients at risk for AF onset or progression. Once the invasively constructed gold-standard electrical signal fingerprint has been established, it might stimulate the development of less invasive and even non-invasive signal fingerprints for the accurate staging of AF.
The electrical signal fingerprint might be complemented with a biological signal fingerprint. Several clinical studies have demonstrated a positive correlation between proteostasis, DNA damage and inflammatory biomarkers in human atrial tissue and/or blood samples and AF incidence and onset of post-operative AF (TABLE 2). Potential mechanism-based markers for biological signal fingerprinting include blood levels of mitochondrial DNA, the oxidative stress marker 8-hydroxy-2′-deoxyguanosine (8-OHdGX), HSPs, and inflammatory markers such as C-reactive protein (CRP), IL-6, IL-1β, myeloperoxidase (MPO) and tumour necrosis factor (TNF)123,130–135. Most of these markers correlate with the progression of AF and also have value in predicting the outcome of AF ablation and onset of post-operative AF130. Although fibrosis makers have been proposed as biological markers for AF diagnostics and therapeutics, a study showed no correlation between the degree of fibrosis and the severity and extensiveness of conduction inhomogeneity in atrial appendages of patients in various stages of AF136. This observation suggests that fibrosis is of limited value as a biomarker for electropathology, AF staging and signal fingerprinting136. These findings indicate that, in addition to electrical signal fingerprinting, mechanism-based biological signal fingerprinting might also have value in identifying patients at risk for AF onset or progression. Whether a single or a combination of biomarkers is needed to accurately diagnose and stage AF remains to be elucidated.
Table 2 |.
Clinical studies showing a correlation between mechanism-b ased markers and AF
| Markera | Material | Correlation with AF | Refs |
|---|---|---|---|
| Proteostasis or protein quality control | |||
| HSPB1 (HSP27) | Atrial tissue | Levels inversely correlated with duration of AF and extent of structural damage (myolysis) | 83 |
| Blood | Concentration predicts AF recurrence after PVI ablation | 131 | |
| Concentration correlates with LAD, LAV and fractionated intervals | 328 | ||
| HSPB7 | Blood | Concentration does not correlate with PAF, PeAF or AF recurrence after PVI ablation | 131 |
| HSPA1A (HSP70) | Atrial tissue | Low levels correlate with high incidence of POAF | 329,330 |
| Blood | Concentration does not correlate with PAF, PeAF or AF recurrence after ablation | 131 | |
| No correlation with incidence of POAF | 329 | ||
| HSPD1/E1 (HSP60/10) | Atrial tissue | Reduced levels in AF with spontaneous SR restoration | 331 |
| Increased in PeAF versus SR | 332 | ||
| No correlation AF | 131,333 | ||
| Blood | Levels of anti-H SPD1 antibodies correlate with POAF | 334 | |
| Levels do not correlate with PAF, PeAF or AF recurrence after ablation | 131 | ||
| Autophagy or mitophagy | |||
| mtDNA (ND1, COX3) | Blood | Increase in PAF in men Increase in AF recurrence after PVI ablation and EC |
123 |
| Increase in POAF | 335 | ||
| DNA damage | |||
| mtDNA (ND1, COX3) | Blood | Increase in PAF in men Increase in AF recurrence after PVI ablation and EC |
123 |
| 8-OHdG | Atrial tissue | Increase in AF vs SR | 130 |
| Blood | Gradual increase in PAF, PeAF, LSPeAF Increase in AF recurrence after PVI ablation Increase in POAF |
130 | |
| Cytoskeletal proteins | |||
| cTnT, hsTnT | Atrial tissue | Gradual decrease in PAF and PeAF vs SR | 84 |
| Blood | Increase correlates with AF onset, recurrence, POAF | 336 | |
| Increase correlates with systemic embolic events in AF | 135 | ||
| cTnI | Atrial tissue | Gradual decrease in PAF and PeAF vs SR | 84 |
| Blood | Increase correlates with AF onset, recurrence, POAF | 336 | |
| cTnC | Atrial tissue | Gradual decrease in PAF and PeAF vs SR | 84 |
| Inflammation | |||
| CRP | Blood | Gradual increase in PAF, PeAF | 132,337 |
| Association with new-o nset AF in CAD | 133 | ||
| Increase correlated with successful EC | 132 | ||
| IL-2 | Blood | Increase in AF recurrence after PVI ablation | 338 |
| IL-6 | Blood | Gradual increase in PAF, PeAF | 339 |
| Correlation with AF and new- onset AF in CKD | 134 | ||
| Correlation with POAF | 340 | ||
| IL-17A | Blood | Increased risk for AF | 341 |
| IL-18 | Blood | Gradual increase in PAF, PeAF | 342 |
| TNF | Blood | Increase in AF versus SR | 337,343 |
| MPO | Blood | Increase in AF versus SR, especially left atrial blood | 344 |
| Levels correlate with AF recurrence after PVI ablation | 345 | ||
8-OHdG, 8-hydroxy-2’-d eoxyguanosine; AF, atrial fibrillation; CAD, coronary artery disease; CKD, chronic kidney disease; CRP, C-r eactive protein; cTnC, cardiac troponin C; cTnI, cardiac troponin I; cTnT, cardiac troponin T; EC, electrical cardioversion; HSP, heat shock protein; hsTnT, high-s ensitivity troponin T; LAD, left atrial diameter; LAV, left atrial voltage; LSPeAF, long-standing persistent atrial fibrillation; MPO, myeloperoxidase; mtDNA, mitochondrial DNA; PAF, paroxysmal AF; PeAF, persistent AF; POAF, post-o perative AF; PVI, pulmonary vein isolation; SR, sinus rhythm; TNF, tumour necrosis factor.
Table provides a selection of biomarkers related to derailed proteasome and protein quality control pathways, degradation of the cytoskeletal proteins, and induction of inflammation, all of which are mechanisms found to drive AF.
Management
Management strategies of AF: towards a holistic approach
Ideally, the goal of AF therapy is to eliminate AF episodes, restore sinus rhythm, re-establish atrio-ventricular asynchrony and improve atrial contribution (‘atrial kick’) to the stroke volume. In the 2020 ESC guidelines for AF diagnosis and treatment, the proposed patient management pathway requires confirmation of the arrhythmia, followed by characterization of patients using the 4S-AF scheme, which includes stroke risk (CHA2DS2VASc score), symptom severity (EHRA symptom score), severity of AF burden (self-terminating, paroxysmal, persistent, permanent) and substrate severity (ageing, comorbidities, structural heart disease)137, as well as a holistic or integrated care approach based on the Atrial fibrillation Better Care (ABC) holistic pathway.
The ABC pathway includes ‘A’ (Avoid stroke/Anticoagulation), ‘B’ (Better symptom management with patient-centred symptom-directed decisions on rate or rhythm control) and ‘C’ (Cardiovascular risk and comorbidity optimization, including attention to lifestyle changes, patient psychological morbidity and patient values/preferences), and is recommended by various guidelines1,138–141. These general principles (‘Easy as ABC’) involve shared decision-making and can be followed by any health-care professional at any step of the patient experience.
The ABC pathway approach is supported by an increasing number of evidence-based findings. The mAFA-II trial compared a mobile health App based on the ABC pathway to usual care142. This prospective cluster-randomized trial showed that rates of the composite outcome of “ischemic stroke/systemic thromboembolism, death, and rehospitalization” were lower with the mAFA intervention than with usual care (1.9% versus 6.0%; HR 0.39; 95% CI 0.22–0.67; P < 0.001). Rates of rehospitalization were also lower with the mAFA intervention (1.2% versus 4.5%; HR 0.32; 95% CI 0.17–0.60; P < 0.001)142. The long-term extension of the mAFA-II trial reported that the benefits endured, with high adherence (>70%) and persistence (>90%) of use143. In the multimorbidity subgroup of the mAFA-II trial, the benefits of mAFA intervention were also clearly evident compared with usual care144. The ABC pathway has also been tested in post hoc analyses of clinical trial cohorts145,146, prospective cohort studies147,148 and nationwide cohort data149, all showing consistency in reporting better outcomes for patients with AF who adhere to the ABC pathway (that is, integrated care) compared with those who do not, irrespective of the region of the world where the study was conducted. A systematic review of the ABC pathway found a lower risk of all-cause death (OR 0.42, 95% CI 0.31–0.56), cardiovascular death (OR 0.37, 95% CI 0.23–0.58), stroke (OR 0.55, 95% CI 0.37–0.82) and major bleeding (OR 0.69, 95% CI 0.51–0.94) with management adherent to the ABC pathway compared with non-compliance150. Compliance with the ABC pathway has also been associated with improved outcomes in patients with clinical complexity146 and with a lower risk of dementia in patients with AF151.
‘A’ Avoid stroke: the role of anticoagulation therapy
Overall, AF increases the risk of stroke fivefold but this risk is not homogeneous and depends on the presence of various stroke risk factors152. The more common and validated stroke risk factors have been used to formulate schemes to stratify stroke risk, with the CHA2DS2VASc score being the most commonly used scheme in guidelines153; this score and the CHADS2 and ABC scores offer the best prediction for stroke events154. Given the limitations of all clinical stroke risk stratification schemes for identifying patients at high risk and the dynamic nature of stroke risks (which changes with ageing and incident comorbidities), recent guidelines have simplified the decision-making process so that the ‘default’ should be to offer stroke prevention, which is oral anticoagulation unless the patient is at ‘low risk’, in which case no antithrombotic therapy is recommended1. Such patients at ‘low risk’ can be defined as those with a CHA2DS2VASc score of 0 in men or 1 in women and the stroke event rate is <1% per year (considered the threshold for oral anticoagulant (OAC) treatment).
As stroke prevention requires oral anticoagulation, assessment of bleeding risk is also important, and guidelines from the ESC and APHRS recommend that the HAS-BLED score155 be used to draw attention to the modifiable bleeding risks (such as uncontrolled blood pressure, labile International normalized ratios (INRs; if on warfarin), concomitant use of NSAIDs and aspirin, and alcohol excess) for mitigation and to flag up patients at high risk of bleeding for early review and follow-up. In a Patient-Centered Outcomes Research Institute (PCORI) systematic review and evidence appraisal, the HAS-BLED score provided the best prediction of bleeding risk154. In the prospective cluster-randomized mAFA-II trial, appropriate use of the HAS-BLED score as part of the ABC pathway intervention resulted in a lower major bleeding rate at 1-year follow-up as well as in an increase in OAC use compared with more bleeds and a decline in OAC use in the usual care clusters156. Stroke and bleeding risk stratification schemes in AF management approaches have been reviewed in detail elsewhere157,158.
Stroke prevention has long been the cornerstone of AF management, whereby the historical trials showed that vitamin K antagonists (VKA; for example, warfarin), compared with placebo or control, reduced the risk of stroke or systemic embolism by 64% and all-cause mortality by 26%159. Most guidelines1,139 have given preference to non-VKA OACs (sometimes referred to as direct OACs) given their efficacy, safety and convenience relative to VKAs, hence their increasing use in clinical practice160,161.
‘B’ Better symptom management: rate and rhythm control
Currently, pharmacological anti-arrhythmic therapy of AF is aimed at either rate or rhythm control. Anti-arrhythmic drugs target ion channels and their efficacy is low. In addition, most of these drugs have severe and potentially life-threatening adverse effects (reviewed elsewhere1,162).
