

Abstract
The human Son of Sevenless (SOS) activates the signal-transduction protein Ras by forming the complex SOS·Ras and accelerating the GTP exchange in Ras. Inhibition of SOS·Ras could regulate the function of Ras in cells and has emerged as an effective strategy for battling Ras related cancers. A key factor to the success of this approach is to understand the conformational change of Ras during the GTP exchange process. In this study, we perform an extensive molecular dynamics simulation to characterize the specific conformations of Ras without and with guanine nucleotide exchange factors (GEFs) of SOS, especially for the substates of State 1 of HRasGTP·Mg2+. The potent binding pockets on the surfaces of the RasGDP·Mg2+, the S1.1 and S1.2 substates in State 1 of RasGTP·Mg2+ and the ternary complexes with SOS are predicted, including the binding sites of other domains of SOS. These findings help to obtain a more thorough understanding of Ras functions in the GTP cycling process and provide a structural foundation for future drug design.
Keywords: conformations, Ras complexes, binding pockets, REMD simulation
Graphical Abstract
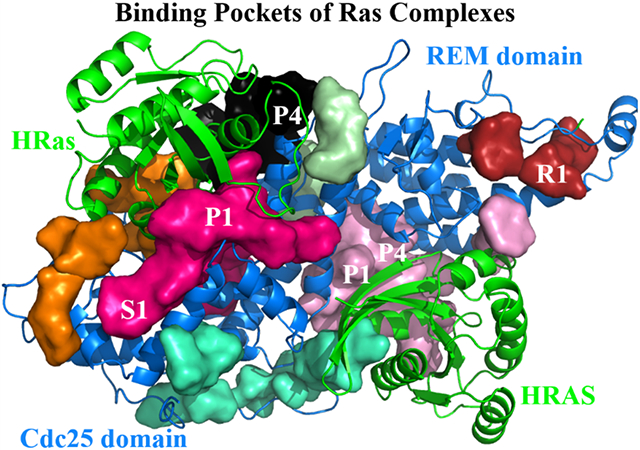
The catalytic HRas, the allosteric HRAS and the SOS domains in the Ras complexes possess a number of binding pockets of P1, P4, S1 and R1 for small molecules.
Introduction
The small GTPase Ras1-4 regulates the vital cellular processes of proliferation, growth and differentiation in signaling pathways. In cells, an inactive Ras complexes with the guanosine diphosphate (GDP) and Mg2+ to form the complex RasGDP·Mg2+.5 The activation of RasGDP·Mg2+ is regulated by the guanine nucleotide exchange factors (GEFs)6. They accelerate the GDP exchange with the guanosine triphosphate (GTP) in plasma to yield the pre-hydrolysis state RasGTP·Mg2+.7 The active conformation of RasGTP·Mg2+ interacts with effectors such as Raf to transmit the signals downstream. Malfunction of Ras often leads to various tumor-related cancers.8 So far, 19% tumor-caused diseases8-10 in human are related to the three Ras isoforms of KRas, HRas and NRas.
The family of Ras-specific GEFs includes a diverse set of homologous domains such as CDC2511 and SDC2512. The human Son of Sevenless (SOS)13 is a multi-domain protein with a catalytic module (residues 550-1050) composed of a Ras exchange motif (REM) (residues 550-750) and a CDC25 homology domain (residues 750-1050) of GEFs. In 1998, Boriack-Sjodin et al.14 reported the crystal structure (PDB: 1BKD) of the nucleotide-free binary complex SOS·HRas, where the SOS refers to both the catalytic REM and CDC25 domains. Subsequently, Margarit et al.13 obtained the ternary complex crystal structure 1NVW formed with SOS and HRas, denoted as HRAS·SOS·HRas herein, where the uppercase “HRAS” represents the whole RasGTP·Mg2+ bound at the allosteric site of SOS, while the notation “HRas” denotes the nucleotide-free one at the catalytic site of SOS.
In the past few decades, Ras has emerged as a major drug target for cancer treatment.8,15,16 For example, it was reported that Amgen’s covalent inhibitor of the KRasG12C mutant has succeeded in curing lung cancers.17 Others have pursued to target the SOS·Ras complexes18-21and to inhibit the GTP exchange.22 Burns et al.23 reported the ternary crystal structures HRAS·SOS·HRas in which a few small molecules bound at the CDC25 domains (PDB IDs: 4NYI, 4NYJ and 4NYM). These bound molecules were observed to increase the GTP exchange in vitro and modulate the Ras signaling pathways in cells. Further, Winter et al.19 identified three fragment binding sites on the SOS·HRas complexes. Among them, one site of SOS (PDB: 4URU) is the same as that found in 4NYI. The other two binding sites in 4URY and 4USO are located at the functional regions of Switch I (SW1) and Switch II (SW2) for Ras, corresponding to the P1 and the P4 sites24,25 reported previously.
Among the successful cases of inhibitor discovery, it is worth noting that structure-based design has succeeded in identifying potent inhibitors. For instance, Zheng and co-workers18,26 carried out a virtual screening of small molecules based on the crystal structure 1XD2 of SOS·HRas and found the lead inhibitor NSC-658497 that suppressed the GEF and HRas interaction. Fesik and co-workers20,27,28 employed structure-based methods and discovered a series of inhibitors for the HRAS·SOS·HRas ternary complex. Thus, the SOS and Ras complexes appear to be good targets for regulating the functions of Ras in cells. Regulating the conformation-related functions29 of Ras in solution has been the goal of NMR investigations30-41 in the past decades. As a complementary approach, molecular dynamics (MD) simulation studies focused on discriminating the different functional states of the wild-type and the mutant Ras,42-51 characterizing the structure features of the Ras complexes,52-56 and simulating the conformational change of Ras during the GTP exchange process.57-61
Recently, we have identified the major substates of HRasGTP·Mg2+ during the GTP hydrolysis cycle.48,49 The State 2 of HRasGTP·Mg2+, the HRasGDP·Pi·Mg2+S2 and HRasGDP·Mg2+S1.1 act as the most stable substates along the energy-favorable path in the GTP hydrolysis. These stable conformations could serve as the potent targets of drug design in inhibiting the functions of Ras in the GTP hydrolysis. In this work, we resort to the MD and replica-exchange MD (REMD)62 simulations to characterize the major conformations of HRas and HRAS·SOS·HRas complexes in the GTP exchange process. These refined conformations represent the stable substates along the paths of the GTP exchange from the inactive HRasGDP·Mg2+ state to the active HRasGTP·Mg2+ state. Meanwhile, we summarize the potent binding sites on the surfaces of the HRas and the HRAS·SOS·HRas complexes and suggest new binding sites at the allosteric sites of SOS.
