

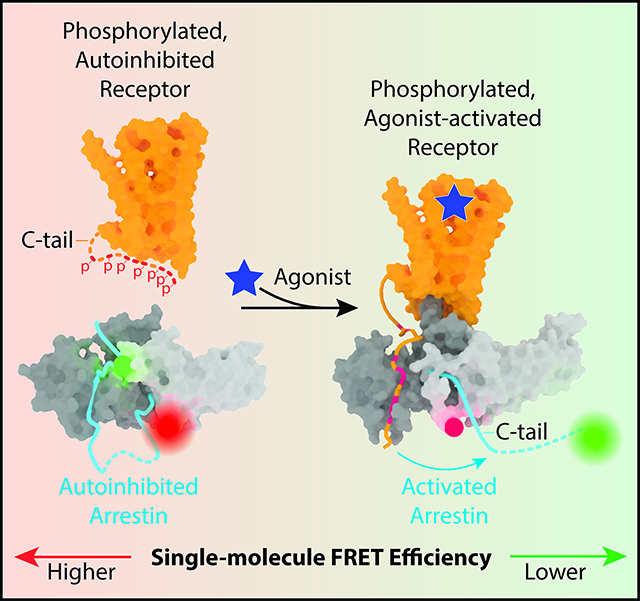
SUMMARY
β-arrestins bind G protein-coupled receptors to terminate G-protein signaling and to facilitate other downstream signaling pathways. Using single-molecule fluorescence resonance energy transfer imaging, we show that β-arrestin is strongly autoinhibited in its basal state. Its engagement with a phosphopeptide mimicking the phosphorylated receptor tail efficiently releases the β-arrestin tail from its N domain to assume distinct conformations. Unexpectedly, we find that β-arrestin binding to phosphorylated receptor, with a phosphorylation barcode identical to the isolated phosphopeptide, is highly inefficient and that agonist-promoted receptor activation is required for β-arrestin activation, consistent with release of a sequestered receptor C tail. These findings, together with focused cellular investigations, reveal that agonism and receptor C-tail release are specific determinants of the rate and efficiency of β-arrestin activation by phosphorylated receptor. We infer that receptor phosphorylation patterns, in combination with receptor agonism, synergistically establish the strength and specificity with which diverse, downstream β-arrestin-mediated events are directed.
Keywords: β-arrestin, arrestin, G protein-coupled receptor, GPCR, Single-Molecule FRET, conformational dynamics, phosphorylation, phosphorylation barcode, agonist, receptor signaling
In Brief
Receptor-mediated β-arrestin activation requires relief of autoinhibition mediated by the respective C-terminal tails of the β2-adrenergic receptor and β-arrestin1. These processes depend not only on the degree of receptor tail phosphorylation but also on concurrent agonist-mediated receptor activation
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCRs), the largest family of cell surface receptors, regulate a multitude of physiological processes and are the most common targets for marketed drugs (Sriram and Insel, 2018). The nonvisual arrestins, β-arrestin (βarr) 1 and 2 (also known as arrestin 2 and 3, respectively) (Attramadal et al., 1992; Lohse et al., 1990), are recruited by hundreds of different GPCRs (Gurevich and Gurevich, 2019) to desensitize G protein signaling and to promote clathrin-mediated receptor endocytosis (Figure 1A) (Hilger et al., 2018). By scaffolding kinases and other molecules, receptor-activated β-arrestins also can directly facilitate diverse cellular signaling pathways distinct from those initiated by G proteins (Jean-Charles et al., 2017; Smith et al., 2018). There is great interest in identifying therapeutics that preferentially promote or block βarr signaling to allow for more efficacious and specific targeting with the potential of reduced side effects. Structural and kinetic insights into the mechanism by which ligand-mediated GPCR activation enables βarr coupling and activation are required to achieve this goal.
Figure 1. β-arrestin1 C-tail dynamic behavior in the basal state.
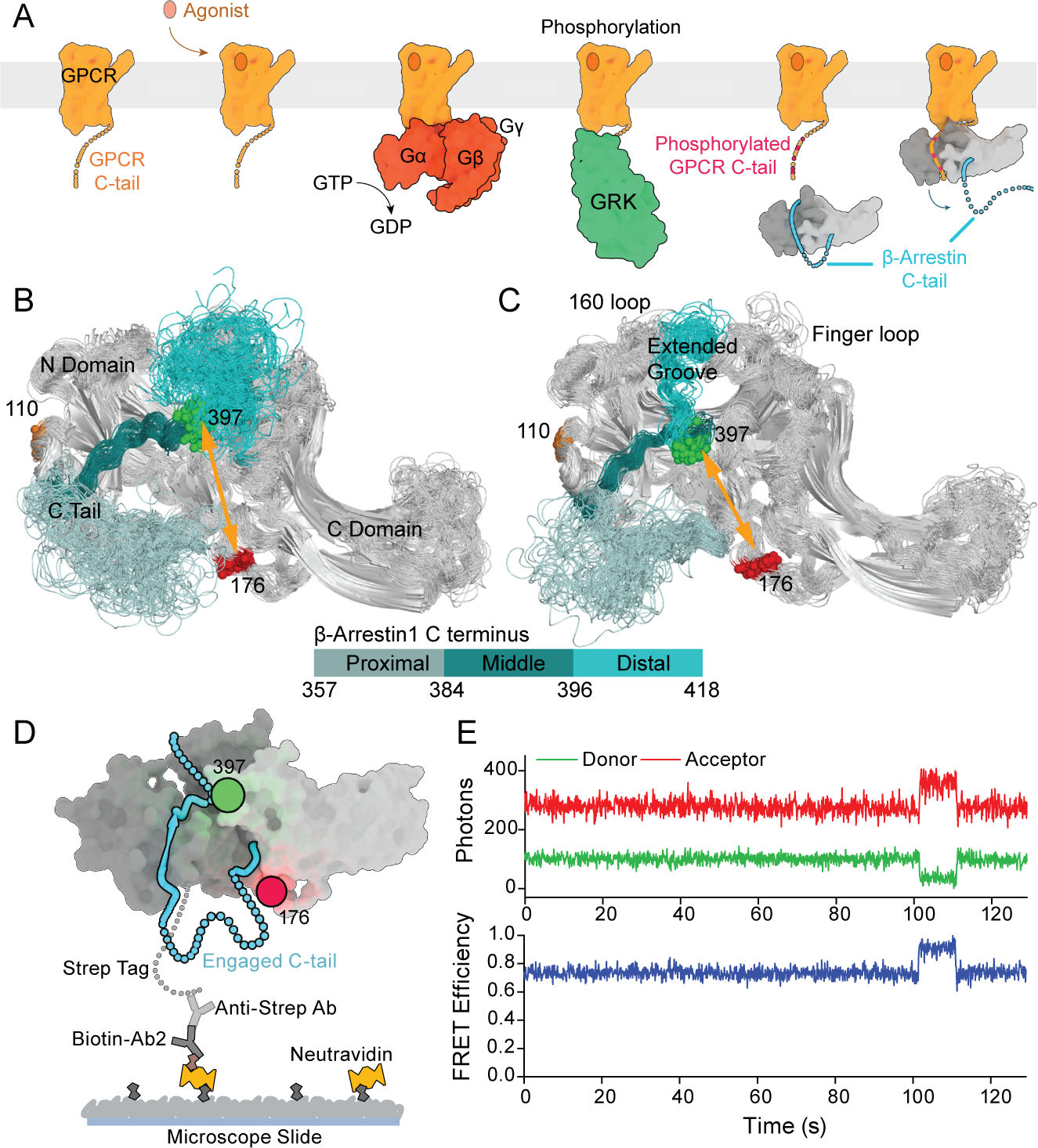
A, Schematic of βarr binding to an agonist-activated GPCR in the plasma membrane. B, Representative MD frames of simulated βarr1 in the inactive state showing the distal βarr1 tail outside the extended groove. C, MD frames from a subset of these simulations showing the distal tail can dock within the extended groove, bringing the mean dye positions closer together. D, Schematic of the smFRET experiment with fluorophore (green and red circles)-labeled βarr1 tail sensor tethered to an imaging surface. E, Representative single-molecule fluorescence (top; donor in green, acceptor in red) and FRET (bottom; blue) time traces of surface-tethered βarr1 recorded with 100 ms time resolution in the absence of ligands, with a rare excursion out of the basal state to a ~0.89 FRET state.
Previous investigations have established that receptors interact with βarr at two distinct interfaces. The GPCR kinase (GRK)-phosphorylated receptor carboxy (C)-terminal tail (Rp tail), or phosphorylated intracellular loops in some receptors, binds a positively charged groove within the βarr amino (N)-terminal domain (N domain). For the Rp tail to engage the groove, β-arrestin’s C-terminal tail (βarr tail) must be displaced from this N-domain groove (Figure 1A). The receptor transmembrane (TM) helices and cytoplasmic loops (receptor core) can also interact with the central crest loops of βarr, including the finger loop (Kahsai et al., 2018; Scheerer and Sommer, 2017; Shukla et al., 2014a).
Structural studies have shown that the receptor can engage βarr solely through its Rp tail (“tail-only” engagement) or through more extensive contacts involving both the receptor core and the Rp-tail (“tight” engagement) (Figure 1A) (Shukla et al., 2014a). These modes are thought to confer distinct biological functions (Cahill et al., 2017). Crystal and cryo-electron microscopy structures of active βarr have revealed rearrangements in the central crest loops and rotation of the C-terminal domain (C domain) relative to the N domain (Kang et al., 2015; Shukla et al., 2013a; Staus et al., 2020a). However, the conformational changes and dynamics of the βarr tail associated with its release from the N-domain groove, and how receptor engagement affects these events, have yet to be delineated.
In basal (inactive)-state βarr structures, only a short segment of the βarr tail anchored to the N domain is resolved (Han et al., 2001; Milano et al., 2002). The position and conformation of the βarr tail in the active state are not known, as the entire βarr tail is either removed or unresolved in these structures (Gurevich et al., 2018). The βarr tail contains regions that bind clathrin and adaptin, and it may also be directly involved in, or expose N domain regions important for, scaffolding signaling kinases including a direct interaction with EKR2 (Gurevich and Gurevich, 2015; Perry-Hauser et al., 2022). Thus, the timing and nature of βarr tail release, as it relates to receptor engagement and other structural rearrangements in βarr, are critical for understanding βarr-mediated signaling. In addition, a deeper understanding of βarr activation and how it may vary for different receptors and different ligands is necessary to provide insight into the mechanisms underlying biased agonism (Xiao and Sun, 2018).
Here, we use total internal reflection fluorescence (TIRF)-based single-molecule fluorescence resonance energy transfer (smFRET) imaging (Juette et al., 2014) to investigate the β-arrestin1 (βarr1) activation mechanism by examining release of its C-terminal tail region (βarr1 tail) upon binding of phosphorylated receptor or known mimics of the Rp tail. These measurements enabled direct observation of stochastic, dynamic processes in the βarr1 tail, largely inaccessible to ensemble methods, associated with βarr1’s interaction with receptor and of how these processes are regulated by GPCR activation.
We leveraged our insights from smFRET imaging for targeted, hypothesis-driven studies in living cells to show that agonist stimulated, full-length wild type β2-adrenergic receptor (β2AR) is unable to recruit βarr2 in the absence of GRK expression, whereas truncation of the receptor’s distal C tail reveals measurable GRK-independent βarr2 recruitment. These findings provide evidence for receptor autoinhibition mechanisms that contribute to regulation of agonist-induced βarr engagement, downstream signaling and receptor internalization.
RESULTS
The basal state of β-arrestin1 is strongly autoinhibited
We first sought to investigate the structure and dynamics of the βarr1 tail in the inactive conformation of βarr1 using extended molecular dynamics (MD) simulations (see STAR Methods) (Table S1), where the βarr1 tail is defined as the segment from K357 to the C-terminal residue R418 (Figure 1B). This analysis revealed a stable interaction of the βarr1 tail segment consisting of residues D384–L396, which we define as the middle segment, with the N-domain groove (Figures 1B and 1C). We refer to the βarr1 tail segments before and after the middle segment as the proximal and distal βarr1 tail, respectively. These simulations suggested logical donor and acceptor fluorophore attachment points for smFRET imaging of βarr1 tail dynamics: K397, at the juncture of the middle and distal βarr1 tail segments, and E176, a relatively static and centrally located position in the inter-domain hinge region (Figures 1B and 1C). Cysteine substitutions were introduced at these positions into fully functional cysteine-less βarr1 (Hanson et al., 2007) and the protein was labeled with LD555p and LD655, the donor and acceptor fluorophores, to yield what we refer to as the βarr1 tail sensor (see STAR Methods) (Figures 1D and S1A).
For TIRF imaging, the βarr1 tail sensor was tethered to the surface of a polyethylene-glycol passivated and biotinylated quartz slide, using a NeutrAvidin bridge to biotinylated anti-mouse antibody, a mouse StrepMAB-Immo antibody and N-terminal Strep tagged βarr1 tail sensors (see STAR Methods) (Figure 1D and S1B). In the basal state, individual smFRET trajectories of βarr1 tail sensor displayed a highly stable FRET efficiency centered at ~0.72 (referred to as the high-FRET, inactive state) (Figures 1E and S1C). Analogous results were also observed using confocal microscopy of individual, freely diffusing βarr1 tail sensor molecules (see STAR Methods) (Figure S1D). Stable high-efficiency FRET is consistent with an inactive, autoinhibited βarr1 conformation in which the βarr1 tail middle segment (near position 397) remains tightly anchored within the N-domain groove, as observed throughout our MD simulations of the basal state (~50 μs) (Figure 1B). Implicit solvent simulations of fluorophore dynamics (Noel et al., 2016) starting from a representative MD frame estimated a distance of ~53 Å between the dyes at positions 176 and 397, consistent with ~53 Å inter-dye distance estimated from smFRET (see STAR Methods for calculations and assumptions). Notably, rare excursions out of the basal state to a ~0.89 FRET efficiency state (Figure 1E) were observed. This finding is consistent with MD simulations in which the distal βarr1 tail occasionally docked within the “extended groove” between the finger loop and the 160-loop of the N domain, bringing the mean dye positions closer together (Figure 1C).
Receptor phospho-tail mimetics differentially activate β-arrestin1
Having validated the βarr1 tail sensor as a reliable reporter of the inactive conformation, we next examined the effects of activation in the presence of a synthetic phosphorylated peptide mimetic of the human V2 vasopressin receptor tail (V2Rpp; Table 1). V2Rpp binds to, and functionally activates, βarr1 (Shukla et al., 2013a; Xiao et al., 2004) and thus serves as a model for the tail-only receptor engagement mode. When V2Rpp was injected into the microfluidic imaging chamber in which βarr1 tail sensor molecules were surface tethered, we observed reversible transitions to a distinct state, with a mean FRET efficiency value of ~0.46 (mid-FRET, active state) (Figures 2A–2C). Occupancy of the mid-FRET, active state increased with higher V2Rpp concentrations, while that of the high-FRET, inactive states was depleted (Figure 2D). The EC50 of this binding process was 2.6±0.36 μM, in accordance with the binding affinity of V2Rpp for unlabeled wild-type βarr1 measured by isothermal titration calorimetry (see STAR Methods) (2.4±0.2 μM; Figure 2E), indicating negligible effects of the cysteine-substitutions, fluorophore attachment, or surface tethering of βarr1. Similar results were obtained from confocal smFRET measurements of the same binding reaction (Figure S1D). Measurement of smFRET dynamics by TIRF of a different βarr1 tail sensor variant labeled at positions 110 (at the end of the N-domain helix; Figure 1B) and 397, in the presence of increasing V2Rpp concentrations, yielded similar outcomes from a distinct structural perspective (Figure S2).
Table 1.
Amino acid sequences of receptor variants and related phosphopeptides.
| Construct | Amino acid Sequence |
|---|---|
| pβ2V2R | F336QELLC-ARGRT1PPS2LGPQDES3CT4T5AS6S7S8LAKDTSS |
| bpβ2V2R | F336QELLC-ARGRT1PPS2LGPQDES3CT4T5AS6S7S8LAKDTSS |
| V2Rpp | ARGRT1PPS2LGPQDES3CT4T5AS6S7S8LAKDTSS |
| V2Rbp | ARGRT1PPS2LGPQDES3CT4T5AS6S7S8LAKDTSS |
| V2Rnp | ARGRT1PPS2LGPQDES3CT4T5AS6S7S8LAKDTSS |
Amino acid sequence of the two receptor variants and three phosphopeptide variants. The receptor sequences are shown starting from residue F336 though C341 of β2AR, followed by the V2 receptor C tail residues. The phosphorylated residues in the receptor V2 tail and phosphopeptides are shown in bold and underlined. The positions of the sites of phosphorylation have the assigned numbering P1 to P8 denoted as a subscript for each residue.
Figure 2. Activation of β-arrestin1 by V2Rpp.