Non-pharmacological rate control therapy consists of His bundle ablation followed by cardiac pacing. As this approach will not stop AF, continuation of anticoagulation therapy is required. In addition, continuous right ventricular pacing is needed after interruption of atrio-ventricular conduction by His bundle ablation and might cause deterioration of cardiac function1. Non-pharmacological rhythm control therapy consists of electrical cardioversion and catheter or surgical ablation. Unfortunately, electrical cardioversion does not prevent the development of new AF episodes. Moreover, ablative therapy is only moderately effective, particularly in patients with persistent types of AF. The recurrence rate is high and repeat procedures might be required52,163. Rhythm control might be a reasonable option in those with recent-onset AF. In the EAST-AF trial164, a strategy of early rhythm control was associated with a lower risk of adverse cardiovascular outcomes compared with usual care (HR 0.79; 96% CI 0.66–0.94; P = 0.005) among patients with early AF (median time since diagnosis, 36 days) and cardiovascular conditions.
In the EORP-AF registry, only 34% of participants met eligibility criteria for early rhythm control according to the EAST trial criteria, and although use of an early rhythm control strategy was associated with a lower rate of major adverse events, this difference was non-significant on multivariate analysis, being mediated by differences in baseline characteristics and clinical risk profile165. Additionally, the beneficial effects of early rhythm control on clinical outcomes might gradually decline if rhythm control was delayed166. In the past years, many substrate-based ablation approaches have been tested, including targeting of low-voltage areas, fractionated potentials or rotational activity in the atria. Unfortunately, the effectiveness of these substrate-based ablation approaches is also suboptimal167.
The main reason for the moderate effectiveness of current anti-arrhythmic therapies is the inadequate knowledge of the underlying mechanisms driving electropathology and AF in individual patients. Consequently, mechanism-based AF therapies are in their infancy. Neuromodulation by low-level vagus nerve stimulation (LLVNS) is a recently introduced novel therapy for AF and involves stimulation of the sensible auricle branch of the vagus, located at the skin of the external acoustic meatus and the auricle168,169, below the patient-specific bradycardia threshold, which might have anti-arrhythmogenic effects170. LLVNS is effective in reducing both the incidence and burden of AF. Attenuation of the inflammation response by activation of the cholinergic anti-inflammatory pathway might contribute to the anti-arrhythmic effect of LLVNS, although the exact mechanisms remain to be elucidated.
‘C’ Cardiovascular risk factor and comorbidity management
As many of the wear-and-tear risk factors for AF are reversible, addressing these modifiable risks might be effective in primary and secondary AF prevention. Several lifestyle changes hold promise in attenuating AF. Although subject to methodological safeguard issues171, in the PREDIMED trial, a Mediterranean diet enriched with extra-virgin olive oil reduced the incidence of AF, and the follow-up PREDIMAR trial is currently testing a similar intervention in secondary prevention172,173. While research into the effect of plant-based diets in those with AF is limited, these diets reduce the risk and prevalence of hypertension174–176, diabetes177–181, obesity182–185, inflammation186–188, and obstructive sleep apnoea189 and, in addition, prevent and reverse atherosclerosis and coronary artery disease events190–191. Owing to these health effects, this diet is likely to decrease AF risk by reducing the traditional AF risk factors192–193. Furthermore, a low-carbohydrate, high-fat ketogenic diet might be detrimental for patients with AF, especially those with diabetes, as high levels of ketone bodies increase cardiac fibrosis and are related to diabetic ketoacidosis and end-stage heart failure194–197.
The potential ability of magnesium supplementation to prevent and/or treat AF has been recognized by the community of patients with AF. Many patients with AF reported via patient platforms that magnesium glycinate and magnesium taurate are beneficial in managing their AF. Magnesium is an abundant cation in the human body and a prevalent intracellular cation in heart tissue198. The primary physiological roles of Mg2+ include enzyme activity and protein transport and it is an essential component of all ATP-utilizing systems199. As such, magnesium has a crucial role in cardiac function, and low dietary intake of magnesium has been associated with a 50% higher risk of new-onset AF200–202. In addition, a meta-analysis of 20 randomized controlled trials indicates prophylactic magnesium supplementation to prevent post-operative AF onset203, although uncertainly exists regarding the optimal dose, timing and type of magnesium to provide a protective effect198. Although debate continues about the efficiency of magnesium in attenuating AF, magnesium glycinate and magnesium taurate, both identified as beneficial from patient experience, have not been tested in clinical trials to date.
Emerging evidence indicates that regular moderate-intensity exercise (up to 150 min weekly) reduces AF burden and improves symptoms and quality of life (QOL) of patients, while extreme exercise might increase the risk of developing AF. The exact pathogenetic mechanisms underlying this increased AF risk from excess exercise are unknown but might include atrial enlargement, inflammation and autonomic imbalance37,204,205. In addition, studies have shown that mind–body exercises, including yoga, tai chi and qigong, have beneficial effects on cardiac autonomic function, normalize biomarkers for AF, and enhance healthy ageing206 and might therefore improve symptoms in patients with AF37,207. Furthermore, cognitive behavioural therapy via mindfulness and interoceptive exposure therapy might help to reduce anxiety sensitivity during AF as well as AF symptoms208–211. To obtain a better insight into the effect of lifestyle changes in enhancing the management of AF, additional randomized trials in defined patients with AF as well as mechanistic studies are needed.
In a single-centre, partially blinded, randomized controlled study of ambulatory patients with overweight and obesity (n = 150; 15 months follow-up) and with symptomatic AF, weight reduction with intensive risk factor management resulted in a reduction in AF symptom burden and severity as well as changes in cardiac remodelling212. This benefit was reaffirmed in a small cohort study of 281 consecutive patients undergoing AF ablation (ARREST-AF), which showed that aggressive (proactive) risk factor management improved the long-term success of AF ablation213. This study was followed by the LEGACY cohort study, which showed that long-term sustained weight loss was associated with a significant reduction of AF burden and maintenance of sinus rhythm in individuals with obesity and AF who were undergoing rhythm control214.
Novel mechanism-based pharmaceutical and nutraceutical therapies
A promising new approach to treat AF is to restore proteostasis using a number of compounds, so-called proteoceuticals, directed at repair of PQC, DNA damage and mitochondrial function, as well as compounds directed at inflammation suppression (TABLE 3). First, securing adequate HSP levels might limit the expansion of AF electropathology and, as such, might prevent AF induction and progression82,83. In experimental settings, the HSP-inducing compound geranylgeranylacetone (GGA) reduces cardiomyocyte proteotoxic stress by decreasing ROS production215, reversing damage to the sarcomeres, stimulating refolding of damaged proteins and assisting in their clearance18,82,93,216. Consistent with this result, oral GGA treatment consistently prevents electrical and contractile dysfunction and AF promotion in atrial tachypaced and (acute) ischaemia-induced canine models of AF, suggesting that the induction of HSPs by GGA might have potential clinical value in the treatment of AF18,83,216–218. Moreover, 3 days of oral GGA administration upregulated HSP27 and HSP70 expression levels in atrial tissue of patients undergoing cardiac surgery. In patients treated with GGA, HSP27 was more abundant on myofilaments than in patients treated with placebo. This finding suggests protection of this network, which might have beneficial effects during periods of stress such as during an AF episode218. As GGA has not been registered in several countries, l-glutamine might represent an alternative to GGA in these regions. l-Glutamine also induces HSP expression and reduces oxidative stress, and oral intake alters HSP levels and normalizes the metabolic signature in blood samples of patients with AF219–222. In addition, compounds directed at the prevention of proteotoxic ER stress and subsequent protein degradation might also represent interesting candidates to treat AF. Among the available compounds, (sodium) 4-phenylbutyric acid (4PBA) is seemingly promising because it has been approved for clinical use to treat urea cycle disorders. As 4PBA acts as a chemical chaperone, it can alleviate ER stress in cardiomyocytes and protect against AF promotion in tachypaced atrial cardiomyocytes, Drosophila melanogaster and a canine model of AF86. As most proteins (at least one-third) are synthesized and folded in the ER223, this compartment is highly susceptible to proteotoxic stress induced by AF. In a phase I clinical study, 4PBA was safe and displayed only minor adverse effects224. Because of its protective effect on ER integrity, 4PBA is currently being tested in several clinical trials for misfolded protein diseases, including cystic fibrosis, amyotrophic lateral sclerosis and Huntington disease. Results of these studies might inform us on the effectiveness of 4PBA with respect to AF.
Table 3 |.
Novel, potentially beneficial pharmaceuticals and nutraceuticals targeting molecular causes of AF
| Druga | Mechanism of action | Condition | Trial phase | Refs or clinical trial identifier |
|---|---|---|---|---|
| Nicotinamide riboside | Increase in NAD+ and NADH levels, decrease in oxidative damage to proteins and DNA | Heart failure | I, II | NCT02689882, NCT03423342, NCT03727646 |
| Obesity, insulin resistance | II | NCT02835664 | ||
| Parkinson disease, neurodegenerative diseases | II | NCT03816020, NCT03568968 | ||
| SARS-CoV-2 infection in older individualsb, AKI | II | NCT04818216c, NCT04407390c | ||
| Immunity | II | NCT02812238 | ||
| Atrial fibrillation | Preclinical | 19,21 | ||
| l-Glutamine | Fuels TCA cycle, reduces ROS formation and ER stress, and increases HSP expression and nucleotide, protein and fatty acid synthesis | Heart failure | II | NCT01534663 |
| Pulmonary hypertension in SCD | II | NCT01048905, NCT01794884 | ||
| Coronary heart disease | II | NCT04019184 | ||
| AKI after cardiac surgery | II | NCT02838979 | ||
| CKD | II | NCT03113240 | ||
| Critical illness | III | NCT02998931 | ||
| Atrial fibrillation | II | 219 | ||
| Geranylgeranylacetone | Induces HSP production and reduces ROS formation | Gastric ulcers | IV | NCT01190657 |
| Gastritis | IV | NCT01547559 | ||
| Gastric lesion | IV | NCT01397448 | ||
| Atrial fibrillation | Preclinical | 18,82 | ||
| Cardiac bypass surgery | II | 218 | ||
| 4-Phenylbutyrate | Chemical chaperone | Cystic fibrosis | II | NCT00590538 |
| Amyotrophic lateral sclerosis | II | NCT00107770 | ||
| Inhibitor of ER stress | Huntington disease | II | NCT00212316 | |
| Pulmonary tuberculosis | II | NCT01580007 | ||
| HDAC inhibitor | Maple syrup urine disease | III | NCT01529060 | |
| Diabetes | IV | NCT00533559 | ||
| Urea cycle disorder | III | NCT00947544 | ||
| Atrial fibrillation | Preclinical | 86 | ||
| Tubastatin, ACY-1215 | HDAC6 inhibitor | Diabetic peripheral neuropathy | II | NCT03176472 |
| Lymphoma | I | NCT02091063 | ||
| Breast cancer | I | NCT02632071 | ||
| Atrial fibrillation | Preclinical | 19 | ||
| ABT-888 | PARP1 inhibitor | Metastatic breast cancer | II | NCT01009788 |
| Hepatocellular carcinoma | II | NCT01205828 | ||
| Adult solid neoplasm | I | NCT01154426 | ||
| Ovarian cancer | II | NCT01113957 | ||
| Colorectal cancer | II | NCT01051596 | ||
| Atrial fibrillation | Preclinical | 21 | ||
| SP600125 | JNK inhibitor | Atrial fibrillation | Preclinical | 45,96,117 |
| JNKI-IX | JNK2 inhibitor | Atrial fibrillation | Preclinical | 71–73 |
AF, atrial fibrillation; AKI, acute kidney injury; CKD, chronic kidney disease; ER, endoplasmic reticulum; JNK, JUN N-terminal kinase; PARP1, poly(ADP-r ibose) polymerase 1; ROS, reactive oxygen species; SCD, sickle cell disease; TCA, tricarboxylic acid.