Methods
System Setup
The crystal structures 4Q2163 and 4EFL64 were chosen to construct the product state HRasGDP·Mg2+ and the State 1 of HRasGTP·Mg2+, respectively. The 4Q21 structure represents a typical product state of HRasGDP·Mg2+ with a hydrolyzed GDP bound at the catalytic site. The 4EFL structure represents State 1 of HRasGTP·Mg2+, which features the broken coordination of the Thr35 residue to the central Mg2+. Water molecules at the catalytic sites of HRas in these crystal structures were maintained in the computational models to establish the correct six-coordination of the Mg2+ ion. The analogue GppNHp in 4EFL was substituted with the GTP molecule. To simulate the nucleotide-free intermediate state of Ras, the Mg2+ ion and GDP were removed from the structure of 4Q21.
For the setup of GEF·Ras models, the ternary nucleotide-free complex 1NVW13 was used to construct the intermediate state, denoted as HRAS·SOS·HRas, where the uppercase “HRAS” represents the HRasGTP·Mg2+ complex at the allosteric site and the “HRas” denotes the nucleotide-free one at the catalytic site. For the GDP- and the GTP-bound ternary complexes, we performed a least-square fitting of the catalytic HRas in 1NVW to the structures of 4Q21 and 4EFL, respectively. The Mg2+ ion and GDP/GTP were added into 1NVW to obtain the GDP-bound HRAS·SOS·HRasGDP·Mg2+ and the GTP-bound HRAS·SOS·HRasGTP·Mg2+ complexes, respectively. The binding sites of HRasGTP·Mg2+ and the ternary complexes were analyzed using the sitemap module65 in the Schrödinger software and the free version of IChem66 package, respectively. The volumes of the binding sites of the ternary complexes were calculated using the Volsite module in the IChem package.
Details of MD Simulation
The MD simulation package AMBER1867 is used for all the simulations. The Amber14SB68 force field is used to describe the systems and the substrates adopt the parameters contributed from the Amber developers.69 All the systems were immersed in truncated octahedron boxes filled with the TIP3P70 water molecules under the periodic boundary condition, where the counter ions Na+ and Cl− were added to neutralize the system charges. The cutoff of the van der Waals and the real-space electrostatic interactions is set to 8 Å. The long-range electrostatic interactions are treated using the Particle Mesh Ewald method.71 Langevin dynamics72 was carried out with a collision frequency of 2.0 ps at the temperature of 300 K and 1 atmosphere pressure. Each system was first energy-minimized and then heated to 300 K within 2 ns in the NPT ensemble. All the systems were pre-equilibrated for 50 ns and then 1 μs simulations were performed with a step size of 2 fs.
REMD Simulation
For the REMD simulation for State 1 of HRasGTP·Mg2+, a snapshot was extracted from its normal MD trajectory as the initial structure. A set of distance restraints (see Figure S1 of the supporting information (SI)) was added to the atom pairs at the catalytic site to maintain the six-coordination of Mg2+ during the REMD simulation, as was done previously for HRas.49 A total number of 64 replicas spanning the temperature range of 278-429 K was employed for enhanced sampling. The SHAKE algorithm73 was utilized to constrain the bonds with hydrogen atoms. The system was pre-equilibrated at each temperature for 200 ps with the step size of 2 fs. Then, the simulation trajectories of each replica were extended to 350 ns. The two-dimensional free energy landscape (2D-FEL)48,49 was constructed from the 100-350 ns trajectory of 278 K using the weighted histogram analysis method,74 and the reaction coordinates are the root-mean-square deviation (RMSD) relative to the heavy atoms of 3L8Z and gyration (Rg). The calculated RMSD and Rg values every 50 ns indicate a good convergence in the 100-350 ns REMD trajectory (Figure S1 of SI). The single linkage method with the RMSD cutoff of 1.0 Å was used to cluster the conformations from trajectories and the CPPTRAJ75 module was used to analyze the data.
Results and Discussion
Conformations of HRas in the GTP Exchange
Our previous REMD simulation49 of the hydrolysis product state HRasGDP·Mg2+ revealed that it featured at least two major stable substates, HRasGDP·Mg2+S1.1 and HRasGDP·Mg2+S1.2. The S1.1 substate represents the most populated conformation in solution, and the S1.2 substate might be the specific one that interacts with GEFs. In order to get a stable structure for docking, we perform a 1 μs MD simulation of the S1.1 substate (Figure S2 of SI) and present the representative structure from cluster analysis in Figure 1a. Since the central Mg2+ ion plays an important role in stabilizing the active center, it is necessary to get an insight into the atomistic details for the coordination of the Mg2+ ion.
Figure 1.

(a) The coordination of Mg2+ with four H2O molecules, Ser17, Asp57 and GDP at the catalytic site of the representative structure of HRasGDP·Mg2+, where OW1-OW4, OD1, OD2 and OG denote the oxygen atom types of H2O, Asp57 and Ser17, respectively. (b) The distances of Mg2+-OG, OD2-OW1 and OD1-OG as shown in panel (a) during the 1 μs trajectory of HRasGDP·Mg2+. The inset shows the calculated root-mean square fluctuation of Cα atoms for all protein residues. (c) The least-square fitting of the heavy atoms of the representative structure of HRas to the crystal structure of 4Q21. The inset shows the H-bond network formed by Ser17, Asp57 and Ala59. (d) The distances of OD1-OG between Asp57 and Ser17 and OD2-N between Asp57 and Ala59 during the simulation.