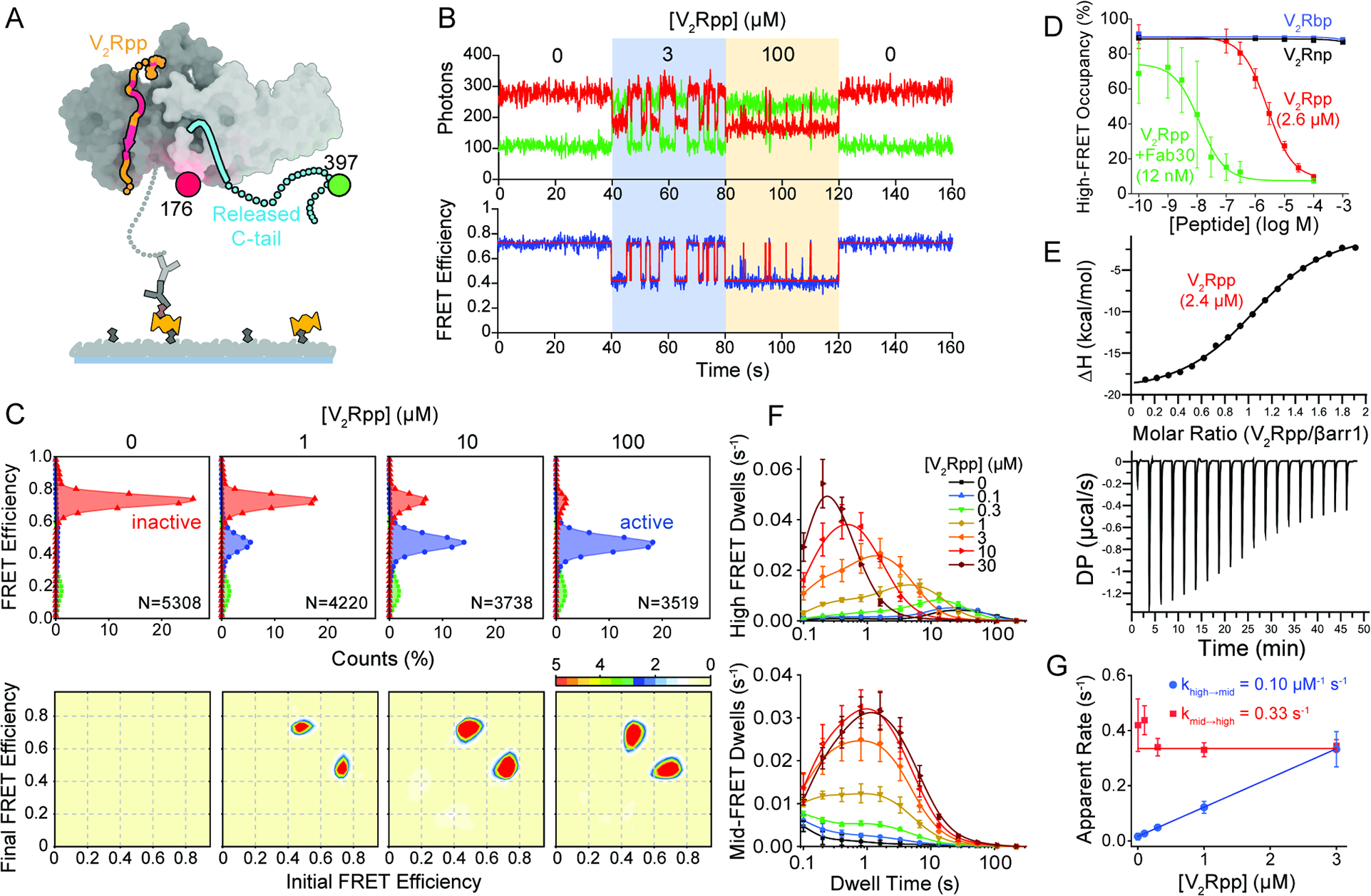
A, Schematic of the smFRET experiment where V2Rpp (orange with red for phosphorylation sites) binding induces βarr1 tail (cyan) disengagement. B, Representative single-molecule traces with state assignment (bottom, red line);surface-tethered βarr1 tail sensor was sequentially exposed to buffer containing the indicated V2Rpp concentrations (shaded areas). C, Population smFRET histograms for inactive (red) and active (blue and green) states (top; N, number of traces) and corresponding transition density plots displaying the mean FRET values before (x axis) and after (y axis) each transition (bottom; scale bar, 10−3 transitions per bin per second) in the presence of the indicated V2Rpp concentrations. D, Ensemble average high FRET inactive state occupancy induced by increasing concentrations of various peptides: V2Rpp (red), V2Rpp with 0.1 μM Fab30 (green), V2Rbp (blue), and V2Rnp (black). Lines are fits to dose-response functions with Hill slope of 1.0 and EC50 of 2.6 μM for V2Rpp alone and 0.012 μM for V2Rpp with Fab30, respectively. E, Analysis of V2Rpp interaction with wildtype βarr1 by isothermal titration calorimetry (ITC). Representative binding isotherm (top) and thermogram (bottom), with the best titration curve fit are shown. Summary of thermodynamic parameters obtained by ITC: binding affinity (KD = 2.4 ± 0.2 μM), stoichiometry (N = 1.1 ± 0.1 sites), enthalpy (ΔH = −20.3 ± 0.4 kcal/mol), and entropy (−TΔS = 13.9 ± 2.0 kcal/mol). F, Dwell time histograms (symbols) in the inactive (top) and active (bottom) states from experiments corresponding to panels C-D with spline fits (lines). G, Apparent rate constants for V2Rpp binding (blue) and unbinding (red) of 0.10 μM−1 s−1 and 0.33 s−1, respectively. Error bars, mean ± S.D. of at least two repeats with at least 3,500 traces for each condition and a total of more than 33,000.
In line with the ability of V2Rpp to act as a phosphorylated receptor surrogate for activation of βarr1, the corresponding non-phosphorylated peptide (V2Rnp; Table 1) had no effect on the FRET efficiency distribution even at millimolar concentrations (Figure 2D). Thus, as expected, V2Rpp binding leads, in a phosphorylation-dependent manner, to release of the βarr1 tail middle segment from the N domain. In addition, the observed changes in FRET efficiency suggest a movement of the middle and distal segments of the tail away from both positions 176 and 110.
To investigate the kinetics of V2Rpp binding and dissociation from βarr1, we calculated the transition rates between the high-FRET inactive and mid-FRET active states using hidden Markov modeling (HMM) and likelihood maximization (see STAR Methods) (Juette et al., 2016). Increasing V2Rpp concentrations reduced the duration of high-FRET inactive state dwell times, while having no effect on mid-FRET active state dwell times (Figure 2F), consistent with concentration-dependent V2Rpp binding and concentration-independent unbinding processes, respectively. The apparent rate constant for V2Rpp binding was ~0.10 μM−1 s−1 (Figure 2G), more than three orders of magnitude slower than expected for a diffusion-controlled bimolecular interaction between structured macromolecules (on the order of 100–1000 μM−1 s−1) (Schreiber et al., 2009). Stopped-flow V2Rpp addition and washout studies revealed apparent binding and unbinding rates similar to those evidenced by HMM analysis of the equilibrium data (kon: ~0.27 μM−1 s−1; koff: ~0.21 s−1) (Figures S1E and S1F). Thus, V2Rpp-induced βarr1 tail release, while appreciably faster than might be expected by autoinhibited apo βarr1, is rate-limited by one or more unresolved conformational events. We speculate that an initial interaction of V2Rpp with the βarr1 “extended groove” could facilitate βarr1 tail release through active mechanisms. However, we cannot rule out a mechanism involving spontaneous tail release that is either too rapid or small in scale to be detected by our smFRET analysis.
An antibody fragment (Fab30) against βarr1, developed for structural determination purposes, is known to stabilize a βarr1 conformation that preferentially binds activated GPCRs (Shukla et al., 2013b). Fab30 induced a > 200-fold increase in apparent V2Rpp binding affinity (12 nM vs. 2.6 μM; Figure 2D). Kinetic dissociation experiments confirmed that the βarr1 tail sensor remained bound to V2Rpp in the presence of Fab30 for T1/2 ≈ 33 min (Figures S3A–S3D), suggesting that the increase in affinity imparted by Fab30 is due to a dramatic decrease of the V2Rpp dissociation rate.
Prior studies have suggested that heparin (Figure 3A), a naturally occurring glycosaminoglycan that has been used to activate arrestins (Xiao et al., 2004; Zhuang et al., 2010), may stabilize βarr1 tail conformations distinct from those stabilized by V2Rpp (Xiao et al., 2004). In striking contrast to V2Rpp, heparin induced βarr1 tail dynamics between high-FRET inactive and mid-FRET active states that were roughly two orders of magnitude more rapid (Figure 3B), as well as transitions between mid-FRET and a distinct low-FRET active state with a mean FRET value of ~0.16 (Figure 3C). Moreover, heparin exhibited an approximately 17 nM affinity for βarr1, roughly 180-fold higher than V2Rpp (Figures 3D and E). Given the observation that a similar low-FRET, active state was also observed, albeit rarely, with V2Rpp (Figure 2C), we speculate that heparin facilitates a βarr1 switch to an alternative active state that exhibits distinct C-tail positioning and dynamics. Consistent with this model, switching events between both activated modes were directly evidenced by smFRET, albeit rarely (Figure 3F). These data suggest that the βarr1 tail can adopt at least two distinct active-state conformations, where the “released” βarr1 tail may exhibit alternative residual structured elements or docking sites.
Figure 3. Activation of β-arrestin1 by heparin, IP6 and PIP2.
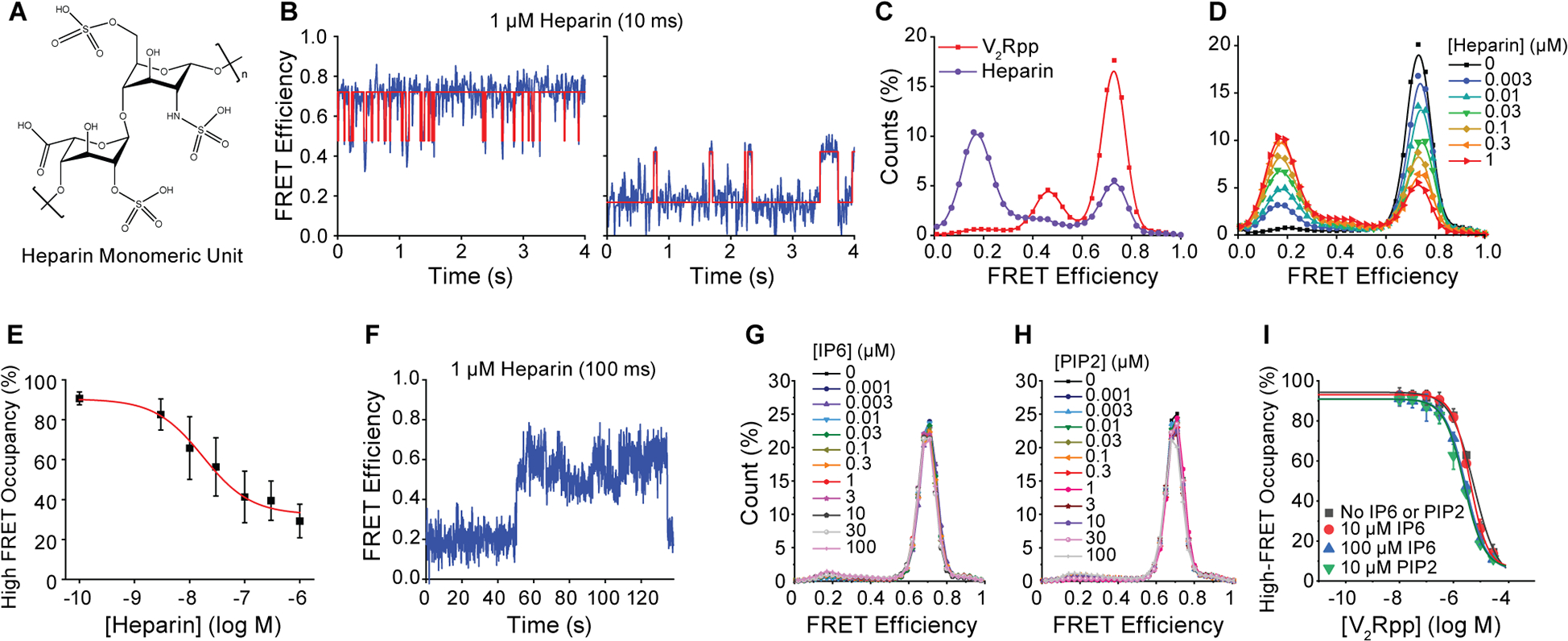
A, Structure of heparin. B, Representative smFRET traces from experiments imaging βarr1 tail sensor in the presence of 1 μM heparin with 10 ms time resolution. C, Overlay of FRET histograms experiments with 1 μM heparin (purple) and V2Rpp (red). D, FRET histograms and E, high-FRET state occupancy (symbols) from experiments imaging βarr1 tail sensor in the presence of the indicated concentrations of heparin. Line is a fit to a dose-response function (red line) with Hill slope of 1.0 and EC50 of 17 nM. F, Example trace recorded at 100 ms time resolution in the presence of 1 μM heparin showing rare switching between dynamic modes. Error bars, mean ± S.D. of two repeats. G, Population FRET histograms of βarr1 C tail sensor, imaged at 100 ms time resolution, with indicated concentrations of IP6 and H, PIP2. I, Ensemble average high-FRET inactive state occupancy as a function of V2Rpp concentrations in the absence and presence of indicated concentrations of IP6 or PIP2 (symbols) fit to dose-response functions (lines), revealing no substantial change in EC50 values.
βarrs also bind phosphoinositides such as phosphatidylinositol 4,5-bisphosphate, PIP2 (Gaidarov et al., 1999) and the soluble inositol hexaphosphate, IP6 (Chen et al., 2017, 2021a; Milano et al., 2006a; Zhuang et al., 2010). PIP2 has been implicated in membrane trafficking (De Matteis and Godi, 2004; Roth, 2004) and recruitment of various components of clathrin-coated structures (Kadlecova et al., 2017; Keen et al., 1991). Likewise, IP6 regulates several intracellular processes including agonist-induced receptor desensitization (Sasakawa et al., 1994). In contrast to heparin and V2Rpp, IP6 and the soluble PIP2 derivative, diC8-PI(4,5)P2, failed to induce any detectible transitions out of the high-FRET, inactive state in the βarr1 tail sensor (Figure 3G and 3H). The binding affinity of βarr1 for V2Rpp was also unaffected by either IP6 or PIP2 over the range of physiological concentrations tested (Figure 3I). These findings are consistent with the effects of IP6 being specific to βarr2 and not βarr1 activation (Chen et al., 2017, 2021a) and PIP2 requiring proximal membranes rather than being mediated by direct association with βarr1.
Activating mutations promote β-arrestin1 tail disengagement
We next sought to investigate the molecular determinants that enforce βarr1 autoinhibition and to explore possible roles of allostery in βarr1 activation. Mutations in distinct structural domains of βarr have been described as constitutively activating due to their propensity to induce receptor desensitization and internalization even in the absence of agonist-induced receptor activation (Eichel et al., 2018; Latorraca et al., 2018). Constitutively activating mutations include those that disrupt the three-element interaction (3-EI) region that anchors the βarr tail middle segment to the N-domain groove in the inactive state (Gurevich, 1998; Ostermaier et al., 2014; Scheerer and Sommer, 2017) (Figure 4A, left).
Figure 4. Effect of activating mutations on β-arrestin1 tail disengagement.
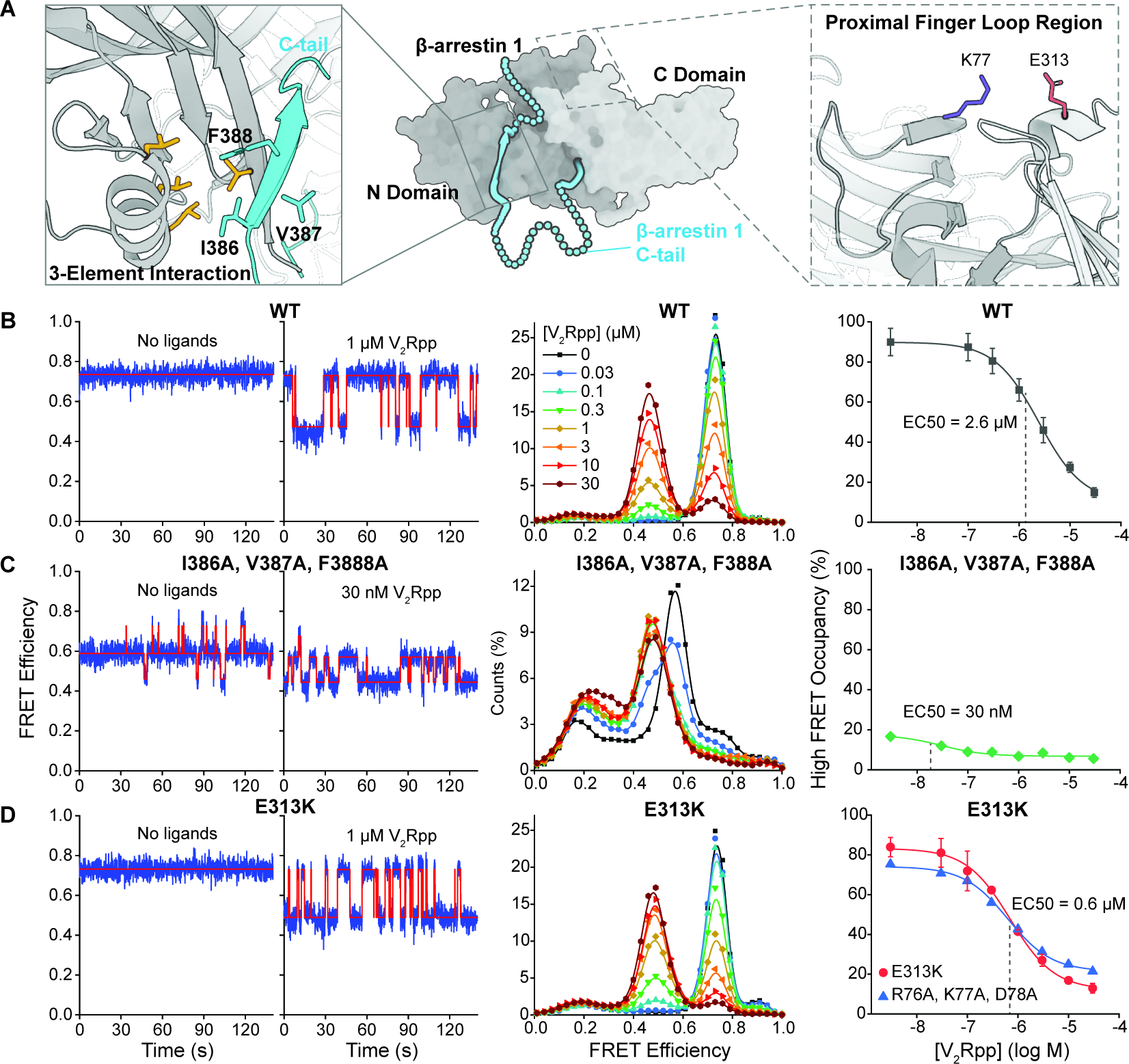
A, Basal structure of βarr1 (PDB: 1G4M) showing regions and residues associated with the three-element interaction (left) and finger-loop proximal (right) mutants. B, WT-, C, I386A/V387A/F388A- and D, E313K-βarr1 tail sensor was imaged in the absence and presence of varying concentrations of V2Rpp. Left, Representative smFRET traces in the absence (left) and presence (right) of V2Rpp. Middle, population FRET histograms (symbols) with spline fits (lines). Right, ensemble average occupancy in the high-FRET inactive state as a function of V2Rpp concentration (symbols) fit to dose-response functions (lines) with Hill slope of 1.0 and EC50 values of 2.6(WT, black), 0.030 (3-EI mutant, green), 0.070 (E313K, red), and 0.063 μM (R76A/K77A/D78A, blue). Error bars, mean ± S.D. of at least two repeats.