Drugs are presented in order of how close they are to clinical use in AF.
Age >70 years.
Currently recruiting.
Second, specific inhibition of HDACs that have been implicated in AF might be beneficial. Among various HDACs, HDAC6 emerges as a key regulator in AF progression as it induces α-tubulin deacetylation, and consequently calpain-induced microtubule disruption, and might therefore represent a druggable target in AF87,225 (BOX 1). Two potent HDAC6 inhibitors, tubastatin A and ricolinostat (ACY-1215), have shown beneficial effects against microtubule disruption in mouse models of neurodegenerative diseases and cancer226–228. In addition, tubastatin A protects against electrical (ion channel, calcium handling and APD) and contractile remodelling and subsequent AF promotion in a canine model of AF87. Because specific inhibition of HDAC6 has not been associated with any serious toxicity to date229,230, clinical trials might be initiated to evaluate the possible beneficial effects of these inhibitors in AF.
The third aspect in normalizing proteostasis is to prevent NAD+ depletion and the subsequent cardio-myocyte dysfunction. PARP1 inhibitors are therefore interesting compounds as PARPs consume NAD+ in response to DNA damage. The first PARP1 inhibitors developed, such as 3-AB, compete with NAD+ for binding to PARPs and consequently inhibit PARP1 and other PARP family members as well as the cardiac-protective enzymes mono-ADP-ribosyl-transferases and sirtuins231. Therefore, early PARP1 inhibitors are probably not ideal to treat AF. However, more recently developed PARP inhibitors, such as ABT-888 and olaparib, have greater potency and specificity than earlier inhibitors. For example, ABT-888 directly inhibits PARP1 and PARP2 but has no effect on sirtuins232, and is currently in phase I and II clinical studies in cancer233. Olaparib has been tested in phase III clinical trials for the treatment of metastatic breast cancers and had no effect on QT/QTc interval, and is therefore likely safe to treat cardiac diseases234,235. Replenishing the NAD+ pool is another therapeutic approach to protect against AF-related electropathology, which can be achieved by supplementation with NAD+ or its precursors such as nicotinamide and nicotinamide riboside. Interestingly, nicotinamide is both a PARP1 inhibitor as well as a NAD+ precursor, thereby protecting against oxidative protein and DNA damage19,21,222. In experimental heart failure and dilated cardiomyopathy model systems, nicotinamide displayed a protective effect, demonstrating a clear benefit of normalizing NAD+ levels in failing hearts236–238. An open-label pharmacokinetics study with nicotinamide riboside in healthy volunteers showed that nicotinamide riboside stably increased circulating NAD+ levels in the blood and was well tolerated239. In addition, in a long-term human cohort study, high dietary intake of naturally occurring NAD+ precursors, including nicotinamide, was associated with a reduction in risk factors for AF and reduced the risk of cardiac mortality240, substantiating the possible beneficial effect in AF. Collectively, these results support the intake of NAD+ precursors, and especially nicotinamide riboside, either from dietary sources or by nutraceuticals as a potential therapeutic approach in treating AF.
Furthermore, as Ca2+ handling abnormalities can initiate AF, suppression of the pro-arrhythmic molecule CaMKII might represent a target for therapy and inspired the development of CAMKII inhibitors45,63,241. Unfortunately, CAMKII inhibitors have off-target effects that hinder their clinical application242. As JNK2 regulates CAMKII activity, compounds that target JNK2 might be more specific. To date, the JNK2 inhibitors SP600125 (REFS45,96,117) and JNKI-IX71–73 revealed protective effects against AF onset in experimental model systems (TABLE 3). More research is warranted to explore JNK2 as an anti-arrhythmic drug target in clinical AF.
Blocking inflammation can be achieved through the administration of a variety of drugs, including steroids and NSAIDs. Unfortunately, clinical trials with dexamethasone and the corticosteroid prednisone were without effect on post-operative AF243 and recurrence of AF after ablation244, respectively. These results suggest that non-specific and non-selective anti-inflammatory therapies have minimal or no impact on the prevention of AF onset and progression. Therefore, a specific and selective NLRP3 inhibitor might be desirable. Short hairpin RNA-mediated knockdown of Nlrp3 or the NLRP3-selective inhibitor MCC950 (REF.245) both reduce AF inducibility in NLRP3 gain-of-function mice, which provides a proof of concept for NLRP3 inhibition in AF prevention.
Quality of life
AF is associated with adverse health outcomes, poor health-related QOL (HRQOL) and high health-care costs246 (BOX 2). The goals of health-care providers are to alleviate AF symptoms, reduce the risk of long-term sequelae (including systemic embolism, stroke and heart failure), reduce mortality and improve QOL of the patient. Interestingly, physicians rate patient QOL higher than patients with AF do. The discordance is most significant in patients with depression, sleep disorders and physical inactivity247. To overcome this discordance, recent global initiatives provide, for the first time, standardized approaches to report on outcomes of quality of care (QOC) and QOL based on patient-reported outcome measures (PROMs)248. PROMs facilitate the assessment of QOL of patients with AF, which serves to inform their health-care providers on the QOC. The outcomes that matter most to patients with AF include HRQOL, emotional functioning, physical functioning, exercise tolerance, symptom severity, ability to work and cognitive functioning248 (BOX 1). In addition, the patient-perceived treatment burden is an independent predictor of decreased QOL249,250. By implementing PROMs in the QOC, a holistic standardized outcome set in AF for integration into routine clinical practice is realized.
Box 2 |. Spoken from the heart: the story of a patient with AF.
Unless you’ve had atrial fibrillation (AF), it’s hard to understand the impact of living with it. Thus, doctors may underestimate how dramatically it affects us. It’s hard to work or sleep with your heart flopping around in your chest like a fish. You feel dizzy and lightheaded as your heart races and you can’t catch your breath. You feel like you’re running a marathon 24 hours a day. AF can be totally debilitating, leaving you drained of energy and yet unable to rest. That’s just the physical part.
In the emotional part, you worry about making commitments for fear of disappointing family, friends and co-workers if you don’t feel up to what you committed to doing. You can’t think or focus between the AF and the medications that cause brain fog and memory loss. AF hijacks patients’ lives, and we may live in fear of a stroke, heart failure, dementia, or even early death.
Then, there’s the financial toll AF takes as we’re frequently in the emergency department or admitted to the hospital, which insurance may not fully cover. Patients with AF may be seen as unreliable because they’re in the hospital yet again, so some lose their jobs. They may also lose their cars, houses, and even their families. AF takes a terrible toll on patients and families living with it. Far too many patients with AF are being treated by generalists who do not take AF seriously. They take a watch-and-wait stance rather than referring patients to specialists who understand the condition more. The patient tends to have been on rate control only, so that often means trying rhythm control, either anti-arrhythmic drugs or catheter ablation. By then, it’s usually too late for lifestyle changes to make a difference, and something more is needed.
If our doctors really understood the impact AF has on us and how it takes over our lives, they might treat our AF in line with the patient’s values and preferences. We want our doctors to understand what is important to us and work with us to find a solution quickly. We just want to get our lives back and do the things we love with the people we love.
AF symptoms are considerably more severe in women than in men246. Negative emotions might increase the risk of developing AF, whereas, conversely, positive emotions might be protective. Women with AF have higher stress, although, after adjusting for other factors, only traumatic life events are associated with developing AF251. Mindfulness-based programmes have reduced psychological distress and improved QOL211. In addition to PROMs for QOL assessment, patients might also be encouraged to improve QOL by implementing personalized AF management programmes in their daily life. In a study to characterize AF triggers, nearly three-quarters of patients with paroxysmal AF reported having triggers for their AF episodes, with the most common being alcohol (35%), caffeine (28%), exercise (23%) and lack of sleep (21%). Vagally mediated triggers tended to cluster together within individuals252. Knowledge of individual triggers might help to reduce AF episodes and improve QOL. For example, smartphone apps based on photoplethysmography technology, which detects pulse pressure signals resulting from the propagation of blood pressure pulses along arterial blood vessels253,254, and one-lead ECG detection for managing health and wellness (including monitoring of medication adherence, diet, exercise, sleep and other lifestyle areas) could be implemented. The Individualized Studies of Triggers of Paroxysmal Atrial Fibrillation (I-STOP-AFib, NCT03323099) study aims to improve the QOL of patients with AF by identifying their specific triggers so that they can be avoided255. Study outcomes show that ‘N-of-1’ trigger testing did not enhance AF-related QOF but reduced the number of AF episodes; alcohol but not caffeine increased the risk of AF events. To ensure co-creation, the I-STOP-Afib study has a patient principal investigator and a patient advisory board to define study needs, advise on how to carry out the study, recruit patients for defining specific AF triggers to study and as study participants, and help to interpret the results (BOX 3). Additionally, smart devices can be used for screening to detect undiagnosed AF and for monitoring in already diagnosed patients to help them manage their condition and monitor for AF following a procedure256. These smart devices provide a platform to leverage ‘big data’, for example, the Apple Heart Study, with almost 420,000 adult participants enrolled to identify possible AF using an Apple Watch257; the current Heartline Study (NCT04276441), with the goal to recruit 150,000 people to determine if early diagnosis by an Apple Watch can reduce the risk of stroke; and the Huawei Heart Study (mAFA-II trial discussed in detail above) in China142.
Box 3 |. A new scientific paradigm: empowering patients with AF to co-create AF studies.
Implementation of personalized atrial fibrillation (AF) treatments is impossible without the participation of patients in research. Participation involves education about AF, communication of research results, and support for patients to become self-aware and record episodes along with AF triggers. This empowerment of patients with AF as individuals who contribute to research and who, as such, might control their disease more effectively is a prerequisite to create positive health. To accomplish this mission, non-profit organizations have been established such as StopAfib.org in the USA. This organization was founded by Mellanie True Hills, a patient with AF who became AF-free owing to a minimally invasive surgical ablation procedure. To facilitate communication and understanding between patients and their doctors, Mellanie shares the patient perspective with health-care professionals within the scientific community. In the Netherlands, the AF-Innovation-Platform (AFIP) was founded by AF scientists, medical doctors, engineers and patients. AFIP provides a platform on which patients are encouraged to share their experience of AF with the community, which might help to uncover unrecognized AF triggers and ways to stop the arrhythmia. The exchange of information and ideas between patients and researchers has already resulted in the co-creation of several translational research projects. As such, these organizations help to empower and improve the quality of life of patients with AF and their family members and friends worldwide.
In 2014, the Health eHeart Alliance Patient-Powered Research Network, composed of researchers at the University of California, San Francisco (UCSF) and patient organizations, convened the Patient-Powered Research Summit to bring heart patients and cardiovascular researchers from many institutions together to identify research needs and develop proposals for funding. The patients with AF at the summit identified the need for research into AF triggers; thus, an ‘N-of-1’ trigger study was proposed using a smartphone app to allow patients with AF to test their individual triggers255. Additional big data studies utilizing smart devices are ongoing and their findings might elucidate personalized triggers for AF onset.
