

Abstract
Chromatin is a DNA-protein polymer that represents the functional form of the genome. The main building block of chromatin is the nucleosome, a structure that contains 147 base pairs of DNA and two copies each of the histone proteins H2A, H2B, H3 and H4. Previous work has shown that magic angle spinning (MAS) NMR spectroscopy can capture the nucleosome at high resolution although studies have been challenging due to low sensitivity, the presence of dynamic and rigid components, and the complex interaction networks of nucleosomes within the chromatin polymer. Here, we use dynamic nuclear polarization (DNP) to enhance the sensitivity of MAS NMR experiments of nucleosome arrays at 100 K and show that well-resolved 13C-13C MAS NMR correlations can be obtained much more efficiently. We evaluate the effect of temperature on the chemical shifts and linewidths in the spectra and demonstrate that changes are relatively minimal and clustered in regions of histone-DNA or histone-histone contacts. We also compare samples prepared with and without DNA and show that the low temperature 13C-13C correlations exhibit sufficient resolution to detect chemical shift changes and line broadening for residues that form the DNA-histone interface. On the other hand, we show that the measurement of DNP-enhanced 15N-13C histone-histone interactions within the nucleosome core is complicated by the natural 13C abundance network in the sample. Nevertheless, the enhanced sensitivity afforded by DNP can be used to detect long-range correlations between histone residues and DNA. Overall, our experiments demonstrate that DNP-enhanced MAS NMR spectroscopy of chromatin samples yields spectra with high resolution and sensitivity and can be used to capture functionally relevant protein-DNA interactions that have implications for gene regulation and genome organization.
Keywords: Nucleosome arrays, Dynamic nuclear polarization, Magic angle spinning, Protein-DNA interactions
Graphical Abstract
1. Introduction
Chromatin is a complex biological polymer that packages DNA and regulates access to the genetic information stored in eukaryotic cells. The fundamental building block of chromatin is the nucleosome, a structure that comprises 147 bp of DNA wrapped around an octamer core of the histone proteins H2A, H2B, H3 and H4 [1, 2]. Each nucleosome core contains one H3-H4 tetramer and two H2A-H2B dimers, with regular contacts to the wrapped DNA mediated by positively charged residues [2, 3]. Inside the nucleosome core, each histone adopts a relatively rigid helix-turn-helix motif while the dynamic and unstructured N- and C-terminal histone tails extend away from the core where they are subjected to numerous post-translational modifications (PTMs) [4, 5].
The presence of both rigid and dynamic components and the importance of chromatin interactions on many different length and timescales have presented a challenge for structural biologists. While there are numerous cryo-electron microscopy (cryo-EM) and X-ray crystallography structures of nucleosomes in complex with different chromatin interacting proteins, these structural models often lack electron density for the dynamic components of the assembly [6–8]. Solution NMR spectroscopy can be used to investigate the dynamic histone tails and the interactions of single nucleosomes with other proteins, however, it has been difficult to extend these studies to chromatin polymers [9–12]. While magic angle spinning (MAS) NMR spectroscopy can capture both the dynamic and rigid components in the chromatin polymer context, resonance assignments and structural constraints have typically required large amounts of sample and long acquisition times [13–18]. The sample requirement can be particularly limiting in this case as the assembly of homogeneous chromatin polymers requires the recombinant production of two DNA pieces and four histone proteins in both isotopically labeled and natural abundance variations. The structural and mechanistic studies of histone PTMs can further exacerbate this problem as the preparation of such samples is often laborious and inefficient, especially if isotopic labeling is required.[13, 19]
To address these limitations, here we turn to dynamic nuclear polarization (DNP) and take advantage of its unprecedented ability to increase the sensitivity of MAS NMR experiments of complex biological samples [20–27]. In a DNP-enhanced MAS NMR experiment, polarization is transferred from unpaired electron spins to nuclei, resulting in significant gains in nuclear polarization [28–30]. The source of unpaired electron spins is usually a small stable nitroxide-based biradical such as AMUPol that is mixed into the sample [31]. MAS DNP NMR experiments are then performed at 100 K where the sample is irradiated with high-power, high-frequency microwaves that excite the electron spin transitions and facilitate polarization transfer [32]. Typical DNP enhancements for biological systems are in the 10 – 150 range, which can reduce data acquisition times from weeks to days and enable the structural investigation of μg amounts of material [23, 25, 33]. Therefore, DNP-enhanced MAS NMR spectroscopy can potentially provide tremendous benefits for chromatin structural studies and allow the capture of functionally important but elusive interactions such as those between the nucleosomes within a chromatin polymer, between histone tails and chromatin effector proteins, or among remodelers, transcription factors and nucleosomal DNA.
While DNP can offer much needed sensitivity to characterize precious amounts of biological material, the low temperatures required for efficient polarization transfer often lead to severe line broadening [34, 35]. Potential sources of line broadening at low temperatures include sample heterogeneity, the presence of dynamic and unstructured regions in the protein of interest, the rearrangement of crystallographic water molecules, the temperature dependent motions of specific side-chains, or paramagnetic relaxation enhancement effects due to close interactions with the polarization agent [36–42]. As these issues can hamper the analysis and interpretation of data, it is important to understand the effects of low temperatures on chromatin samples and to evaluate the information content of DNP-enhanced MAS NMR chromatin spectra. In particular, we wondered whether there are structural differences between nucleosomes studied at 295 K and 100 K, whether the resolution of the low temperature spectra would allow the detection of meaningful structural interactions within the nucleosome, and whether tailored labeling schemes and DNP would enable the investigation of functionally relevant protein-protein and protein-DNA long-distance contacts. Our results show that nucleosome arrays compacted in the presence of Mg2+ display high resolution 13C-13C spectra at low temperatures and that histone-DNA contacts within the nucleosome can be identified through chemical shift perturbation and linewidth analysis. Long-range histone-DNA contacts can be captured through 15N-13C experiments such as TEDOR, while the measurement of specific protein-protein interactions is complicated by the multicomponent nature of the array polymer and the presence of a large natural abundance 13C network.