We hypothesized that such mutations should increase βarr1 tail disengagement in the absence of, and/or increase sensitivity to, V2Rpp. Consistent with this hypothesis, we observe that in the absence of V2Rpp, the βarr1 tail sensor bearing the 3-EI mutations, I386A, V387A, and F388A (Celver et al., 2002) (3EI-βarr1 tail sensor, see STAR Methods), showed a nearly complete loss of high-FRET inactive state occupancy, and instead predominantly exhibited a distinct ~0.57 FRET state not present in the parent βarr1 tail sensor (Figures 4B, 4C, and S4A). MD simulations of basal βarr1 with the 3-EI mutations showed that disrupted hydrophobic interactions between the βarr1 tail middle segment and the N-domain helix propagated to allosterically perturb R393, which dissociated from the polar core. Consistent with the decreased FRET efficiency in the absence of V2Rpp in the 3EI-βarr1 tail sensor, these reconfigurations affected the position of residue 397, lengthening the distance between 176 and 397 (Figures S4B and S4C). Weaker βarr1 tail anchoring was also evidenced by an 83-fold increase in apparent V2Rpp binding affinity (EC50: V2Rpp with the 3EI mutant, 30 nM vs. V2Rpp with the parent construct, 2.6 μM) (Figure 4C). In contrast to the parent sensor, the 3EI-βarr1 tail sensor displayed increased occupancy in the low-FRET, active state, which increased as a function of V2Rpp concentration (Figures 4C and S4D).
Mutations proximal to the finger loop region of βarr, which is responsible for engaging the receptor core (Eichel et al., 2018; Latorraca et al., 2018) (Figure 4A, right), were also shown to constitutively activate βarr, possibly by disrupting a salt bridge that stabilizes the inactive state conformation with respect to inter-domain rotation (Eichel et al., 2018). In contrast to the 3EI-βarr1 tail sensor, we observed that a finger loop-proximal point mutation (E313K; 313K-βarr1 tail sensor) (see STAR Methods) did not substantially perturb the anchored βarr1 tail under basal conditions. However, V2Rpp bound with a 4-fold higher apparent affinity (EC50 of ~0.63 μM for E313K vs. 2.6 μM for the parent sensor) (Figure 4D), an effect arising predominantly from a faster apparent rate of V2Rpp binding (Figures S4E and S4F). Similar results were also observed for a distinct proximal finger loop mutant (R76A, K77A, D78A) (Figure S4G). These results suggest that mutations in both the finger loop proximal and 3-EI regions can promote βarr1 tail disengagement from the βarr1 N-domain groove, implicating both regions in a bidirectional allosteric process important for regulating βarr1 activation.
β-arrestin1 activation by phosphorylated receptor is agonist dependent
Building on these observations, we set out to examine how GRK-phosphorylated receptor engages and activates βarr1. More specifically, we asked if the activation mechanism is identical in the context of the full receptor and to what extent βarr1 engagement with the core of an activated GPCR may alter this mechanism. To this end, we expressed and purified a β2AR chimera with its C terminus replaced by that of the V2 vasopressin receptor (β2V2R) (see STAR Methods). Such receptor-V2R C-terminal tail chimeras (Cahill et al., 2017; Lee et al., 2020; Staus et al., 2018, 2020a) are useful models to study βarr1 activation, due to their higher affinity for βarr compared to the wild-type receptors (Oakley et al., 2000). The β2V2R chimera is phosphorylated in vivo by GRK2 (Shukla et al., 2014a) to yield a pattern of phosphorylated residue positions (P1 to P8) (Latorraca et al., 2020) identical to that of V2Rpp (Table 1). Importantly, β2V2R exhibits a normal β2AR pharmacological profile (Oakley et al., 2000), allowing exploration of the effects of well-characterized β2AR-orthosteric ligands and allosteric modulators. Notably, this approach also allowed us to deconvolute the roles of phosphorylation and agonist binding, two mechanistic steps that are conflated in cellular systems, where agonist binding promotes receptor phosphorylation.
We first imaged surface-tethered βarr1 tail sensor dynamics in the presence of increasing concentrations of GRK2-phosphorylated β2V2R (pβ2V2R) (see STAR Methods) (Figure 5A). Surprisingly, in the presence of 1 μM pβ2V2R in the unliganded (apo) state or bound to the inverse agonist carazolol (cz), the βarr1 tail sensor stably occupied the high-FRET, inactive state, exhibiting only rare, transient excursions to the mid-FRET active state (Figures 5B–5C), which were not observed in the absence of receptor (Figure 1E). This result was unexpected, as similar concentrations of the free V2Rpp peptide were strongly activating (Figure 2D). Strikingly, the βarr1 tail sensor remained in a predominantly inactive conformation even in the presence of 5 μM pβ2V2R (Figure 5D). We infer from these observations that the phosphorylated V2R tail, when attached to β2AR, is relatively inaccessible to βarr1, indicating that context-specific effects sequester the Rp tail. While the receptor context appeared to significantly reduce the frequency and duration of phosphorylated V2R tail binding events to βarr1, Fab30 nevertheless promoted receptor engagement and βarr1 tail release (Figure S3E), suggesting that rare and transient interactions of the inactive-state receptor tail with βarr1 can be stabilized by Fab30.
Figure 5. Activation of β-arrestin1 by phosphorylated, agonist-bound receptor.
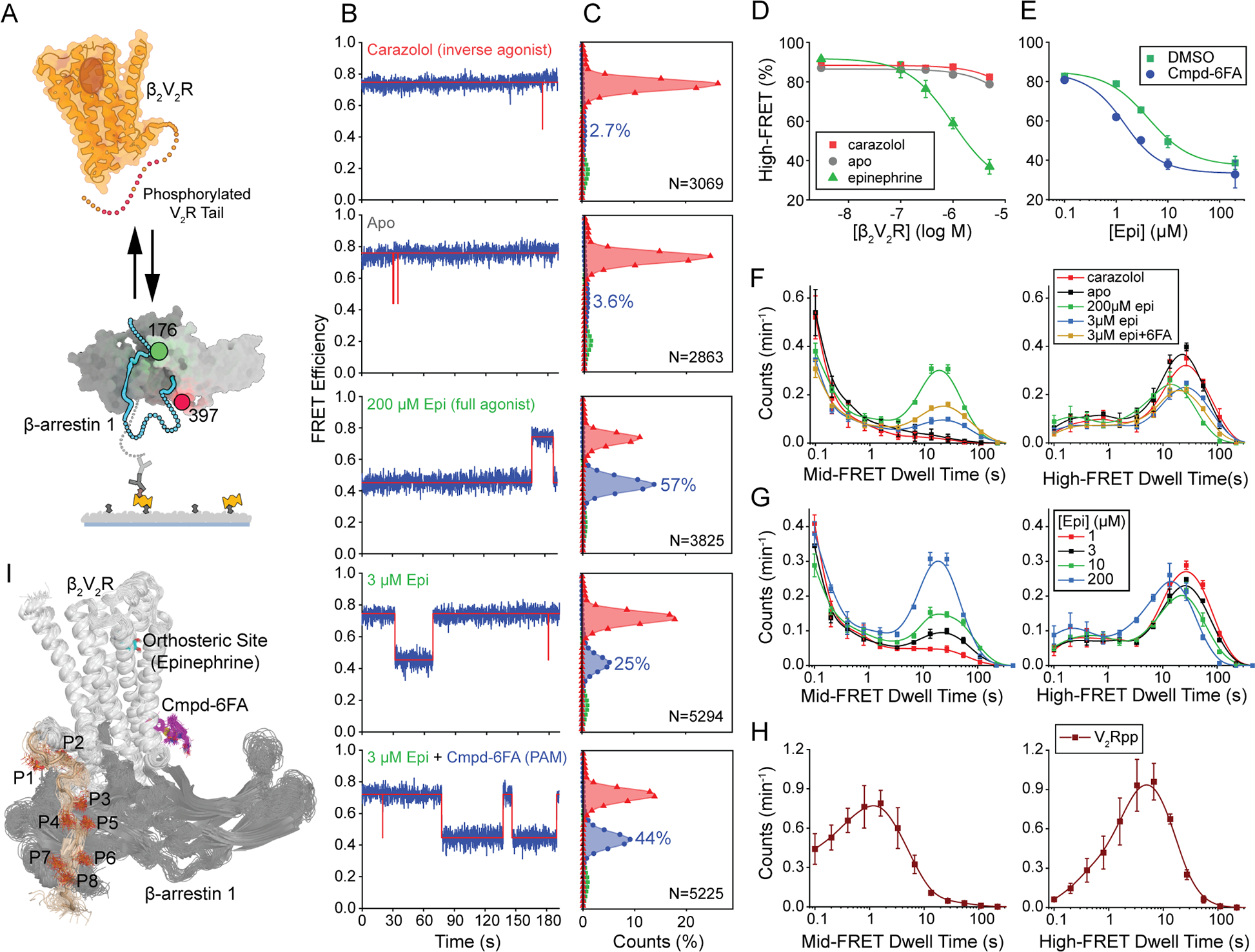
A, Schematic of the experiment showing βarr1 tethered to an imaging surface interacting with pβ2V2R free in solution and bound to ligands of varying efficacy. B, Representative FRET (blue) and state assignment (red) traces and C, population FRET histograms from smFRET imaging of βarr tail sensor conducted in the presence of 1 μM pβ2V2R and 10 μM carazolol, no ligands, 200 μM epi, 3 μM epi, or 3 μM epi with 10 μM Cmpd-6FA, respectively. D, High-FRET inactive state occupancy as a function of pβ2V2R concentration in the presence of saturating concentrations of the indicated receptor-binding ligand (symbols) fit to dose response functions (lines) with a Hill slope of 1 and EC50 values of 44 (carazolol; red), 30 (apo; gray), and 1.0 μM (epi; green). E, High-FRET inactive state occupancy from experiments conducted in the presence of 1 μM pβ2V2R and the indicated concentration of epinephrine in the absence (blue) and presence (green) of 10 μM Cmpd-6FA fit to dose response functions with Hill slope of 1.0 and EC50 values of 3.7 and 1.0 μM, respectively. F-H, Histograms of dwell times in the mid-FRET active (left) and high-FRET inactive (right) states: F, from experiments corresponding to panels B-C, G, from experiments with 1 μM pβ2V2R and the indicated concentrations of epinephrine, and H, from experiments with 1 μM V2Rpp duplicated from Figure 2 for comparison. I, Representative MD frames of the simulated pβ2V2R-βarr complex model bound to epinephrine (cyan) and Cmpd-6FA (magenta) showing the positions and dynamics of P1 to P8 in V2Rpp ( wheat color). Error bars, mean ± S.D. of two repeats.
In contrast to apo and inverse agonist-bound receptor, in the presence of pβ2V2R with saturating concentrations of the full agonist epinephrine (epi), βarr1 displayed mid-FRET active state dwells that were more than 100 times longer than in the absence of agonist, resulting in substantial mid-FRET active state occupancy (Figures 5B–G). The concentration dependence of epi-activated pβ2V2R binding to βarr1 tail sensor revealed an apparent affinity of ~1 μM (Figure 5D). Thus, agonist binding to pβ2V2R promotes much stronger Rp tail engagement and βarr1 activation, resulting in apparent affinity comparable to that of free V2Rpp.
However, in contrast to V2Rpp, the epi-activated pβ2V2R induced multi-modal dynamics of the βarr1 tail, consisting of both transient (~0.1 s) and long-lived (~30–60 s, limited by photobleaching) engagement durations (Figures 5F and 5G). We speculate that the transient dwells correspond to configurations in which only the Rp tail is engaged (tail-only). Notably, however, as for the apo and inverse agonist-bound receptor, we found the mean dwell times of tail-only engagement to be shorter than those observed with V2Rpp (Figure 5H), suggesting a less optimal binding of the phosphorylated V2R tail in the context of the receptor compared to that of the free peptide. In considering the basis for this impaired binding mode, we noted that the first two phosphorylated residues (P1 and P2) of the V2Rpp peptide interact with residues near the finger loop region of βarr1 (Shukla et al., 2013a) (Figure S5A) (see STAR Methods). In contrast, the cryo-EM structure of βarr1 in tail-only engagement with the receptor (Nguyen et al., 2019), P1 and P2 are unresolved, and the residue before P3 that is resolved points away from the extended groove in βarr1 unlike in the V2Rpp bound βarr1 structure (Figure S5B). Thus, in the context of the receptor, P1 and P2 cannot optimally bind to βarr1 due to steric constraints imposed by the linkage of the phosphorylated V2R tail to helix 8 (H8) of the receptor. Cryo-EM structures of muscarinic acetylcholine receptor 2 and β1 adrenergic receptor complexed with βarr1 (Lee et al., 2020; Staus et al., 2020b) also show P1 and P2 to be unresolved, suggesting these regions remain highly dynamic.
In contrast, the longer dwells suggest a substantially more stable complex of βarr1 with pβ2V2R than with V2Rpp. Consistent with a highly stable complex, the mid-FRET active state persisted for several minutes after free receptor was washed from the imaging chamber (Figures S6A and S6B). We speculate that this stability is driven by “tight” receptor engagement inducing global conformational changes in βarr that further weaken its propensity for autoinhibition and contribute stabilizing contacts between βarr1 and receptor (Lee et al., 2020). Indeed, despite incomplete engagement of the P1 and P2 positions in the Rp-tail with βarr1, MD simulations of pβ2V2R revealed extensive and stable interactions of activated βarr1 with the receptor core (Figure 5I). We note in this context that since P2 makes an important contribution to V2Rpp binding affinity (Latorraca et al., 2020), the stability of the long-lived complex observed in smFRET may also reflect more effective engagement enabled by conformational changes in the activated complex.
β-arrestin1 tail release is modulated by the extent of receptor activation and pattern of phosphorylation
We next explored the impact of β2AR positive allosteric modulators (PAMs) on βarr1 tail release and βarr1 activation. Compound 6FA (Cmpd-6FA) exhibits positive cooperativity with epi and binds β2AR near intracellular loop 2 in a pocket between TMs 3 and 4 to promote βarr coupling (Ahn et al., 2018; Liu et al., 2019). Consistent with previous cellular investigations, Cmpd-6FA (see STAR Methods) enhanced the potency of epi to promote long-lived mid-FRET active state dwells of βarr1 in the presence of 1 μM pβ2V2R (Figures 5B–5C and 5E–F). These observations are consistent with a model in which PAMs increase agonist binding affinity for the receptor, promoting long-lived activating interactions with βarr1.
To explore the contribution of receptor tail phosphorylation to βarr1 tail disengagement and βarr1 activation, we examined β2V2R prepared without agonist treatment and in the absence of GRK2 (see STAR Methods). Under these conditions, β2V2R is basally phosphorylated at positions P1, P2, P7, and P8 but not the other four positions (P3 to P6) (Table 1) (Nguyen et al., 2019). In the presence of 10 μM Cmpd-6FA and saturating epi, we observed no substantial effect on βarr1 tail release with the basally phosphorylated receptor (bpβ2V2R) (Figure S6C), nor any evidence of stable receptor-βarr1 complex formation by fluorescence correlation spectroscopy (FCS) of freely diffusing molecules (Figures S6D and S6E). Similarly, the corresponding basally phosphorylated peptide (V2Rbp; Table 1) showed no effect on βarr1 tail release or dynamics at concentrations as high as 1 mM (Figure 2D). The finger-loop proximal mutation E313K, which increases the affinity of V2Rpp engagement (Figure 4D), was unable to rescue βarr1 tail activation by bpβ2V2R (Figure S6F). These findings indicate that: 1) receptor core engagement is transient in the absence of adequately phosphorylated Rp tail, 2) core engagement alone, if it occurs, is insufficient to induce appreciable βarr1 tail release, and 3) P3 to P6 are critical to the βarr1 activation mechanism.
To investigate whether it is possible to capture the tail-only mode of Rp tail-bound βarr1 without engaging the receptor core, we used nanobody Nb6B9, a G protein mimetic expected to compete with βarr1 for core engagement. In these experiments, epi was insufficient to induce βarr1 tail release, and Fab30 was required to capture βarr1 engaged with the Rp tail in the tail-only configuration (Figure S3E). These findings suggest that the tail-only mode is transient in the absence of Fab30 stabilization and that both core and Rp tail engagement are required to induce efficient βarr1 tail release in the context of the full receptor.
The β2AR C-tail inhibits β-arrestin recruitment in living cells
The experiments described above were conducted using purified receptor of defined phosphorylation state and purified βarr1, making it possible for us to deconvolute the effects of agonist binding from those of receptor phosphorylation. This cannot be achieved in the context of living cells since agonist stimulation of receptor is required for efficient GRK phosphorylation. We therefore sought to examine the implications of our findings in a more physiological, cellular context. To do so, we used HEK293 cells in which GRK 2/3/5/6 were simultaneously knocked out (HEK GRK KO) via CRISPR-Cas9 methods (Kawakami et al., 2022) to explore core-mediated βarr recruitment in the absence of GRK-mediated receptor tail phosphorylation. To generalize our findings to a more native scenario, we used wild-type β2AR with its native C tail (Figure S7A) as well as βarr2, which is more commonly associated with the physiological effects of β2AR activation in vivo. For these investigations, we used a bystander bioluminescence resonance energy transfer (BRET) assay that detects recruitment of βarr to the plasma membrane upon activation of receptors (Donthamsetti et al., 2015; Perry-Hauser et al., 2021) (see STAR Methods) (Figure 6A). The bystander-BRET assay avoids receptor C tail modifications that might perturb receptor function or interactions. Notably, we were unable to detect βarr2 surface recruitment by isoproterenol (ISO)-activated β2AR in the absence of GRK (Figures 6B–C), whereas robust βarr2 recruitment was rescued by expression of GRK2 (Figures 6D–E), which mediates Rp tail phosphorylation. These results are consistent with inferences from our smFRET and FCS data that core engagement of βarr is weak in the absence of Rp tail phosphorylation.