Figure 1a illustrates that the Mg2+ ion at the catalytic site maintains a stable six-coordination with four water molecules OW1-OW4, as well as Ser17, and GDP; the sidechain of Asp57 is also involved in positioning the hydroxyl of Ser17 to coordinate with the Mg2+ ion. The interactions with the γ-group of GTP and Thr35 in the pre-hydrolysis state are substituted by water molecules (OW2 and OW4) following the GTP hydrolysis. The Mg2+ ion and the surrounding ligands form a stable H-bond network, as observed in the MD simulations of HRasGTP·Mg2+ 48,54 and GAP·HRasGDP·Pi·Mg2+.53,55 The distance between the Mg2+ ion and OG of Ser17 maintains stably around 2.1 Å (Figure 1b), suggesting a strong interaction between them. The H-bond distance between OD2 of Asp57 and OW1 varies in the range of 2-3 Å, while the OD1-OG pair of Asp57 and Ser17 fluctuates between 2.5 and 4.5 Å. The current simulated results are consistent with the kinetic analysis,76 showing that mutations of Ser17 and Asp57 to Ala reduced the dissociation constant of Mg2+ by 30- and 16-fold, respectively, whereas the T35A mutation had little effect. In addition, the calculated RMSF in the inset indicates large fluctuations of the SW1 and the SW2 regions, in line with the experimental 15N spin relaxation measurement.77
The dissociation of GDP from the product state was observed to be a slow process in vitro.78 According to the dissociation scheme of HRasGDP·Mg2+ ⇔ HRas + GDP·Mg2+ proposed by John et al.76 at a low concentration of Mg2+, we constructed the nucleotide-free state denoted as HRas and simulated it for 1 μs with the RMSD around 4.7 Å (Figure S2 of SI). The representative clusters of HRas in Figure 1c show that the positions of the SW1 and the SW2 regions in solution derivate largely from those in the crystal structure 4Q21. The loop of the SW1 region (30-37 residues) becomes more flexible than that in the S1.1 substate due to the lack of a bound GDP in the pocket. The inset of Figure 1c shows that Ser17, Asp57 and Ala59 constitute a triplet H-bond interaction pattern, where the H-OG group of Ser17 of α1-helix forms a H-bond interaction with the OD1 atom of Asp57 of the β3-sheet, while another carboxylate OD2 atom of Asp57 interacts with the amide group of Ala59. The two groups of H-bonds fluctuate with distances below 5 Å in the simulation, as shown in Figure 1d.
The association constant of GTP measured in experiments was nearly two times greater than that of GDP.78 The reloading of GTP·Mg2+ into Ras yields State 164 of the HRasGTP·Mg2+ complex. Figure 2a shows that the representative structure of HRasGTP·Mg2+ from the 1 μs MD simulation (Figure S2 of SI) is similar to the State 1 structure of 4EFL, with the coordination between Mg2+ and Thr35 disrupted. At the catalytic center, the Mg2+ ion maintains the six-coordination with three H2O molecules, Asp56, Ser17 and GTP. The OW2 in Figure 1a is replaced with the γ-O atom of GTP. The inset in Figure 2b shows that the H-bond interactions of Mg2+, Ser17 and Asp57 remain stable during the 1 μs MD simulation. In experiments, the S17A and D57A mutations76 lowered the affinity of GTP to HRas.
Figure 2.

(a) Least-square fitting of the representative structure of HRasGTP·Mg2+ from 1 μs MD trajectory to the State 1 structure 4EFL. The inset shows the six-coordination of Mg2+ with three H2O molecules, Ser17, Asp57 and GTP. (b) Constructed 2D-FEL with respect to the RMSD (relative to 3L8Z) and Rg from the 1 μs MD trajectory of HRasGTP·Mg2+, where the energy scale bar is in kcal/mol. The insert shows the distances of OD1-OW1, OD2-OG and Mg2+-OG at the catalytic site of HRasGTP·Mg2+ during the simulation.
Feature of the 2D-FEL for State 1 of HRasGTP·Mg2+
The 2D-FEL projection of 1 μs trajectory of HRasGTP·Mg2+ on Figure 2b leads to an elliptical free energy basin, which is more localized in nature than State 1 of HRasGTP·Mg2+. Our previous REMD simulation48 of the HRasGTP·Mg2+ complex initiating from the State 2 structure showed that it featured State 2 and State 1 on its 2D-FEL, where State 1 spans the region with RMSD of 3.5-4.0 Å and Rg of 15.0-15.5 Å, respectively. A previous NMR study30,79 on the conformational conversion of State 2 and State 1 of HRasGTP·Mg2+ indicated a high barrier existing between them, which made us realized that the sampling of REMD initiated from State 2 might be insufficient for State 1. Thus, further enhanced sampling is required to explore the more extensive conformational space of State 1 of HRasGTP·Mg2+. Here, we have performed REMD simulations initiated from the State 1 structure of 4EFL for 350 ns. As shown by the 2D-FEL in Figure 3a constructed from 100-350 ns trajectory, State 1 spans a large region with the RMSD and Rg of 3.0-7.0 Å and 15.1-16.1 Å, respectively, much broader than that obtained in our previous REMD simulations that initiated from structure 3L8Z.48 Within the RMSD values of 3.0-5.0 Å, the highly populated region appears to exhibit five energy minima as marked with the five triangles. To characterize the structural features of these minima, we performed clustering analysis on the conformations extracted from these regions, denoted as C1-C5 in Figure 3a.
Figure 3.

(a) Constructed 2D-FEL of State 1 of HRasGTP·Mg2+ with respect to the RMSD (relative to the reference structure 3L8Z) and Rg of heavy atoms. C1-C5 denote the representative conformations of the five free energy minima marked with the orange triangles on the 2D-FEL. The C1-C5 conformations represent the two substates HRasGTP·Mg2+S1.1 (C1, C2, C3) and HRasGTP·Mg2+S1.2 (C4, C5), respectively. The red pentagram and square denote the projection of the crystal structures 4EFL and Ras in 1NVW. The unit of the energy scale bar is kcal/mol. (b) The least-square fitting of C1-C5 conformations to 1NVW shows that the loops of C1, C2 and C3 overlap with the α-helix of GEF. The inset shows the distances between Thr35 and Mg2+ in the C1 and C2 conformations.
Figure 3b shows the least-square fitted conformations C1-C5 (Figure S3 of SI) overlaid with the HRas in the crystal structure of the complex 1NVW. The NMR experiments80 suggested that State 1 is responsible for the interaction with GEFs. However, the C1-C5 conformations exhibit significant variations in their secondary structures, especially for the loops of the SW1 region. Figure 3b shows that the loops of the C1-C3 conformations overlap with the inserted α-helix of GEF at the catalytic center, while the loops of C4 and C5 are as open as that in 1NVW. The first two conformations C1 and C2 have similar RMSD values compared to 4EFL on the 2D-FEL. The inset in Figure 3b shows that no coordination forms between Thr35 and Mg2+, which supports that they represent State 1 of HRasGTP·Mg2+ in solution. The C3 conformation has a larger RMSD than the first two, thus representing a more flexible conformation. The C4 and C5 conformations with large RMSDs have exposed catalytic sites, which allow the insertion of an α-helix of GEF to form the complex. Based on the structural comparison, the five representative conformations C1-C5 can be divided into two categories according to their biological functions. As shown in Figure 3a, the C1-C3 conformations are categorized into the substate S1.1 denoted as HRasGTP·Mg2+S1.1, while the C4 and C5 conformations belong to the substate S1.2 HRasGTP·Mg2+S1.2, which interact with GEFs. In other words, State 1 of HRasGTP·Mg2+ also features two functionally distinct substates S1.1 and S1.2.