2. Experimental details
2.1. Materials
All reagents and solvents were purchased from Sigma Aldrich or Fisher Scientific. Isotopically enriched reagents were purchased from Cambridge Isotope Laboratories. Dialysis kits and protein concentrators were obtained from Thermo Fisher Scientific and Sartorius, respectively. For size-exclusion chromatography, a Superdex 200 10/300 column was used with an ÄKTA pure protein purification system (GE Healthcare Life Sciences/Cytiva). For ion-exchange, we used a 5 mL HiTrap SP HP column on the ÄKTA pure protein purification system (GE Healthcare Life Sciences/Cytiva). Reverse-phase high performance liquid chromatography (RP HPLC) was performed on a 2545 Binary Gradient Module Waters system equipped with a 2484 UV/vis detector. For prep scale RP-HPLC purification, we relied on a Waters XBridge BEH C18 19 mm × 250 mm, 10 μm particle size column, while analytical measurements were performed using a Symmetry300 C18 4.6 mm × 150 mm, 5 μm particle size column. HPLC solvent A contained 100% H2O + 0.1% trifluoroacetic acid (TFA), while solvent B contained 100% acetonitrile + 0.1 % TFA. Mass spectrometry analysis was conducted on an Agilent 6230 Accurate-Mass TOF-MS. Gel images were acquired using a camera and light box from Fotodyne Incorporated. Protein absorbances were measured using a Nanodrop One Spectrophotometer by Thermo Scientific.
2.2. Histones and DNA expression and purification
Histones H4, H2A, H3 and H2B were expressed and purified according to ref. [43]. Briefly, histone plasmids were expressed in BL21(DE3)-Rosetta cells in either Luria Bertani media (natural abundance histones) or M9 media supplemented with 15N ammonium chloride (15N-labeled H3), 13C6-glucose (13C-labeled H4) or both (15N,13C-labeled H4). The cells were grown at 37 °C with shaking until OD600 = 0.6 and induced with 0.5 mM IPTG. Cells were collected 3 hours later by centrifugation at 5,000 xg for 20 minutes. The cells pellet was lysed, and the inclusion body was washed with lysis buffer (20 mM Tris pH = 7.6 at 4 °C, 200 mM NaCl, 1 mM EDTA, 1 mM 2-mercaptoethanol) containing 0.1% v/v of Triton-X. The inclusion body pellet was then resuspended in suspension buffer (6 M guanidine, 20 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 7.5 at 4 °C), incubated at 4 °C for 120 minutes, and centrifuged for 30 min at 27,000 xg. The supernatant was dialyzed against buffer containing 7 M urea, 10 mM Tris, 1 mM EDTA, 100 mM NaCl, 1 mM DTT, pH 7.5 at 4°C, and then purified by cation exchange chromatography. Pure fractions were collected and loaded onto a Waters Xbridge BEH C18 prep-size column and fractions were collected using 30%–70% solvent B gradient. SDS-PAGE, analytical RP-HPLC and mass spectrometry were used to assess protein purity (Fig S1).
12×601 DNA (12 repeats of the 601 DNA sequence separated by 30 bp linkers, ref. [44]) was prepared according to ref. [43, 45]. Briefly, the DNA plasmid encoding the 12×601 DNA construct was transformed into DH5α cells and cells were grown in Terrific Broth media for 18 hr at 37 °C. Large scale plasmid purification was performed by first suspending the bacterial cells in alkaline lysis buffer I (50 mM glucose, 25 mM Tris-HCl, 10 mM EDTA, pH 8.0). Later, the sample was mixed quickly in alkaline lysis buffer II (0.2 M NaOH, 1% sodium dodecyl sulfate), and the cellular debris was precipitated using alkaline lysis buffer III (4 M potassium acetate, 2 N acetic acid). The plasmid DNA was precipitated using isopropanol precipitation at 25 °C. The DNA pellet was then suspended in TE buffer (10 mM Tris-HCl, 50 mM EDTA, pH 8.0), containing 20 mg/mL RNase A, and incubated at 37 °C for 24 hours. To remove proteins from nucleic acids, phenol chloroform extraction was performed three times. To remove the digested RNA, the plasmids were precipitated by PEG precipitation (10% (w/v) PEG 6000, 0.5 M NaCl). The DNA plasmids were then digested with the Eco-RV restriction enzyme (New England Biosciences), the undesired small fragments were removed by another PEG purification, and the pure 12×601 DNA fragment was precipitated from solution by ethanol extraction. The DNA pellets were re-dissolved in TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0), and stored at −20 °C.
2.3. Histone octamers and nucleosome arrays assembly
Histone octamers and nucleosome arrays were assembled according to ref. [43]. Briefly, lyophilized histones were dissolved separately in buffer containing 6 M guanidinium HCl, 20 mM Tris, 5 mM dithiothreitol, pH 7.6 at 4 °C. The concentration of each solution was determined and the histones were combined in a 1.1:1.1:1.0:1.0 ratio of H2A:H2B:H3:H4. The mixture was dialyzed against buffer containing 2 M NaCl, 10 mM Tris, 1 mM EDTA, 1 mM dithiothreitol, pH 7.6 at 4 °C. The refolded histone octamers were purified by size exclusion chromatography, and stored in 50% glycerol at −20 °C. The octamer purity was assessed by 18% SDS-PAGE gel electrophoresis. Nucleosome arrays were reconstituted by combining 12-mer DNA and histone octamers in a 1.6:1 ratio in 2M TEK buffer (2 M KCl, 10 mM Tris pH 7.5 at 4 °C, 0.1 mM EDTA, 1 mM dithiothreitol). 0.3 eq of the 155 bp MMTV DNA was added to the mixture to bind excess octamers. This was followed by salt gradient dialysis against 10 mM TEK buffer (10 mM KCl, 10 mM Tris pH 7.5 at 4 °C, 0.1 mM EDTA, 1 mM DTT), for ~24 hr at 4 °C. The assembly and purity of nucleosome arrays was visualized using APAGE (2% acrylamide, 1% Biorad agarose) gel electrophoresis (Fig. S2).
2.4. Sample preparation for conventional NMR and DNP-enhanced NMR spectroscopy
For MAS NMR experiments at 275 K, purified nucleosome arrays (2.5 μM, ~20 ml) were concentrated to a final volume of 5 mL using 10,000 molecular weight cutoff concentrators. To precipitate the arrays, 6 mM MgCl and 0.01% NaN3 were added to the solution. The arrays were incubated on ice for 10 min and centrifuged at 5000 xg for 1–2 hr at 4 °C using a swinging bucket centrifuge. The sample was then scooped into a thin-wall 3.2 mm rotor and centrifuged gently to distribute equally inside the rotor. The final amount of sample packed into the NMR rotor was determined by measuring the A260 absorbance of DNA of the leftover supernatant and subtracting it from the A260 absorbance before centrifugation. The reported temperature reflects the temperature of the cooling gas, while we estimate that the sample temperature is 10 – 15 °C higher.