Figure 6. The β2AR C-tail inhibits β-arrestin recruitment in living cells.
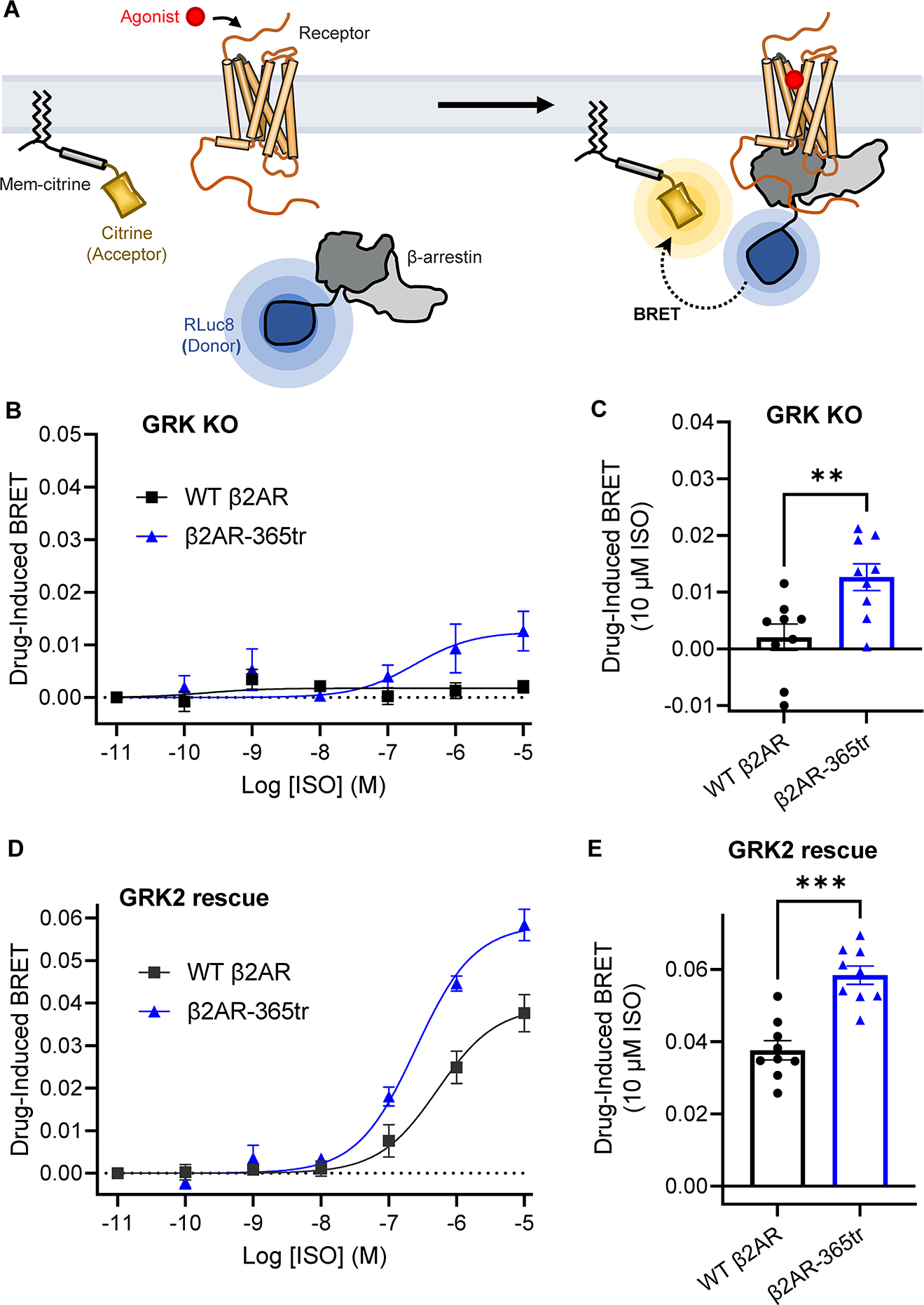
A, Schematic of the bystander BRET-based βarr membrane recruitment assay performed in living cells where the N-terminus of βarr is fused to the donor Rluc8 and a plasma membrane marker is fused to the acceptor citrine. Upon agonist stimulation, receptors with unmodified C-tails recruit βarr to the plasma membrane, resulting in an increase of BRET between the donor and acceptor molecules. B, Dose-response curves of βarr2 recruitment to the plasma membrane by wildtype (WT) β2AR (black squares) and β2AR-365tr (blue triangles) in HEK293 GRK2/3/5/6 knockout cells in the absence (GRK KO) or D, presence of GRK2 expression via transient transfection (GRK2 rescue). Dose-response curves of three independent experiments performed with triplicate samples (mean ± S. E.). C, βarr2 membrane recruitment by WT β2AR and β2AR-365tr in response to10 μM isoproterenol stimulation in the absence and E, presence of GRK2 transfection. Bars represents the mean ± S. E. of three independent experiments performed in triplicate. Symbols represent the measured drug-induced BRET value. **, p = 0.0056; ***, p = 0.0002, unpaired, two-tailed Mann-Whitney test.
Given that the Rp tail of β2V2R does not efficiently activate βarr1 in the absence of agonist, pointing to receptor C tail sequestration and resulting inhibition of βarr binding, we wondered whether the wild-type β2AR C tail in living cells might also inhibit βarr interaction with receptor even in the presence of agonist. If so, this autoinhibition might be relieved via removal of the receptor C tail. An extensive truncation of the β2AR C tail after palmitoylated C341 (β2AR-341tr) led to very limited surface expression, but another previously described β2AR construct truncated after residue 365 (β2AR-365tr) was expressed at levels only slightly lower than wildtype β2AR (Figure S7A and S7B). This construct preserves residues T360 and S364, which have been shown to be robustly phosphorylated by GRK2 (Nobles et al., 2011). While a prior study suggested that β2AR-365tr does not interact with βarr (Nuber et al., 2016), a more recent study showed that β2AR-365tr can still mediate βarr activation (Eichel et al., 2018). Interestingly, in our bystander-BRET assay, unlike for wild-type β2AR, we observed significant βarr2 recruitment to the plasma membrane by ISO-activated β2AR-365tr in the HEK GRK KO cells, despite the absence of GRK-mediated Rp tail phosphorylation (Figure 6B, 6C). This finding suggests that removal of the distal β2AR C tail leads to more efficient core engagement. Moreover, β2AR-365tr also recruited βarr2 more efficiently than wild-type receptor upon co-expression of GRK2 (Figure 6D, 6E), suggesting the possibility that an autoinhibitory role of the receptor C tail might still be operative even with Rp tail phosphorylation. Notably, wild-type β2AR and β2AR-365tr both were capable of surface recruitment of a G protein surrogate in an analogous bystander assay (Figure S7C), suggesting that the receptor autoinhibition is somehow selective for βarr recruitment and activation. This assay was further leveraged to show that the dopamine D2 receptor (D2R), which natively terminates after H8 and thus has no Rp tail, is able to recruit βarr2 in the HEK GRK KO cells in a GRK-independent manner similar to β2AR-365tr; βarr2 recruitment was also dramatically enhanced by co-expression of GRK2 (Figure S7A, S7D), likely due to GRK phosphorylation of sites in the 3rd cytoplasmic loop of D2R (Clayton et al., 2014).
DISCUSSION
Single-molecule imaging has previously provided unprecedented insight into receptor dynamics and the receptor-mediated G protein activation mechanism (Asher et al., 2021; Gregorio et al., 2017; Quast and Margeat, 2019; Zhu et al., 2017). Here, smFRET investigations of βarr1 activation reveal that βarr1 resides in a highly stable, autoinhibited basal state, in which the middle βarr1 tail segment is tightly bound to its N-domain groove, thereby occluding receptor C-tail engagement. An important functional consequence of this autoinhibition is that phosphorylated receptor tail binding releases βarr1’s tail at a rate 2–3 orders of magnitude slower than diffusion. Moreover, substantial βarr1 tail displacement with pβ2V2R was only observed when the receptor was activated by agonist. These findings suggest that the Rp tail may be inaccessible or sterically constrained in the absence of receptor activation. Thus, in addition to agonist activation promoting receptor phosphorylation, we infer that efficient βarr engagement likely arises from agonist-induced accessibility of the Rp tail, stabilizing contacts facilitated by core engagement, and conformational changes in this “tight” complex. Such a mechanism would limit βarr engagement only to agonist-bound receptor, decreasing the potential impact of bystander phosphorylation. The receptor autoinhibition mechanism, combined with the autoinhibition of βarr itself, is likely to enhance the fidelity of βarr-mediated downstream signaling. Since βarr recruitment is impaired while G protein engagement seems unaffected, receptor C-tail autoinhibition may also play a role in biased agonism.
While agonist binding and receptor phosphorylation cannot be deconvoluted in living cells, our cell-based experiments are consistent with an autoinhibitory role of the wild-type β2AR C tail for βarr recruitment that was relieved, at least in part, by partial truncation of the receptor tail. Indeed, D2R, which natively terminates after H8, can measurably recruit βarr2 in the GRK KO cells in the absence of GRK phosphorylation. Recent reports also suggest an inhibitory role of the metabotropic glutamate 2 receptor C tail in βarr2 recruitment (Abreu et al., 2021). Notably, the C-terminal region beyond H8 is unresolved in nearly all reported GPCR structural models. However, select structures of rhodopsin (Nakamichi et al., 2007; Palczewski et al., 2000; Teller et al., 2001) have resolved a portion of the C-terminus bound to both helix 8 and the intracellular face of the receptor, a position occupied by GRK1 in a recent structure of a GRK1:rhodopsin complex (Chen et al., 2021b). Binding of the receptor C-terminus to this location would also likely inhibit βarr binding to the receptor core. Thus, a receptor C tail-dependent autoinhibition of βarr recruitment may be a mechanism shared by multiple GPCRs.
Structural studies of Fab30-stabilized receptor:βarr1 complexes have revealed βarr1 in both tail-only and tail plus core conformations. We find that Fab30 enables epi-activated phosphorylated receptor-mediated βarr1 tail release even when the receptor core is occupied by Nb6B9. Such a tail-only configuration of the receptor:βarr1:Nb6B9 complex would be analogous to the “megaplex” in which receptor is bound simultaneously to Gs in its TM core and to βarr1 in a tail-only conformation (Nguyen et al., 2019). These findings suggest that tail-only engagement between receptor and βarr1 is transient in the absence of Fab30. Interestingly, a finger loop deletion mutation in βarr2 with reduced propensity to engage the receptor core—and that, in the presence of Fab30, promotes the tail-only interaction—is still able to mediate receptor internalization and signaling (Cahill et al., 2017). Similarly, a receptor mutant reported to be incapable of core engagement can also mediate receptor internalization (Kumari et al., 2017). These findings in living cells suggest that other effectors, such as adaptin, clathrin and/or lipid (Eichel et al., 2018; Staus et al., 2020a) contribute to the βarr-receptor interaction.
As the agonist-bound bpβ2V2R containing only four phosphorylated C tail residues does not trigger appreciable βarr1 activation, or promote stable complex formation, despite the expectation that the receptor core should be able to engage βarr1, we infer that efficient βarr1 coupling to activated receptor requires a critical extent and/or pattern of phosphorylation within the Rp tail. Moreover, our cellular studies of βarr2 recruitment by distal C-tail truncated receptor support the broader conclusion that receptor-βarr core only interactions are quite weak, even when autoinhibition is partially relieved.
Cellular investigations implicate the βarr C tail in internalization as well as signaling processes downstream of receptor engagement (Gurevich and Gurevich, 2015; Perry-Hauser et al., 2022). When βarr is activated, its C tail may sample an ensemble of positions on a sub-millisecond time scale or adopt a more limited set of positions that are partially stabilized by residual contacts with the βarr N- and C-terminal domains. Both models are consistent with increased βarr1 tail and/or fluorophore mobility upon activation as inferred from ensemble fluorescence anisotropy measurements (Table S2).
Previous DEER measurements of βarr1 bound to light-activated, phosphorylated rhodopsin also predicted that position 392 in the middle βarr1 tail segment adopts a specific position (Zhuo et al., 2014), in contrast to the C tail in visual arrestin, which was disordered and dynamic (Hanson et al., 2006). From our observed time-averaged FRET efficiencies, we can estimate distance constraints to show that the βarr1 tail labeling site (residue 397) in the mid-FRET, active state should reside somewhere along a ring enclosing the intersection of the N and C domains of βarr1 (Figure S5C). DEER measurements for 12/392 and 192/392 also predict a location on a ring enclosing this intersection (Figure S5C). Combining these distance constraints limit the positions of residues 392 and 397 to a region between the N and C domains on the side of βarr1 that interacts with the third intracellular loop (IL3) of the receptor. In this context, we note that the low-FRET, active state observed in the presence of heparin likely reflects an extended tail conformation that positions 397 even further from both 176 and 110. We speculate that a potential interaction between the βarr tail and IL3, which is highly divergent among receptors, could also play a role in fine-tuning βarr activation and signaling.
Alternative βarr1 tail conformations could be stabilized and/or differentially recognized by effectors such as clathrin, adaptin and various kinases, providing an important step towards a mechanistic understanding of the barcode hypothesis (Dwivedi-Agnihotri et al., 2020; Kim et al., 2005; Srivastava et al., 2015). By extension, distinct extents and patterns of receptor phosphorylation could differentially impact βarr activity (Mayer et al., 2019; Sente et al., 2018; Urs et al., 2016). Additional studies may reveal how the barcode is interpreted by βarr at the level of both conformational states and dynamics.
Limitations of the study
Inferring structural constraints from FRET efficiencies involves many assumptions and approximations (Hellenkamp et al., 2018; Lerner et al., 2021). As such, additional experiments and simulations of βarr1 bound to V2Rpp or receptor are necessary to further define the structure of the distinct βarr1 active states that we have identified here using smFRET – and of the observed autoinhibitory mechanisms. The receptor utilized in the smFRET experiments was solubilized in detergent to facilitate comparison with existing structures (Huang et al., 2020; Nguyen et al., 2019), yet yielded results highly consistent with our cell-based studies. Future in vitro receptor studies in reconstituted membrane environments will likely be needed to recapitulate cellular regulation mechanisms that require βarr:membrane interactions, such as those with PIP2 (Eichel et al., 2018). We focused here on GRK-mediated phosphorylation of the receptor C tail. Future studies should also address a potential role for other kinases, as well as for phosphorylation of intracellular loops, and of differential roles of phosphorylation of distinct sets of sites by different GRKs (Nobles et al., 2011).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be filled by the lead contact, Jonathan A. Javitch (Jonathan.Javitch@nyspi.columbia.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availably
Source data generated and analyzed that support the findings of this study will be shared by the lead contact upon request.
This paper does not report original code. All Software used to collect and analyze data for this work was either published previously or is commercially available.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
β-arrestin1 expression in E coli
βarr variants were expressed in BL21-Gold competent E. coli cells (Agilent Technologies) and subsequently purified and labeled with fluorophores for TIRF- and confocal-based smFRET experiments. The cells were transformed with 1 ng of expression plasmid per manufacturer’s protocol and 100 μg/mL ampicillin was used for selection. Next, 70 mL Luria-Bertani (LB) medium was inoculated with a single bacterial colony and grown at 37 °C at 250 rpm until the OD600 reached 0.1–0.2. 15 mL of starter growth was then transferred to 1 L of LB and grown at 30 °C overnight at 250 rpm, followed by induction with 35 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 6 hours.
β2V2R expression in Sf9 insect cells
The N-terminal FLAG-tagged human β2V2R was co-expressed with or without GRK2-CAAX in Sf9 insect cells at 27 °C using Baculovirus Expression System as described previously (Kahsai et al., 2016; Shukla et al., 2014b). Sixty-six hours post-infection, cells without GRK2-CAAX were immediately harvested for preparation of bpβ2V2R, and cells with coexpressed GRK2-CAAX were stimulated with isoproterenol at 37 °C to generate pβ2V2R, and then subsequently harvested.
HEK293 GRK2/3/5/6 knockout cell culture
The HEK293A GRK2/3/5/6 Knockout cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco), 10% fetal bovine serum (FBS; corning), 1 X MEM non-essential amino acids (Gibco), 1 X sodium pyruvate (Gibco), 1 X penicillin-streptomycin (Gibco) at 37 °C in 5% CO2. 2.5 million cells were seeded in 10 cm tissue culture dishes 24 hours prior to transfection. The cells are female in origin and were authenticated by the Inoue lab using restriction enzyme-based genotyping and sequencing of the targeted loci and western blot analysis.
