

Abstract
A novel analogue of psilocybin was produced by hybrid chemoenzymatic synthesis in sufficient quantity to enable bioassay. Utilizing purified 4-hydroxytryptamine kinase from Psilocybe cubensis, chemically synthesized 5-methylpsilocin (2) was enzymatically phosphorylated to provide 5-methylpsilocybin (1). The zwitterionic product was isolated from the enzymatic step with high purity utilizing a solvent–antisolvent precipitation approach. Subsequently, 1 was tested for psychedelic-like activity using the mouse head-twitch response assay, which indicated activity that was more potent than the psychedelic dimethyltryptamine, but less potent than that of psilocybin.
Graphical Abstract
The psychedelic natural product psilocybin has recently shown efficacy in phase II clinical trials for depression and anxiety, with effects that appear to be rapid and enduring.1–3 Psilocybin as a prodrug to psilocin has been proposed as a potential modulator of brain plasticity with the ability to modify functional connectivity between brain regions, providing potential therapeutic applications in many neuro-psychiatric diseases.4–6 While the mechanism of action of psilocybin and psychedelics in general remains unclear, activation of the serotonin 5-HT2A receptor subtype likely plays a key role in their effects in humans7–9 and other species.10 The potential for psilocybin analogues to similarly influence and enhance functional connectivity, stimulate neurogenesis, reduce inflammation, increase neural plasticity, or enhance cognition provides impetus to explore new therapeutic targets and is a compelling argument for further investigation into derivatives with similar structure to psilocybin.
Previous structure-activity relationship studies in humans with N,N-dialkyltryptamines have indicated that sterically small substitutions, such as hydroxy, methoxy, methylthio, or methylenedioxy, at the C4 and/or C5 positions of the indole core are typically tolerated while retaining psychedelic activity.11,12 A similar general trend has been observed in animal behavioral experiments.13–15 The compounds 5-methyl-N,N-dimethyltryptamine (5-Me-DMT), N,N-dimethyltryptamine (DMT), and psilocin were previously assayed in rats trained to discriminate the psychedelic 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT) from saline (Figure 1).16 5-Me-DMT substituted for 5-MeO-DMT with a median effective dose (ED50) of 2.32 μmol/kg and was approximately equipotent to psilocin (ED50: 2.36 μmol/kg) in the assay.While individual substitutions at either the C4 or C5 position of the N,N-dialkyltryptamine core have been explored for potential psychedelic-like activity, few examples exist where compounds with combined C4 and C5 substitutions were explored. Here we report a scalable chemoenzymatic route to a new 5-methylated derivative of psilocybin, 5-methylpsilocybin (1), and subsequently explore its activity in the mouse head-twitch response (HTR) assay, a 5-HT2A receptor-mediated behavior that serves as a behavioral proxy for psychedelic effects in humans.17
Figure 1.

Structural comparison of several known psychedelics bearing C4 or C5 substitutions with the prototypical psychedelic tryptamine N,N-dimethyltryptamine (DMT) and the corresponding novel 5-methylated analogues, 5-methylpsilocybin (1) and 5-methylpsilocin (2).
A unique property inherent to psilocin is its oral activity in humans, whereas DMT, the analogous unsubstituted tryptamine lacking C4 hydroxylation, does not demonstrate bioavailability when administered orally without a monoamine oxidase inhibitor.18 Despite their pharmacological utility, in vivo assays of 4-hydroxylated tryptamines may be complicated by their chemical instability and tendency toward oxidation with formation of oligomeric degradants.19 The C4 hydroxy group on these compounds is therefore frequently modified to provide the corresponding prodrugs bearing a metabolically labile phosphate or acetate substitution.20 Aphosphorylated prodrug tryptamine, such as psilocybin, is particularly attractive because the resulting chemically stable polar zwitterionic species is not expected to cross the blood–brain barrier with intraperitoneal administration or absorb and distribute into the bloodstream prior to dephosphorylation with oral administration, whereas other nonzwitterionic prodrugs, such as acetates, may potentially introduce multiple central nervous system (CNS)-active drugs, complicating their study in vivo.
The advantages of hybrid chemoenzymatic approaches to complex natural products have recently become recognized, especially when applied to late-stage modifications where the regioselectivity and efficiency of enzymes may be particularly useful.21 While the synthesis of 4-hydroxylated tryptamines is relatively straightforward, the subsequent 4-O-phosphorylation step has been challenging to accomplish by chemical synthesis until recently.22 Therefore, we sought to apply a hybrid synthetic/biocatalytic procedure developed for psilocybin gram-scale production.23 The process circumvents chemical phosphorylation by an enzymatic transfer with adenosine triphosphate (ATP), catalyzed by the 4-hydroxytryptamine kinase (PsiK) of the psilocybin biosynthesis intrinsic to Psilocybe cubensis.24 Prior work to produce psilocybin congeners 6-methylbaeocystin and 7-phosphoryloxytryptamine enzymatically pointed to PsiK as a somewhat flexible enzyme.25,26 Therefore, a small-scale reaction was set up first to test if 5-methylpsilocin (2) served as a phosphate acceptor substrate and could be converted to 1.