To perform DNP experiments at 100 K, the sample needs to be cryoprotected with a glassy matrix before freezing. We cryo-protected the nucleosome arrays with “DNP juice” which consists of glycerol, D2O and H2O in a 60/30/10 w/v ratio. We used glycerol-d8 (D8, 99%), or when available, 13C-depleted deuterated glycerol (12C3, 99.95%; D8, 98%). The solution also contained 10 mM AMUPol (Cortecnet). To prepare sample A, we prepared nucleosome arrays (2.5 μM, 3 mL), concentrated the sample to a final volume of 1 mL, and precipitated the arrays using 6 mM MgCl. The precipitated pellet was impregnated with DNP juice and scooped into 1.9 mm zirconia MAS rotor (Fig. 1a). The rotor was closed with a top and bottom silicone plug and an SP1 drive cap. Sample B was prepared by concentrating nucleosome arrays (2.5 μM, ~3 mL) into DNP juice to a final volume of 15 μL. 10 μL of the sample were then pipetted into the rotor (Fig. 1b). Octamer samples were prepared by concentrating 13C, 15N-labled H4 histone octamers in a solution that contained 60% w/v 12C glycerol-d8, 40% D2O, 10% H2O, 800 mM NaCl, 4 mM Tris, and 10 mM AMUPol. The high salt concentration is necessary to prevent octamer dissociation into H2A/H2B dimers and H3-H4 tetramers in the absence of DNA.
Figure 1.
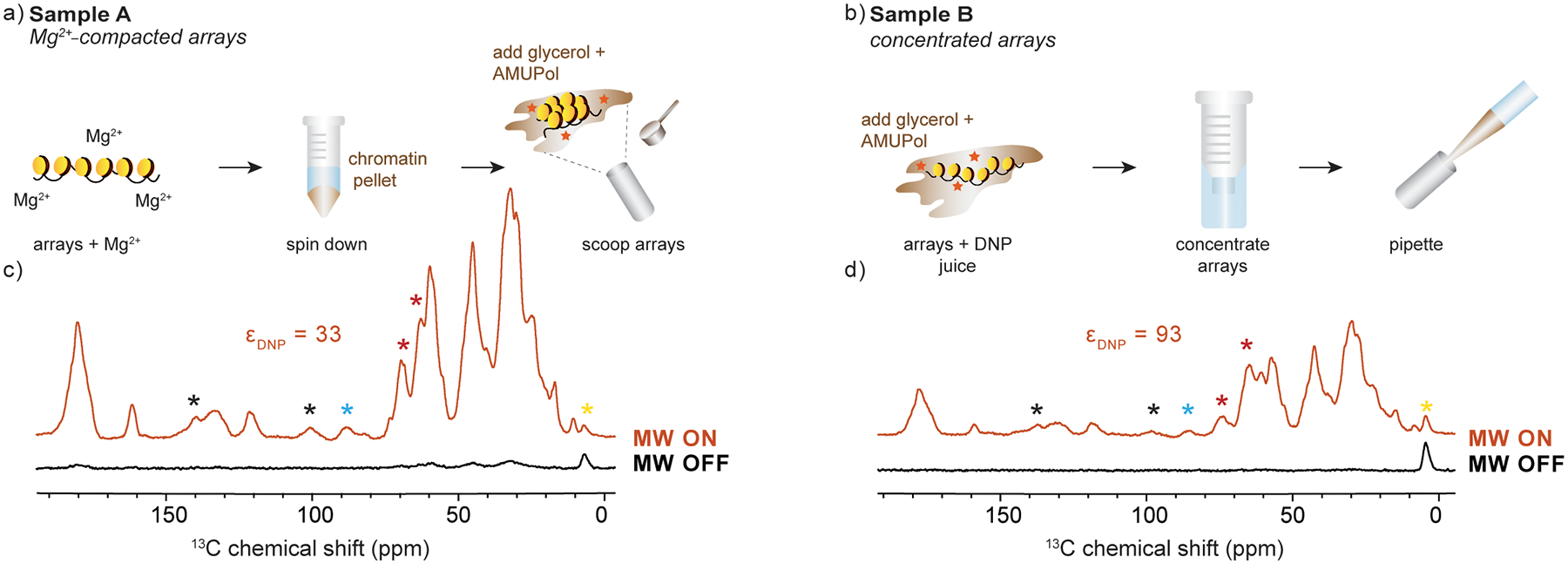
(a-b) Sample preparation and DNP MAS rotor packing methods for Sample A and Sample B, respectively. (c-d) Low temperature 1D 13C spectra with DNP (MW on, orange) and without DNP (MW off, black) for Sample A and Sample B, respectively. Each spectrum was acquired at ω1H = 600 MHz and 100 K with 64 scans and ωr =12 kHz. Red asterisks denote natural abundance glycerol peaks, black asterisks denote spinning sidebands, yellow asterisks denote a peak arising from the DNP rotor silicon plug, and blue asterisks denote natural abundance DNA signals. εDNP – representative DNP enhancement for each sample.
2.5. Solid-state NMR spectroscopy
Solid-state NMR experiments at ambient temperatures were conducted on a 750 MHz NMR spectrometer, equipped with an AVANCE II Bruker console and a triple resonance (1H, 13C, 15N) 3.2 mm E-free Bruker probe. The spinning frequency was set to 11.1 kHz. Spectra were referenced to the 40.49 ppm 13C peak of adamantane [46]. 2D 13C-13C DARR experiments [47] were recorded at 275 K with the following parameters: 1024 points and 9 ms acquisition in the direct dimension, 256 points with 3.2 ms acquisition in the indirect dimension, 64 scans, SPINAL64 decoupling, 20 ms mixing, and an interscan delay of 4 s. 1H and 13C and 15N pulses were set to 3 μs and 5 μs, respectively.