Binding Pockets of HRasGDP·Mg2+ and HRasGTP·Mg2+
Actually, the stable conformations of HRasGDP·Mg2+ and HRasGTP·Mg2+ in the GTP exchange could be used for inhibitor design. The conformations of HRas exhibit a diversity of binding pockets on their surfaces including the P1-P4 sites as reported in the previous work.24,81 For example, Rosnizeck et al.82 has reported the Zn2+-cyclen complex in which the Zn2+ cyclen bound at the P3 site stabilizes the inactive State 1 and thus impairs the binding affinity of Raf to the SW1 region of HRas. Shima et al.83 reported that the small molecule Kobe0065 bound at the P1 site has antitumor activity. Motivated by these considerations, we predict the binding pockets of the low-energy S1.1 substate of HRasGDP·Mg2+ and both the substates of HRasGTP·Mg2+ using the sitemap65 module in the Schrödinger software. The representative conformation of HRasGDP·Mg2+S1.1 is chosen from Section 3.1 and the representative structures of C1-C5 conformations are generated by clustering their respective 500 ns MD trajectories (Figure S4 of SI).
The predicted binding pockets of HRasGDP·Mg2+S1.1 and HRasGTP·Mg2+ are shown in Figure 4a-4f, respectively. The conformation of HRasGDP·Mg2+S1.1 features the two binding sites of P2 and P3. The P2 site is a typically covalent inhibition site84-86 that has been widely identified in the KRas G12C mutants, while P3 is an allosteric site distant from the catalytic site.82 The conformations of C1-C3 of HRasGTP·Mg2+ feature three sites: P1, P3 and P4. The P1 site83 was located between the β1-3 sheets. Figure 4b shows that the C1 conformation with the smallest RMSD only has a P4 site around the loop region of SW1 due to its compactness, while the more flexible C2 conformation in Figure 4c exhibits three sites. As for the C4-C5 conformations in Figure 4d-4f, two or more binding sites are found on the surfaces of HRas as expected because of their high degree of flexibility. We expect that the binding pockets predicted from the simulated structures in solution are likely more accessible than the ones predicted based on the crystal structure 4EFL. Thus, they could be used for inhibitor design for the purpose of stabilizing the inactive State 1.
Figure 4.

The predicted binding pockets of the S1.1 substate of (a) HRasGDP·Mg2+ and the (b)-(f) C1-C5 conformations of HRasGTP·Mg2+ using the sitemap module. The P1 site is located between the α2-helix and the β1-3 sheets. The P2 site is located at the cleft of two α-helices. The P3 site is located at the cavity formed by the α5-helix and the L7 loop. The P4 site is located at the cleft between the α1-helix and the loop of SW1.
Conformations of GEF·HRas in the GTP Exchange
The three ternary complexes, HRAS·SOS·HRasGDP·Mg2+, HRAS·SOS·HRas and HRAS·SOS·HRasGTP·Mg2+, were constructed from the nucleotide-free crystal structure 1NVW. The three complexes represent the crucial intermediate states in the GTP exchange and have been each simulated with 1 μs MD simulations (Figure S5 of SI). Clustering analysis was then carried out on the simulated trajectories to extract the representative conformations of the ternary complexes. The conformations of the catalytic HRas in the complexes are denoted as HRasGEFGDP·Mg2+S1, HRasGEF and HRasGEFGTP·Mg2+S1, respectively.
It has been pointed out previously that the conformation of HRasGEFGDP·Mg2+S1 is more similar to that of HRasGDP·Mg2+S1.2 than HRasGDP·Mg2+S1.1. Figure 5a shows more details about the coordination of the catalytic HRas in the HRAS·SOS·HRasGDP·Mg2+. The insertion of the α-helix into the active site causes a severe distortion of the original coordination. Different from the isolated HRasGDP·Mg2+S1.1 in Figure 1a, the coordination of Ser17 to Mg2+ is destabilized by the α-helix of SOS so that it is disrupted at the 600 ns in the simulation, as indicated by the black curve in Figure 5a. Ser17 and Asp57 still form a stable H-bond interaction with the OD1-OG distance around 4.0 Å in the 1 μs trajectory, although both of them gradually drift away from the Mg2+ ion. The Mg2+ ion thus only coordinates to the four H2O molecules and the GDP. The observed instability caused by GEF from our simulation is consistent with the experimental observation that GEF accelerates the dissociation of GDP from Ras by 105-fold.87
Figure 5.

(a) The distances of OD1-OG and Mg2+-OG at the catalytic site of HRAS·SOS·HRasGDP·Mg2+ during the MD simulations. The inset shows that Ser17 and Asp57 drift away from the Mg2+ ion. (b) The distances of OD1-OG and O-N at the catalytic site of HRAS·SOS·HRas during the MD simulation. The inset shows the H-bond network formed by Ser17, Asp57 and Ala59. (c) The distances of OD1-OG, OD2-OW1 and Mg2+-OG at the catalytic site of HRAS·SOS·HRasGTP·Mg2+ during the MD simulation. The inset shows the stable coordination of Mg2+ to Ser17, Asp57, GTP and the H2O molecules. (d) The least-square fitted conformations of the catalytic HRasGDP·Mg2+, HRas and HRasGTP·Mg2+ in complexes to the reference structure 3L8Z, with the calculated RMSD values of 0.672, 0.666 and 0.827 Å, respectively.