Outlook
To date, AF continues to be the most common cardiac tachyarrhythmia in the Western world. In the past decade, important discoveries have been made in the fields of epidemiology, genetics, electrophysiology and molecular biology, which have provided a deeper understanding of AF incidence and pathophysiology. These findings reveal that the AF aetiology, electropathology severity and corresponding underlying molecular pathways might differ between individual patients or groups of patients with AF. This observation calls for the development of patient-tailored diagnostic testing and treatment strategies. To achieve this goal, patient participation in studies and data and biomaterial sharing are prerequisites, although these efforts are currently still in their infancy. To realize such a holistic concept of AF management, several steps are needed.
First, we foresee that patient participation in co-design and co-creation of studies will boost research on effective and personalized AF diagnostics and therapeutics. As such, patients, researchers and health-care professionals need a platform where all stakeholders can participate and collaborate in ‘citizen science’ and scientific research to increase scientific knowledge and achieve AF solutions. The AF-Innovation-Platform (AFIP) in the Netherlands is a platform aimed at the co-creation of AF clinical trials and research studies. As millions of people worldwide suffer with AF, patient platforms might use the collective strength of the community to identify research questions, collect and analyse data, interpret results, test the efficacy of lifestyle interventions, make new discoveries (for example, personal experience with triggers of AF) and co-develop technologies and applications, all to enhance understanding of AF, improve the QOL of patients and reduce the AF burden for the society.
Second, investing in the dissection of molecular pathways and corresponding electrical conduction abnormalities driving AF is needed to improve AF therapy and diagnostics. As such, non-invasive or minimally invasive approaches are needed to determine the severity of electropathology in individual patients and correlate the findings with underlying environmental and genetic risk factors and treatment outcomes. In addition, further research into mechanism-based electropathology biomarkers might aid in creating a personal AF fingerprint and in the selection of a patient-tailored treatment approach.
Third, a personal AF fingerprint might be complemented with a genetic risk score. However, several research challenges need to be considered before a genetic risk score can be implemented in routine clinical care. Research should progress from association studies towards the identification of causative gene variants with a mechanistic link to AF258. In addition, more insight into the genetics of AF should be obtained for different ethnic groups. A consensus should be reached on the use of a genetic risk score and testing to identify the risk of AF development and predict the stage and progression of the arrhythmia.
Finally, emerging key pathways have been identified in which molecular changes drive electropathology in AF, including derailment of protein homeostasis, stress signalling and inflammasome activation. Importantly, key modulators in these pathways also represent potential druggable targets. Currently marketed drugs are available for repurposing and exploration of their role to treat AF in the clinical setting; public and private funding programmes are needed to support these translational studies from bench to bedside.
Supplementary Material
Acknowledgements
The authors thank the Dutch Heart Foundation (2020–2020B003 to B.J.J.M.B.), CVON-STW2016–14728 AFFIP (to N.M.S.d.G. and B.J.J.M.B.), NWO-Vidi (2016–91717339 to N.M.S.d.G.), KNAW Science Communication grant (to B.J.J.M.B., N.M.S.d.G. and M.F.K.), National Institution of Health (AA024769 and HL146744 to X.A.), and Medical Delta (to N.M.S.d.G. and B.J.J.M.B.) for financial support and Leonoor Wijdeveld for graphic design support. The authors thank the patients, who remain anonymous, for sharing their experiences as outlined in Box 2.
Footnotes
Competing interests
G.Y.H.L. is a consultant and speaker for BMS/Pfizer, Boehringer Ingelheim and Daiichi-Sankyo. No fees are received personally. M.T.H. is an employee of a non-profit, StopAfib.org, and a for-profit speaking and consulting company, True Hills, Inc. Both G.Y.H.L. and M.T.H. receive funds from industry; however, no fees are received personally. B.J.J.M.B., X.A., M.F.K. and N. M. S. d. G. declare no competing interests.
Peer review information
Nature Reviews Disease Primers thanks N. Li, H. Oral and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41572-022-00347-9.
References
- 1.Hindricks G et al.2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 42, 373–498 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Lip GYH et al. Atrial fibrillation. Nat. Rev. Dis. Prim. 2, 16016 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Waldmann V, Laredo M, Abadir S, Mondesert B & Khairy P Atrial fibrillation in adults with congenital heart disease. Int. J. Cardiol. 287, 148–154 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Teuwen CP & de Groot NMS Atrial fibrillation: the next epidemic for patients with congenital heart disease. J. Am. Coll. Cardiol. 70, 2949–2950 (2017) [DOI] [PubMed] [Google Scholar]
- 5.Teuwen CP et al. Frequent atrial extrasystolic beats predict atrial fibrillation in patients with congenital heart defects. Europace 20, 25–32 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Darbar D et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J. Am. Coll. Cardiol. 41, 2185–2192 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Ellinor PT, Yoerger DM, Ruskin JN & MacRae CA Familial aggregation in lone atrial fibrillation. Hum. Genet. 118, 179–184 (2005) [DOI] [PubMed] [Google Scholar]
- 8.Palatinus JA & Das S Your father and Grandfather’s atrial fibrillation: a review of the genetics of the most common pathologic cardiac dysrhythmia. Curr. Genomics 16, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker NR, Clauss S & Ellinor PT Common variation in atrial fibrillation: navigating the path from genetic association to mechanism. Cardiovasc. Res. 109, 493–501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zoni-Berisso M, Lercari F, Carazza T & Domenicucci S Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 6, 213–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burdett P & Lip GYH Atrial fibrillation in the United Kingdom: predicting costs of an emerging epidemic recognising and forecasting the cost drivers of atrial fibrillation-related costs. Eur. Heart J. Qual. Care Clin. Outcomes 10.1093/ehjqcco/qcaa093 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Krittayaphong R et al. A randomized clinical trial of the efficacy of radiofrequency catheter ablation and amiodarone in the treatment of symptomatic atrial fibrillation. J. Med. Assoc. Thai. 86 (Suppl. 1), S8–S16 (2003). [PubMed] [Google Scholar]
- 13.Stabile G et al. Catheter ablation treatment in patients with drug-refractory atrial fibrillation: a prospective, multi-centre, randomized, controlled study (Catheter Ablation for the Cure of Atrial Fibrillation Study). Eur. Heart J. 27, 216–221 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Pappone C et al. A randomized trial of circumferential pulmonary vein ablation versus antiarrhythmic drug therapy in paroxysmal atrial fibrillation: the APAF Study. J. Am. Coll. Cardiol. 48, 2340–2347 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Calkins H et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm 14, e275–e444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blomstrom-Lundqvist C et al. Effect of catheter ablation vs antiarrhythmic medication on quality of life in patients with atrial fibrillation: The CAPTAF randomized clinical trial. JAMA 321, 1059–1068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovan R et al. Relationship of quality of life with procedural success of atrial fibrillation (AF) ablation and postablation AF burden: substudy of the STAR AF randomized trial. Can. J. Cardiol. 29, 1211–1217 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Brundel BJJM et al. Induction of heat-shock response protects the heart against atrial fibrillation. Circ. Res. 99, 1394–1402 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Zhang D et al. Activation of histone deacetylase-6 (HDAC6) induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 129, 346–358 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Yao C et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat. Commun. 10, 1307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boriani G & Pettorelli D Atrial fibrillation burden and atrial fibrillation type: clinical significance and impact on the risk of stroke and decision making for long-term anticoagulation. Vasc. Pharmacol. 83, 26–35 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Charitos EI, Purerfellner H, Glotzer TV & Ziegler PD Clinical classifications of atrial fibrillation poorly reflect its temporal persistence: insights from 1,195 patients continuously monitored with implantable devices. J. Am. Coll. Cardiol. 63, 2840–2848 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Schnabel RB et al. 50 Year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet 386, 154–162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li N & Brundel B Inflammasomes and proteostasis novel molecular mechanisms associated with atrial fibrillation. Circ. Res. 127, 73–90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Marion DMS et al. Atrial heat shock protein levels are associated with early postoperative and persistence of atrial fibrillation. Heart Rhythm 18, 1790–1798 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Li J et al. Blood-based 8-hydroxy-2′-deoxyguanosine level: a potential diagnostic biomarker for atrial fibrillation. Heart Rhythm 18, 271–277 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Johnsen SP, Guo Y & Lip GYH Epidemiology of atrial fibrillation: geographic/ecological risk factors, age, sex, genetics. Card. Electrophysiol. Clin. 13, 1–23 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Kornej J, Borschel CS, Benjamin EJ & Schnabel RB Epidemiology of atrial fibrillation in the 21st century: novel methods and new insights. Circ. Res. 127, 4–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lubitz SA et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 304, 2263–2269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lippi G, Sanchis-Gomar F & Cervellin G Global epidemiology of atrial fibrillation: an increasing epidemic and public health challenge. Int. J. Stroke 16, 217–221 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Allan V et al. Are cardiovascular risk factors also associated with the incidence of atrial fibrillation? A systematic review and field synopsis of 23 factors in 32 population-based cohorts of 20 million participants. Thromb. Haemost. 117, 837–850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allan V et al. Are cardiovascular risk factors also associated with the incidence of atrial fibrillation? A systematic review and field synopsis of 23 factors in 32 population-based cohorts of 20 million participants. Thromb. Haemost. 117, 837–850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rowan CJ et al. Very low prevalence and incidence of atrial fibrillation among Bolivian forager-farmers. Ann. Glob. Health 87, 18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rix TA et al. A U-shaped association between consumption of marine n-3 fatty acids and development of atrial fibrillation/atrial flutter-a Danish cohort study. Europace 16, 1554–1561 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Shen J et al. Dietary factors and incident atrial fibrillation: the Framingham Heart Study. Am. J. Clin. Nutr. 93, 261–266 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung MK et al. Lifestyle and risk factor modification for reduction of atrial fibrillation: a scientific statement from the American Heart Association. Circulation 141, e750–e772 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Staerk L et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: cohort study based on longitudinal data from the Framingham Heart Study. BMJ 361, k1453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SR et al. Association between clustering of unhealthy lifestyle factors and risk of new-onset atrial fibrillation: a nationwide population-based study. Sci. Rep. 10, 19224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tse HF et al. Stroke prevention in atrial fibrillation — an Asian stroke perspective. Heart Rhythm 10, 1082–1088 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Olson EN Gene regulatory networks in the evolution and development of the heart. Science 313, 1922–1927 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fatkin D, Santiago CF, Huttner IG, Lubitz SA & Ellinor PT Genetics of atrial fibrillation: state of the art in 2017. Heart Lung Circ. 26, 894–901 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Ellinor PT et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat. Genet. 44, 670–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai W et al. A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice. eLife 8, e41814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao X et al. Transcriptional regulation of stress kinase JNK2 in pro-arrhythmic CaMKIIdelta expression in the aged atrium. Cardiovasc. Res. 114, 737–746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan J et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J. Mol. Cell Cardiol. 114,105–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roselli C et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 50, 1225–1233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen JB et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 50, 1234–1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teuwen CP et al. Time course of atrial fibrillation in patients with congenital heart defects. Circ. Arrhythm. Electrophysiol. 8, 1065–1072 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Teuwen CP, Ramdjan TT & de Groot NM Management of atrial fibrillation in patients with congenital heart defects. Expert Rev. Cardiovasc. Ther. 13, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Haissaguerre M et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Kottkamp H et al. Time courses and quantitative analysis of atrial fibrillation episode number and duration after circular plus linear left atrial lesions: trigger elimination or substrate modification: early or delayed cure? J. Am. Coll. Cardiol. 44, 869–877 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Yamada T et al. Incidence, location, and cause of recovery of electrical connections between the pulmonary veins and the left atrium after pulmonary vein isolation. Europace 8, 182–188 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Teuwen CP et al. Relevance of conduction disorders in Bachmann’s bundle during sinus rhythm in humans. Circ. Arrhythm. Electrophysiol. 9, e003972 (2016). [DOI] [PubMed] [Google Scholar]