2.6. DNP NMR spectroscopy
DNP experiments were performed on a 600 MHz Bruker DNP NMR spectrometer equipped with a NEO console and a 395 GHz gyrotron for high-power microwave irradiation. All spectra were recorded using a triple resonance (1H, 13C, 15N) 1.9 mm low temperature MAS probe. The MAS frequency was set to 12 kHz and spectra were referenced to the 40.49 ppm 13C peak of adamantane at room temperature or to the silicon plug signal the 3.2 ppm at 100 K. The DNP enhancement was measured by recording spectra with and without microwave irradiation at 5 W. 2D 13C-13C DARR experiments were recorded at 100 K with the following parameters: 1536 points and 16 ms acquisition in the direct dimension, 640 points with 8 ms acquisition in the indirect dimension, interscan delay of 4 s, 10 ms mixing, 16 scans for the nucleosome arrays sample, or 32 scans for the octamer sample. 1D TEDOR experiments were recorded with 4096 scans with the following parameters: 1024 points, 10 ms acquisition, interscan delay of 4 s, and 1.3, 5.3, 10.6, 16 or 20 ms TEDOR mixing [48]. A 2D TEDOR experiment was recorded with 5.3 ms mixing, 32 scans, 1024 points and 10 ms acquisition in the direct dimension, and 128 points with 5 ms acquisition in the indirect dimension. 1H, 13C and 15N pulses were set to 2.5 μs, 3 μs and 6.25 μs, respectively.
2.7. EPR spectroscopy
EPR experiments to measure the concentration of radical in the rotor were performed on a 9.3 GHz Bruker EMXplus X-band CW EPR spectrometer equipped with a standard high sensitivity resonator. To perform the measurements, the rotor was placed into a standard X-band 707-SQ-250M EPR tube (Wilmad Labglass), the EPR spectrum was measured, double integration was performed, and the integrated intensity was compared to a standard curve prepared with rotors containing known concentrations of AMUPol (Fig. S3).
2.8. Data analysis
NMR spectra were processed with TopSpin 4.0.5 and analyzed using NMRFAM-SPARKY and CCPNMR 3.0.2 [49, 50]. Nucleosome images were rendered using the Chimera software [51]. Resonance assignments for H4 were transferred from ref. [16]. EPR analysis was performed using EasySpin [52].
3. Results and Discussion
3.1. Preparation and DNP enhancements of chromatin samples
Homogeneous chromatin polymer samples are typically reconstituted using a long piece of DNA that contains twelve repeats of the strong nucleosome positioning sequence known as Widom “601” DNA [44]. In our case, we used 30 base pair linkers between the nucleosomes, resulting in a DNA sequence of 12×177 bp repeats. Histone octamers were reconstituted separately at high salt by mixing the four histones in equimolar ratios. Since room temperature resonance assignments are available for histone H4 [16], we decided to focus on this component of the assembly and to characterize its interactions with DNA. We therefore prepared a 13C,15N-labeled version of H4, while all other histones and DNA were used at natural abundance (Fig. S1). After preparation, the histone octamers and DNA were mixed, and the salt concentration was gradually reduced so that the octamers could position on the DNA through electrostatic interactions. To ensure homogeneous sample preparation and uniform distribution of the nucleosomes in the polymer, a small amount of a short piece of DNA with low propensity for nucleosome formation (“buffer DNA”) was added to remove excess octamers. The resulting 12-mer nucleosome arrays were purified using Mg2+ precipitation (Fig. S2). It should be noted that labeled H4 represents only 12% of the final nucleosome array (by weight), thus presenting a significant sensitivity challenge for structural analysis with NMR.
The DNP sample preparation protocol and the rotor packing technique can be crucial in ensuring good DNP enhancements and optimal sensitivity [53]. In this case, we used 1.9 mm MAS rotors with an effective volume of 10 μL which required an efficient protocol for packing a large amount of sample into a small volume. To determine the best strategy for mixing chromatin, polarization agent and “DNP juice” (glycerol, D2O and H2O in a 60/30/10 w/v ratio), we prepared two different samples (Fig. 1a,b). In Sample A, chromatin polymers were precipitated with ~ 6 mM Mg2+, producing a compact gel. The gel pellet was impregnated with DNP juice and 10 mM AMUPol and the mixture was scooped into the rotor. In this case, the effective concentration of biradical inside the rotor was ~ 6 mM as determined by EPR spectroscopy (Fig. S3). In sample B, chromatin arrays were pre-mixed with 10 mM AMUPol and DNP juice, and then concentrated without Mg2+ using a standard centrifuge tube concentrator. In this case, the sample remained as a viscous liquid and could be pipetted into the rotor. This type of preparation resulted in ~ 15 mM of AMUPol inside the rotor (Fig. S3).
The resulting enhancements and signal-to-noise (SNR) ratios differed depending on the rotor packing method (Fig. 1c, d). Packing chromatin by concentration (Sample B) resulted in greater enhancement, as this strategy appears to concentrate more biradical into the rotor, and likely yields much more uniform distribution of the polarization agent around chromatin polymers. On the other hand, precipitation of chromatin directly into the rotor (Sample A) afforded the packing of larger amounts of chromatin. Overall, Sample A produced a higher SNR ratio despite the lower enhancement. We, therefore, proceeded with this packing strategy for the nucleosome array samples described below.
3.2. Comparison of DNP-enhanced and room temperature chromatin spectra
To investigate the resolution of the DNP-enhanced low temperature chromatin spectra, we prepared two new nucleosome array samples containing 13C,15N-labeled H4, and proceeded to record 2D 13C-13C correlations with DARR mixing. One of the samples was packed into a 1.9 mm rotor with 10 mM AMUPol and was investigated at 100 K and in a 600 MHz spectrometer with DNP. This sample had a reproducible DNP enhancement of ~ 33. The second sample was prepared into a 3.2 mm rotor and experiments were performed at 275 K in a conventional 750 MHz NMR spectrometer. Despite the lower amount of sample used in the DNP experiments (3 mg vs 10 mg total array sample), DNP allowed us to shorten the acquisition time from 18 hr to 11 hr. While it is possible to obtain high sensitivity DARR data of chromatin with DNP in less time, here we opted to collect a large number of points in the indirect dimension (256 vs 640) in order to maximize the resolution of the spectrum.