The release of Mg2+ and GDP from the active site of HRAS·SOS·HRasGDP·Mg2+ yields the nucleotide-free intermediate state HRAS·SOS·HRas. In Figure 5b, it is observed that the three residues Asp57, Ser17 and Ala59 form stable H-bond interactions at the catalytic center of HRasGEFGDP. The O-N atom distance of Asp57 and Ala59 is stable around 4 Å, while the OD1-OG distance of Asp57 and Ser17 fluctuates within a large range of 3-7 Å. Such H-bond interaction network between Ser17-Asp57-Ala59 is also observed in the isolated HRas in Figure 1d. Figure 6c shows the simulation results of the HRasGEFGTP·Mg2+S1 in the HRAS·SOS·HRasGTP·Mg2+ complex. It is found that the three oxygen atoms in the triphosphate group of GTP chelate with the Mg2+ ion, leading to a stable six-coordination pattern in the entire 1 μs MD simulation. Comparing the coordination pattern to that in Figure 6a, it is observed that the bi-chelated GDP does not sustain the stability of the catalytic site, while the tri-chelated GTP does. Such a difference from the MD simulations is in line with the kinetic measurements, showing that the association constant of GTP is nearly two times greater than that of GDP.87
Figure 6.

(a) The S1, P1 and P4 binding pockets in 1BKD are predicted using the IChem package. The inset shows the small molecules bound at S1, P1 and P4 in the crystal strucutres 4UR4, 4URZ and 4US0, respectively. The plots (b)-(d) show all the binding sites of the CDC25 domains, the catalytic HRas, the REM domains and the allosteric HRAS in the tertariy complexes, respectively. The REM domains have the R1 binding sites. The allesteric HRAS have the P1 and P4 sites similar to the catalytic HRas.
Thus, the reloaded GTP chelates with the Mg2+ ion in the three-coordination pattern to rescore the coordination of Mg2+ to Ser17 in HRasGTP·Mg2+ after the dissociation of GEF from HRas. As soon as the dissociation occurs, the conformation of HRasGTP·Mg2+ transforms to State 1. The calculated RMSD of HRasGTP·Mg2+ in HRAS·SOS·HRasGTP·Mg2+ relative to the reference 3L8Z is 4.9 Å, falling in the region of the S1.2 substate of HRasGTP·Mg2+ as shown in Figure 3a. This further supports the view that Ras selects S1.2 to interact with GEFs. Comparing the overlaid conformations of the catalytic HRas in the three complexes with each other in Figure 6d, we notice that the major difference in their secondary structures comes from the loop regions of SW1 and SW2. As is presented in Figure 6a-6c, it can be seen that the different coordinations of the Mg2+ ions and the substrates at the catalytic centers would lead to the different conformations of the SW1 and the SW2 regions, as well as the shapes of the binding pockets at the interfaces of HRas and SOS.
Binding Pockets of the Ternary Complexes
The stable complex structures could serve as targets of the drug design for Ras. Winter et al.19 have identified three binding sites on the crystalized structures of the binary complexes SOS·HRas. The sitemap module was used to predict the binding pockets of the SOS·HRas complexes, although it was unable to predict the S1 site reported in the crystal structure 4UR4 (Figure S6 of SI). Thus, the IChem66 software was used to predict the three binding pockets S1, P1 and P4 based on the crystal structure 1NVW, as shown in Figure 6a. The inset shows that the three small molecules identified in experiments bound at the sites of S1, P1 and P4 in the crystal structures 4US0, 4URU and 4URZ, respectively. The S1 site is a concave cavity formed by a few helices of the CDC25 domain. The P1 and the P4 sites are located around the regions of SW1 and SW2 of HRas at the active site, similarly to that of KRas reported previously.25
Figure 6b-6d present the predicted binding sites of the three ternary complexes. It is seen that the ternary complexes possess not only the binding sites as the binary complexes, but also additional binding sites at the allosteric sites and the REM domains. To characterize these binding pockets quantitatively, we present the data of the calculated volumes in Table 1, including the results of 1BKD14 for comparison. In 1BKD, the three pockets of S1, P1 and P4 are separated and their volume values are 28, 117 and 157 Å3, respectively. In HRAS·SOS·HRasGDP·Mg2+, the three pockets merge with each other and form a large cleft with the volume of 162 Å3. In the intermediate HRAS·SOS·HRas, the total volume of S1 and P1 is 170 Å3, much larger than 145 Å3 of the corresponding counterpart in 1BKD. The volume of the P1 site in HRAS·SOS·HRasGTP·Mg2+ is up to 211 Å3, suggesting that the cleft between the active HRas and CDC25 is a good targeting site.
Table 1.
The predicted S1, P1 and P4 binding sites of the CDC25 domain and the catalytic HRas in 1BKD and the three ternary complexes using the IChem software, with the background colored in gray. The R1 binding site of the REM domain as well as the P1 and the P4 sites of the allosteric HRAS are also reported. The volumes of the separated and the merged binding pockets are calculated in the units of Å3.
| Complex Structuresa | CDC25 | HRas | REM | HRAS | |
|---|---|---|---|---|---|
| S1 | P1 | P4 | R1 | P1+P4 | |
| 1BKD | 28 | 117 | 157 | ||
| HRAS·SOS·HRasGDP·Mg2+ | 162 (S1+P1+P4) | 49 | 74 | ||
| HRAS·SOS·HRas | 170 (S1+P1) | 111 | 70 | 112 | |
| HRAS·SOS·HRasGTP·Mg2+ | 13 | 211 | 36 | 63 | 80 |
HRAS denotes the whole HRasGTP·Mg2+ at the allosteric site of SOS and HRas denotes the nucleotide-free one at the catalytic sites of SOS.
Based on these structures, binding pockets at the allosteric HRAS and the REM domains are also predicted using IChem, as shown in Table 1. Compared to the binary structure 1BKD, which lacks the REM domain and the allosteric HRAS, the ternary complexes also have the P1 and the P4 binding sites in the allosteric HRAS, while a R1 binding site at the REM domain, colored in pink and brown, respectively. The calculated volumes of the R1 sites in the three complexes are 49, 70 and 63 Å3, respectively, larger than 28 Å3 of the S1 site in the CDC25 domain of 1BKD. The calculated total volumes of P1 and P4 in the allosteric HRAS are 74, 112 and 80 Å3, smaller than the counterparts in the catalytic HRas. These pockets are generated due to the large conformational changes caused by the GTP exchange at the interface of HRas and CDC25, as indicated from the comparison of structures in Figure 5. The diverse pockets in REM and HRAS suggest that it is possible to design small molecules to bind at these sites to regulate the SOS-mediated GTP exchange process.