- 55.van der Does L, Kik C, Allessie M & de Groot N Endo-epicardial dissociation in conduction. Eur. Heart J. 38, 1775 (2017). [DOI] [PubMed] [Google Scholar]
- 56.de Groot NM et al. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation 122, 1674–1682 (2010). [DOI] [PubMed] [Google Scholar]
- 57.de Groot N et al. Direct proof of endo-epicardial asynchrony of the atrial wall during atrial fibrillation in humans. Circ. Arrhythm. Electrophysiol. 9, e003648 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Allessie MA et al. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ. Arrhythm. Electrophysiol. 3, 606–615 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Christ T et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Nattel S, Maguy A, Le Bouter S & Yeh YH Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Nattel S, Heijman J, Zhou L & Dobrev D Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ. Res. 127, 51–72 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bers DM Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Ai X, Curran JW, Shannon TR, Bers DM & Pogwizd SM Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Respress JL et al. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walden AP, Dibb KM & Trafford AW Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J. Mol. Cell Cardiol. 46, 463–473 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Venetucci LA, Trafford AW, O’Neill SC & Eisner DA The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Chelu MG et al. Calmodulin kinase UU-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neef S et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 106, 1134–1144 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Voigt N et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voigt N et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129, 145–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yan J et al. Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ. Res. 122, 821–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Yan J et al. JNK2, a newly-identified SERCA2 enhancer, augments an arrhythmic [Ca2+]SR leak-load relationship. Circ. Res. 128, 455–470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan J et al. JNK2, a newly-identified SERCA2 enhancer, augments an arrhythmic [Ca2+]SR leak-load relationship. Circ. Res. 128, 455–470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan J et al. Role of stress kinase JNK in binge alcohol-evoked atrial arrhythmia. J. Am. Coll. Cardiol. 71, 1459–1470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Litvinukova M et al. Cells of the adult human heart. Nature 588, 466–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hartl FU, Bracher A & Hayer-Hartl M Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Balch WE, Morimoto RI, Dillin A & Kelly JW Adapting proteostasis for disease intervention. Science 319, 916–919 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Neef DW et al. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 9, 955–966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kampinga HH & Bergink S Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 15, 748–759 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Balchin D, Hayer-Hartl M & Hartl FU In vivo aspects of protein folding and quality control. Science 353, aac4354 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Vilchez D, Saez I & Dillin A The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Morimoto RI & Cuervo AM Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 69 (Suppl. 1), S33–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu X, Li J, van Marion DMS, Zhang D & Brundel B Heat shock protein inducer GGA*−59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental atrial fibrillation. J. Mol. Cell Cardiol. 134, 86–97 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Brundel BJ et al. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell Cardiol. 41,555–562 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Ke L et al. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 45, 685–693 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Brundel BJJM et al. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 54, 380–389 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Wiersma M et al. Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. J. Am. Heart Assoc. 6, e006458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang D et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 129, 346–358 (2014). [DOI] [PubMed] [Google Scholar]
- 88.Ravikumar B et al. Mammalian macroautophagy at a glance. J. Cell Sci. 122, 1707–1711 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoyer-Hansen M et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Kroemer G, Marino G & Levine B Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakai A et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Noda NN & Inagaki F Mechanisms of autophagy. Annu. Rev. Biophys. 44, 101–122 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Henning RH & Brundel BJJM Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 14, 637–653 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Li J, Zhang D, Wiersma M & Brundel BJJM Role of autophagy in proteostasis: friend and foe in cardiac diseases. Cells 7, 279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis RJ Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000). [DOI] [PubMed] [Google Scholar]
- 96.Yan J et al. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc. Res. 97, 589–597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chiang DY et al. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ. Arrhythm. Electrophysiol. 7, 1214–1222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bare DJ, Yan J & Ai X Evidence of CaMKII-regulated late INa in atrial fibrillation patients with sleep apnea: one-step closer to finding plausible therapeutic targets for atrial fibrillation? Circ. Res. 126, 616–618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lebek S et al. Enhanced CaMKII-dependent late INa induces atrial proarrhythmic activity in patients with sleep-disordered breathing. Circ. Res. 126, 603–615 [DOI] [PubMed] [Google Scholar]
- 100.Erickson JR et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raman M, Chen W & Cobb MH Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Pogoda K, Kameritsch P, Retamal MA & Vega JL Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. BMC Cell Biol. 17 (Suppl. 1), 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kelley N, Jeltema D, Duan Y & He Y The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiu H et al. Chronic kidney disease increases atrial fibrillation inducibility: involvement of inflammation, atrial fibrosis, and connexins. Front. Physiol. 9, 1726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fender AC et al. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res. Cardiol. 115, 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chelu MG et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heijman J et al. Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ. Res. 127, 1036–1055 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Molina CE et al. Profibrotic, electrical, and calcium-handling remodeling of the atria in heart failure patients with and without atrial fibrillation. Front. Physiol. 9, 1383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fakuade FE et al. Altered atrial cytosolic calcium handling contributes to the development of postoperative atrial fibrillation. Cardiovasc. Res. 117, 1790–1801 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Salvo TG Holiday heart: some sobering mechanistic insights. J. Am. Coll. Cardiol. 71, 1471–1473 (2018). [DOI] [PubMed] [Google Scholar]
- 111.Wakili R et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ. Arrhythm. Electrophysiol. 3, 530–541 (2010). [DOI] [PubMed] [Google Scholar]
- 112.Li N et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129, 1276–1285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wijffels MC, Kirchhof CJ, Dorland R & Allessie MA Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92, 1954–1968 (1995). [DOI] [PubMed] [Google Scholar]
- 114.Janse MJ & Wit AL Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 69, 1049–1169 (1989). [DOI] [PubMed] [Google Scholar]
- 115.McCauley MD et al. Ion channel and structural remodeling in obesity-mediated atrial fibrillation. Circ. Arrhythm. Electrophysiol. 13, e008296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Igarashi T et al. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 125, 216–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan J et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J. Mol. Cell Cardiol. 114, 105–115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Platonov PG, Mitrofanova LB, Orshanskaya V & Ho SY Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J. Am. Coll. Cardiol. 58, 2225–2232 (2011). [DOI] [PubMed] [Google Scholar]
- 119.Yan J et al. Novel methods of automated quantification of gap junction distribution and interstitial collagen quantity from animal and human atrial tissue sections. PLoS ONE 9, e104357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kauppila TES, Kauppila JHK & Larsson NG Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Kujoth GC et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Schumacher B, Pothof J, Vijg J & Hoeijmakers JHJ The central role of DNA damage in the ageing process. Nature 592, 695–703 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wiersma M et al. Cell-free circulating mitochondrial DNA: a potential blood-based marker for atrial fibrillation. Cells 9, 1159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wiersma M et al. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 8, 1202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ramos KS & Brundel B DNA damage, an innocent bystander in atrial fibrillation and other cardiovascular diseases? Front. Cardiovasc. Med. 7, 67 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Konings KT, Smeets JL, Penn OC, Wellens HJ & Allessie MA Configuration of unipolar atrial electrograms during electrically induced atrial fibrillation in humans. Circulation 95, 1231–1241 (1997). [DOI] [PubMed] [Google Scholar]
- 127.van Schie MS et al. Classification of sinus rhythm single potential morphology in patients with mitral valve disease. Europace 22, 1509–1519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van Schie MS, Starreveld R, Bogers A & de Groot NMS Sinus rhythm voltage fingerprinting in patients with mitral valve disease using a high-density epicardial mapping approach. Europace 23, 469–478 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ye Z, van Schie MS & de Groot NMS Signal fingerprinting as a novel diagnostic tool to identify conduction inhomogeneity. Front. Physiol. 12, 652128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J et al. Blood-based 8-hydroxy-2′-deoxyguanosine level: a potential diagnostic biomarker for atrial fibrillation. Heart Rhythm 18, 271–277 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Marion D et al. Evaluating serum heat shock protein levels as novel biomarkers for atrial fibrillation. Cells 9, 2105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dernellis J & Panaretou M C-reactive protein and paroxysmal atrial fibrillation: evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 56, 375–380 (2001). [DOI] [PubMed] [Google Scholar]
- 133.Nortamo S et al. Association of sST2 and hs-CRP levels with new-onset atrial fibrillation in coronary artery disease. Int. J. Cardiol. 248, 173–178 [DOI] [PubMed] [Google Scholar]