The overlay of the two spectra (Fig. 2a) provides an excellent opportunity to evaluate the resolution and the information content of the low temperature chromatin experiments. While the DNP spectrum contains several broad regions, there are also many well resolved peaks exhibiting 13C linewidths in the range 1 – 1.6 ppm (Fig. 2b). In comparison, the room-temperature spectrum obtained at the slightly higher magnetic field exhibited linewidths in the range of 0.7 – 1 ppm. The nucleosome array samples used in this study are a highly heterogeneous system that contains both rigid and dynamic components. The dynamic H4 tail (residues 1 – 24) is not visible in dipolar experiments at room temperature [16], and likely freezes out in different conformations at low temperature and thus remains undetectable. The nucleosome core, on the other hand, is relatively rigid and is expected to adopt a more uniform conformation at low temperature. The rigidity is further improved by the presence of Mg2+ ions which compact chromatin and further reduce dynamics and heterogeneity. Previous work has shown a correlation between the rigidity of a biological system and the linewidths observed in DNP-enhanced low temperature experiments [21, 27, 34, 54]. For example, the T3SS needle MxiH protein, a highly rigid system, displays DNP linewidths on the order of 1 ppm at 600 MHz [54], while other systems of more heterogeneous nature may exhibit low temperature linewidths greater than 2 ppm [22, 23, 37, 55].
Figure 2.
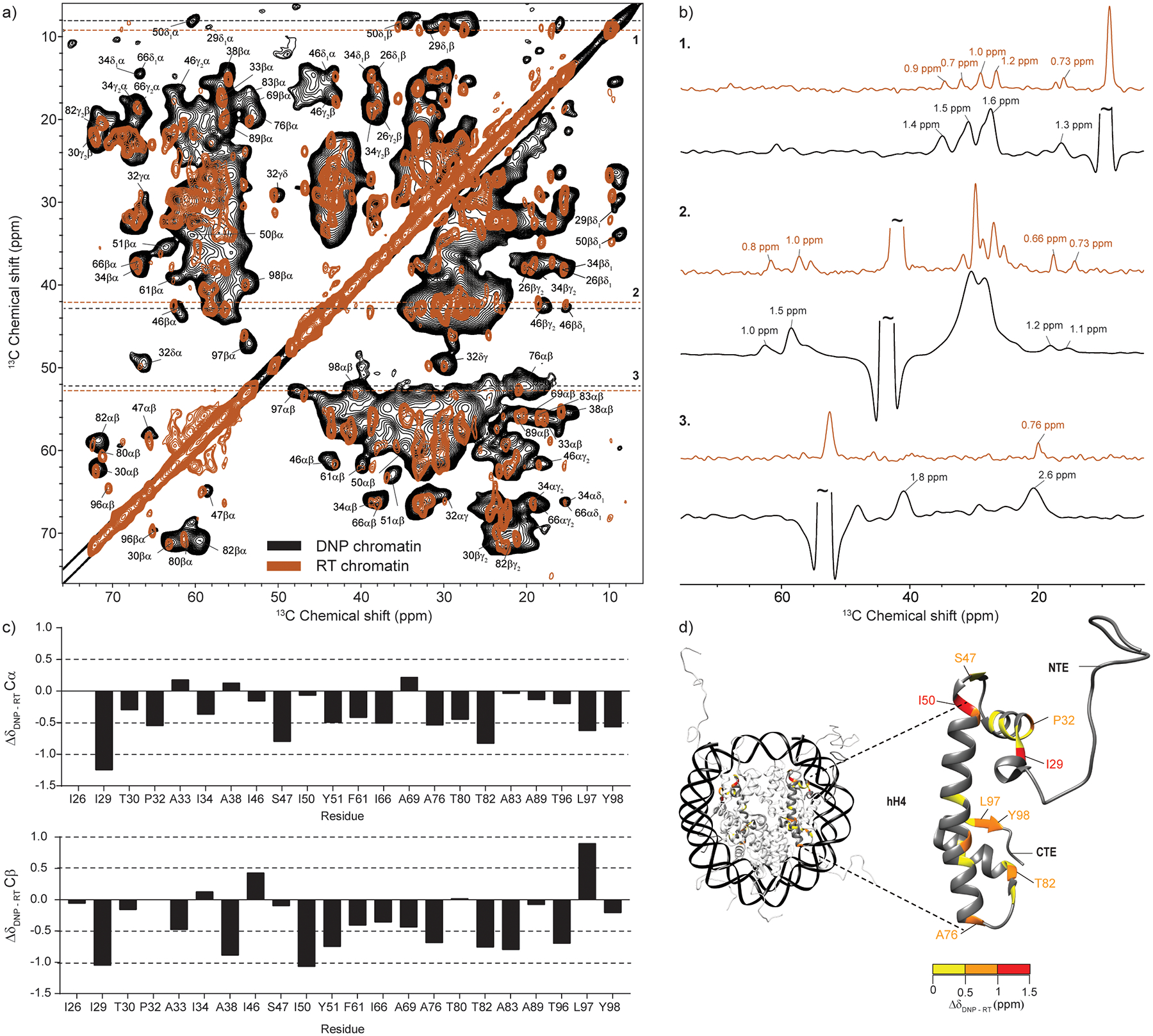
(a) Overlay of DNP-enhanced and room-temperature 13C-13C DARR correlation spectra of 13C,15N-labeled H4 nucleosome array samples. The DNP spectrum (black) was acquired at 100 K and ω1H = 600 MHz with 16 scans, ωr =12.5 kHz and 10 ms DARR mixing time. Sample contained 0.4 mg of labeled protein. The room temperature spectrum (brown) was acquired at 275 K and ω1H = 750 MHz with 64 scans, ωr =11.1 kHz and 20 ms DARR mixing time. Sample contained 1 mg of labeled protein. The labels correspond to assignments obtained at 275 K according to ref. [16]. (b) Representative slices and linewidths in the two spectra. (c) Analysis of the Cα and Cβ chemical shift changes for resolved correlations in the spectra. (d) Chemical shift changes for histone H4 mapped on the nucleosome surface (PDB ID: 1KX5, ref. [4]). H4 residues with no resolved Cα and Cβ correlations in the DNP spectrum are shown in dark grey. The color coding reflects the largest of either the Cα or Cβ chemical shift change.