To supply the enzymatic phosphotransfer reaction with the phosphate acceptor substrate, 2 was synthesized (Scheme 1). Acetyl protection of commercial 5-methyl-1H-indol-4-ol with acetic anhydride and NaHCO3 in toluene provided 5-methyl-1H-indol-4-ol acetate (3) in 97% yield and was accomplished using a described general method.27 To complete the synthesis of 2, another two steps were required that followed a published protocol based on a Speeter-Anthony tryptamine synthesis in modified form.28,29 Indole 3 was reacted with oxalyl chloride in tetrahydrofuran (THF) overnight followed by direct amide formation by addition of excess dimethylamine to provide ketoamide 4, which, after purification, was reduced by lithium aluminum hydride to provide 2 in 84% yield.
Scheme 1.
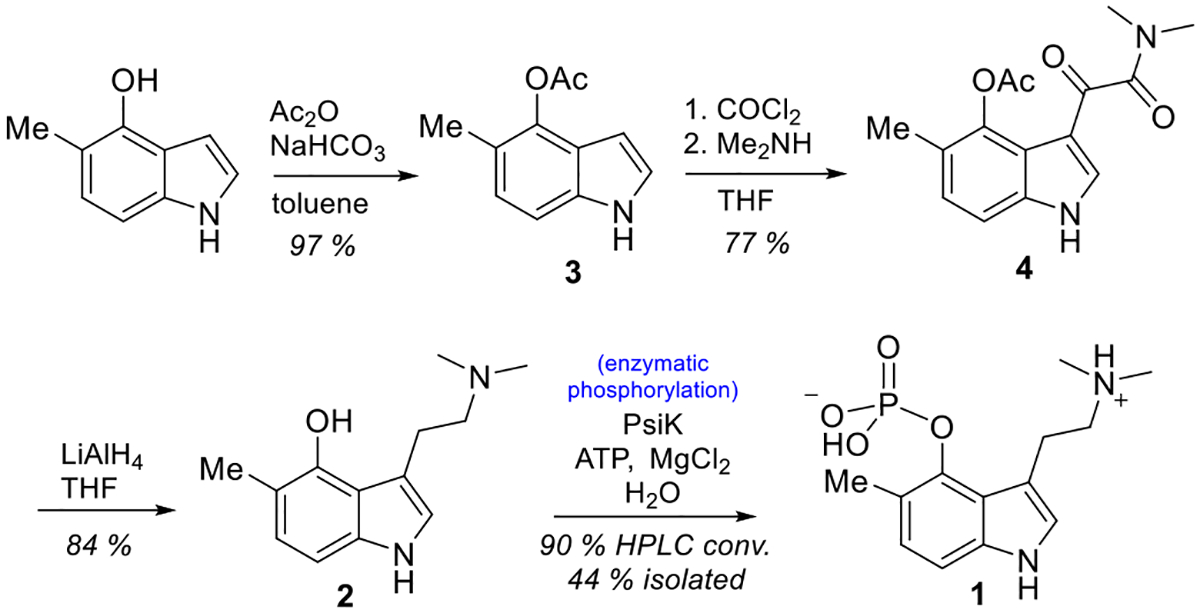
Speeter-Anthony Tryptamine Synthesis of 2 and Chemoenzymatic Phosphorylation to 1
Chemically synthesized 2 was incubated with purified PsiK kinase and ATP in 100 μL reactions. In parallel controls, heat-denatured enzyme was used. LC/ESIMS analysis of the enzymatic reaction showed a new peak at tR = 2.8 min (Figure 2A). Its UV–vis spectrum was near-identical to that of psilocybin, but its mass was determined to be m/z 299.1155 [M + H]+, which is consistent with a methylated psilocybin (Figure 2B). The new signal was absent in the chromatograms of the control reactions employing heat-denatured enzyme. Subsequently, in a scaled-up reaction 49.7 mg of 2 was used as substrate. After 16 h, about 90% conversion was observed according to UHPLC peak areas. The reaction was lyophilized, and a portion of the residue was used to purify the new compound by semipreparative HPLC, which yielded 2 mg of analytical reference compound. Subsequent 1D and 2D NMR spectroscopy verified that 1 had been produced. Compared to previously reported NMR spectra for psilocybin,20,22,28,30 the spectra for 1 were nearly identical, but marked by an additional carbon-13 signal (δ 15.8 ppm) and an additional proton signal (δ 2.36), which integrated to 3H, both consistent with the presence of a C5 aromatic-substituted methyl group on a psilocybin core. Additionally, HMBC provided correlations between the novel 5-methyl group and neighboring aromatic carbons on the indole core (Table 1, Figures S1–S4).
Figure 2.