METHOD DETAILS
Reagents
LD555p-MAL and LD655-MAL (Lumidyne Technologies) were reconstituted in DMSO at ~10 mM concentration and stored at −20° C. NeutrAvidin (Thermo Scientific) was resuspended to a final concentration of 80 μM in T50 buffer with 10% glycerol, 100 μM 25 nucleotide duplex DNA (IDT), and 100 μM bovine serum albumin (EMD). The biotinylated rabbit anti-mouse IgG secondary antibody (biotin-AB) (ThermoFisher Scientific) was reconstituted in 50% glycerol to a final concentration of 10 μM and stored at −20° C, as recommended by the manufacturer. The mouse high-affinity anti-Strep tag IgG1 monoclonal antibody (StrepMAB-Immo, IBA GmbH) was reconstituted in PBS at 1 μM concentration, flash frozen, and stored at −80° C as single use aliquots. V2Rpp, biotin-V2Rpp, V2Rnp, and V2Rbp were synthesized by the Tufts University peptide synthesis core facility. The peptides were resuspended in 20 mM HEPES, 100 mM NaCl at 2 mM final concentration, adjusted with 1M NaOH to pH ~7, flash frozen, and stored at −80° C. Heparin (Sigma) was resuspended in water to 50 mg/mL (~2.8 mM), filtered at 0.22 micron, and stored at 4°C. IP6 (Sigma) was resuspended in water to 1 mM and stored at −80°C. PIP2 (Echelon Biosciences) was resuspended in 20 mM HEPES-NaOH (pH 7.5), 100 mM NaCl to 2 mM and stored at −80°C. Carazolol (Sigma) was prepared as 100 μM stocks in DMSO. Fab30, Nb6B9, and Cmpd-6FA were prepared as previously described (Liu et al., 2019; Shukla et al., 2013b). Epinephrine (Sigma) was prepared immediately before use at 200 mM in water and stored at 4° C. Glucose oxidase (Sigma) and catalase (Sigma) were each resuspended in T50 buffer with 20 mM β-mercaptoethanol, purified via size exclusion chromatography, and concentrated, and stored at −20°C with 50% glycerol. Isoproterenol hydrochloride (Tocris Biosciences) was dissolved immediately before use in DPBS at 10 mM final concentration. Dopamine hydrochloride was dissolved immediately before use in ultrapure water at 22.22 mM with 40 mg/L sodium bisulfite.
Plasmid construction
Plasmids were constructed using standard techniques in molecular biology and confirmed by DNA sequencing (Psomagen). Briefly, a DNA insert flanked by Nco I and Avr II sites and coding for the Strep-tag followed by a linker region and then by the N-terminal region (residues 2 – 24) of bovine βarr1 was prepared by PCR amplification from two overlapping single-stranded oligonucleotides synthesized by ThermoFisher Scientific. The insert was cloned into the Nco I and Avr II sites of a previously described pTrcHisB vector coding cysteine-less βarr1, yielding the plasmid containing the complete coding region for Strep-tagged cysteine-less βarr1. Cysteine point mutations introduced for fluorophore labeling and the E313K mutation were prepared in the plasmid described above using QuickChange site-directed mutagenesis (Agilent). The 3-EI mutations and the R76A, K77A, D78A proximal finger loop mutations were introduced using a modified two-stage PCR protocol for site-directed mutagenesis (Wang and Malcolm, 1999).
For the live cell assays, the coding region of full length human β2AR, β2AR-365tr, and β2AR-341tr were cloned into the pcDNA3.1 (Thermo Fisher Scientific) expression vector with a signal peptide and SNAPfast tag at the receptor N-terminus.
Purification of β-arrestin1 variants
Bacterial cells containing overexpressed βarr1 variants were harvested by centrifugation at 8000 rpm for 10 minutes at 4 °C and resuspended in 15 mL of lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM glucose, 5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 mM benzamidine (BA), 2 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM dithiothreitol (DTT), and 1 mg/mL protease cocktail inhibitor (Sigma Aldrich), 10 mg/mL lysozyme (EMD Millipore), incubated on ice for 40 min, and frozen at −80 °C until further use.
After thawing, the cell suspension was supplemented with the following (final concentrations): 24 U/mL DNAse I (Sigma), 32 U/mL DNAse II (Sigma), 0.16 mg/mL RNAse A (Sigma), and 7 mM MgCl2 and incubated on ice for 60 min. The cells were then lysed for five minutes at 500–1000 psi at 4°C using an EmulsiFlex-C3 Homogenizer (Avastin) per manufacturer’s protocol. Cell debris was removed by centrifugation at 17,000 rpm for 90 min and the lysate was collected, followed by the addition 0.32 g ammonium sulfate per mL of lysate. The precipitated protein was pelleted by centrifugation at 17,000 rpm for 90 minutes and frozen at −80°C until further use. Protein pellets were thawed on ice, dissolved in 10 mL of resuspension buffer (50 mM Tris-HCl pH 7.5, 400 mM NaCl, 2 mM EGTA, 2 mM ethylenediaminetetraacetic acid (EDTA), 1 mM PMSF, 1 mM BA, and 2 mM DTT), and centrifuged at 17,000 rpm for 90 minutes at 4°C to remove insoluble material. The supernatant was syringe filtered (0.8 μm, MilliPore) immediately before affinity purification.
For affinity purification, the supernatant described above was added to 3 mL of Strep-Tactin Superflow High Capacity resin (IBA GmbH) equilibrated with resuspension buffer and purified by gravity flow following a modified version of the manufacturer’s protocol. Briefly, after loading filtered supernatant, the resin was washed with 30 mL resuspension buffer followed by elution with 20 mL of the same buffer containing 5 mM D-desthiobiotin (IBA GmbH). Eluted and purified βarr1 variants were concentrated using Amicon® Ultra-15 Centrifugal Filter Units (Millipore Sigma) to a final volume of 250 μL and quantified using the Bradford assay (Bio Rad). Purity was confirmed by SDS-PAGE analysis using standard protocols. Before use, βarr1 aliquots were thawed on ice and centrifuged for 1 hr at 100,000 × g and the top ~25% of volume was extracted for use.
Labeling of purified β-Arrestin1 samples
0.25 mg of the purified βarr1 variants in a total volume of ~350 μL (~15 μM protein) were labeled with 15.2 μM LD555p-MAL and 17.6 μM LD655-MAL in 100 mM sodium phosphate pH 7.0, 750 mM NaCl at 4°C for 3 hours in the dark, followed by addition of 1 μL of 1 M DTT. To remove free dye, the solution was then loaded onto a Zeba™ 2 mL 7K MWCO spin desalting column (ThermoFisher) equilibrated in the buffer described above, and the resulting flow-through was loaded onto a second desalting column equilibrated in PBS, pH 7.0. The flow through containing pure labeled samples was centrifuged at 100,000 × g for 1 hr at 4°C to remove insoluble aggregates. Labeled βarr1 variants were aliquoted and frozen at −80°C until further use. Fluorescence anisotropy measurements were also conducted using single-cysteine βarr1 variants labeled with LD555 or LD655 to verify that the fluorophores are not conformationally restricted (Table S2).
Purification of β2V2R
Insect cells containing overexpressed pβ2V2R or bpβ2V2R were lysed using a lysis buffer [10 mM Tris, pH 7.4, benzamidine (10 mg/mL), leupeptin (10 mg/mL)] and subsequently solubilized in an n-Dodecyl-β-D-maltoside (DDM; Anatrace) containing solubilization buffer [1.0% DDM, 500 mM NaCl, 20 mM Tris, pH 7.4, benzamidine (10 mg/mL), leupeptin (10 mg/mL), PMSF (200 μmol)]. Functional bpβ2V2R and pβ2V2R were obtained in 0.01% (w/v) maltose neopentyl glycol (MNG) containing 20 mM HEPES, pH 7.5, 100 mM NaCl buffer using a three-step affinity-chromatographic procedure: involving a first M1 anti-FLAG-antibody affinity column, followed by alprenolol-ligand column and a second M1 anti-FLAG antibody-affinity column. Purified receptors samples were either used fresh in the experiments or flash-frozen in small aliquots after addition of 15% glycerol and stored at −80 °C until further use. Mass spectrometry analysis confirmed the eight phosphorylated residues in the V2 tail of the pβ2V2R, and the four such positions in bpβ2V2R,to be robustly (≥84%) phosphorylated (Nguyen et al., 2019).
TIRF-based single-molecule FRET imaging
Microfluidic imaging chambers were prepared by passivating quartz glass slides and coverslips with polyethylene glycol (PEG) and biotin-PEG (Blanchard et al., 2004). For direct immobilization of βarr1 variants, the imaging surface was first exposed to 0.8 μM NeutrAvidin, followed by 100 nM biotin-AB and then 10 nM StrepMAB-Immo in T50 buffer (10 mM Tris-Acetate, 50 mM KCl). The surface was then exchanged into imaging buffer (50 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.8% w/v glucose). Strep-tagged βarr1 variants were incubated at 200 pM within the imaging chamber for 5 minutes in imaging buffer with 2 μM 25-nucleotide DNA duplex (IDT) and 2 μM BSA (EM-2930, VWR) as surface blocking agents. For experiments tethering βarr1 to the surface via V2Rpp interactions, a complex of biotin-V2Rpp and βarr1 was formed at a 1:2 molar ratio with 100 nM Fab30 for 10 min prior to injection into the imaging chamber treated with NeutrAvidin only. Imaging was performed in imaging buffer containing glucose oxidase (2.5 U/mL) and catalase (1.75 U/μL), which was incubated for 5 minutes in a glass syringe and 5 minutes in the microfluidic chamber, unless otherwise noted. Where specified, MNG-solubilized receptor was diluted to the final concentration in imaging buffer with any receptor ligands for 5 min to form a stable complex and this mixture was injected into the imaging chamber at the specified concentrations without additional detergent.
TIRF-based smFRET imaging experiments were performed at 25°C with a custom-built TIRF microscope (Juette et al., 2016). LD555p fluorophores attached to βarr1 variants near the surface were illuminated with a 532 nm diode-pumped solid-state laser (Opus, LaserQuantum). Fluorescence emission from LD555p and LD655 was collected by a 60x, 1.27 NA super resolution water immersion objective (Nikon), passed through a ET550lp filter (Chroma) to remove excitation light, spectrally split in a MultiCam Device (Cairn) with a 640lpxr dichroic filter (Chroma), passed through additional bandpass filters (ET585/65 and ET685/50, Chroma), and finally projected onto two synchronized ORCA-Fusion sCMOS cameras (C14440-20UP, Hamamatsu) with 2×2 pixel binning. Unless otherwise specified, recordings were made at a 10 s−1 frame rate (100 ms time resolution) in the “standard” readout mode. Instrument control was performed with in-house developed software written in LabVIEW.
Analysis of TIRF-based single-molecule data
Wide-field movies were analyzed with SPARTAN version 3.7 (Juette et al., 2016). Briefly, molecules were detected as local intensity maxima in an image composed of the sum of donor and acceptor channels (aligned using the iterative closest points algorithm) averaged over the first 10 frames and background subtracted. Molecules closer than 3.5 pixels were excluded from analysis. Traces were extracted from the selected intensity maxima by summing the 9 most intense pixels on each fluorescence channel. The arbitrary pixel intensity units were converted to approximate photon counts using the conversion factor provided by the manufacturer with each camera. Each trace was then baseline subtracted by using the intensity levels in each channel after donor photobleaching. Finally, traces were corrected for donor to acceptor channel crosstalk, unequal apparent brightness of donor and acceptor fluorophores, and acceptor direct excitation (Hellenkamp et al., 2018). Corrected FRET values (E) were then used for estimating inter-dye distances (R) using the following equation and an R0 of 62 Å (Girodat et al., 2020).
Inherent in this calculation are many assumptions, including rapid isotropic tumbling of the fluorophores. As such, calculated inter-dye distance estimates are approximations to the true inter-dye distances. One important assumption is that the fluorophores are freely tumbling (Lakowicz, 2006). Along these lines, we would like to point out that the LD555p and LD655 fluorophores have long (13 atoms) linkers to minimize any possible restrictions on fluorophore tumbling and a number of polar groups to minimize any possible hydrophobic interactions with the protein. Explicit solvent, full potential MD simulations with these fluorophores attached to a protein of similar size to βarr1 showed minimal protein-fluorophore interactions and estimated κ2 to be near to the ideal value (Girodat et al., 2020).
Traces were selected for further analysis if they met the following criteria for experiments recorded with 100 ms (10 ms) time resolution: SNRBG > 20 (15), FRET lifetime > 50 (20) frames (using a threshold of 0.08 FRET), donor acceptor correlation coefficient < 0.5, donor fluorophore blinking events < 4, single step photobleaching, and mean total intensity (donor + acceptor) over the first 10 frames within two standard deviations from the mean. State assignment from smFRET traces was accomplished using the segmental K-means algorithm (Qin, 2004) using a 4-state linear model with the following mean (± S.D.) starting values: 0 (0.06), 0.16 (0.06), 0.46 (0.06), 0.72 (0.06) and rate constants set at 1 s−1. For dwell-time histograms and maximum likelihood analysis of the dynamic sampling of mid- and high-FRET states, traces were selected if their mean FRET values were within 0.1 of the ensemble average and there were at least 100 frames (10 seconds) in the mid- and high-FRET states. Dwell-time histograms are normalized as the number of dwells within each duration bin divided by the total imaging time, giving units of dwells per second. This approach ensures that (1) the histogram does not over-emphasize spurious events in conditions for which conformational transitions are very rare, (2) that the y axis height is directly related to the frequency of each transition type, and (3) that the histogram is minimally sensitive to any variation in the number of molecules contributing to the experiment and the photobleaching rate.
Transition density plots and dwell-time histograms represent data from full length traces. Mean state occupancies and FRET histograms are obtained from the first 100 frames. Rate constants were estimated using the maximum interval likelihood algorithm (Qin et al., 2000).
Confocal-based single-molecule FRET
Confocal-based single-molecule FRET measurements were performed using a MicroTime 200 equipped with a HydraHarp 400 counting module (PicoQuant). The donor and acceptor dyes were alternatively excited with a pulsed interleaved excitation scheme (Müller et al., 2005) with light from a 531 nm (LDH-P-FA-530L, PicoQuant) and a 639 nm (D-C-640, PicoQuant) diode laser, each operated at 20 MHz frequency, with an average power of 50 and 25μW at the sample, respectively. Emitted photons were collected by the microscope objective (Olympus UPlanSApo 60x/1.20 W), focused onto a 100-μm pinhole, and then separated into four channels with a polarizing beam splitter and two dichroic mirrors (T635lpxr, Chroma). Emission was additionally filtered by bandpass filters (ET585/65M and H690/70, Chroma) before being focused onto one of four single-photon avalanche detectors (Excelitas SPCM-AQRH-TR).
FRET efficiency histograms of doubly labelled and freely diffusing βarr1 were acquired at a concentration of 50–100 pM in the presence of 0.01% Tween 20 (Pierce) to minimize surface adhesion (Schuler, 2007). The data were collected for 1–8 hrs. To ensure consistency with TIRF experiments, confocal FRET experiments were carried out in the same microfluidic channels used for wide-field TIRF measurements.
Fluorescence bursts were identified by combining successive photons separated by 100 μs or less (Eggeling et al., 2001), and events comprising 40 to 60 or more photons were kept for analysis. Average FRET efficiencies were obtained from in which and are the numbers of donor (D) and acceptor (A) photons, respectively, in each burst after donor excitation (d), corrected for background, channel crosstalk α and acceptor direct excitation δ, defined as and , where is the average apparent FRET efficiency (E) of molecules containing only an active donor fluorophore, and is the average apparent stoichiometry ratio (S) of molecules containing only an active acceptor fluorophore, with S defined as , in which represents the emission of acceptor photons after direct acceptor excitation (a) at 639 nm (Lee et al., 2005). Cross-talk- and direct excitation-corrected average Eapp and Sapp need to be further corrected to account for the relative excitation efficiency of the two fluorophores, β, and the relative quantum yields and detection efficiencies for donor and acceptor emission γ: with β = IaεA/IdεD, where Ia,d are the relative powers of the acceptor and donor excitation lasers, and εA,D are the extinction coefficients of the acceptor and donor fluorophores at their respective excitation wavelengths; and γ = ϕAηA/ϕDηD, where ϕA,D are the quantum yields of acceptor and donor, and ηA,D are the detection efficiencies for acceptor and acceptor photons, respectively. The values of β and γ have been determined from the dependence of Sapp on Eapp using three subpopulations with different donor–acceptor distances, according to Sapp = (1 + γβ + β(1 − γ)Eapp)−1 (Holmstrom et al., 2018; Lee et al., 2005). These two correction factors were then used to calculate the average FRET efficiency E and stoichiometry ratio S as expressed above.
Fluorescence correlation spectroscopy
FCS data were acquired at a concentration of 0.5–2 nM of the freely diffusing labeled βarr1 and an excess of the partner to saturate binding. Donor and acceptor fluorescence emission after continuous-wave excitation at 532 nm was focused onto a 50-μm pinhole, recorded with two detectors each and summed before being cross-correlated between 1 μs and 1 s with logarithmically spaced time steps. The resulting fluorescence intensity cross-correlations, G(τ), between donor and acceptor signal were fitted with a model including translational diffusion and triplet blinking to determine the diffusion time, τD, through the confocal volume:
where N is the average number of labeled molecules in the confocal volume, s is the ratio of the lateral to the axial radii of the confocal volume, and τT is the correlation time of triplet blinking.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed in a MicroCal PEAQ-ITC (Malvern) at 25°C. The plasmid coding for wild type βarr1 and its purification were described previously (Nobles et al., 2007). Both wildtype βarr1 and V2Rpp were dialyzed in 20 mM HEPES pH 7.4, 125 mM NaCl, TCEP (10 μM). βarr1 (25 μM) was loaded into the sample cell and V2Rpp (250 μM) into the injection syringe. Titrations were initiated by a 0.4 μL injection from syringe, followed by 19 injections of 2.0 μL at 150 s intervals. During the experiment, the reference power was set to 7 μcal·s−1 and the sample cell was stirred continuously at 750 rpm. Data analysis was performed by nonlinear regression using the MicroCal PEAQ-ITC analysis software (Malvern).
Molecular modeling
A βarr1 model in the basal state matching the sequence of the bovine cysteine-less βarr1 construct used in the experimental studies was constructed by homology modeling with Modeller (v10.0) (Eswar et al., 2006), using a selected set of crystal structures of βarr1 as templates. These structures include βarr1 (PDB 1JSY) (Milano et al., 2002) (the main template for residues up to 399), βarr1 (PDB 1ZSH) (Milano et al., 2006b) (which includes the key interaction R7-E389 missing in 1JSY), βarr1 in complex with M2 muscarinic receptor PDB 6U1N (Staus et al., 2020a) (the template for loop residues 331 to 339 and 356 to 364), visual arrestin (PDB 1CF1) (Hirsch et al., 1999) (the homologous template for residues 400 to 404). Parts without any template, i.e., residues 1 to 5, 365 to 382, and 405 to 418 were ab initio modeled with Modeller.