Analysis of the DNP-enhanced DARR spectrum shows many correlations that can be identified based on their room temperature assignments. For example, the spectrum contains resolved correlations for I26, which is the first visible core residue typically identified in room temperature spectra. Comparison of the chemical shifts obtained at cryogenic and ambient temperature for 22 resolved Cα-Cβ correlations reveals that most chemical shift changes are within 1 ppm (Fig. 2c). The largest chemical shift changes are observed for I29 Cα and Cβ atoms and the I50 Cβ atom. These residues also experience chemical shift changes for their side-chain atoms. I29 and I50 are positioned at the top of the nucleosome (Fig 2d), close to the DNA entry and exit site, and make important contacts with the H3 α-helices in the H3/H4 dimer. This region also includes S47 and P32 that show changes in the 0.5 – 1.0 ppm range. Closer to the C-terminus, changes in this range are experienced by A76, T82, L97 and Y98, residues that make strategic contacts with H3 or DNA. It should be pointed out that biological systems at low temperature often exhibit 13C chemical shift and linewidth changes on the order of 1 ppm or larger [27, 37]. These changes are often more severe for residues that are in close contact with the surrounding water molecules [37]. This is attributed to rearrangements in the water hydrogen bond networks upon slow freezing that change the chemical environment surrounding the protein [37]. Modeling of water molecules has suggested that extensive water networks play an important role in DNA-histone and histone-histone interactions in the nucleosome [4]. Therefore, residues at these interfaces could be particularly sensitive to the environmental changes induced by freezing. Solid-state NMR at ambient temperature has also shown that some of the detected residues are part of a histone network that can sense changes in dynamics and interactions with DNA [18]. Despite these local detectable perturbations in chemical shifts, however, the overall nucleosome structure appears intact and is not dramatically rearranged by the freezing process required for DNP.
Protein spectra acquired near 100 K often exhibit weak methyl group correlations and strong peaks in the aromatic region [34, 38, 39]. At 100 K, for example, the methyl group three-fold symmetry rotations typically slow down to 10−3 – 10−6 s−1, rates of motion that can interfere with the pulse frequencies used for recoupling and decoupling under magic angle spinning conditions [38]. This can cause the broadening or disappearance of methyl group signals in cross polarization-based 1D and 2D experiments. Surprisingly, the low temperature DNP 13C-13C DARR nucleosome array spectrum contains strong and resolved methyl group correlations for many assignable methyl-bearing sidechains, including Thr, Ala and Ile. While most of the relevant cross-peaks fall within the average 1 – 1.6 ppm linewidth (Fig. 2b), there are some methyl bearing residues that experience line-broadening consistent with interfering motions, e.g. the Cα-Cβ correlation of A76 broadens to 2.6 ppm (Fig. 2b). This residue also presents a large chemical shift change upon freezing as described above.
Aromatic sidechains, on the other hand, experience two-fold flip motions that interfere with recoupling and decoupling at ambient temperature [39]. However, at the cryogenic temperatures used for DNP, these motions can slow down enough to produce strong correlations [34, 38, 56]. We observed many strong aromatic correlations in our DNP spectrum while the room temperature spectrum has no intensity in this region (Fig. 4S). We took advantage of this phenomenon and obtained low temperature assignments of the aromatic side-chains to complement the published H4 assignments [16] (Table S1).
Figure 4.
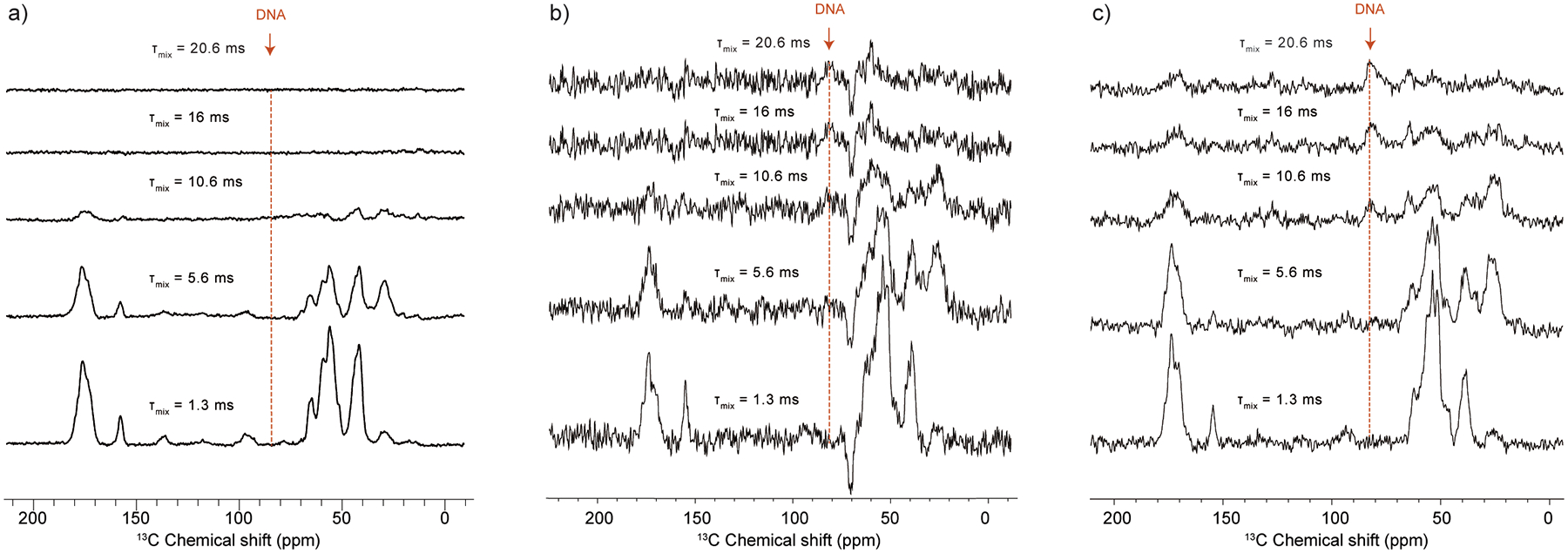
DNP-enhanced 1D ZF-TEDOR experiments of array samples prepared with a) 13C,15N-labeled H4, b) 13C-labeled H4, 15N-labeled H3, and c) 15N-labeled H3. The spectrum in a) was acquired with 16 scans, while the spectra in b) and c) were acquired with 4096 scans. Samples in a) and c) were prepared with 13C-depleted glycerol, while the sample in b) was prepared with natural abundance glycerol, causing the negative peaks in the spectrum. Samples contained ~ 4.5, 4.5 and 2.2 mg of nucleosome arrays, respectively. τmix = TEDOR mixing time.