Conclusion
In this work, we have performed an extensive MD simulation study to characterize the structural features for intermediates of HRas and its complexes in the nucleotide exchange process, with the related conformations summarized in Scheme 1. Release of Pi from the hydrolysis intermediate of Ras yields the product state HRasGDP·Mg2+, which contains two functionally distinct substates S1.1 and S1.2. Release of GDP·Mg2+ from HRasGDP·Mg2+S1.1 yields a nucleotide-free state HRas. The more flexible S1.2 substate interacts with GEF to form the ternary complex HRAS·SOS·HRasGDP·Mg2+. Our MD simulations suggest that the insertion of the GEF α-helix into HRasGEFGDP·Mg2+S1 of the complex would disrupt the stable coordination of Mg2+ and lower its affinity to HRas. Release of GDP·Mg2+ leads to the stable intermediate HRAS·SOS·HRas. The simulations of HRAS·SOS·HRasGTP·Mg2+ reveal that GTP stabilizes the six-coordination of Mg2+ by chelating the three oxygen atoms of the triphosphate group. As soon as the GEF dissociates from HRas, HRasGTP·Mg2+ transforms to State 1. The constructed 2D-FEL shows that State 1 has a number of stable conformations indicated as C1-C5. These stable conformations are categorized into two substates, HRasGTP·Mg2+S1.1 and HRasGTP·Mg2+S1.2. The S1.1 substate represents the most stable conformations of State 1 in solution and is able to convert to State 2 to interact with GAPs and effectors. The S1.2 substate is ready to complex with GEF of SOS and forms the conformation HRasGEFGTP·Mg2+S1 in the ternary complex.
Scheme 1.

An illustration of the conformational change associated with the GTP exchange in HRas and RAS·SOS based on our MD simulations. Pi release from the hydrolysis intermediate state results in the product state HRasGDP·Mg2+, which has two substates HRasGDP·Mg2+S1.1 and HRasGDP·Mg2+S1.2. Release of GDP·Mg2+ from HRasGDP·Mg2+S1.1 yields the nucleotide-free state HRas. Reloading of GTP·Mg2+ into HRas forms the S1.1 substate of State 1 of HRasGTP·Mg2+. HRasGDP·Mg2+ selects the S1.2 substate to interact with the GEF of RAS·SOS and transforms to the complexed conformation HRasGEFGDP·Mg2+S1. Release of GDP·Mg2+ from HRasGEFGDP·Mg2+S1 yields HRasGEF. Reloading of GTP·Mg2+ into RAS·SOS forms the complexed conformation HRasGEFGTP·Mg2+S1. Dissociation of RAS·SOS leads to the S1.2 substate of HRasGTP·Mg2+, which transforms to S1.1 through the coordination of Thr35 to Mg2+, then to State 2.
Based on the refined conformations of Ras, we have analyzed the binding pockets on the surfaces of HRasGDP·Mg2+S1.1, HRasGTP·Mg2+S1.1 and HRasGTP·Mg2+S1.2. The P1-P4 sites revealed on the surfaces of Ras indicate that the strategies of stabilizing the inactive HRasGDP·Mg2+ and State 1 of HRasGTP·Mg2+ are feasible. Also, the binding sites of SOS and the catalytic HRas of ternary complexes are predicted based on the representative structures from the simulations. In particular, the binding sites of the allosteric HRAS and the REM domains in the ternary complexes are revealed, which could be targeted as inhibition sites to regulate the SOS-mediated signaling transduction. This work provides a detailed structural information for future drug designs that aim to regulate the functions of SOS and the theoretical predictions of the inhibition sites for the ternary complexes are expected to be verified further in experiments.
Supplementary Material
Figure S1 shows the added distance restraints at the catalytic site of HRasGTP·Mg2+ and the calculated RMSD and Rg values every 50 ns.
Figure S2 shows the calculated RMSD curves for the HRas conformations in the GTP exchange.
Figure S3 shows the overlaid representative C1-C5 conformations.
Figure S4 shows the calculated RMSD curves for the C1-C5 conformations from trajectories.
Figure S5 shows the calculated RMSD curves for the moieties in the complexes of HRAS·SOS·HRasGDP·Mg2+, HRAS·SOS·HRas and HRAS·SOS·HRasGTP·Mg2+.
Figure S6 shows the active pockets in 1BKD predicted by using the sitemap module.
ACKNOWLEDGMENT
This work was supported by the National Key R&D Program and the National Natural Science Foundation of China (Grants No. 2017YFA0504504, 21688102, 22073029, 21603006, 22025702, 82151211 and 82021003), the Fundamental Research Funds for the Central Universities of China (Grants No. 20720200008) and the Natural Science Foundation of Guangdong Province (Grant No. 2019A1515011079). Q.C. acknowledges support from NIH R01-GM106443. J.Z. acknowledges support from PHD researchers of Guangdong Medical University in 2019. We also acknowledge the support of the NYU-ECNU Center for Computational Chemistry at NYU Shanghai as well as the ECNU Public Platform for Innovation (001) for providing computer time.