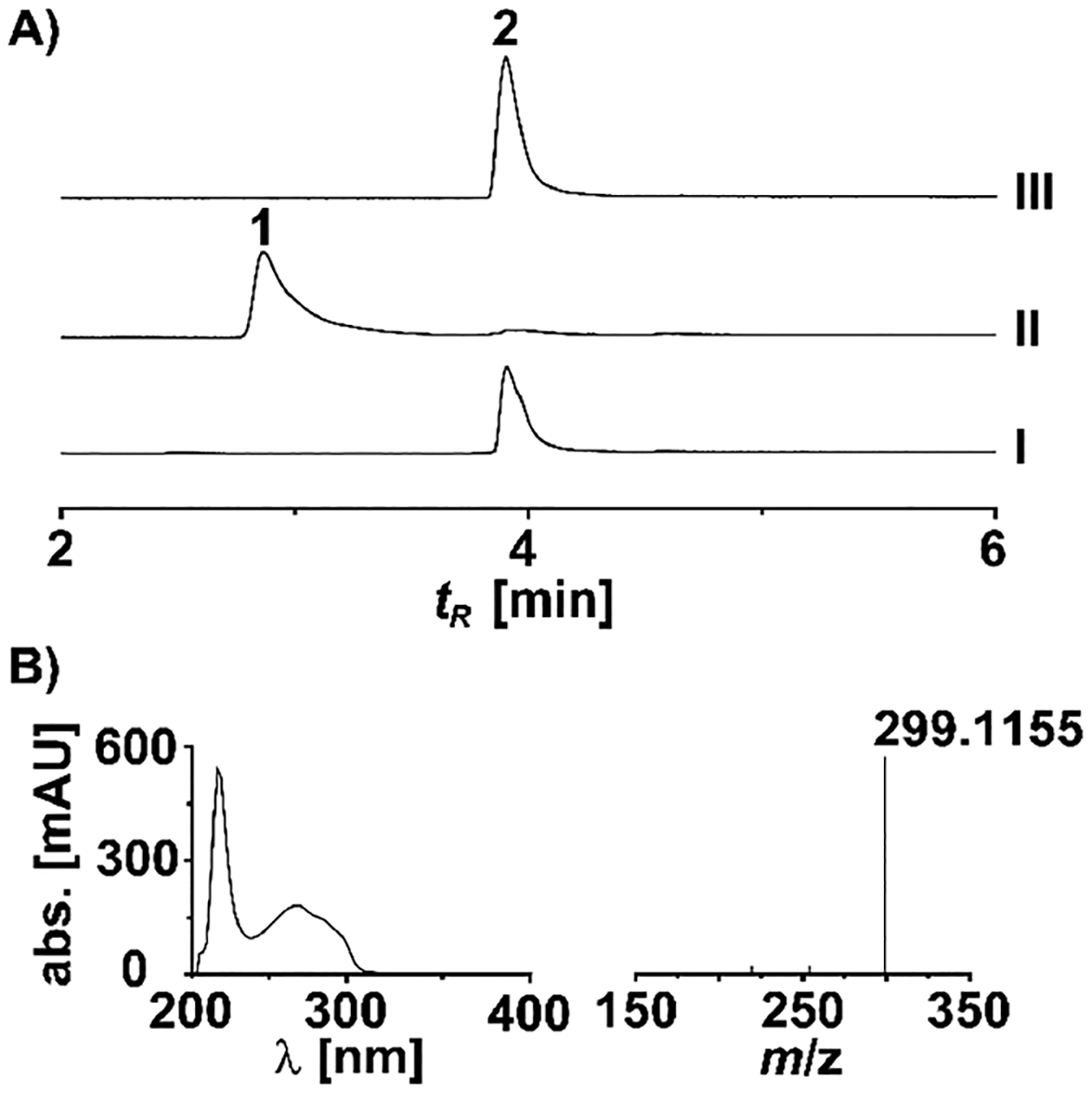
Chromatographic analysis of biocatalytically produced 1. (A) UHPLC traces of the negative control with heat-inactivated enzyme (trace I), the PsiK-catalyzed in vitro reaction (trace II), and a standard of substrate 2 (trace III). Chromatograms were extracted at λ = 280 nm. (B) UV–vis and HRESIMS spectra of 1.
Table 1.
NMR Spectroscopic Data for 5-Methylpsilocybin (1)a
| no. | δC, mult | δH, M (J in Hz) | HMBC |
|---|---|---|---|
| 1 | |||
| 2 | 124.4, CH | 7.12, s | 3, 3a, 7a |
| 3 | 108.0, C | ||
| 3a | 119.8, C | ||
| 4 | 142.8, C | ||
| 5 | 120.2, C | 3a, 7 | |
| 5-CH3 | 15.8 | 2.36, s | 4, 5, 6 |
| 6 | 125.5, CH | 7.01, d (8.4) | 4, 5-CH3, 7a |
| 7 | 108.5, CH | 7.16, d (8.4) | 3a, 5 |
| 7a | 137.3, C | ||
| 1′ | 59.1, CH2 | 3.34 m | 2, 2′, 3a |
| 2′ | 21.6, CH2 | 3.34 m | 1′, 3, N-(CH3)2 |
| N-(CH3)2 | 42.7 | 2.82, s | 1′ |
| N-H | n.o.b |
D2O, 500 MHz for 1H NMR, 125 MHz for 13C NMR.
not observed.
Preparative HPLC was suitable for the purification of small amounts of zwitterionic 1; however, for subsequent scale-up to provide sufficient material for in vivo studies, chromatographic purification was avoided. Traditional approaches, such as liquid–liquid extraction, for the purification of zwitterionic 1 were not suitable, as its aqueous solubility and physical properties are highly variable as a function of pH. Therefore, a nonaqueous solvent-based purification to precipitate the isoelectric solid was desirable.