Overall, the comparison between experiments performed at 100 K and 295 K suggests that nucleosome array samples precipitated with 6 mM Mg2+ produce resolved 13C-13C DNP spectra of the nucleosome core where many correlations could be assigned based on the 295 K spectrum. Low temperature 13C chemical shift changes remain on average below 1 ppm consistent with other samples studied by DNP and at low temperature. The most significant chemical shift changes are experienced by H4 residues that make key contacts with other histones and DNA, and are attributed to rearrangements of the nucleosome water networks upon freezing. The sensitivity enhancement afforded by DNP and the favorable low temperature dynamics of the aromatic sidechains at 100 K have also allowed us to complete the assignments of aromatic correlations that are invisible in spectra acquired at ambient temperature.
3.3. Comparison of DNP-enhanced NMR spectra of nucleosome array and octamer samples
While DNP can provide much needed sensitivity to detect elusive long-range structural correlations in complex biological systems, in practice most studies of this nature typically take advantage of specifically tailored labeling schemes or correlations where the relevant interactions can be resolved and assigned unambiguously. Examples include the strategic placement of 13C and 15N isotopic labels to detect key intermolecular or intramolecular interactions [56, 57], the use of rare nuclei such as 19F [58], or the detection of correlations between proteins, ligands and components with unique chemical shifts [59–61]. While uniformly-13C labeled proteins are relatively straightforward to prepare, structural studies based on changes in the 13C-13C DNP spectra of uniformly labeled large proteins are challenging due to often severe peak crowding and line-broadening [27]. Since the DNP spectra of 13C,15N-H4 nucleosome arrays present many resolved and assigned cross-peaks, we wondered whether they can be used to detect meaningful structural interactions despite the noted complications with uniformly labeled samples. To test this possibility, we prepared a sample containing 13C,15N-labeled histone octamers which represent the intact protein core of the nucleosome but without the wrapped DNA. In this case, we expected that H4 residues in close contact with DNA in the full nucleosome context would experience changes in chemical shifts and/or line broadening. This sample was prepared for DNP following the concentration method, similar to Sample B, as octamers without DNA cannot be precipitated by Mg2+. The observed enhancement was ~ 130, with an effective AMUPol concentration of ~ 15 mM inside the rotor (Fig. S5).
Comparison of the spectra of samples with and without DNA revealed that the overall pattern of 13C-13C correlations remains very similar (Fig. 3a). This is expected since the absence of DNA should not change the overall structure of H4 once it is incorporated into the octamer fold. There is, however, an increase in the linewidth for many resolved cross-peaks suggesting the presence of much more heterogeneity in the octamer when DNA is absent (Fig. 3b). Linewidth changes are particularly prominent for residues I46 and S47 in loop L1 and residues L97 and Y98 at the C-terminal end of H4 (Fig. S6). As expected, there are also noteworthy chemical shift changes (Fig. 3c,d). This includes S47, L97 and Y98, as well as residues I29, P32, and T80. These positions serve as primary H4 contacts with DNA and are already disturbed by the freezing process. Peak doubling is observed for T30 Cα-Cβ and P32 Cβ-Cγ correlations, at the base of the dynamic H4 tail, implying the presence of heterogeneity at this site when DNA is absent.
Figure 3.
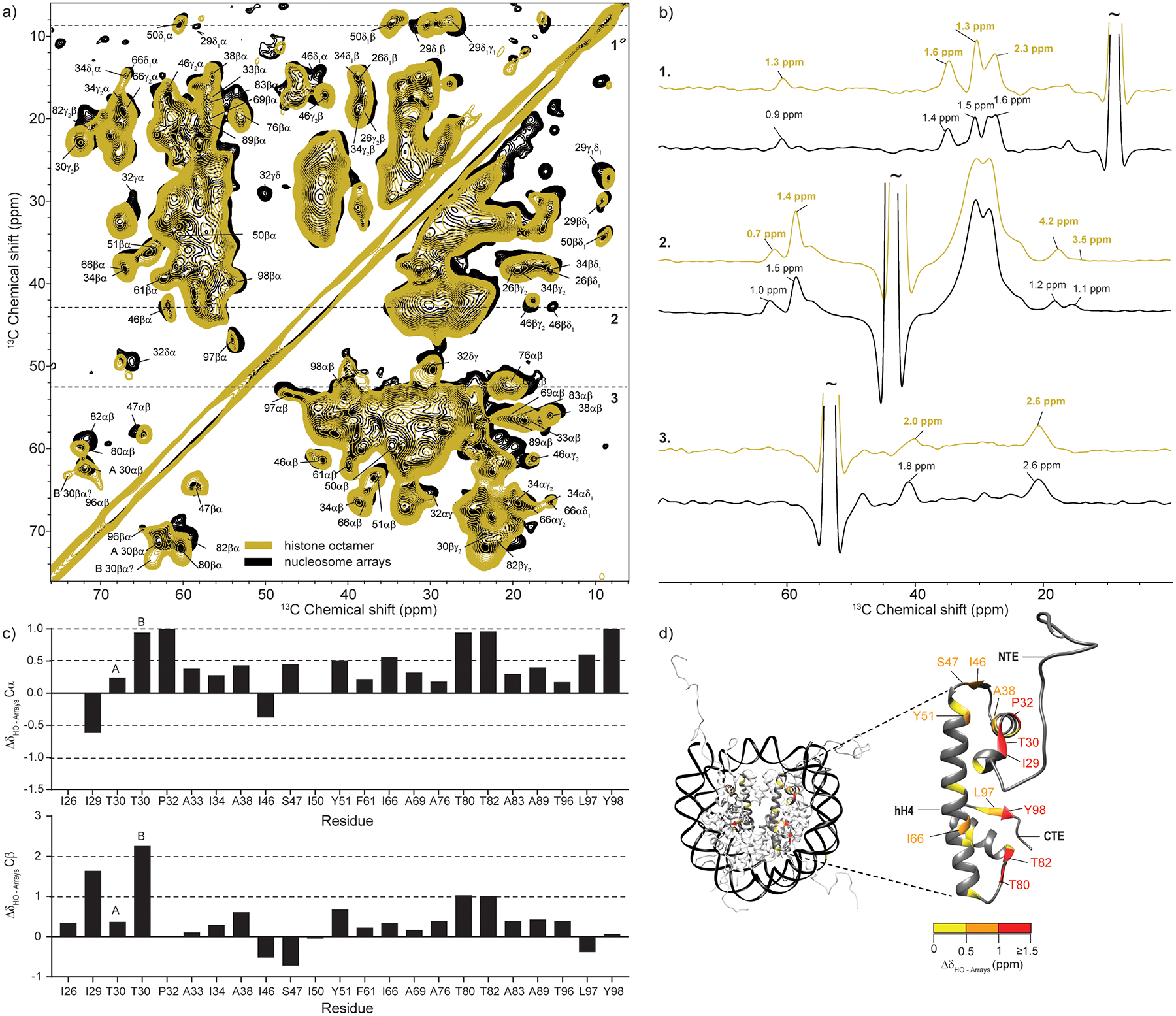
(a) Overlay of DNP-enhanced 13C-13C DARR correlation spectra of 13C,15N-labeled H4 nucleosome array (blue) and octamer (yellow) samples. Both spectra were acquired at 100 K and ω1H = 600 MHz with 16 scans, ωr =12.5 kHz and 10 ms DARR mixing time. (b) Representative slices and linewidths in the two spectra. (c) Analysis of the Cα and Cβ chemical shift changes for resolved correlations in the array and octamer spectra. A and B represent two conformations for T30. (d) Chemical shift changes for histone H4 mapped on the nucleosome surface (PDB ID: 1KX5, ref. [4]). H4 residues with no resolved Cα and Cβ correlations in the DNP spectrum are shown in dark grey. The color coding reflects the largest of either the Cα or Cβ chemical shift change.