References:
- 1.Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A Nature 1989, 341, 209. [DOI] [PubMed] [Google Scholar]
- 2.Vetter IR, Wittinghofer A. Science 2001, 294, 1299. [DOI] [PubMed] [Google Scholar]
- 3.Mott HR, Owen D. Biochem. Soc. Trans 2018, 46, 1333. [DOI] [PubMed] [Google Scholar]
- 4.Vorobyov I, Kim I, Chu ZT, Warshel A. Proteins 2015, 84, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong L, de Vos AM, Milburn MV, Kim SH. J. Mol. Biol 1991, 217, 503. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL, Rehmann H, Wittinghofer A. Cell 2007, 129, 865. [DOI] [PubMed] [Google Scholar]
- 7.Scheidig AJ, Burmester C, Goody RS. Structure 1999, 7, 1311. [DOI] [PubMed] [Google Scholar]
- 8.Prior IA, Lewis PD, Mattos C Cancer Res. 2012, 72, 2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobbs GA, Der CJ, Rossman KL. J. Cell Sci 2016, 129, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prior IA, Hood FE, Hartley JL. Cancer Res. 2020, 80, 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broek D, Toda T, Michaeli T, Levin L, Birchmeier C, Powers S, Wigler M. Cell 1987, 48, 789. [DOI] [PubMed] [Google Scholar]
- 12.Créchet JB, Poullet P, Mistou MY, Parmeggiani A, Camonis J, Boy-Marcotte E, Damak F, Jacquet M. Science 1990, 248, 866. [DOI] [PubMed] [Google Scholar]
- 13.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Cell 2003, 112, 685. [DOI] [PubMed] [Google Scholar]
- 14.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. Nature 1998, 394, 337. [DOI] [PubMed] [Google Scholar]
- 15.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Nat. Rev. Drug Discov 2014, 13, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu S, Jang H, Gu S, Zhang J, Nussinov R. Chem. Soc. Rev 2016, 45, 4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, Lanman BA, Werner J, Rapaport AS, San Miguel T, Ortiz R, Osgood T, Sun JR, Zhu X, McCarter JD, Volak LP, Houk BE, Fakih MG, O'Neil BH, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Hong DS, Ouyang W, Henary H, Arvedson T, Cee VJ, Lipford JR. Nature 2019, 575, 217. [DOI] [PubMed] [Google Scholar]
- 18.Evelyn CR, Duan X, Biesiada J, Seibel WL, Meller J, Zheng Y. Chem. Biol 2014, 21, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winter JJ, Anderson M, Blades K, Brassington C, Breeze AL, Chresta C, Embrey K, Fairley G, Faulder P, Finlay MR, Kettle JG, Nowak T, Overman R, Patel SJ, Perkins P, Spadola L, Tart J, Tucker JA, Wrigley G. J. Med. Chem 2015, 58, 2265. [DOI] [PubMed] [Google Scholar]
- 20.Hodges TR, Abbott JR, Little AJ, Sarkar D, Salovich JM, Howes JE, Akan DT, Sai J, Arnold AL, Browning C, Burns MC, Sobolik T, Sun Q, Beesetty Y, Coker JA, Scharn D, Stadtmueller H, Rossanese OW, Phan J, Waterson AG, McConnell DB, Fesik SW. J. Med. Chem 2018, 61, 8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, Werbeck ND, Briem H, Boemer U, Weiske J, Badock V, Mastouri J, Petersen K, Siemeister G, Kahmann JD, Wegener D, Böhnke N, Eis K, Graham K, Wortmann L, von Nussbaum F, Bader B. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu S, Jang H, Zhang J, Nussinov R. ChemMedChem 2016, 11, 814. [DOI] [PubMed] [Google Scholar]
- 23.Burns MC, Sun Q, Daniels RN, Camper D, Kennedy JP, Phan J, Olejniczak ET, Lee T, Waterson AG, Rossanese OW, Fesik SW. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy M, Prakash P, Gorfe AA. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 48, 3. [DOI] [PubMed] [Google Scholar]
- 25.Gupta AK, Wang X, Pagba CV, Prakash P, Sarkar-Banerjee S, Putkey J, Gorfe AA. Chem. Biol. Drug Des 2019, 94, 1441. [DOI] [PubMed] [Google Scholar]
- 26.Evelyn CR, Biesiada J, Duan X, Tang H, Shang X, Papoian R, Seibel WL, Nelson S, Meller J, Zheng Y. J. Biol. Chem 2015, 290, 12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akan DT, Howes JE, Sai J, Arnold AL, Beesetty Y, Phan J, Olejniczak ET, Waterson AG, Fesik SW. ACS Chem. Biol 2019, 14, 325. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar D, Olejniczak ET, Phan J, Coker JA, Sai J, Arnold A, Beesetty Y, Waterson AG, Fesik SW. J. Med. Chem 2020, 63, 8325. [DOI] [PubMed] [Google Scholar]
- 29.Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, Zhang J. Chem. Rev 2016, 116, 6607. [DOI] [PubMed] [Google Scholar]
- 30.Geyer M, Schweins T, Herrmann C, Prisner T, Wittinghofer A, Kalbitzer HR. Biochemistry 1996, 35, 10308. [DOI] [PubMed] [Google Scholar]
- 31.Rohrer M, Prisner TF, Brügmann O, Käss H, Spoerner M, Wittinghofer A, Kalbitzer HR. Biochemistry 2001, 40, 1884. [DOI] [PubMed] [Google Scholar]
- 32.Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spoerner M, Nuehs A, Ganser P, Herrmann C, Wittinghofer A, Kalbitzer HR. Biochemistry 2005, 44, 2225. [DOI] [PubMed] [Google Scholar]
- 34.Ye M, Shima F, Muraoka S, Liao J, Okamoto H, Yamamoto M, Tamura A, Yagi N, Ueki T, Kataoka T. J. Biol. Chem 2005, 280, 31267. [DOI] [PubMed] [Google Scholar]
- 35.Liao J, Shima F, Araki M, Ye M, Muraoka S, Sugimoto T, Kawamura M, Yamamoto N, Tamura A, Kataoka T. Biochem. Biophys. Res. Commun 2008, 369, 327. [DOI] [PubMed] [Google Scholar]
- 36.Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, Matsumoto K, Yamamoto N, Sugimoto T, Yoshikawa Y, Kumasaka T, Yamamoto M, Tamura A, Kataoka T. J. Biol. Chem 2010, 285, 22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Araki M, Shima F, Yoshikawa Y, Muraoka S, Ijiri Y, Nagahara Y, Shirono T, Kataoka T, Tamura A. J. Biol. Chem 2011, 286, 39644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalbitzer HR, Rosnizeck IC, Munte CE, Narayanan SP, Kropf V, Spoerner M. Angew. Chem. Int. Ed. Engl 2013, 52, 14242. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Yao H, Wang H, Mao Y, Liu D, Long D. Angew. Chem. Int. Ed. Engl 2019, 58, 2730. [DOI] [PubMed] [Google Scholar]
- 40.Liu D, Chen X, Long D. J. Phys. Chem. Lett 2020, 11, 3642. [DOI] [PubMed] [Google Scholar]
- 41.Liu D, Mao Y, Gu X, Zhou Y, Long D. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2024725118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi C, Saito S. Biophys. J 2010, 99, 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clausen R, Ma B, Nussinov R, Shehu A. PLoS Comput. Biol 2015, 11, e1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapoor A, Travesset A. Proteins 2015, 83, 1091. [DOI] [PubMed] [Google Scholar]