In modeling the β2V2R-βarr1 complex, we used the cryo-EM structure of β1V2R-βarr1 (PDB 6TKO) (Lee et al., 2020) as the main template instead of the available β2AR-G protein structures, considering the different orientations of TM5-IL3-TM6 in accommodating G protein versus arrestin and the high homology between β2AR and β1AR. Additional structures were also used for different parts of the model. The structure PDB 6U1N provided the template for the missing residues 331–340 in βarr1 of 6TKO. Two β2AR structures were also included: the structure PDB 4LDO (Ring et al., 2013) was used specifically as the template for TM1, which has noticeable divergence between β2AR and β1AR, while the structure 6TKO misses two extracellular turns in TM1; 4LDO also provided the epinephrine bound in the binding site; the structure PDB 3SN6 (Rasmussen et al., 2011) is five residues longer at the C-terminal end of TM5 than 6TKO. Similar to our βarr1 model in the basal state, we matched the sequence of the receptor and βarr1 in the complex to the β2V2R (Shukla et al., 2014a) and the bovine cysteine-less βarr1 constructs used in the experimental studies. 200 models were generated with Modeller. These models include the complete IL3 that does not have a template for residues 240 to 262, and the complete V2Rpp that does not have a template for residues 342 to 354. The resulting model with the lowest DOPE score was selected for the following steps.
Based on the selected Modeller model, for the IL3 portion lacking any template and its nearby residues (residues 236 to 263) and the V2Rpp portion (residues 342 to 355) that has no template, we remodeled them with the next-generation kinematic closure (NGK) protocol implemented in ROSETTA (version 2019.47.61047) (Stein and Kortemme, 2013), which considers the context of conformational sampling of local regions, in this case, the nearby regions of receptor and the bound βarr1. We used the NGK protocol with default options to generate 20,000 models for each modelled loop and integrated the lowest-scoring models in the final complex model to build the simulation system.
Molecular dynamic simulations and analysis
The selected βarr1 model was processed by the Protein Preparation Wizard of Schrodinger Suite (version 2021–1). The first and last residues of this model are in positively and negatively charged states, respectively, without capping, as assumed in their natural condition. The prepared model was then immersed in a simulation water box using the System Builder of Schrodinger Suite (version 2021–1). Na+ and Cl− ions were added to neutralize the system, and to make the salt concentration of the system 0.15 M. The total system size was ~143,000 atoms.
The selected β2V2R-βarr1 complex model was processed by the Protein Preparation Wizard. The sidechains of D792.50 and D1303.49 of the receptor were protonated to their neutral forms as assumed in the active state of aminergic GPCRs (Dror et al., 2011). In addition, E1223.41, a residue facing lipids, was also protonated to its neutral form. The resulting model was then placed into explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid bilayer (POPC) using the orientation of the structure 4LDO from the Orientation of Proteins in Membranes database (Borroto-Escuela and Fuxe, 2019; Lomize et al., 2006). Simple point charge (SPC) water model was used to solvate the system, charges were neutralized, and 0.15M NaCl was added. The total system size was ~179,000 atoms.
MD simulations were carried out using Desmond MD System (version 6.1; D.E. Shaw Research, New York, NY) (Bowers et al., 2006) with the OPLS4 force field (Lu et al., 2021). The simulation systems were minimized and equilibrated with restraints on the ligand heavy atoms and the protein backbone. For both the equilibrations and the following production runs, the constant temperature 310 K was maintained by Langevin dynamics, 1 atm constant pressure was achieved with the Langevin piston method (Feller et al., 1995). A cutoff distance of 9 Å was used for the nonbonded interactions, and the particle-mesh Ewald summation method was used for the electrostatics interactions. The integration timestep was set to 2.5 fs. For the βarr1 simulations in a water box, the isothermal-isobaric (NPT) ensemble was used in a periodic boundary condition. For the β2V2R-βarr1 simulations in a lipid bilayer, The NPγT ensemble (the pressure treatment on the z direction (membrane normal axis) is decoupled from those of the x and y directions) was used on an anisotropic flexible periodic cell with a constant ratio of the x-y plane. The production runs of βarr simulations are without any restraints, those of β2V2R-βarr1 complex are restrained in the C domain of arrestin (Cα atoms of residue 231–239) and near ligand binding site on the extracellular side of the receptor TM domain (Cα atoms of 30 residues), to retain the orientation between the receptor and βarr1 observed in the structure 6TKO, without any restraints near the receptor-arrestin interface.
The analysis and visualization were performed with VMD (Humphrey et al., 1996) and PyMol (Schrodinger).
Estimating inter-dye distances
Representative frames from the above MD simulations corresponding to the two distinct basal state βarr1 conformations (Figures 1B,C) were selected for estimating inter-dye distances. Residues 176 and 397 of βarr1 were substituted for cysteine and LD555p and LD655 fluorophore models with 13-atom linkers were attached at these respective positions via the thiol-maleimide bond using PyMol version 1.4.1. Fluorophore models were constructed and geometries optimized with Avogadro version 1.2.0 (Hanwell et al., 2012). Coordinate and topology files for the structure-based models were constructed using SMOG version 2.2 (Noel et al., 2016) with default parameters. Simulations were performed using GROMACS version 2019 (Hess et al., 2008). Following steepest-descent equilibration with 10 million timesteps, the simulation was run for 95 million timesteps of size 0.005 with frames saved every 1,000 timesteps. Frames were aligned using the protein portion and the inter-dye distances were measured between the center carbon of each fluorophore’s polymethine chain, and these distances were then averaged across all frames.
With this approach, the conformation of βarr1 is maintained by a harmonic potential on native contacts within the structure. By contrast, the fluorophores and linkers are not subject to these constraints, enabling them to explore all accessible volume. The resulting ensemble of fluorophore positions serves as an approximation to the experimental ensemble that gives rise to the observed FRET efficiencies. While these simplified, structure-based potential simulations do not capture all contributors to fluorophore positions and dynamics, they have been shown to provide good correspondence to smFRET experiments with a protein of similar size to βarr1 (Girodat et al., 2020).
Bystander BRET and Live-Cell Assays
All live-cell assays were carried out in the HEK293A GRK2/3/5/6 knockout cells. For the bystander BRET assay, cells were measured 24 hours post transient transfection using lipofectamine 2000 (Thermo Fisher Scientific) per manufacturer’s protocol. For this assay, plasmids coding for receptor (2 μg), Rluc8-βarr2 (0.25 μg), mem-citrine (13 μg), and either non-coding plasmid (pcDNA5 FRT, 5 μg) or GRK2 (5 μg) were transfected. The transfected cells were prepared in a 96-well microplate format and assayed as described previously using a Pherastar FS plate reader (BMG Labtech) (Donthamsetti et al., 2015). After incubation with coelenterazine H (5 μM, Dalton Pharma Services) for 8 minutes, ISO or dopamine was injected to stimulate the β2AR variants or D2R, respectively. BRET measurements were acquired 10 minutes post injection of agonists. Data analysis was carried out as described previously (Donthamsetti et al., 2015).
To determine receptor surface expression in the bystander BRET assay, live cells transfected as described above were incubated with 1 μM SNAP-Surface Alexa Fluor 647 in complete tissue culture media for 30 min at 37 °C under 5% CO2 to label the N-terminal SNAP tags of the β2AR variants. After labeling, the cells were washed three times with media, twice with DPBS, then harvested and resuspended in DPBS supplemented with 5 mM glucose. The number of cells in the resulting suspension was measured using the Countess II cell counter (Thermo Fisher Scientific). Using a Pherastar FS plate reader (BMG Labtech), the same cell suspension transferred to black bottom 96-well plates (Greiner Bio-One) excited with 640 nm light to detect fluorescence (680 nm) from labeled receptors. Surface expression was determined by calculating the ratio of fluorescence from Alexa Fluor 647-labeled receptors by the measured number of cells.
For the bystander miniGs assay, cells were propagated in plastic flasks and on 6-well plates 24 h prior to transfection. Cells were transiently transfected with plasmids coding for NES-NanoLuc-mGs (Wan et al., 2018), venus-kras (Lan et al., 2011), β2AR receptors variants, and GRK2 (or pcDNA3.1(+) control) in a (1:20:4:10) ratio using linear polyethyleneimine MAX (Polysciences) and were used for experiments 24 h later. To measure BRET signals, cells were washed with DPBS, harvested by trituration, and transferred to opaque black 96-well plates containing diluted drug solutions. Furimazine (Nano-Glo; 1:1000, Promega Corporation) was added to all wells immediately prior to making measurements. BRET measurements were made using a Mithras LB940 photon-counting plate reader (Berthold Technologies GmbH, Bad Wildbad, Germany). Raw BRET signals were calculated as the emission intensity at 520–545 nm divided by the emission intensity at 475–495 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
For TIRF smFRET experiments, each replicate (n) corresponds to a single movie recorded in a distinct microfluidic chamber, typically on distinct days using a distinct aliquot of each reagent. Data are displayed as the mean across replicates and error bars represent the standard deviation across replicates. Inclusion criteria for smFRET traces is described above in the section titled “Analysis of TIRF-based single-molecule recordings”. Plotting, distribution fitting and statistics for all TIRF smFRET data were carried out using OriginPro 2020b (OriginLab).
Confocal single-molecule FRET and FCS data were analyzed with Fretica, a custom WSTP software package for Mathematica (Wolfram Research).
Data from the BRET-based live cell assays were plotted and fit using GraphPad Prism (GraphPad Software). To determine p values, an unpaired two-sided Mann-Whitney test was used, where comparison with p > 0.05 were considered statistically significant. Statistical details of experiments can be found in the figure legends.
Supplementary Material
Figure S1. Site-specific fluorophore labeling and surface tethering of β-arrestin1, related to Figure 1. A, Purified βarr1 “cys-less” background (left) and with cysteines introduced (E176C/ K397C, right) were analyzed with SDS-PAGE and imaged to detect LD555p fluorescence (top), LD655 fluorescence (middle), and Coomassie stain (bottom). B, Total fluorescence intensity monitored over time after fluorophore-labeled βarr1 was introduced into an imaging chamber prepared without (non-specific, red) and with (specific, black) StrepMAB-Immo antibody. C, Representative smFRET traces of βarr1 tail sensor from TIRF experiments performed in the absence (top) and presence (bottom) of 3 μM V2Rpp and recorded with 10 ms time resolution. D, Single-molecule confocal FRET efficiency histograms of freely diffusing βarr1 tail sensor alone (top) and in the presence of saturating concentrations of V2Rpp (middle) or heparin (bottom), fitted with Gaussian functions (colored areas). E, Ensemble average FRET efficiency and F, example smFRET traces from TIRF experiments in which 3 μM V2Rpp was added (vertical dotted lines; top row) or removed (bottom row) from solution during imaging.
Figure S2. Alternative labeling site for measuring β-arrestin1 C-tail dynamics, related to Figure 2. βarr1 E110C/K397C was purified and labeled with LD555p and LD655 and tethered to the imaging surface for smFRET in the absence and presence of varying concentrations of V2Rpp. A, FRET histograms, B, representative smFRET traces, and C, high FRET inactive state occupancy (red symbols) fit to a dose-response function with Hill slope of 1.0 and EC50 of 4.2 μM (red line). Data from βarr1 E176C/K397C labeling variant is shown for comparison (green).
Figure S3. β-arrestin1 complexes with V2Rpp and pB2V2R are stabilized by Fab30, related to Figures 2 and 5. A, Schematic of the experiment in which a complex was formed of βarr1 (gray), Fab30 (pink), and biotin-V2Rpp (orange) and tethered to a NeutrAvidin treated imaging surface. B, Population FRET histogram. C, transition density plot. D, Relative number of fluorescent βarr1 particles detected on the surface as a function of time (symbols) fit to a decay curve with a half-life (T1/2) of 33 min (line). E, Population FRET histograms of the inactive (red) and active (blue and green) states from experiments imaging βarr1 in the presence of 1 μM pβ2V2R and saturating concentrations of the indicated ligands and in the absence (top row) and presence (bottom row) of Fab30.
Figure S4. Supporting data for the constitutively activating mutants, related to Figure 4. A, Population FRET histograms of 3EI-βarr1 imaged at 10 ms camera integration time in the presence of the indicated concentrations of V2Rpp (left) and a representative FRET trace in the absence of V2Rpp (right). B, In the MD simulations, I386, V387, and F388 in the middle segment of the βarr1 tail are tightly packed with three Leu residues of the N-domain helix. In addition, three charged residues, E389, D390, and R393 form ionic interactions with residues from both the N terminus (R7 and K10) and the polar core (D26 and D297). Together, these interactions anchor the middle tail stably within the N-domain groove in the inactive state. C, In the MD simulations of the βarr1/3-EI mutation construct, the mutations, I386A/V387A/F338A, were found to be able to disrupt the hydrophobic interactions of these residues with the N-domain helix, including loss of the interaction with L108 entirely, resulting in an altered orientation of the middle βarr1 tail segment. This change propagated to allosterically perturb R393, which dissociated from the polar core and reoriented to interact with E389. Correspondingly, the E389-R7 and D390-K10 interactions are weakened. These reconfigurations in the middle loop affect the position of residue 397 and its distance from residue 176, likely underlying the altered basal FRET state in the 3EI-mutation. D, Example smFRET trace exhibiting low FRET efficiency from experiments imaging 3EI-βarr1 at 100 ms integration time. E, Rate constants derived from maximum likelihood model optimization using idealized smFRET traces from experiments imaging E313K-βarr1 in the presence of indicated V2Rpp concentrations (symbols); lines are linear fits with apparent V2Rpp on (blue) and off (red) rate constants of 0.17 μM−1 s−1 and 0.24 s−1, respectively. F, Overlay of dwell time histograms (symbols; lines are spline fits for clarity) from experiments in panel E with 1 μM V2Rpp and E313K (red) and wild type for comparison (black). G, FRET histograms (left) and representative traces (middle and right) from experiments imaging R76A,K77A,D78A-βarr1 with 100 ms camera integration time in the presence of the indicated concentrations of V2Rpp. Error bars, mean ± S.D. of two repeats.
Figure S5. MD simulations of β-arrestin1:receptor complexes, related to Figure 5. A, Representative MD frames of the simulated β2V2R-βarr1 complex model, showing the dynamics of the V2Rpp region between H8 of the receptor and the P2 position. The V2Rpp attached to H8 is in wheat color. Due to the strain from H8 as well as to steric clash with the finger loop from the β2V2R-βarr1 complex in its core-engagement orientation, the phosphorylated residues T346 (P1) and S349 (P2) cannot dock at the same locations in the extended groove as their aligned residues in the 4JQI structure. B, In the structure of βarr1 from the megacomplex (PDB 6NI2) (Nguyen et al., 2019), the V2Rpp peptide attached to H8 (green) shows a potentially different orientation before the P3 position compared to V2Rpp bound to βarr1 in the 4JQI structure (light pink). In the β2V2R-βarr1 complex, V2Rpp attached to H8 is likely in an orientation more similar to that of the 6NI2 structure. The black ribbons are the βarr1 from the simulated β2V2R-βarr1 complex of which the finger loop occupies or disrupts the P1 and P2 anchoring positions in the 4JQI structure. C, Representative MD frames of the β2V2R-βarr1 complex. Two distance constraints derived from DEER data (Zhuo et al., 2014), the 192–392 distance at 63 Å and the 12–392 distance at 55 Å would position 392 on rings encircling the intersection of N and C domains (yellow) of βarr1. Similarly, the Cα-Cα distances of the 176–397 and 110–397 pairs from the current smFRET study at 43.5 and 50 Å, respectively, would position 397 on another set of rings encircling the intersection (green). We inferred these Cα-Cα distances from their inter-dye distances, 63.5 and 69.9 Å, assuming a ~20 Å difference between the Cα-Cα and inter-dye distances. The close proximity of the 392 (yellow) and 397 (green) rings on the opposite side of the N-domain groove is insensitive to substantial variation (± 5 Å) of the estimated Cα-Cα distances. Notably, the pairs of points from the 392 and 397 rings that are 9 Å from each other, the estimated range of distance between the two residues, are indicated with larger spheres and connected by cyan lines. Such an intersection of the two sets of rings would position the βarr1 distal tail in proximity to IL3 of the receptor on the opposite side from the N-domain groove. This finding suggests a dramatic relocation of position 397 in the βarr1 distal tail from near the N-domain groove in the inactive state (indicated by the small dotted green circle in the inset) to the proximity of IL3 of the receptor in the active state. This relocation is indicated by green dotted arrows in both the main panel and the inset, and may be related, at least in part, to a potential electrostatic repulsion between the competing Rp tail and βarr tail, both of which are highly enriched with negatively charged residues.
Figure S6. Evaluating the stability of β-arrestin1:receptor complexes, related to Figure 5. A, Population FRET histograms from experiments imaging βarr1 in the presence of 1 μM pβ2V2R (left) and after exchanging to solution without receptor with a 5 min incubation (right) with 200 μM epi present throughout. B, Example trace after receptor washout. C, Population FRET histograms from experiments imaging βarr1 in the presence of 1 μM pβ2V2R (left) or bpβ2V2R (right) with 200 μM epi and 10 μM Cmpd-6FA. D, Diffusion times τD through the confocal volume of freely diffusing molecules from fluorescence intensity cross-correlation, G(τ), between donor and acceptor signals. A longer diffusion time is indicative of larger particles. Vertical error bars represent the standard deviation from 2 or more independent experiments. E, Representative normalized cross-correlation curves between donor and acceptor fluorescence emission intensity for βarr1 in the absence (light orange) and presence of 5 μM receptor with saturating concentrations of epi and Cmpd-6FA using pβ2V2R (orange) or bpβ2V2R (dark orange). F, As in panel C, but with the finger-loop proximal mutant E313-βarr1.