The remaining lyophilized crude reaction mixture containing product 1, residual protein, adenosine monophosphate (AMP), MgCl2, and trace nonphosphorylated small-molecule impurities including 2 was used to explore a solvent/antisolvent-based precipitation approach to isolate purified 1 with tracking of the mixture components by UHPLC/UV/MS (Figure S11). Briefly, sequential use of methanol, acetonitrile (ACN), and then ethanol was found to leverage the differential solubilities of the individual reaction components (a summary of observed solubility properties is provided in Table 2). With the optimized procedure, ACN was added to a methanol solution of the crude enzymatic reaction residue, which precipitated a solid consisting of protein, AMP, and a small amount of 1, and the resulting suspension was then centrifuged. The methanol/ACN supernatant was found to be enriched in product 1 and free from AMP contamination. The supernatant was concentrated and then slurried in ethanol to wash out a small amount of 2, residual MgCl2, and other minor late-eluting impurities to provide a white solid. The suspension was filtered and dried to initially provide 22 mg of purified 1 (98% peak area by UHPLC, Figure S11). This process was repeated on the precipitated solid pellet from the first step to isolate an additional 8 mg of 1 (>95% peak area by UPLC, Figure S11) to give a combined 44% isolated recovery after drying. The isolated solid proved to be spectroscopically consistent with the material purified by preparative HPLC. Although the recovered yield from the process (44%) was lower than UHPLC product conversion (90%), some losses were attributed to transfers and accumulation given the relatively small scale of the process. Additionally, with future larger scale reactions we envisage that a final recrystallization of the isolated solid may be possible to further increase product purity and uniformity.
Table 2.
Summary of Crude Chemoenzymatic Reaction Components and Their Observed Properties with Select Solvents
| component/solvent | methanol | acetonitrile | ethanol |
|---|---|---|---|
| 1 | solvent | partial antisolvent | antisolvent |
| AMP | solvent | antisolvent | n/a |
| protein residue | solvent | antisolvent | n/a |
| 2, minor impurities | solvent | solvent | solvent |
| MgCl2 | solvent | solvent | solvent |
To determine whether compound 1 induces the HTR, purified material was administered to mice. In the experiment, head movement was recorded using a head-mounted magnet and a magnetometer coil,31 and then head-twitches were detected in the recordings using a published method based on artificial intelligence.32 Intraperitoneal administration of 1 produced a dose-dependent increase in head-twitch counts (F(4,20) = 9.61, p = 0.0002), with an ED50 of 1.23 (95% CI 0.76–1.98) mg/kg. Based on molar mass, the ED50 for 1 was equivalent to 4.11 μmol/kg. As positive controls, the known psychedelics DMT and psilocybin induced the HTR with ED50 values of 5.05 μmol/kg17 and 1.40 μmol/kg,33 respectively, when tested using identical methods by our laboratory. In summary, activity in the HTR assay was retained when psilocybin was methylated at the C5 position but there was a 3-fold reduction in potency. These data suggest that small substitutions at the C5 position of 4-hydroxylated tryptamines may be tolerated, retaining psychedelic activity, although an overall reduction of in vivo potency may be observed. Additional studies are required to determine whether disubstitution at the C4 and C5 positions reduces the potency of tryptamines at the receptor level. Alternatively, since psilocybin acts as a prodrug, adding a C5 methyl group could potentially reduce the potency of psilocybin by altering its enzymatic dephosphorylation.
In summary, the novel C4–C5-disubstituted tryptamine 1 was efficiently synthesized by leveraging the enzymatic flexibility of P. cubensis kinase, PsiK, in the final step of the reaction sequence. The utility of hybrid chemoenzymatic synthesis was demonstrated by providing sufficient material to enable in vivo bioassay of 1 in rodents. The in vivo data supported the hypothesis that combined C4–C5-substituted tryptamines would retain psychedelic-like activity. Subsequent studies should examine additional substrates amenable to phosphorylation by PsiK as well as psychedelic structure–activity relationships with combined C4 and C5 disubstituted tryptamines.
EXPERIMENTAL SECTION
General Experimental Procedures.
To record 1D and 2D NMR spectra, 1 was dissolved in D2O. 1H and 13C chemical shifts were referenced relative to residual nondeuterated solvent traces (water and MeOH) at δH 4.71 and 48.9 ppm, respectively. NMR spectra were recorded on Bruker Avance III 500 and 600 MHz spectrometers, at 300 K. Semipreparative HPLC of 1 was performed using an Agilent Infinity 1260 chromatograph equipped with a ThermoFisher Hypercarb column (150 × 10 mm, 5 μm particle size). For UHPLC/MS, an Agilent Infinity II 1290 instrument, interfaced with an Agilent 6130 mass detector and fitted to a Phenomenex Luna Omega Polar C18 100 A column (100 × 2.1 mm, 1.6 μm), was used. HRESIMS data were recorded with a Thermo Fisher Accela chromatograph with a Grom-Sil 100 ODS-0 AB (250 × 4.6 mm, 3 μm) C18 column and an Exactive Orbitrap mass spectrometer, operated in positive ESI mode. Chromatograms were recorded at λ = 280 nm. Solvent-based purification process development was performed with a Waters Acquity I-Class UHPLC utilizing a Waters HSS T3 column (2.5 μm, 2.1 mm × 30 mm) run in gradient mode with H2O (0.1% formic acid, FA) and ACN (0.1% FA) mobile phases at 0.6 mL/min. Samples were diluted in ACN or water to approximately 1 mg/mL, and 0.1 μL was injected. Chromatographic peaks were detected by a diode array detector at 269 nm. High-resolution mass spectra were acquired in line with UV detection on a Waters Xevo G2-XS QTof in ESI-positive mode. Low and high collision energy mass spectra were acquired using a Waters MSe experiment. Reagents, buffer components, and solvents were purchased from Deutero, Sigma-Aldrich, Roth, and VWR.