- 134.Amdur RL et al. Interleukin-6 is a risk factor for atrial fibrillation in chronic kidney disease: findings from the CRIC Study. PLoS ONE 11, e0148189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Oyama K et al. Serial assessment of biomarkers and the risk of stroke or systemic embolism and bleeding in patients with atrial fibrillation in the ENGAGE AF-TIMI 48 trial. Eur. Heart J. 42, 1698–1706 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ramos KS et al. Degree of fibrosis in human atrial tissue is not the hallmark driving AF. Cells 11, 427 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Potpara TS et al. The 4S-AF scheme (stroke risk; symptoms; severity of burden; substrate): a novel approach to in-depth characterization (rather than classification) of atrial fibrillation. Thromb. Haemost. 121, 270–278 (2021). [DOI] [PubMed] [Google Scholar]
- 138.Chao TF et al. 2021 Focused update of the 2017 consensus guidelines of the Asia Pacific Heart Rhythm Society (APHRS) on stroke prevention in atrial fibrillation. J. Arrhythm. 37, 1389–1426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lip GYH et al. Antithrombotic therapy for atrial fibrillation: CHEST guideline and expert panel report. Chest 154, 1121–1201 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Lip GYH The ABC pathway: an integrated approach to improve AF management. Nat. Rev. Cardiol. 14, 627–628 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Chao TF et al. 2021 Focused update consensus guidelines of the Asia Pacific Heart Rhythm Society on stroke prevention in atrial fibrillation: executive summary. Thromb. Haemost. 122, 20–47 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo Y et al. Mobile health technology to improve care for patients with atrial fibrillation. J. Am. Coll. Cardiol. 75, 1523–1534 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Guo Y et al. Mobile health technology-supported atrial fibrillation screening and integrated care: a report from the mAFA-II trial long-term extension cohort. Eur. J. Intern. Med. 82, 105–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yao Y, Guo Y & Lip GYH mAF-App II trial investigators. the effects of implementing a mobile health-technology supported pathway on atrial fibrillation-related adverse events among patients with multimorbidity: the mAFA-II randomized clinical trial. JAMA Netw. Open 4, e2140071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Proietti M, Romiti GF, Olshansky B, Lane DA & Lip GYH Improved outcomes by integrated care of anticoagulated patients with atrial fibrillation using the simple ABC (Atrial Fibrillation Better Care) Pathway. Am. J. Med. 131, 1359–1366.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Proietti M, Romiti GF, Olshansky B, Lane DA & Lip GYH Comprehensive management with the ABC (Atrial Fibrillation Better Care) pathway in clinically complex patients with atrial fibrillation: a post hoc ancillary analysis from the AFFIRM Trial. J. Am. Heart Assoc. 9, e014932 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pastori D, Pignatelli P, Menichelli D, Violi F & Lip GYH Integrated care management of patients with atrial fibrillation and risk of cardiovascular events: the ABC (Atrial fibrillation Better Care) pathway in the ATHERO-AF study cohort. Mayo Clin. Proc. 94, 1261–1267 (2019). [DOI] [PubMed] [Google Scholar]
- 148.Proietti M et al. Relation of outcomes to ABC (Atrial Fibrillation Better Care) pathway adherent care in European patients with atrial fibrillation: an analysis from the ESC-EHRA EORP Atrial Fibrillation General Long-Term (AFGen LT) Registry. Europace 23, 174–183 (2021) [DOI] [PubMed] [Google Scholar]
- 149.Yoon M et al. Improved population-based clinical outcomes of patients with atrial fibrillation by compliance with the simple ABC (Atrial Fibrillation Better Care) pathway for integrated care management: a nationwide cohort study. Thromb. Haemost. 119, 1695–1703 (2019). [DOI] [PubMed] [Google Scholar]
- 150.Romiti GF et al. Adherence to the ‘Atrial Fibrillation Better Care’ pathway in patients with atrial fibrillation: impact on clinical outcomes — a systematic review and meta-analysis of 285,000 patients. Thromb. Haemost. 10.1055/a-1515-9630 (2021). [DOI] [PubMed] [Google Scholar]
- 151.Yang PS et al. The effect of integrated care management on dementia in atrial fibrillation. J. Clin. Med. 9, 1696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pisters R, Lane DA, Marin F, Camm AJ & Lip GY Stroke and thromboembolism in atrial fibrillation. Circ. J. 76, 2289–2304 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Lip GY, Nieuwlaat R, Pisters R, Lane DA & Crijns HJ Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest 137, 263–272 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Borre ED et al. Predicting thromboembolic and bleeding event risk in patients with non-valvular atrial fibrillation: a systematic review. Thromb. Haemost. 118, 2171–2187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pisters R et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 138, 1093–1100 (2010). [DOI] [PubMed] [Google Scholar]
- 156.Guo Y, Lane DA, Chen Y, Lip GYH & mAF-App II Trial Investigators. Regular bleeding risk assessment associated with reduction in bleeding outcomes: the mAFA-II randomized trial. Am. J. Med. 133, 1195–1202.e2 (2020). [DOI] [PubMed] [Google Scholar]
- 157.Lip GYH et al. Stroke prevention in atrial fibrillation. Trends Cardiovasc. Med. 10.1093/eurheartj/suaa180 (2021). [DOI] [PubMed] [Google Scholar]
- 158.Chao TF, Nedeljkovic MA, Lip GYH & Potpara TS Stroke prevention in atrial fibrillation: comparison of recent international guidelines. Eur. Heart J. Suppl. 22 (Suppl. O), O53–O60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hart RG, Benavente O, McBride R & Pearce LA Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann. Intern. Med. 131, 492–501 (1999). [DOI] [PubMed] [Google Scholar]
- 160.Hohmann C et al. Oral anticoagulants in comparison to phenprocoumon in geriatric and non-geriatric patients with non-valvular atrial fibrillation. Thromb. Haemost. 119, 971–980 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Hohnloser SH, Basic E & Nabauer M Changes in oral anticoagulation therapy over one year in 51,000 atrial fibrillation patients at risk for stroke: a practice-derived study. Thromb. Haemost. 119, 882–893 (2019). [DOI] [PubMed] [Google Scholar]
- 162.De Vecchis R et al. High prevalence of proarrhythmic events in patients with history of atrial fibrillation undergoing a rhythm control strategy: a retrospective study. J. Clin. Med. Res. 11, 345–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nery PB et al. Relationship between pulmonary vein reconnection and atrial fibrillation recurrence: a systematic review and meta-analysis. JACC Clin. Electrophysiol. 2, 474–483 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Kirchhof P et al. Early rhythm-control therapy in patients with atrial fibrillation. N. Engl. J. Med. 383, 1305–1316 (2020). [DOI] [PubMed] [Google Scholar]
- 165.Proietti M et al. Real-world applicability and impact of early rhythm control for European patients with atrial fibrillation: a report from the eSc-EHRA EORP-AF long-term general registry. Clin. Res. Cardiol. 111, 70–84 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim D et al. Comparative effectiveness of early rhythm control versus rate control for cardiovascular outcomes in patients with atrial fibrillation. J. Am. Heart Assoc. 10, e023055 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Calkins H et al. 2017 HRS/EHRA/ECAS/APHRS/ SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. Heart Rhythm 14, e445–e494 (2017). [DOI] [PubMed] [Google Scholar]
- 168.Stavrakis S et al. Low-level vagus nerve stimulation suppresses post-operative atrial fibrillation and inflammation: a randomized study. JACC Clin. Electrophysiol. 3, 929–938 (2017). [DOI] [PubMed] [Google Scholar]
- 169.Stavrakis S et al. Low-level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. J. Am. Coll. Cardiol. 65, 867–875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Takei M et al. Vagal stimulation prior to atrial rapid pacing protects the atrium from electrical remodeling in anesthetized dogs. Jpn. Circ. J. 65, 1077–1081 (2001). [DOI] [PubMed] [Google Scholar]
- 171.Agarwal A & Ioannidis JPA PREDIMED trial of Mediterranean diet: retracted, republished, still trusted? BMJ 364, l341 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Martinez-Gonzalez MA et al. Extravirgin olive oil consumption reduces risk of atrial fibrillation: the PREDIMED (Prevencion con Dieta Mediterranea) trial. Circulation 130, 18–26 (2014). [DOI] [PubMed] [Google Scholar]
- 173.Barrio-Lopez MT et al. PREvention of recurrent arrhythmias with Mediterranean diet (PREDIMAR) study in patients with atrial fibrillation: Rationale, design and methods. Am. Heart J. 220, 127–136 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Appleby PN, Davey GK & Key TJ Hypertension and blood pressure among meat eaters, fish eaters, vegetarians and vegans in EPIC-Oxford. Public Health Nutr. 5, 645–654 (2002). [DOI] [PubMed] [Google Scholar]
- 175.Alexander S, Ostfeld RJ, Allen K & Williams KA A plant-based diet and hypertension. J. Geriatr. Cardiol. 14, 327–330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yokoyama Y et al. Vegetarian diets and blood pressure: a meta-analysis. JAMA Intern. Med. 174, 577–587 (2014). [DOI] [PubMed] [Google Scholar]
- 177.Yokoyama Y, Barnard ND, Levin SM & Watanabe M Vegetarian diets and glycemic control in diabetes: a systematic review and meta-analysis. Cardiovasc. Diagn. Ther. 4, 373–382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tonstad S et al. Vegetarian diets and incidence of diabetes in the Adventist Health Study-2. Nutr. Metab. Cardiovasc. Dis. 23, 292–299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Orlich MJ & Fraser GE Vegetarian diets in the Adventist Health Study 2: a review of initial published findings. Am. J. Clin. Nutr. 100 (Suppl. 1), 353S–358S (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McMacken M & Shah S A plant-based diet for the prevention and treatment of type 2 diabetes. J. Geriatr. Cardiol. 14, 342–354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee YM et al. Effect of a brown rice based vegan diet and conventional diabetic diet on glycemic control of patients with type 2 diabetes: a 12-week randomized clinical trial. PLoS ONE 11, e0155918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Appleby PN & Key TJ The long-term health of vegetarians and vegans. Proc. Nutr. Soc. 75, 287–293 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Barnard ND, Levin SM & Yokoyama Y A systematic review and meta-analysis of changes in body weight in clinical trials of vegetarian diets. J. Acad. Nutr. Diet. 115, 954–969 (2015). [DOI] [PubMed] [Google Scholar]
- 184.Tonstad S, Butler T, Yan R & Fraser GE Type of vegetarian diet, body weight, and prevalence of type 2 diabetes. Diabetes Care 32, 791–796 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Turner-McGrievy G, Mandes T & Crimarco A A plant-based diet for overweight and obesity prevention and treatment. J. Geriatr. Cardiol. 14, 369–374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shah B et al. Anti-inflammatory effects of a vegan diet versus the American Heart Association-recommended diet in coronary artery disease trial. J. Am. Heart Assoc. 7, e011367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Craddock JC, Neale EP, Peoples GE & Probst YC Vegetarian-based dietary patterns and their relation with inflammatory and immune biomarkers: a systematic review and meta-analysis. Adv. Nutr. 10, 433–451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Franco-de-Moraes AC et al. Worse inflammatory profile in omnivores than in vegetarians associates with the gut microbiota composition. Diabetol. Metab. Syndr. 9, 62 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cao Y et al. Nutrient patterns and chronic inflammation in a cohort of community dwelling middle-aged men. Clin. Nutr. 36, 1040–1047 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Ornish D et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA 280, 2001–2007 (1998). [DOI] [PubMed] [Google Scholar]
- 191.Ornish D Avoiding revascularization with lifestyle changes: the Multicenter Lifestyle Demonstration Project. Am. J. Cardiol. 82, 72T–76T (1998). [DOI] [PubMed] [Google Scholar]
- 192.Storz MA & Helle P Atrial fibrillation risk factor management with a plant-based diet: a review. J. Arrhythm. 35, 781–788 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lau DH, Nattel S, Kalman JM & Sanders P Modifiable risk factors and atrial fibrillation. Circulation 136, 583–596 (2017). [DOI] [PubMed] [Google Scholar]
- 194.Xu S et al. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal. Transduct. Target. Ther. 6, 54 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tao J et al. Ketogenic diet suppressed t-regulatory cells and promoted cardiac fibrosis via reducing mitochondria-associated membranes and inhibiting mitochondrial function. Oxid. Med. Cell. Longev. 2021,5512322 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Aubert G et al. The failing heart relies on ketone bodies as a fuel. Circulation 133, 698–705 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bedi KC Jr. et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 133, 706–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Baker WL Treating arrhythmias with adjunctive magnesium: identifying future research directions. Eur. Heart J. Cardiovasc. Pharmacother. 3, 108–117 [DOI] [PubMed] [Google Scholar]