Figure S7. Supporting data for β-arrestin recruitment experiments in living cells, related to Figure 6. A, Amino acid sequence of the C-terminal region of receptors used in this study. The C-tail of receptors is shown in orange, and the palmitoylated cysteine residue is in bold. B, Relative surface expression of the indicated β2AR variants as determined by fluorescence intensity measurements of cells containing N-terminal SNAPfast-tagged receptors labeled with fluorescent dye. Bars represents the mean ± S. E. of three independent experiments performed in triplicate. Symbols represent the measured fluorescence intensity per the number of cells used for each measurement. C, Dose-response curves of mini-Gs recruitment to the plasma membrane by wildtype (WT) β2AR (black squares) and β2AR-365tr (blue triangles) in HEK293 GRK2/3/5/6 knockout cells in the (left plot) absence (GRK KO) or (right plot) presence of GRK2 expression via transient transfection (GRK2 rescue). Dose-response curves of four independent experiments (mean ± S. E.). D, Dose-response curves of βarr2 recruitment to the plasma membrane by the dopamine 2 receptor (D2R) in HEK293 GRK2/3/5/6 knockout cells in the absence (GRK KO) or presence of GRK2 expression via transient transfection (GRK2 rescue). Dose-response curves are of three independent experiments performed with triplicate samples (mean ± S. E.).
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| biotinylated rabbit anti-mouse IgG secondary antibody | ThermoFisher Scientific | Cat#31813 |
| mouse high-affinity anti-Strep tag IgG1 monoclonal antibody | IBA GmbH | Cat#2-1517-001 |
| Bacterial and virus strains | ||
| Escherichia coli: BL21-Gold competent cells | Agilent Technologies | Cat#230130 |
| Baculovirus Expression System | Expression Systems | Cat#91-200 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| LD555p-MAL | Lumidyne Technologies | Cat#03/04 |
| LD655-MAL | Lumidyne Technologies | Cat#09/10 |
| SNAP-Surface Alexa Fluor 647 | New England Biolabs | Cat#S9136S |
| V2Rpp | Tufts University peptide synthesis core facility | N/A |
| V2Rnp | Tufts University peptide synthesis core facility | N/A |
| V2Rbp | Tufts University peptide synthesis core facility | N/A |
| Biotin-V2Rpp | Tufts University peptide synthesis core facility | N/A |
| Heparin | Sigma-Aldrich | Cat# H3149-10KU |
| IP6 | Sigma-Aldrich | Cat#407125 |
| diC8-PI(4,5)P2 | Echleon Biosciences | Cat#P-4508 |
| Carazolol | Sigma | Cat#53787 |
| Epinephrine | Sigma | Cat#E4642 |
| Dopamine hydrochloride | Sigma | Cat#H8502 |
| Sodium bisulfite | Thermo Fisher Scientific | At# S-244 |
| Cmpd-6FA | Ahn S, et. al., 2018 | N/A |
| Isoproterenol hydrochloride | Tocris Bioscience | Cat#1747 |
| Fab30 | Shukla AK et. al., 2013 | N/A |
| Nb6B9 | Liu, et. al., 2019 | N/A |
| NeutrAvidin | Thermo Fisher Scientific | Cat#31000 |
| Bovine Serum Albumin | VWR | Cat#EM-2930 |
| Glucose oxidase | Sigma | Cat#G2133 |
| Catalase | Sigma | Cat#C40 |
| pβ2V2R | Kahsai, et. al., 2016; Shukla, et. al., 2014b | N/A |
| bpβ2V2R | Kahsai, et. al., 2016; Shukla, et. al., 2014b | N/A |
| Strep-β-arrestin1 E176C, K397C (βarr1 tail sensor) | This paper | N/A |
| Strep-β-arrestin1 E110C, K397C | This paper | N/A |
| Strep-β-arrestin1 E176C, K397C; 3-EI mutations: I386A, V387A, F388A (3EI-βarr1 tail sensor) | This paper | N/A |
| Strep-β-arrestin1 E176C, K397C; E313K finger loop proximal mutation (313K-βarr1 tail sensor) | This paper | N/A |
| Strep-β-arrestin1 E176C, K397C; finger loop proximal mutations: R76A, K77A, D78A | This paper | N/A |
| Strep-β-arrestin1 E176C | This paper | N/A |
| Strep-β-arrestin1 K397C | This paper | N/A |
| Wildtype β-arrestin 1 | Nobles KN et. al., 2007 | N/A |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668019 |
| linear polyethyleneimine MAX (MW 40,000) | Polysciences | Cat#24761-1 |
| Coelenterazine h | Dalton Pharma Services | Cat#20-0770 |
| Nano-Glo Substrate | Promega | Cat# N113A |
| Critical commercial assays | ||
| Deposited data | ||
| Active-state crystal structure of β-arrestin 1 bound to the phosphorylated V2R C-tail phosphopeptide and Fab30 | Shukla et al., 2013a | PDB: 4JQI |
| Inactive-state crystal structure of β-arrestin 1 | Han et al., 2001 | PDB: 1G4M |
| Inactive-state crystal structure of β-arrestin 1 | Milano et al., 2002 | PDB: 1JSY |
| Crystal structure of bovine arrestin-2 in complex with inositol hexakisphosphate (IP6) | Milano et al., 2006a | PDB: 1ZSH |
| Inactive-state crystal structure of visual arrestin | Hirsch et al., 1999 | PDB: 1CF1 |
| Inactive-state crystal structure of bovine rhodopsin | Palczewski et al., 2000 | PDB: 1F88 |
| Inactive-state crystal structure of bovine rhodopsin | Teller et al., 2001 | PDB: 1HZX |
| Crystal structure of 9-cis-rhodopsin | Nakamichi et al., 2007 | PDB: 2PED |
| Structure of β2 adrenoceptor bound to adrenaline and an engineered nanobody | Ring et al., 2013 | PDB: 4LDO |
| Crystal structure of the β2 adrenergic receptor-Gs protein complex | Rasmussen et al., 2011 | PDB: 3SN6 |
| Cryo-EM structure of β-arrestin 1 bound to M2 muscarinic acetylcholine receptor containing the V2R C-tail (M2V2R) | Staus et al., 2020a | PDB: 6U1N |
| Phosphorylated turkey β1 adrenoceptor with bound agonist formoterol coupled to arrestin-2 in lipid nanodisc | Lee et al., 2020 | PDB: 6TKO |
| Stabilized β-arrestin 1-V2T subcomplex of a GPCR-G protein-β-arrestin mega-complex | Nguyen et al., 2019 | PDB: 6NI2 |
| GRK1-rhodopsin complex | Chen et al., 2021b | PDB: 7MTA |
| Experimental models: Cell lines | ||
| Human: Passage 4-15 HEK293A GRK2/3/5/6 knockout cells | Kawakami et. al., 2022 | N/A |
| Sf9 Insect cells | Expression Systems | Cat#94-001F |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| 25-nucleotide DNA duplex as blocking agent sequence: GTGCTTGGGACCCCACGTGTAAACG | IDT | N/A |
| Recombinant DNA | ||
| pTrcHisB Cysless β-arrestin1 | Hanson et. al., 2007 | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E176C, K397C (βarr1 tail sensor) | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E110C, K397C | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E176C, K397C; 3-EI mutations: I386A, V387A, F388A (3EI-βarr1 tail sensor) | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E176C, K397C; E313K finger loop proximal mutation (313K-βarr1 tail sensor) | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E176C, K397C; finger loop proximal mutations: R76A, K77A, D78A | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 E176C | This paper | N/A |
| pTrcHisB Cysless Strep-β-arrestin1 K397C | This paper | N/A |
| Plasmid: wildtype β-arrestin 1 | Nobles KN et. al., 2007 | N/A |
| Plasmid: GRK2-CAAX | Kahsai et. al., 2016; Shukla et. al., 2014b | N/A |
| pcDNA3.1 SNAPf-β2AR | This paper | N/A |
| pcDNA3.1 SNAPf-β2AR-365tr | This paper | N/A |
| pcDNA3.1 FLAG-D2L | Donthamsetti et. al., 2015 | N/A |
| pcDNA3.1 mem-citrine | Donthamsetti et. al., 2015 | N/A |
| pcDNA3.1 Rluc8-β-arrestin2 | Donthamsetti et. al., 2015 | N/A |
| Plasmid: GRK2 | Donthamsetti et. al., 2015 | N/A |
| pcDNA5 FRT | Invitrogen | Cat#601020 |
| pcDNA3.1(+) | Thermo fisher Scientific | Cat#V79020 |
| Plasmid: NES-NanoLuc-mGs | Wan et. al., 2018 | N/A |
| Plasmid: Venus-kras | Lan et. al., 2011 | N/A |
| Software and algorithms | ||
| LabVIEW | National Instruments | https://www.ni.com/ |
| SPARTAN | Juette et. al., 2016 | https://www.scottcblanchardlab.com/spartan-download |
| MATLAB 2018a | Mathworks | https://www.mathworks.com/ |
| Fretica (WSTP for Wolfram Mathematica) | Wolfram Research | https://schuler.bioc.uzh.ch/programs |
| Confocal data acquisition - SymPhoTime 64 V2.5 | Picoquant | https://www.picoquant.com/products/category/software/symphotime-64-fluorescence-lifetime-imaging-and-correlation-software |
| MicroCal PEAQ-ITC analysis software | Malvern | https://www.malvernpanalytical.com |
| Modeller v10.0 | Eswar et. al., 2006 | https://salilab.org/modeller/ |
| ROSETTA v2019.47.61047 | Stein et. al., 2013 | https://www.rosettacommons.org/software |
| Schrodinger Suite v2021-1 | Schrodinger | https://www.schrodinger.com/ |
| Desmond MD System v6.1 | D.E. Shaw Research | https://www.deshawresearch.com/resources_desmond.html |
| VMD v1.9.3 | Humphrey et. al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| PyMol v1.4.1 | Schrödinger | https://pymol.org/ |
| Avogadro v1.2.0 | Hanwell, et. al., 2012 | http://avogadro.openmolecules.net/ |
| SMOG v2.2 | Noel, 2016 | https://smog-server.org/smog2/ |
| GROMACS v2019 | Hess et. al., 2008 | https://www.gromacs.org/ |
| OriginPro v2020b | OriginLab | https://www.originlab.com/ |
| GraphPad Prism v9.3.1 | GraphPad Software, LLC | https://www.graphpad.com/ |
| Other | ||
| Microfluidic imaging chambers for TIRF-based smFRET | Blanchard et. al., 2004 | N/A |
Highlights.
β-arrestin1 is highly autoinhibited by its tightly bound C tail.
Phosphopeptide mimics of receptor C tail facilitate β-arrestin1 C tail release.
Phosphorylated receptor must be agonist bound to efficiently activate β-arrestin1.
The receptor C tail plays an autoinhibitory role in β-arrestin1 recruitment.
ACKNOWLEDGEMENTS
This work was supported by NIH grants R21NS102694 (S.C.B.), R01MH054137 (J.A.J.), R01HL016037 (R.J.L.), R35 GM122491 (V.V.G.), R01 GM130142 (N.A.L.), the Hope for Depression Research Foundation (J.A.J.), the Brain and Behavior Research Foundation NARSAD Young Investigator Award (W.B.A.), the National Institute on Drug Abuse – Intramural Research Program (Z1A DA000606 (LS), the PhRMA Foundation (W.J.), and the Single-Molecule Imaging Center at St. Jude Children’s Research Hospital. R.J.L. is an HHMI Investigator. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We thank Michael Berne (Tufts University Core Facility) for preparing the synthetic peptides and related discussions. We thank Harel Weinstein and Giulia Morra for helpful discussion about βarr dynamics, Xuanzhi Zhan and Sergey Vishnivetskiy for helpful discussions concerning βarr expression, and Marina Dawoud, Lisa Lin, and Mel Nelson for technical assistance. We thank Kuo-Hao Lee for help with Rosetta modeling. We thank Ines Chen for editing the manuscript and Zhaowen Luo for assistance rendering protein structures and schematics. This work is dedicated to the life and memory of Miriam Javitch, who loved hearing the story and looking at the figures as they emerged.
Footnotes
DECLARATION OF INTERESTS
Competing interests
S.C.B has an equity interest in Lumidyne Technologies. The other authors have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abreu N, Acosta-Ruiz A, Xiang G, and Levitz J (2021). Mechanisms of differential desensitization of metabotropic glutamate receptors. Cell Rep. 35, 109050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Pani B, Kahsai AW, Olsen EK, Husemoen G, Vestergaard M, Jin L, Zhao S, Wingler LM, Rambarat PK, et al. (2018). Small-Molecule Positive Allosteric Modulators of the β2-Adrenoceptor Isolated from DNA-Encoded Libraries. Mol. Pharmacol 94, 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher WB, Geggier P, Holsey MD, Gilmore GT, Pati AK, Meszaros J, Terry DS, Mathiasen S, Kaliszewski MJ, McCauley MD, et al. (2021). Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 18, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, and Lefkowitz RJ (1992). Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem 267, 17882–17890. [PubMed] [Google Scholar]
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, and Puglisi JD (2004). tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol 11, 1008–1014. [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, and Fuxe K (2019). Oligomeric Receptor Complexes and Their Allosteric Receptor-Receptor Interactions in the Plasma Membrane Represent a New Biological Principle for Integration of Signals in the CNS. Front. Mol. Neurosci 12, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Chow E, Xu H, Dror R, Eastwood M, Gregersen B, Klepeis J, Kolossvary I, Moraes M, Sacerdoti F, et al. (2006). Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In ACM/IEEE SC 2006 Conference (SC’06), (IEEE), pp. 43–43. [Google Scholar]
- Cahill TJ, Thomsen ARB, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang L-Y, Kahsai AW, Bassoni DL, Gavino BJ, et al. (2017). Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, and Gurevich VV (2002). Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem 277, 9043–9048. [DOI] [PubMed] [Google Scholar]
- Chen Q, Perry NA, Vishnivetskiy SA, Berndt S, Gilbert NC, Zhuo Y, Singh PK, Tholen J, Ohi MD, Gurevich EV, et al. (2017). Structural basis of arrestin-3 activation and signaling. Nat. Commun 8, 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhuo Y, Sharma P, Perez I, Francis DJ, Chakravarthy S, Vishnivetskiy SA, Berndt S, Hanson SM, Zhan X, et al. (2021a). An Eight Amino Acid Segment Controls Oligomerization and Preferred Conformation of the two Non-visual Arrestins. J. Mol. Biol 433, 166790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Plasencia M, Li Z, Mukherjee S, Patra D, Chen C-L, Klose T, Yao X-Q, Kossiakoff AA, Chang L, et al. (2021b). Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, and Neve KA (2014). Mutation of three residues in the third intracellular loop of the dopamine D2 receptor creates an internalization-defective receptor. J. Biol. Chem 289, 33663–33675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donthamsetti P, Quejada JR, Javitch JA, Gurevich VV, and Lambert NA (2015). Using Bioluminescence Resonance Energy Transfer (BRET) to Characterize Agonist-Induced Arrestin Recruitment to Modified and Unmodified G Protein-Coupled Receptors. Curr. Protoc. Pharmacol 70, 2.14.1–2.14.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011). Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi-Agnihotri H, Chaturvedi M, Baidya M, Stepniewski TM, Pandey S, Maharana J, Srivastava A, Caengprasath N, Hanyaloglu AC, Selent J, et al. (2020). Distinct phosphorylation sites in a prototypical GPCR differently orchestrate β-arrestin interaction, trafficking, and signaling. Sci. Adv 6, eabb8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling C, Berger S, Brand L, Fries JR, Schaffer J, Volkmer A, and Seidel CA (2001). Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J. Biotechnol 86, 163–180. [DOI] [PubMed] [Google Scholar]
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita J-B, Dror RO, and von Zastrow M (2018). Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M-Y, Pieper U, and Sali A (2006). Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5, Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SE, Zhang Y, Pastor RW, and Brooks BR (1995). Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 103, 4613–4621. [Google Scholar]
- Gaidarov I, Krupnick JG, Falck JR, Benovic JL, and Keen JH (1999). Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J 18, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girodat D, Pati AK, Terry DS, Blanchard SC, and Sanbonmatsu KY (2020). Quantitative comparison between sub-millisecond time resolution single-molecule FRET measurements and 10-second molecular simulations of a biosensor protein. PLoS Comput. Biol 16, e1008293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV (1998). The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J. Biol. Chem 273, 15501–15506. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, and Gurevich EV (2015). Arrestins: critical players in trafficking of many gpcrs. Prog Mol Biol Transl Sci 132, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, and Gurevich EV (2019). GPCR signaling regulation: the role of grks and arrestins. Front. Pharmacol 10, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV, and Uversky VN (2018). Arrestins: structural disorder creates rich functionality. Protein Cell 9, 986–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, and Schubert C (2001). Crystal Structure of β-Arrestin at 1.9 Å. Structure 9, 869–880. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, and Gurevich VV (2006). Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. USA 103, 4900–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, Nair KS, Slepak VZ, Klug CS, and Gurevich VV (2007). Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J. Mol. Biol 368, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, and Hutchison GR (2012). Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenkamp B, Schmid S, Doroshenko O, Opanasyuk O, Kühnemuth R, Rezaei Adariani S, Ambrose B, Aznauryan M, Barth A, Birkedal V, et al. (2018). Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat. Methods 15, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, and Lindahl E (2008). GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Hilger D, Masureel M, and Kobilka BK (2018). Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, and Sigler PB (1999). A Model for Arrestin’s Regulation: The 2.8 Å Crystal Structure of Visual Arrestin. Cell 97, 257–269. [DOI] [PubMed] [Google Scholar]
- Holmstrom ED, Holla A, Zheng W, Nettels D, Best RB, and Schuler B (2018). Accurate Transfer Efficiencies, Distance Distributions, and Ensembles of Unfolded and Intrinsically Disordered Proteins From Single-Molecule FRET. Meth. Enzymol 611, 287–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, et al. (2020). Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, , 27. [DOI] [PubMed] [Google Scholar]
- Jean-Charles P-Y, Kaur S, and Shenoy SK (2017). G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol 70, 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Zhou Z, Altman RB, Zheng Q, and Blanchard SC (2014). The bright future of single-molecule fluorescence imaging. Curr. Opin. Chem. Biol 20, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Altman RB, Zhou Z, Zhao H, and Blanchard SC (2016). Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat. Methods 13, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlecova Z, Spielman SJ, Loerke D, Mohanakrishnan A, Reed DK, and Schmid SL (2017). Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J. Cell Biol 216, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Wisler JW, Lee J, Ahn S, Cahill Iii TJ, Dennison SM, Staus DP, Thomsen ARB, Anasti KM, Pani B, et al. (2016). Conformationally selective RNA aptamers allosterically modulate the β2-adrenoceptor. Nat. Chem. Biol 12, 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Pani B, and Lefkowitz RJ (2018). GPCR signaling: conformational activation of arrestins. Cell Res 28, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Yanagawa M, Hiratsuka S, Yoshida M, Ono Y, Hiroshima M, Ueda M, Aoki J, Sako Y, and Inoue A (2022). Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying β-arrestin transducer bias. Nat. Commun 13, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen JH, Beck KA, Kirchhausen T, and Jarrett T (1991). Clathrin domains involved in recognition by assembly protein AP-2. J. Biol. Chem 266, 7950–7956. [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren X-R, Whalen EJ, Reiter E, Wei H, and Lefkowitz RJ (2005). Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 102, 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P, Srivastava A, Ghosh E, Ranjan R, Dogra S, Yadav PN, and Shukla AK (2017). Core engagement with β-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR (2006). Principles of Fluorescence Spectroscopy (Springer US; ). [Google Scholar]