Heterologous Enzyme Production.
Production of the P. cubensis kinase PsiK followed a published procedure.23 The enzyme was stored at −80 °C. Prior to use, the protein was rebuffered, and glycerol was removed by gel filtration chromatography, using an ÄktaPure FPLC (GE Healthcare) equipped with a Superdex 200 Increase 10/300 GL column. The eluent was reaction buffer (50 mM NaH2PO4, pH 7). The protein concentration was determined by Bradford’s assay.34
Synthesis of 2 and Its Precursors.
5-Methyl-1H-indol-4-ol Acetate (3).
The product was synthesized from 5-methyl-1H-indole-4-ol (500 mg, 3.4 mmol) analogously to Lugemwa et al.27 in dry toluene to provide an off-white solid (623 mg, 97%): mp 145–146 °C; 1H NMR (acetone-d6, 600 MHz) δ 10.26 (1H, br, NH), 7.26 (1H, dd, J1 = J2 = 2.7 Hz, H-2), 7.22 (1H, d, J = 8.2 Hz, H-7), 6.97 (1H, d, J = 8.2 Hz, H-6), 6.33 (1H, m, H-3), 2.36 (3H, s, 4-OAc), 2.20 (3H, s, 5-CH3); 13C NMR (acetone-d6, 150 MHz) δ 169.5, 143.1, 138.1, 126.3, 125.3, 123.6, 120.3, 110.30, 99.4, 21.1, 16.2 (Figures S5 and S6); HRESIMS m/z 190.08597 [M + H]+ (calcd for C11H12NO2+ 190.08626).
5-Methyl-3-(2-dimethylamino-2-oxoacetyl)-1H-indol-4-yl Acetate (4).
Indole 3 (100 mg, 0.53 mmol) dissolved in dry THF (1.75 mL) was added dropwise under an inert atmosphere to a stirred solution of (COCl)2 (200 μL, 2.33 mmol, 4.4 equiv) in dry THF (400 μL), cooled on ice. After stirring for 16 h at 0 °C, the reaction was diluted with dry THF (2 mL), and dimethylamine (2 M solution in THF) was added very slowly under continued cooling, until moist pH paper gave a basic reaction. The volatile components were removed under reduced pressure, and the residue was purified by silica column chromatography (CH2Cl2/MeOH, 40:1 to 20:1) to provide 4 as a white solid (118 mg, 77%): mp 215–216 °C; 1H NMR (acetone-d6, 600 MHz) δ 11.42 (1H, s br, NH), 8.02 (1H, s, H-2), 7.35 (1H, d, J = 8.2 Hz, H-7), 7.18 (1H, d, J = 8.2 Hz, H-6), 3.00 (3H, s, N-CH3) 2.94 (3H, s, N-CH3), 2.42 (3H, s, 4-OAc) 2.26 (3H, s, 5-CH3); 13C NMR (acetone-d6, 150 MHz) δ 187.4, 170.1, 169.3, 144.0, 139.6, 139.4, 127.8, 124.9, 120.3, 114.8, 111.2, 37.6, 34.2, 21.8, 16.6 (Figures S7 and S8); HRESIMS m/z 289.11775 [M + H]+ (calcd for C15H17N2O4+ 289.11828).
5-Methylpsilocin (2).
Ketoamide 4 (117 mg, 0.41 mmol) was reduced by lithium aluminum hydride (1.23 mmol) by adapting the published method developed for psilocin.28 After quenching (“Procedure B” of Sherwood et al.,28 200 μL of THF/H2O, 27:100), anhydrous NaSO4 (400 mg), silica gel (200 mg), and CH2Cl2 (1.2 mL) were added. Solids were filtered off and washed with 4.5 mL of CH2Cl2/MeOH, 9:1. Combined fractions yielded a gray solid after solvent evaporation (74 mg, 84%): mp 156–159 °C (dec); 1H NMR (acetone-d6, 600 MHz) δ 12.13 (1H, br, 4-OH), 9.67 (1H, br, NH), 6.91 (1H, s, H-2), 6.79 (1H, d, J = 8.1 Hz, H-6) 6.72 (1H, d, J = 8.1 Hz, H-7), 2.93 (2H, m, CH2), 2.70 (2H, m, CH2), 2.35 (6H, s, N(CH3)2) 2.23 (3H, s, 5-CH3); 13C NMR (acetone-d6, 150 MHz) δ 150.7, 139.7, 126.2, 122.9, 122.7, 119.1, 113.8, 103.3, 63.2, 46.0, 26.3, 16.7 (Figures S9 and S10); HRESIMS m/z 219.14926 [M + H]+ (calcd for C13H19N2O+ 219.14919).
5-Methylpsilocybin (1).