- 199.Kolte D, Vijayaraghavan K, Khera S, Sica DA & Frishman WH Role of magnesium in cardiovascular diseases. Cardiol. Rev. 22, 182–192 (2014). [DOI] [PubMed] [Google Scholar]
- 200.Misialek JR et al. Serum and dietary magnesium and incidence of atrial fibrillation in whites and in African Americans — Atherosclerosis Risk in Communities (ARIC) study. Circ. J. 77, 323–329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Khan AM et al. Low serum magnesium and the development of atrial fibrillation in the community: the Framingham Heart Study. Circulation 127, 33–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Markovits N et al. Database evaluation of the association between serum magnesium levels and the risk of atrial fibrillation in the community. Int. J. Cardiol. 205, 142–146 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Chaudhary R et al. Role of prophylactic magnesium supplementation in prevention of postoperative atrial fibrillation in patients undergoing coronary artery bypass grafting: a systematic review and meta-analysis of 20 randomized controlled trials. J. Atr. Fibrillation 12, 2154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Buckley BJR, Lip GYH & Thijssen DHJ The counterintuitive role of exercise in the prevention and cause of atrial fibrillation. Am. J. Physiol. Heart Circ. Physiol. 319, H1051–H1058 (2020). [DOI] [PubMed] [Google Scholar]
- 205.Malmo V et al. Aerobic interval training reduces the burden of atrial fibrillation in the short term: a randomized trial. Circulation 133, 466–473 (2016). [DOI] [PubMed] [Google Scholar]
- 206.Tolahunase M, Sagar R & Dada R Impact of yoga and meditation on cellular aging in apparently healthy individuals: a prospective, open-label single-arm exploratory study. Oxid. Med. Cell. Longev. 2017, 7928981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kanmanthareddy A et al. Alternative medicine in atrial fibrillation treatment-Yoga, acupuncture, biofeedback and more. J. Thorac. Dis. 7, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Oser M, Khan A, Kolodziej M, Gruner G, Barsky AJ & Epstein L Mindfulness and interoceptive exposure therapy for anxiety sensitivity in atrial fibrillation: a pilot study. Behav. Modif. 45, 462–479 (2021). [DOI] [PubMed] [Google Scholar]
- 209.Reavell J, Hopkinson M, Clarkesmith D & Lane DA Effectiveness of cognitive behavioral therapy for depression and anxiety in patients with cardiovascular disease: a systematic review and meta-analysis. Psychosom. Med. 80, 742–753 (2018). [DOI] [PubMed] [Google Scholar]
- 210.Dossett ML et al. A SMART approach to reducing paroxysmal atrial fibrillation symptoms: results from a pilot randomized controlled trial. Heart Rhythm O2 2, 326–332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Malm D et al. Effects of brief mindfulness-based cognitive behavioural therapy on health-related quality of life and sense of coherence in atrial fibrillation patients. Eur. J. Cardiovasc. Nurs. 17, 589–597 (2018). [DOI] [PubMed] [Google Scholar]
- 212.Abed HS et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: a randomized clinical trial. JAMA 310, 2050–2060 (2013). [DOI] [PubMed] [Google Scholar]
- 213.Pathak RK et al. Aggressive risk factor reduction study for atrial fibrillation and implications for the outcome of ablation: the ARREST-AF cohort study. J. Am. Coll. Cardiol. 64, 2222–2231 (2014). [DOI] [PubMed] [Google Scholar]
- 214.Pathak RK et al. Long-term effect of goal-directed weight management in an atrial fibrillation cohort: a long-term follow-up study (LEGACY). J. Am. Coll. Cardiol. 65, 2159–2169 (2015). [DOI] [PubMed] [Google Scholar]
- 215.Kim YJ, Kim JY, Kang SW, Chun GS & Ban JY Protective effect of geranylgeranylacetone against hydrogen peroxide-induced oxidative stress in human neuroblastoma cells. Life Sci. 131,51–56 (2015). [DOI] [PubMed] [Google Scholar]
- 216.van Marion DM et al. Screening of novel HSP-inducing compounds to conserve cardiomyocyte function in experimental atrial fibrillation. Drug Des. Devel Ther. 13, 345–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sakabe M et al. Effects of heat shock protein induction on atrial fibrillation caused by acute atrial ischemia. Cardiovasc. Res. 78, 63–70 (2008). [DOI] [PubMed] [Google Scholar]
- 218.van Marion DMS et al. Oral geranylgeranylacetone treatment increases heat shock protein expression in human atrial tissue. Heart Rhythm 17, 115–122 [DOI] [PubMed] [Google Scholar]
- 219.Starreveld R, Ramos KS, Muskens A, Brundel B & de Groot NMS Daily supplementation of l-glutamine in atrial fibrillation patients: the effect on heat shock proteins and metabolites. Cells 9, 1729 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang H et al. Glutamine promotes Hsp70 and inhibits a-Synuclein accumulation in pheochromocytoma PC12 cells. Exp. Ther. Med. 14, 1253–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yang J et al. Heat shock protein 70 induction by glutamine increases the alpha-synuclein degradation in SH-SY5Y neuroblastoma cells. Mol. Med. Rep. 12, 5524–5530 (2015). [DOI] [PubMed] [Google Scholar]
- 222.Pool L, Wijdeveld L, de Groot NMS & Brundel B The role of mitochondrial dysfunction in atrial fibrillation: translation to druggable target and biomarker discovery. Int. J. Mol. Sci. 22, 8463 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Li J, Zhang D, Brundel BJ & Wiersma M Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells 8 1617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Carducci MA et al. A phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin. Cancer Res. 7, 3047–3055 (2001). [PubMed] [Google Scholar]
- 225.Zhang D et al. Converse role of class I and class IIa HDACs in the progression of atrial fibrillation. J. Mol. Cell Cardiol. 125, 39–49 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Butler KV et al. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 132, 10842–10846 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.d’Ydewalle C et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974 (2011). [DOI] [PubMed] [Google Scholar]
- 228.Santo L et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579–2589 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Vogl DT et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23, 3307–3315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Witt O & Lindemann R HDAC inhibitors: magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 280, 123–124 (2009). [DOI] [PubMed] [Google Scholar]
- 231.Hassa PO & Hottiger MO The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 13, 3046–3082 (2008). [DOI] [PubMed] [Google Scholar]
- 232.Donawho CK et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 13, 2728–2737 (2007). [DOI] [PubMed] [Google Scholar]
- 233.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH & Poirier GG PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 10, 293–301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Swaisland H et al. Olaparib does not cause clinically relevant QT/QTc interval prolongation in patients with advanced solid tumours: results from two phase I studies. Cancer Chemother. Pharmacol. 78, 775–784 (2016) [DOI] [PubMed] [Google Scholar]
- 235.Robson M et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017). [DOI] [PubMed] [Google Scholar]
- 236.Diguet N et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 137, 2256–2273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lee CF et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134, 883–894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Walker MA & Tian R Raising NAD in heart failure: time to translate? Circulation 137, 2274–2277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Airhart SE et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 12, e0186459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Abdellatif M et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 13, eabd7064 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhang R et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417 (2005). [DOI] [PubMed] [Google Scholar]
- 242.Anderson ME Calmodulin kinase and L-type calcium channels; a recipe for arrhythmias? Trends Cardiovasc. Med. 14, 152–161 (2004). [DOI] [PubMed] [Google Scholar]
- 243.Yared JP et al. Effect of dexamethasone on atrial fibrillation after cardiac surgery: prospective, randomized, double-blind, placebo-controlled trial. J. Cardiothorac. Vasc. Anesth. 21, 68–75 (2007). [DOI] [PubMed] [Google Scholar]
- 244.Iskandar S et al. Use of oral steroid and its effects on atrial fibrillation recurrence and inflammatory cytokines post ablation — the steroid AF study. J. Atr. Fibrillation 9, 1604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21,248–255 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.von Eisenhart Rothe A et al. Depressed mood amplifies heart-related symptoms in persistent and paroxysmal atrial fibrillation patients: a longitudinal analysis — data from the German Competence Network on Atrial Fibrillation. Europace 17, 1354–1362 (2015). [DOI] [PubMed] [Google Scholar]
- 247.von Eisenhart Rothe AF et al. Depression in paroxysmal and persistent atrial fibrillation patients: a cross-sectional comparison of patients enroled in two large clinical trials. Europace 16, 812–819 (2014). [DOI] [PubMed] [Google Scholar]
- 248.Seligman WH et al. Development of an international standard set of outcome measures for patients with atrial fibrillation: a report of the International Consortium for Health Outcomes Measurement (ICHOM) atrial fibrillation working group. Eur. Heart J. 41, 1132–1140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Potpara TS et al. Self-reported treatment burden in patients with atrial fibrillation: quantification, major determinants, and implications for integrated holistic management of the arrhythmia. Europace 22, 1788–1797 (2020). [DOI] [PubMed] [Google Scholar]
- 250.Eton DT et al. Development and validation of the Patient Experience with Treatment and Selfmanagement (PETS): a patient-reported measure of treatment burden. Qual. Life Res. 26, 489–03 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Westcott SK et al. Relationship between psychosocial stressors and atrial fibrillation in women >45 years of age. Am. J. Cardiol. 122, 1684–1687 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Groh CA et al. Patient-reported triggers of paroxysmal atrial fibrillation. Heart Rhythm 16, 996–1002 (2019). [DOI] [PubMed] [Google Scholar]
- 253.Pereira T et al. Photoplethysmography based atrial fibrillation detection: a review. NPJ Digit. Med. 3, 3 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Turakhia MP et al. Rationale and design of a large-scale, app-based study to identify cardiac arrhythmias using a smartwatch: the Apple Heart study. Am. Heart J. 207, 66–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Marcus GM et al. Individualized studies of triggers of paroxysmal atrial fibrillation: the I-STOP-AFib randomized clinical trial. JAMA Cardiol. 7, 167–174 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hills MT Patient perspective: digital tools give afib patients more control. Cardiovasc. Digital Health J. 2, 192–194 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Perez MV et al. Large-scale assessment of a smartwatch to identify atrial fibrillation. N. Engl. J. Med. 381, 1909–1917 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Roselli C, Rienstra M & Ellinor PT Genetics of atrial fibrillation in 2020: GWAS, genome sequencing, polygenic risk, and beyond. Circ. Res. 127, 21–33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Olesen MS, Holst AG, Svendsen JH, Haunso S & Tfelt-Hansen J SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm 9, 770–773 (2012). [DOI] [PubMed] [Google Scholar]
- 260.Watanabe H et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Olesen MS et al. Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793 (2011). [DOI] [PubMed] [Google Scholar]
- 262.Li RG et al. Mutations of the SCN4B-encoded sodium channel beta4 subunit in familial atrial fibrillation. Int. J. Mol. Med. 32, 144–150 (2013). [DOI] [PubMed] [Google Scholar]
- 263.Makiyama T et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J. Am. Coll. Cardiol. 52, 1326–1334 (2008). [DOI] [PubMed] [Google Scholar]
- 264.Li Q et al. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 380, 132–137 (2009). [DOI] [PubMed] [Google Scholar]
- 265.Benito B et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 5, 1434–1440 (2008). [DOI] [PubMed] [Google Scholar]
- 266.Jabbari J et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ. Cardiovasc. Genet. 8, 64–73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Macri V et al. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm 11, 1055–1062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Weigl I et al. The C-terminal HCN4 variant P883R alters channel properties and acts as genetic modifier of atrial fibrillation and structural heart disease. Biochem. Biophys. Res. Commun. 51 9, 141–147 (2019). [DOI] [PubMed] [Google Scholar]