- Lan T-H, Kuravi S, and Lambert NA (2011). Internalization dissociates β2-adrenergic receptors. PLoS One 6, e17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, and Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Masureel M, Hollingsworth SA, Heydenreich FM, Suomivuori C-M, Brinton C, Townshend RJL, Bouvier M, Kobilka BK, and Dror RO (2020). How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 183, 1813–1825.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, and Weiss S (2005). Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J 88, 2939–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehmé R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, Edwards PC, García-Nafría J, Leslie AGW, Shukla AK, et al. (2020). Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 583, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner E, Barth A, Hendrix J, Ambrose B, Birkedal V, Blanchard SC, Börner R, Sung Chung H, Cordes T, Craggs TD, et al. (2021). FRET-based dynamic structural biology: Challenges, perspectives and an appeal for open-science practices. Elife 10, e60416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Masoudi A, Kahsai AW, Huang L-Y, Pani B, Staus DP, Shim PJ, Hirata K, Simhal RK, Schwalb AM, et al. (2019). Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science 364, 1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, and Lefkowitz RJ (1990). beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, and Mosberg HI (2006). OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625. [DOI] [PubMed] [Google Scholar]
- Lu C, Wu C, Ghoreishi D, Chen W, Wang L, Damm W, Ross GA, Dahlgren MK, Russell E, Von Bargen CD, et al. (2021). OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput 17, 4291–4300. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, and Godi A (2004). PI-loting membrane traffic. Nat. Cell Biol 6, 487–492. [DOI] [PubMed] [Google Scholar]
- Mayer D, Damberger FF, Samarasimhareddy M, Feldmueller M, Vuckovic Z, Flock T, Bauer B, Mutt E, Zosel F, Allain FHT, et al. (2019). Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nat. Commun 10, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano SK, Pace HC, Kim Y-M, Brenner C, and Benovic JL (2002). Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 41, 3321–3328. [DOI] [PubMed] [Google Scholar]
- Milano SK, Kim YM, Stefano FP, Benovic JL, and Brenner C (2006a). Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding (vol 281, pg 9812, 2006). J. Bio. Chem 281, 21576–21576. [DOI] [PubMed] [Google Scholar]
- Milano SK, Kim Y-M, Stefano FP, Benovic JL, and Brenner C (2006b). Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J. Biol. Chem 281, 9812–9823. [DOI] [PubMed] [Google Scholar]
- Müller BK, Zaychikov E, Bräuchle C, and Lamb DC (2005). Pulsed interleaved excitation. Biophys. J 89, 3508–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi H, Buss V, and Okada T (2007). Photoisomerization mechanism of rhodopsin and 9-cis-rhodopsin revealed by x-ray crystallography. Biophys. J 92, L106–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AH, Thomsen ARB, Cahill TJ, Huang R, Huang L-Y, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, et al. (2019). Structure of an endosomal signaling GPCR-G protein-β-arrestin megacomplex. Nat. Struct. Mol. Biol 26, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles KN, Guan Z, Xiao K, Oas TG, and Lefkowitz RJ (2007). The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and −2. J. Biol. Chem 282, 21370–21381. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang T-Y, Bressler EA, Hara MR, et al. (2011). Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JK, Levi M, Raghunathan M, Lammert H, Hayes RL, Onuchic JN, and Whitford PC (2016). SMOG 2: A Versatile Software Package for Generating Structure-Based Models. PLoS Comput. Biol 12, e1004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, and Hoffmann C (2016). β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, and Barak LS (2000). Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem 275, 17201–17210. [DOI] [PubMed] [Google Scholar]
- Ostermaier MK, Peterhans C, Jaussi R, Deupi X, and Standfuss J (2014). Functional map of arrestin-1 at single amino acid resolution. Proc. Natl. Acad. Sci. USA 111, 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Perry-Hauser NA, Asher WB, Hauge Pedersen M, and Javitch JA (2021). Assays for detecting arrestin interaction with GPCRs. Methods Cell Biol 166, 43–65. [DOI] [PubMed] [Google Scholar]
- Perry-Hauser NA, Bennett Hopkins J, Zhuo Y, Zheng C, Perez I, Schultz KM, Vishnivetskiy SA, Kaya AI, Sharma P, Dalby KN, et al. (2022). The two non-visual arrestins engage ERK2 differently. J. Mol. Biol 167465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F (2004). Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys. J 86, 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, and Sachs F (2000). A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophys. J 79, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast RB, and Margeat E (2019). Studying GPCR conformational dynamics by single molecule fluorescence. Mol. Cell. Endocrinol 493, 110469. [DOI] [PubMed] [Google Scholar]
- Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, and Kobilka BK (2013). Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MG (2004). Phosphoinositides in constitutive membrane traffic. Physiol. Rev 84, 699–730. [DOI] [PubMed] [Google Scholar]
- Sasakawa N, Ferguson JE, Sharif M, and Hanley MR (1994). Attenuation of agonist-induced desensitization of the rat substance P receptor by microinjection of inositol pentakis-and hexakisphosphates in Xenopus laevis oocytes. Mol. Pharmacol 46, 380–385. [PubMed] [Google Scholar]
- Scheerer P, and Sommer ME (2017). Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol 45, 160–169. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Haran G, and Zhou HX (2009). Fundamental aspects of protein-protein association kinetics. Chem. Rev 109, 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B (2007). Application of single molecule Förster resonance energy transfer to protein folding. Methods Mol. Biol 350, 115–138. [DOI] [PubMed] [Google Scholar]
- Sente A, Peer R, Srivastava A, Baidya M, Lesk AM, Balaji S, Shukla AK, Babu MM, and Flock T (2018). Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol 25, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng W-C, Staus DP, Hilger D, Uysal S, Huang L-Y, et al. (2013a). Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng W-C, Staus DP, Hilger D, Uysal S, Huang L-Y, et al. (2013b). Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang L-Y, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. (2014a). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang L-Y, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. (2014b). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Lefkowitz RJ, and Rajagopal S (2018). Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov 17, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, and Insel PA (2018). G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol 93, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Gupta B, Gupta C, and Shukla AK (2015). Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol. Metab 26, 628–642. [DOI] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC, and Lefkowitz RJ (2018). Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in β-arrestin coupling. Proc. Natl. Acad. Sci. USA 115, 3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, and Skiniotis G (2020a). Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, and Skiniotis G (2020b). Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, and Kortemme T (2013). Improvements to robotics-inspired conformational sampling in rosetta. PLoS One 8, e63090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teller DC, Okada T, Behnke CA, Palczewski K, and Stenkamp RE (2001). Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry 40, 7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, et al. (2016). Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. USA 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, and Lambert NA (2018). Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem 293, 7466–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and Malcolm BA (1999). Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. BioTechniques 26, 680–682. [DOI] [PubMed] [Google Scholar]
- Xiao K, and Sun J (2018). Elucidating structural and molecular mechanisms of β-arrestin-biased agonism at GPCRs via MS-based proteomics. Cell Signal. 41, 56–64. [DOI] [PubMed] [Google Scholar]
- Xiao K, Shenoy SK, Nobles K, and Lefkowitz RJ (2004). Activation-dependent conformational changes in {beta}-arrestin 2. J. Biol. Chem 279, 55744–55753. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zhang L, Zhang XC, and Zhao Y (2017). Structural dynamics of Giα protein revealed by single molecule FRET. Biochem. Biophys. Res. Commun 491, 603–608. [DOI] [PubMed] [Google Scholar]
- Zhuang T, Vishnivetskiy SA, Gurevich VV, and Sanders CR (2010). Elucidation of inositol hexaphosphate and heparin interaction sites and conformational changes in arrestin-1 by solution nuclear magnetic resonance. Biochemistry 49, 10473–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo Y, Vishnivetskiy SA, Zhan X, Gurevich VV, and Klug CS (2014). Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem 289, 20991–21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Site-specific fluorophore labeling and surface tethering of β-arrestin1, related to Figure 1. A, Purified βarr1 “cys-less” background (left) and with cysteines introduced (E176C/ K397C, right) were analyzed with SDS-PAGE and imaged to detect LD555p fluorescence (top), LD655 fluorescence (middle), and Coomassie stain (bottom). B, Total fluorescence intensity monitored over time after fluorophore-labeled βarr1 was introduced into an imaging chamber prepared without (non-specific, red) and with (specific, black) StrepMAB-Immo antibody. C, Representative smFRET traces of βarr1 tail sensor from TIRF experiments performed in the absence (top) and presence (bottom) of 3 μM V2Rpp and recorded with 10 ms time resolution. D, Single-molecule confocal FRET efficiency histograms of freely diffusing βarr1 tail sensor alone (top) and in the presence of saturating concentrations of V2Rpp (middle) or heparin (bottom), fitted with Gaussian functions (colored areas). E, Ensemble average FRET efficiency and F, example smFRET traces from TIRF experiments in which 3 μM V2Rpp was added (vertical dotted lines; top row) or removed (bottom row) from solution during imaging.
Figure S2. Alternative labeling site for measuring β-arrestin1 C-tail dynamics, related to Figure 2. βarr1 E110C/K397C was purified and labeled with LD555p and LD655 and tethered to the imaging surface for smFRET in the absence and presence of varying concentrations of V2Rpp. A, FRET histograms, B, representative smFRET traces, and C, high FRET inactive state occupancy (red symbols) fit to a dose-response function with Hill slope of 1.0 and EC50 of 4.2 μM (red line). Data from βarr1 E176C/K397C labeling variant is shown for comparison (green).
Figure S3. β-arrestin1 complexes with V2Rpp and pB2V2R are stabilized by Fab30, related to Figures 2 and 5. A, Schematic of the experiment in which a complex was formed of βarr1 (gray), Fab30 (pink), and biotin-V2Rpp (orange) and tethered to a NeutrAvidin treated imaging surface. B, Population FRET histogram. C, transition density plot. D, Relative number of fluorescent βarr1 particles detected on the surface as a function of time (symbols) fit to a decay curve with a half-life (T1/2) of 33 min (line). E, Population FRET histograms of the inactive (red) and active (blue and green) states from experiments imaging βarr1 in the presence of 1 μM pβ2V2R and saturating concentrations of the indicated ligands and in the absence (top row) and presence (bottom row) of Fab30.
Figure S4. Supporting data for the constitutively activating mutants, related to Figure 4. A, Population FRET histograms of 3EI-βarr1 imaged at 10 ms camera integration time in the presence of the indicated concentrations of V2Rpp (left) and a representative FRET trace in the absence of V2Rpp (right). B, In the MD simulations, I386, V387, and F388 in the middle segment of the βarr1 tail are tightly packed with three Leu residues of the N-domain helix. In addition, three charged residues, E389, D390, and R393 form ionic interactions with residues from both the N terminus (R7 and K10) and the polar core (D26 and D297). Together, these interactions anchor the middle tail stably within the N-domain groove in the inactive state. C, In the MD simulations of the βarr1/3-EI mutation construct, the mutations, I386A/V387A/F338A, were found to be able to disrupt the hydrophobic interactions of these residues with the N-domain helix, including loss of the interaction with L108 entirely, resulting in an altered orientation of the middle βarr1 tail segment. This change propagated to allosterically perturb R393, which dissociated from the polar core and reoriented to interact with E389. Correspondingly, the E389-R7 and D390-K10 interactions are weakened. These reconfigurations in the middle loop affect the position of residue 397 and its distance from residue 176, likely underlying the altered basal FRET state in the 3EI-mutation. D, Example smFRET trace exhibiting low FRET efficiency from experiments imaging 3EI-βarr1 at 100 ms integration time. E, Rate constants derived from maximum likelihood model optimization using idealized smFRET traces from experiments imaging E313K-βarr1 in the presence of indicated V2Rpp concentrations (symbols); lines are linear fits with apparent V2Rpp on (blue) and off (red) rate constants of 0.17 μM−1 s−1 and 0.24 s−1, respectively. F, Overlay of dwell time histograms (symbols; lines are spline fits for clarity) from experiments in panel E with 1 μM V2Rpp and E313K (red) and wild type for comparison (black). G, FRET histograms (left) and representative traces (middle and right) from experiments imaging R76A,K77A,D78A-βarr1 with 100 ms camera integration time in the presence of the indicated concentrations of V2Rpp. Error bars, mean ± S.D. of two repeats.
Figure S5. MD simulations of β-arrestin1:receptor complexes, related to Figure 5. A, Representative MD frames of the simulated β2V2R-βarr1 complex model, showing the dynamics of the V2Rpp region between H8 of the receptor and the P2 position. The V2Rpp attached to H8 is in wheat color. Due to the strain from H8 as well as to steric clash with the finger loop from the β2V2R-βarr1 complex in its core-engagement orientation, the phosphorylated residues T346 (P1) and S349 (P2) cannot dock at the same locations in the extended groove as their aligned residues in the 4JQI structure. B, In the structure of βarr1 from the megacomplex (PDB 6NI2) (Nguyen et al., 2019), the V2Rpp peptide attached to H8 (green) shows a potentially different orientation before the P3 position compared to V2Rpp bound to βarr1 in the 4JQI structure (light pink). In the β2V2R-βarr1 complex, V2Rpp attached to H8 is likely in an orientation more similar to that of the 6NI2 structure. The black ribbons are the βarr1 from the simulated β2V2R-βarr1 complex of which the finger loop occupies or disrupts the P1 and P2 anchoring positions in the 4JQI structure. C, Representative MD frames of the β2V2R-βarr1 complex. Two distance constraints derived from DEER data (Zhuo et al., 2014), the 192–392 distance at 63 Å and the 12–392 distance at 55 Å would position 392 on rings encircling the intersection of N and C domains (yellow) of βarr1. Similarly, the Cα-Cα distances of the 176–397 and 110–397 pairs from the current smFRET study at 43.5 and 50 Å, respectively, would position 397 on another set of rings encircling the intersection (green). We inferred these Cα-Cα distances from their inter-dye distances, 63.5 and 69.9 Å, assuming a ~20 Å difference between the Cα-Cα and inter-dye distances. The close proximity of the 392 (yellow) and 397 (green) rings on the opposite side of the N-domain groove is insensitive to substantial variation (± 5 Å) of the estimated Cα-Cα distances. Notably, the pairs of points from the 392 and 397 rings that are 9 Å from each other, the estimated range of distance between the two residues, are indicated with larger spheres and connected by cyan lines. Such an intersection of the two sets of rings would position the βarr1 distal tail in proximity to IL3 of the receptor on the opposite side from the N-domain groove. This finding suggests a dramatic relocation of position 397 in the βarr1 distal tail from near the N-domain groove in the inactive state (indicated by the small dotted green circle in the inset) to the proximity of IL3 of the receptor in the active state. This relocation is indicated by green dotted arrows in both the main panel and the inset, and may be related, at least in part, to a potential electrostatic repulsion between the competing Rp tail and βarr tail, both of which are highly enriched with negatively charged residues.
Figure S6. Evaluating the stability of β-arrestin1:receptor complexes, related to Figure 5. A, Population FRET histograms from experiments imaging βarr1 in the presence of 1 μM pβ2V2R (left) and after exchanging to solution without receptor with a 5 min incubation (right) with 200 μM epi present throughout. B, Example trace after receptor washout. C, Population FRET histograms from experiments imaging βarr1 in the presence of 1 μM pβ2V2R (left) or bpβ2V2R (right) with 200 μM epi and 10 μM Cmpd-6FA. D, Diffusion times τD through the confocal volume of freely diffusing molecules from fluorescence intensity cross-correlation, G(τ), between donor and acceptor signals. A longer diffusion time is indicative of larger particles. Vertical error bars represent the standard deviation from 2 or more independent experiments. E, Representative normalized cross-correlation curves between donor and acceptor fluorescence emission intensity for βarr1 in the absence (light orange) and presence of 5 μM receptor with saturating concentrations of epi and Cmpd-6FA using pβ2V2R (orange) or bpβ2V2R (dark orange). F, As in panel C, but with the finger-loop proximal mutant E313-βarr1.
Figure S7. Supporting data for β-arrestin recruitment experiments in living cells, related to Figure 6. A, Amino acid sequence of the C-terminal region of receptors used in this study. The C-tail of receptors is shown in orange, and the palmitoylated cysteine residue is in bold. B, Relative surface expression of the indicated β2AR variants as determined by fluorescence intensity measurements of cells containing N-terminal SNAPfast-tagged receptors labeled with fluorescent dye. Bars represents the mean ± S. E. of three independent experiments performed in triplicate. Symbols represent the measured fluorescence intensity per the number of cells used for each measurement. C, Dose-response curves of mini-Gs recruitment to the plasma membrane by wildtype (WT) β2AR (black squares) and β2AR-365tr (blue triangles) in HEK293 GRK2/3/5/6 knockout cells in the (left plot) absence (GRK KO) or (right plot) presence of GRK2 expression via transient transfection (GRK2 rescue). Dose-response curves of four independent experiments (mean ± S. E.). D, Dose-response curves of βarr2 recruitment to the plasma membrane by the dopamine 2 receptor (D2R) in HEK293 GRK2/3/5/6 knockout cells in the absence (GRK KO) or presence of GRK2 expression via transient transfection (GRK2 rescue). Dose-response curves are of three independent experiments performed with triplicate samples (mean ± S. E.).