Small-scale assays (4 mM ATP, 4 mM MgCl2, 6 mM β-mercaptoethanol, 2 mM acceptor substrate 2, 8 μM PsiK in reaction buffer) were set up in triplicate and in a total volume of 100 μL to evaluate the enzymatic phosphorylation of 2. Incubation was for 16 h at 37 °C. As negative control, heat-inactivated PsiK was used in a parallel reaction. Subsequently, the assays were lyophilized, and the dry residue was dissolved in 100 μL of MeOH, centrifuged (10000g, 10 min, 4 °C), filtered, and analyzed chromatographically by UHPLC/MS and LC/HRMS-MS (below). To biocatalytically produce multi-milligram amounts of 1, the assay volume was scaled up to 50 mL. Intermediate 2 (50 mg, 0.23 mmol) was incubated as described above, but with gentle shaking. The reaction was lyophilized, the residue was dissolved in 50 mL of MeOH and evaporated under reduced pressure, and a portion of the residue was used to purify the new compound by semipreparative HPLC (below). The remaining crude residue (80 mg) was dissolved in water (200 μL); then NaOH (0.1M) was added dropwise to adjust the solution to pH 4–5. The solvent was removed under reduced pressure, and the residue was dissolved in methanol (0.5 mL) and then transferred to a 2 mL microcentrifuge tube. ACN (1.5 mL) was added to form a white precipitate. The suspension was centrifuged (2000g, 5 min) and the supernatant was collected. The solid pellet was redissolved in methanol, and the precipitation process with ACN was repeated twice more. The remaining solid pellet was analyzed by UHPLC and found to contain primarily AMP and 1. UHPLC analysis of the pooled supernatants revealed product 1 absent the AMP peak with several late-eluting byproducts. The supernatant was concentrated to provide a brown residue, which was sonicated in ethanol (1 mL) for 5 min to provide a finely suspended white precipitate. The suspension was cooled by an external ice bath and then centrifuged (2000g, 5 min). The brown-colored supernatant containing 2 and several unidentified late-eluting impurities was removed and discarded. The solid pellet was dried under a gentle stream of nitrogen followed by reduced pressure to provide 1 (22 mg) with 98% peak area by UHPLC. The solid pellet precipitated in the first step was reprocessed using the same procedure to provide an additional 8 mg of 1 as a white solid with 95% peak area by UHPLC: mp 198–200 °C; spectroscopic properties of combined isolated bulk material consistent with material purified by preparative HPLC; UV (H2O/ACN, 4:1) λmax (log ε) 3.00 (280 nm); 1H and 13C NMR data (D2O, 500 and 125 MHz, respectively), see Table 1; HRESIMS m/z 299.1155 [M + H]+ (calcd for C13H19N2O4P+ 299.1155).
Liquid Chromatography and Mass Spectrometry.
UHPLC/ESIMS was performed with 0.1% (v/v) FA in water (solvent A) and ACN (solvent B) under described conditions and settings.35 For LC/HRESIMS, ionization was in positive mode. Solvent A was 0.1% (v/v) FA in water, and solvent B was ACN, at a flow of 1 mL/min. After an initial hold at 5% B for 1 min, a linear gradient to 100% B within 15 min was applied. Semipreparative HPLC was carried out with 0.1% (v/v) trifluoroacetic acid (TFA) in water (solvent A) and 0.1% (v/v) TFA in ACN (solvent B) at a flow of 3 mL/min. The gradient began at 10% B, followed by a linear increase to 30% B within 1 min, to 60% B within a further 9 min, and to 100% B within another 1 min. Fractions containing 1 were combined and dried under reduced pressure.
Assessment of the Head-Twitch Response.
The head-twitch response was assessed using a head-mounted magnet and a magnetometer detection coil.31 HTR experiments were conducted in a well-lit room. Mice (n = 5/group) were injected intraperitoneally (IP, 5 mL/kg) with drug or vehicle (saline), and then head movement was recorded in a glass cylinder surrounded by a magnetometer coil for 30 min. Coil voltage was low-pass filtered (2–10 kHz cutoff frequency), amplified, digitized (20 kHz sampling rate), and saved to a disk using a Powerlab/8SP with LabChart v 7.3.2 (ADInstruments, Colorado Springs, CO, USA). To detect head twitches, events in the recordings were transformed into a visual representation in the time–frequency domain (a scalogram), deep features are extracted using the pretrained convolutional neural network ResNet-50, and then the images were classified using a Support Vector Machine algorithm.32 Total HTR counts were analyzed using a one-way ANOVA. Post hoc pairwise comparisons between selected groups were performed using Tukey’s studentized range method. The median effective dose (ED50) and 95% confidence interval (95% CI) were calculated by nonlinear regression (Prism 7.00, GraphPad Software, San Diego, CA, USA).
Supplementary Material
Figure 3.