- 269.Olson TM et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat. Clin. Pract. Cardiovasc. Med. 4, 110–116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ni H, Adeniran I & Zhang H In-silico investigations of the functional impact of KCNA5 mutations on atrial mechanical dynamics. J. Mol. Cell Cardiol. 111, 86–95 (2017). [DOI] [PubMed] [Google Scholar]
- 271.Christophersen IE et al. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 34, 1517–1525 (2013). [DOI] [PubMed] [Google Scholar]
- 272.Olson TM et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 15, 2185–2191 (2006). [DOI] [PubMed] [Google Scholar]
- 273.Yang Y et al. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J. Hum. Genet. 54, 277–283 (2009). [DOI] [PubMed] [Google Scholar]
- 274.Drabkin M et al. Nocturnal atrial fibrillation caused by mutation in KCND2, encoding pore-forming (alpha) subunit of the cardiac Kv4.2 potassium channel. Circ. Genom. Precis. Med. 11, e002293 (2018). [DOI] [PubMed] [Google Scholar]
- 275.Huang Y et al. A novel KCND3 mutation associated with early-onset lone atrial fibrillation. Oncotarget 8, 115503–115512 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Olesen MS et al. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc. Res. 98, 488–495 (2013). [DOI] [PubMed] [Google Scholar]
- 277.Olesen MS et al. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med. Genet. 13, 24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Voudris KV et al. Genetic diversity of the KCNE1 gene and susceptibility to postoperative atrial fibrillation. Am. Heart J. 167, 274–280.e1 (2014). [DOI] [PubMed] [Google Scholar]
- 279.Yang Y et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 75, 899–905 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Nielsen JB et al. Gain-of-function mutations in potassium channel subunit KCNE2 associated with early-onset lone atrial fibrillation. Biomark. Med. 8, 557–570 (2014). [DOI] [PubMed] [Google Scholar]
- 281.Lundby A et al. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol. Biochem. 21, 47–54 (2008). [DOI] [PubMed] [Google Scholar]
- 282.Mann SA et al. Epistatic effects of potassium channel variation on cardiac repolarization and atrial fibrillation risk. J. Am. Coll. Cardiol. 59, 1017–1025 (2012). [DOI] [PubMed] [Google Scholar]
- 283.Ravn LS et al. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm 5, 427–435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Hong K, Bjerregaard P, Gussak I & Brugada R Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 16, 394–396 (2005). [DOI] [PubMed] [Google Scholar]
- 285.Sinner MF et al. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG). Eur. Heart J. 29, 907–914 (2008). [DOI] [PubMed] [Google Scholar]
- 286.Steffensen AB et al. IKs gain- and loss-of-function in early-onset lone atrial fibrillation. J. Cardiovasc. Electrophysiol. 26, 715–723 (2015). [DOI] [PubMed] [Google Scholar]
- 287.Campbell CM et al. Selective targeting of gain-of-function KCNQ1 mutations predisposing to atrial fibrillation. Circ. Arrhythm. Electrophysiol. 6, 960–966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Abraham RL, Yang T, Blair M, Roden DM & Darbar D Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J. Mol. Cell Cardiol. 48, 181–190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Chen YH et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299, 251–254 (2003). [DOI] [PubMed] [Google Scholar]
- 290.Deo M et al. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc. Natl Acad. Sci. USA 110, 4291–4296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Xia M et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 332, 1012–1019 (2005). [DOI] [PubMed] [Google Scholar]
- 292.Yamada N et al. Mutant KCNJ3 and KCNJ5 potassium channels as novel molecular targets in bradyarrhythmias and atrial fibrillation. Circulation 139, 2157–2169 (2019). [DOI] [PubMed] [Google Scholar]
- 293.Delaney JT et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 14, 1428–1432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Weeke P et al. Whole-exome sequencing in familial atrial fibrillation. Eur. Heart J. 35, 2477–2483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Schrickel JW et al. Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J. Interv. Card. Electrophysiol. 28, 71–80 (2010). [DOI] [PubMed] [Google Scholar]
- 296.van Spaendonck-Zwarts KY et al. Desmin-related myopathy. Clin. Genet. 80, 354–366 (2011). [DOI] [PubMed] [Google Scholar]
- 297.Yokokawa T et al. Case reports of a c.475G>T, p.E159* lamin A/C mutation with a family history of conduction disorder, dilated cardiomyopathy and sudden cardiac death. BMC Cardiovasc. Disord. 19, 298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Han M et al. Lamin A mutation impairs interaction with nucleoporin NUP155 and disrupts nucleocytoplasmic transport in atrial fibrillation. Hum. Mutat. 40, 310–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Glocklhofer CR et al. A novel LMNA nonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family. Europace 20, 2003–2013 (2018). [DOI] [PubMed] [Google Scholar]
- 300.Hoorntje ET et al. Lamin A/C-related cardiac disease: late onset with a variable and mild phenotype in a large cohort of patients with the lamin A/C p. (Arg331Gln) founder mutation. Circ. Cardiovasc. Genet. 10, e001631 (2017). [DOI] [PubMed] [Google Scholar]
- 301.Zhao J et al. A novel nonsense mutation in LMNA gene identified by exome sequencing in an atrial fibrillation family. Eur. J. Med. Genet. 59, 396–400 (2016). [DOI] [PubMed] [Google Scholar]
- 302.Ahlberg G et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat. Commun. 9, 4316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Choi SH et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA 320, 2354–2364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Chalazan B et al. Association of rare genetic variants and early-onset atrial fibrillation in ethnic minority individuals. JAMA Cardiol. 6, 811–819 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gruver EJ et al. Familial hypertrophic cardiomyopathy and atrial fibrillation caused by Arg663His beta-cardiac myosin heavy chain mutation. Am. J. Cardiol. 83, 13H–18HH (1999). [DOI] [PubMed] [Google Scholar]
- 306.Zhang S, Wilson J, Madani M, Feld G & Greenberg B Atrial arrhythmias and extensive left atrial fibrosis as the initial presentation of MYH7 gene mutation. JACC Clin. Electrophysiol. 4, 1488–1490 (2018). [DOI] [PubMed] [Google Scholar]
- 307.Noureldin M, Chen H & Bai D Functional characterization of novel atrial fibrillation-Linked GJA5 (Cx40) mutants. Int. J. Mol. Sci. 19, 977 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lubkemeier I et al. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell Cardiol. 65, 19–32 (2013). [DOI] [PubMed] [Google Scholar]
- 309.Thibodeau IL et al. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 122, 236–244 (2010). [DOI] [PubMed] [Google Scholar]
- 310.Beavers DL et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 62, 2010–2019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Zhang X et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135, 1017–1027 (2008). [DOI] [PubMed] [Google Scholar]
- 312.Tsai CT et al. Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J. Med. Genet. 52, 28–36 (2015). [DOI] [PubMed] [Google Scholar]
- 313.Posch MG et al. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur. J. Med. Genet. 53, 201–203 (2010). [DOI] [PubMed] [Google Scholar]
- 314.Yang YQ et al. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin. Chim. Acta 412, 1825–1830 (2011). [DOI] [PubMed] [Google Scholar]
- 315.Laforest B et al. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J. Clin. Invest. 129, 4937–4950 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Wang XH et al. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 31, 43–50 (2013). [DOI] [PubMed] [Google Scholar]
- 317.Tucker NR et al. Gain-of-function mutations in GATA6 lead to atrial fibrillation. Heart Rhythm 14, 284–291 (2017). [DOI] [PubMed] [Google Scholar]
- 318.Gutierrez-Roelens I et al. A novel CSX/NKX2–5 mutation causes autosomal-dominant AV block: are atrial fibrillation and syncopes part of the phenotype? Eur. J. Hum. Genet. 14, 1313–1316 (2006). [DOI] [PubMed] [Google Scholar]
- 319.Boldt LH et al. Mutational analysis of the PITX2 and NKX2–5 genes in patients with idiopathic atrial fibrillation. Int. J. Cardiol. 145, 316–317 (2010). [DOI] [PubMed] [Google Scholar]
- 320.Wang J et al. NKX2–6 mutation predisposes to familial atrial fibrillation. Int. J. Mol. Med. 34, 1581–1590 (2014). [DOI] [PubMed] [Google Scholar]
- 321.Wang ZC et al. Prevalence and spectrum of TBX5 mutation in patients with lone atrial fibrillation. Int. J. Med. Sci. 13, 60–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Ma JF et al. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc. Res. 109, 442–450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Mechakra A et al. A Novel PITX2c gain-of-function mutation, p.Met207Val, in patients with familial atrial fibrillation. Am. J. Cardiol. 123, 787–793 (2019). [DOI] [PubMed] [Google Scholar]
- 324.Zhou YM, Zheng PX, Yang YQ, Ge ZM & Kang WQ A novel PITX2c lossoffunction mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 32, 827–834 (2013). [DOI] [PubMed] [Google Scholar]
- 325.Muller II et al. Functional modeling in zebrafish demonstrates that the atrial-fibrillation-associated gene GREM2 regulates cardiac laterality, cardiomyocyte differentiation and atrial rhythm. Dis. Model. Mech. 6, 332–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ren X et al. Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clin. Chim. Acta 411, 481–485 (2010). [DOI] [PubMed] [Google Scholar]
- 327.Cheng C et al. Mutation in NPPA causes atrial fibrillation by activating inflammation and cardiac fibrosis in a knock-in rat model. FASEB J. 33, 8878–8891 (2019). [DOI] [PubMed] [Google Scholar]
- 328.Hu YF et al. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 5, 334–340 (2012). [DOI] [PubMed] [Google Scholar]
- 329.Mandal K et al. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 79, 865–871 (2005). [DOI] [PubMed] [Google Scholar]
- 330.St Rammos K et al. Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc. Surg. 10, 228–232 (2002). [DOI] [PubMed] [Google Scholar]
- 331.Cao H et al. Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones 16, 517–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Schafler AE et al. The expression of heat shock protein 60 in myocardium of patients with chronic atrial fibrillation. Basic. Res. Cardiol. 97, 258–261 (2002). [DOI] [PubMed] [Google Scholar]
- 333.Maan A et al. Association between heat shock protein-60 and development of atrial fibrillation: results from the multi-ethnic study of atherosclerosis (MESA). Pacing Clin. Electrophysiol. 39, 1373–1378 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Oc M et al. Heat shock protein 60 antibody. A new marker for subsequent atrial fibrillation development. Saudi Med. J. 28, 844–847 (2007). [PubMed] [Google Scholar]
- 335.Sandler N et al. Mitochondrial DAMPs are released during cardiopulmonary bypass surgery and are associated with postoperative atrial fibrillation. Hear Lung Circ. 27, 122129 (2018). [DOI] [PubMed] [Google Scholar]
- 336.Sepehri Shamloo A et al. Atrial fibrillation: is there a role for cardiac troponin? Diagnosis 8, 295–303 (2007) [DOI] [PubMed] [Google Scholar]
- 337.Cheng T, Wang XF, Hou YT & Zhang L Correlation between atrial fibrillation, serum amyloid protein A and other inflammatory cytokines. Mol. Med. Rep. 6, 581–584 (2012). [DOI] [PubMed] [Google Scholar]
- 338.Cabrera-Bueno F et al. Serum levels of interleukin-2 predict the recurrence of atrial fibrillation after pulmonary vein ablation. Cytokine 73, 74–78 (2015). [DOI] [PubMed] [Google Scholar]
- 339.Stanciu AE, Vatasescu RG, Stanciu MM, Serdarevic N & Dorobantu M The role of pro-fibrotic biomarkers in paroxysmal and persistent atrial fibrillation. Cytokine 103, 63–68 (2018). [DOI] [PubMed] [Google Scholar]
- 340.Gaudino M et al. The 174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 108 (Suppl 1), II 195–II 199 (2003). [DOI] [PubMed] [Google Scholar]
- 341.Wu N et al. Elevated plasma levels of Th17-related cytokines are associated with increased risk of atrial fibrillation. Sci. Rep. 6, 26543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Luan Y et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace 12, 1713–1718 (2010). [DOI] [PubMed] [Google Scholar]
- 343.Li J et al. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 7, 438–444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Holzwirth E et al. Myeloperoxidase in atrial fibrillation: association with progression, origin and influence of renin-angiotensin system antagonists. Clin. Res. Cardiol. 109, 324–330 (2020). [DOI] [PubMed] [Google Scholar]
- 345.Li SB et al. Myeloperoxidase and risk of recurrence of atrial fibrillation after catheter ablation. J. Investig. Med. 61, 722–727 (2013). [DOI] [PubMed] [Google Scholar]
- 346.Yu X et al. MARK4 controls ischaemic heart failure through microtubule detyrosination. Nature 594, 560–565 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Miragoli M et al. Microtubule-dependent mitochondria alignment regulates calcium release in response to nanomechanical stimulus in heart myocytes. Cell Rep. 14, 140–151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.de Brito OM & Scorrano L Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.