- 45.Sayyed-Ahmad A, Prakash P, Gorfe AA. Proteins 2017, 85, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chakrabarti M, Jang H, Nussinov R. J. Phys. Chem. B 2016, 120, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu S, Jang H, Nussinov R, Zhang J. Sci. Rep 2016, 6, 21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Zhang Y, Grosseruschkamp F, Stephan S, Cui Q, Kotting C, Xia F, Gerwert K. J. Phys. Chem. Lett 2018, 9, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng J, Weng JW, Zhang YW, Xia F, Cui Q, Xu X. J. Phys. Chem. B 2021, 125, 8805. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Zhang S, Wang W, Pang L, Zhang Q, Liu X. J. Chem. Inf. Model 2021, 61, 1954. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Zeng Q, Wang W, Hua Q, Bao H. RSC Advances 2022, 12, 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia F, Rudack T, Kotting C, Schlitter J, Gerwert K. Phys. Chem. Chem. Phys 2011, 13, 21451. [DOI] [PubMed] [Google Scholar]
- 53.Rudack T, Xia F, Schlitter J, Kotting C, Gerwert K. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudack T, Xia F, Schlitter J, Kotting C, Gerwert K. Biophys. J 2012, 103, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia F, Rudack T, Cui Q, Kotting C, Gerwert K. J. Am. Chem. Soc 2012, 134, 20041. [DOI] [PubMed] [Google Scholar]
- 56.Khrenova MG, Grigorenko BL, Nemukhin AV. Spectrochim. Acta A Mol. Biomol. Spectrosc 2016, 166, 68. [DOI] [PubMed] [Google Scholar]
- 57.Mori K, Hata M, Neya S, Hoshino T. J. Am. Chem. Soc 2005, 127, 15127. [DOI] [PubMed] [Google Scholar]
- 58.Gorfe AA, Grant BJ, McCammon JA. Structure 2008, 16, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grant BJ, Gorfe AA, McCammon JA. PLoS Comput. Biol 2009, 5, e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lukman S, Grant BJ, Gorfe AA, Grant GH, McCammon JA. PLoS Comput. Biol 2010, 6, e1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao TJ, Jang H, Fushman D, Nussinov R. Biophys. J 2018, 115, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugita Y, Okamoto Y. Chem. Phys. Lett 1999, 314, 141. [Google Scholar]
- 63.Milburn MV, Tong L, deVos AM, Brünger A, Yamaizumi Z, Nishimura S, Kim SH. Science 1990, 247, 939. [DOI] [PubMed] [Google Scholar]
- 64.Muraoka S, Shima F, Araki M, Inoue T, Yoshimoto A, Ijiri Y, Seki N, Tamura A, Kumasaka T, Yamamoto M, Kataoka T. FEBS Lett. 2012, 586, 1715. [DOI] [PubMed] [Google Scholar]
- 65.Schrödinger Suite. Available online: http://www.schrodinger.com. The data of access is from Dec. 3 of 2019 to Dec. 1 of 2024.
- 66.Da Silva F, Desaphy J, Rognan D. ChemMedChem 2018, 13, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TEI, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris R, Homeyer N, Huang Y, Izadi S, Kovalenko A, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein DJ, Merz KM, Miao Y, Monard G, Nguyen C, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Smith J, Salomon-Ferrer R, Swails J, Walker RC, Wang J, Wei H, Wolf RM, Wu X, Xiao L, York DM, Kollman PA. 2018, AMBER 2018, University of California, San Francisco. [Google Scholar]
- 68.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. J. Chem. Theory Comput 2015, 11, 3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meagher KL, Redman LT, Carlson HA. J. Comput. Chem 2003, 24, 1016. [DOI] [PubMed] [Google Scholar]
- 70.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys 1983, 79, 926. [Google Scholar]
- 71.Darden T, York D, Pedersen L. J. Chem. Phys 1993, 98, 10089. [Google Scholar]
- 72.Pastor RW, Brooks BR, Szabo A. Mol. Phys 1988, 65, 1409. [Google Scholar]
- 73.Miyamoto S, Kollman PA. J. Comput.Chem 1992, 13, 952. [Google Scholar]
- 74.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. J. Comput. Chem 1992, 13, 1011. [Google Scholar]
- 75.Roe DR, Cheatham TE. J. Chem. Theory Comput 2013, 9, 3084. [DOI] [PubMed] [Google Scholar]
- 76.John J, Rensland H, Schlichting I, Vetter I, Borasio GD, Goody RS, Wittinghofer A. J. Biol. Chem 1993, 268, 923. [PubMed] [Google Scholar]
- 77.Mao Y, Yao H, Wang H, Cheng P, Long D. Angew. Chem. Int. Ed. Engl 2016, 55, 15629. [DOI] [PubMed] [Google Scholar]
- 78.Feuerstein J, Goody RS, Wittinghofer A. J. Biol. Chem 1987, 262, 8455. [PubMed] [Google Scholar]
- 79.Spoerner M, Wittinghofer A, Kalbitzer HR. FEBS Lett. 2004, 578, 305. [DOI] [PubMed] [Google Scholar]
- 80.Kalbitzer HR, Spoerner M, Ganser P, Hozsa C, Kremer W. J. Am. Chem. Soc 2009, 131, 16714. [DOI] [PubMed] [Google Scholar]
- 81.Buhrman G, O'Connor C, Zerbe B, Kearney BM, Napoleon R, Kovrigina EA, Vajda S, Kozakov D, Kovrigin EL, Mattos C. J. Mol. Biol 2011, 413, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosnizeck IC, Graf T, Spoerner M, Trankle J, Filchtinski D, Herrmann C, Gremer L, Vetter IR, Wittinghofer A, Konig B, Kalbitzer HR. Angew. Chem. Int. Ed. Engl 2010, 49, 3830. [DOI] [PubMed] [Google Scholar]
- 83.Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, Nishiyama Y, Sato C, Muraoka S, Tamura A, Osoda T, Tsuda K, Miyakawa T, Fukunishi H, Shimada J, Kumasaka T, Yamamoto M, Kataoka T. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. Nature 2013, 503, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, Liu Y. Cell 2018, 172, 578. [DOI] [PubMed] [Google Scholar]
- 87.Lenzen C, Cool RH, Prinz H, Kuhlmann J, Wittinghofer A. Biochemistry 1998, 37, 7420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows the added distance restraints at the catalytic site of HRasGTP·Mg2+ and the calculated RMSD and Rg values every 50 ns.
Figure S2 shows the calculated RMSD curves for the HRas conformations in the GTP exchange.
Figure S3 shows the overlaid representative C1-C5 conformations.
Figure S4 shows the calculated RMSD curves for the C1-C5 conformations from trajectories.
Figure S5 shows the calculated RMSD curves for the moieties in the complexes of HRAS·SOS·HRasGDP·Mg2+, HRAS·SOS·HRas and HRAS·SOS·HRasGTP·Mg2+.
Figure S6 shows the active pockets in 1BKD predicted by using the sitemap module.