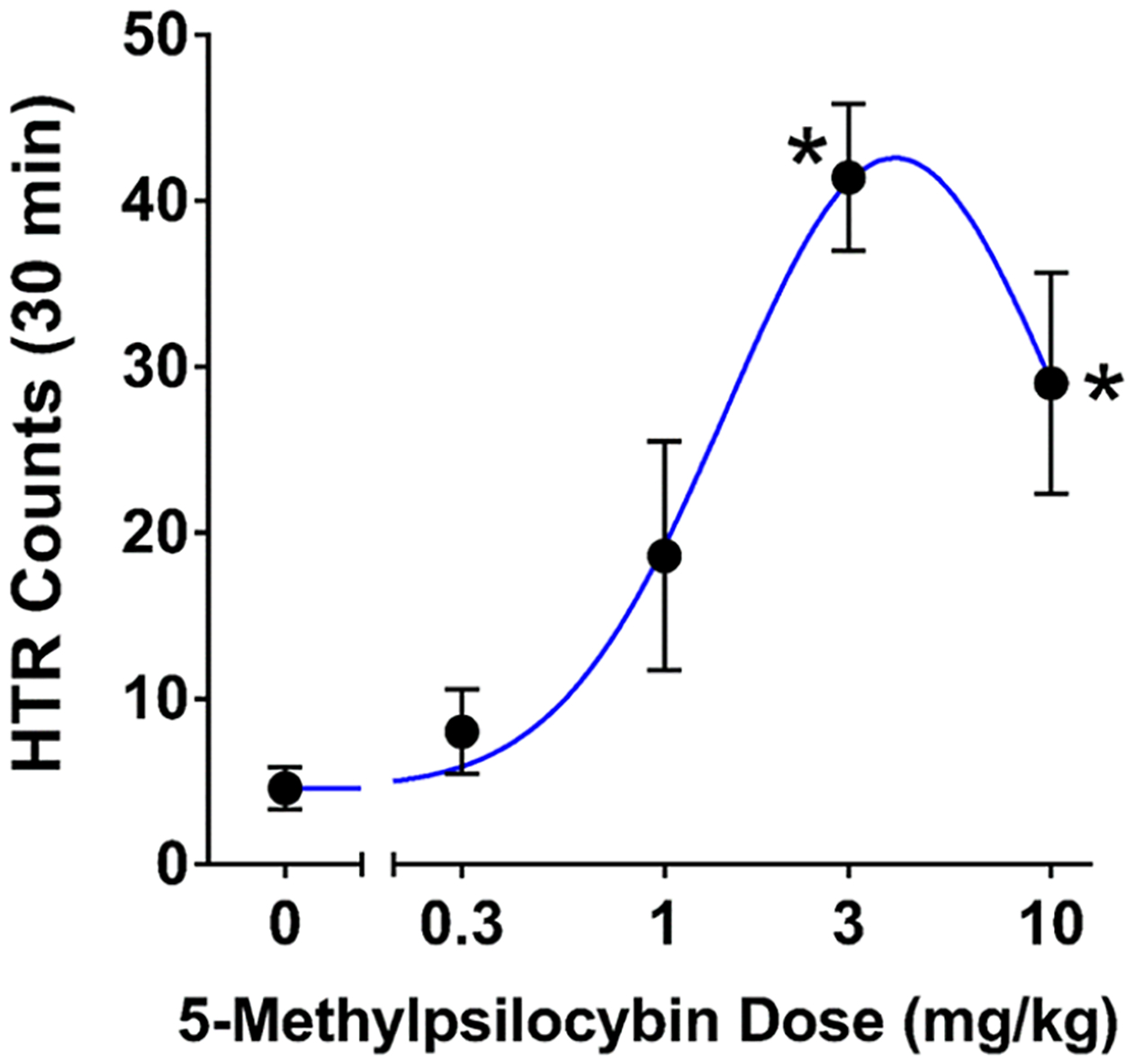
Effect of compound 1 in the mouse head-twitch response (HTR) assay. Mice were injected intraperitoneally, and then HTR behavior was assessed continuously for 30 min. Data are the mean ± standard error of the mean of HTR counts over the entire test session. *p < 0.01 vs vehicle, Dunnett’s test.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, grant HO2515/7-1 to D.H.), the National Institute on Drug Abuse (NIDA, award R01 DA041336 to A.L.H.), and the Usona Institute (Madison, WI, USA). We gratefully acknowledge A. Perner, H. Heinecke, C. Lenz (all Hans Knöll Institute, Jena, Germany), and J. Kimball (Promega Biosciences) for recording high-resolution mass spectra and NMR spectra, additional synthesis and purification of 5-methylpsilocybin, and additional analytical support, respectively. We extend gratitude to P. Stamets for generously providing the image of Psilocybe cubensis used in the TOC graphic and to K. Kaylo for preparing the manuscript.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.1c00087
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.1c00087.
1D and 2D NMR spectra of 5-methylpsilocybin (1), 1D spectra of 5-methylpsilocin (2), 5-methyl-1H-indol-4-ol acetate (3), and 5-methyl-3-(2-dimethylamino-2-oxo acetyl)-1H-indol-4-yl acetate (4), and UPLC chromatograms for isolated 1 (PDF)
The authors declare no competing financial interest.
Contributor Information
Janis Fricke, Department Pharmaceutical Microbiology at the Hans-Knöll-Institute, Friedrich-Schiller-Universität Jena, 07745 Jena, Germany.
Alexander M. Sherwood, Usona Institute, Madison, Wisconsin 53711, United States.
Adam L. Halberstadt, Department of Psychiatry, University of California San Diego, La Jolla, California 92093-0804, United States; Research Service, VA San Diego Healthcare System, San Diego, California 92161, United States.
Robert B. Kargbo, Usona Institute, Madison, Wisconsin 53711, United States
Dirk Hoffmeister, Department Pharmaceutical Microbiology at the Hans-Knöll-Institute, Friedrich-Schiller-Universität Jena, 07745 Jena, Germany.
REFERENCES
- (1).Griffiths RR; Johnson MW; Carducci MA; Umbricht A; Richards WA; Richards BD; Cosimano MP; Klinedinst MA J. Psychopharmacol 2016, 30, 1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ross S; Bossis A; Guss J; Agin-Liebes G; Malone T; Cohen B; Mennenga SE; Belser A; Kalliontzi K; Babb J; Su Z; Corby P; Schmidt BL J. Psychopharmacol 2016, 30, 1165–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Carhart-Harris RL; Bolstridge M; Day CMJ; Rucker J; Watts R; Erritzoe DE; Kaelen M; Giribaldi B; Bloomfield M; Pilling S; Rickard JA; Forbes B; Feilding A; Taylor D; Curran HV; Nutt DJ Psychopharmacology. 2018, 235, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nutt D; Erritzoe D; Carhart-Harris R Cell 2020, 181, 24–28. [DOI] [PubMed] [Google Scholar]
- (5).dos Santos RG; Bouso JC; Alcázar-Córcoles MÁ; Hallak JEC Expert Rev. Clin. Pharmacol 2018, 11, 889–902. [DOI] [PubMed] [Google Scholar]
- (6).Nichols DE Pharmacol. Rev 2016, 68, 264–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Preller KH; Burt JB; Ji JL; Schleifer CH; Adkinson BD; Stämpfli P; Seifritz E; Repovs G; Krystal JH; Murray JD; Vollenweider FX; Anticevic A eLife 2018, 7, e35082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Barrett FS; Preller KH; Herdener M; Janata P; Vollenweider FX Cereb. Cortex 2018, 28, 3939–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kometer M; Schmidt A; Jäncke L; Vollenweider FX J. Neurosci 2013, 33, 10544–10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Halberstadt AL; Geyer MA In Behavioral Neurobiology of Psychedelic Drugs; Springer-Verlag: Berlin, Heidelberg, 2018; Vol. 36, pp 159–199. [Google Scholar]
- (11).Repke DB; Grotjahn DB; Shulgin AT J. Med. Chem 1985, 28, 892–896. [DOI] [PubMed] [Google Scholar]
- (12).Shulgin A; Shulgin A TIHKAL: The Continuation; Transform Press: Berkeley, CA, 1997. [Google Scholar]
- (13).Gatch MB; Hoch A; Carbonaro TM ACS Pharmacol. Transl. Sci 2020, DOI: 10.1021/acsptsci.0c00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Klein LM; Cozzi NV; Daley PF; Brandt SD; Halberstadt AL Neuropharmacology 2018, 142, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Winter JC; Rice KC; Amorosi DJ; Rabin RA Pharmacol., Biochem. Behav 2007, 87, 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Glennon RA; Young R; Rosecrans JA; Kallman MJ Psychopharmacology. 1980, 68, 155–158. [DOI] [PubMed] [Google Scholar]
- (17).Halberstadt AL; Chatha M; Klein AK; Wallach J; Brandt SD Neuropharmacology 2020, 167, 107933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lenz C; Sherwood A; Kargbo R; Hoffmeister D ChemPlusChem 2021, 86, 28–35. [DOI] [PubMed] [Google Scholar]
- (19).Lenz C; Wick J; Braga D; García-Altares M; Lackner G; Hertweck C; Gressler M; Hoffmeister D Angew. Chem., Int. Ed 2020, 59, 1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nichols DE; Frescas S Synthesis 1999, 1999, 935–938. [Google Scholar]
- (21).Li J; Amatuni A; Renata H Curr. Opin. Chem. Biol 2020, 55, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kargbo RB; Sherwood A; Walker A; Cozzi NV; Dagger RE; Sable J; O’Hern K; Kaylo K; Patterson T; Tarpley G; Meisenheimer P ACS Omega. 2020, 5, 16959–16966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fricke J; Kargbo R; Regestein L; Lenz C; Peschel G; Rosenbaum MA; Sherwood A; Hoffmeister D Chem. - Eur. J 2020, 26, 8281–8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fricke J; Blei F; Hoffmeister D Angew. Chem., Int. Ed 2017, 56, 12352–12355. [DOI] [PubMed] [Google Scholar]
- (25).Blei F; Baldeweg F; Fricke J; Hoffmeister D Chem. - Eur. J 2018, 24, 10028–10031. [DOI] [PubMed] [Google Scholar]
- (26).Fricke J; Sherwood A; Kargbo R; Orry A; Blei F; Naschberger A; Rupp B; Hoffmeister D ChemBioChem 2019, 20, 2824–2829. [DOI] [PubMed] [Google Scholar]
- (27).Lugemwa FN; Shaikh K; Hochstedt E Catalysts 2013, 3, 954–965. [Google Scholar]
- (28).Sherwood AM; Meisenheimer P; Tarpley G; Kargbo RB Synthesis 2020, 52, 688–694. [Google Scholar]
- (29).Speeter ME; Anthony WC J. Am. Chem. Soc 1954, 76, 6208–6210. [Google Scholar]
- (30).Shirota O; Hakamata W; Goda Y J. Nat. Prod 2003, 66, 885–887. [DOI] [PubMed] [Google Scholar]
- (31).Halberstadt AL; Geyer MA Psychopharmacology. 2013, 227, 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Halberstadt AL Sci. Rep 2020, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sherwood AM; Halberstadt AL; Klein AK; McCorvy JD; Kaylo KW; Kargbo RB; Meisenheimer P J. Nat. Prod 2020, 83, 461–467. [DOI] [PubMed] [Google Scholar]
- (34).Bradford MA Anal. Biochem 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- (35).Demmler R; Fricke J; Dörner S; Gressler M; Hoffmeister D ChemBioChem 2020, 21, 1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.