Histone proteins contact DNA through several types of interactions (Fig. S7) [4]. The first possibility is the formation of a direct hydrogen bond between a backbone amide and the phosphate oxygen atoms on DNA as exemplified by residues I46 and T80. A second option includes the formation of indirect contacts between the backbone carbonyl atoms and the DNA phosphate oxygen atoms that are mediated through hydrogen bonds with water, e.g. I46. Indirect contacts through water molecules are also possible for some sidechains, e.g. S47. Third, there are direct contacts between lysine and arginine sidechains and DNA. Unfortunately, the lysine and arginine correlations in the low temperature DARR experiments are not well resolved for both the octamer and nucleosome array samples. To gain better insight into the interactions of these sidechains, we recorded a 15N-13C TEDOR spectrum where these residues produce unique cross-peaks (Fig. S8). Analysis of the samples prepared with and without DNA reveals that the arginine Nε-Cζ and Nη1,2 - Cζ correlations are much less intense in the octamer case, while the lysine Nζ-Cε region remains strong. While both arginine and lysine sidechains can form hydrogen bonds with the phosphate atoms of DNA, several H4 arginine sidechains are directly inserted into the minor groove which severely restricts their flexibility and orientation (e.g. R45 in Fig. S7). Therefore, the absence of DNA would likely result in heterogeneity, line-broadening and the observed low intensity for these sites. It is important to note that direct sidechain-DNA interactions are not restricted to lysine and arginine residues. For example, in H4, T80 can also form a direct sidechain hydrogen bond with the phosphate oxygen atom on a cytosine base (Fig. S7), thus substantiating the large chemical shift changes for this residue in the octamer sample. Overall, however, the detected changes in chemical shifts and peak linewidths are mostly restricted to H4 regions that are in close proximity to DNA, leaving the α2 and α3 helices relatively unperturbed (Fig. 3d).
These experiments demonstrate that DNP-enhanced MAS NMR correlations of chromatin can provide sufficient resolution to detect meaningful structural correlations even at the modest MAS frequencies used in this study. As the resolution can no doubt be improved by fast MAS at cryogenic temperatures [62], we expect that this approach can be used to study nucleosome interactions in more complex settings, e.g. in the presence of transcription factors or chromatin remodelers that often peel off the octamer from DNA in order to carry out their biological function [63, 64].
3.4. Observation of long-range intermolecular interactions in chromatin samples
The nucleosome polymers used in this study present a complex system that contains four histone proteins and DNA, with multiple interaction surfaces and points of contact. In the presence of Mg2+, chromatin also adopts a compact state mediated by interactions between the H4 tail and the DNA and/or acidic patch of neighboring nucleosomes [11, 65]. The characterization of these interactions by solid-state NMR methods can be challenging due to the complexity of the system, the presence of dynamic and ordered components at the same time, and the relatively large interaction distances that need to be measured. DNP-enhanced NMR spectroscopy could benefit such studies by providing much needed sensitivity and by capturing dynamic interactions at low temperatures. To evaluate these potential benefits for chromatin, we decided to focus on inter-histone interactions within individual nucleosomes using 15N-13C heteronuclear correlations. Our method of choice was the ZF TEDOR experiment [48], which has been used to measure 15N-13C distances in the 1 – 5 Å range in many complex biological systems with and without DNP [56, 59, 61, 66, 67].
To understand the TEDOR transfer dynamics in chromatin systems, we performed a series of 1D experiments with different mixing times on three different samples. The first sample was a uniformly 13C,15N-labeled H4 nucleosome array sample, similar to Sample A discussed above. This sample served as control and displayed the typical buildup behavior expected of a uniformly labeled protein (Fig. 4a). In this system, the one bond N-Cα and N-CO transfers peak at short mixing times while transfer to the aliphatic carbons of the sidechains reaches a maximum at intermediate mixing times (~ 5 ms) (Fig. 4a), and then rapidly declines due to transverse relaxation processes. The second sample contained 13C-labeled H4 and 15N-labeled H3 within the same octamer, while all other histones and DNA remained at natural abundance. In this case, the shortest possible 15N-13C distances are in the 4 – 5 Å range, representing backbone H3 15N amide correlations to nearby 13C-labeled H4 sidechains, or correlations between nitrogen-bearing sidechains on H3 and nearby H4 13C atoms. Considering the nature of these interactions, we expected that TEDOR buildup will be observed only at long mixing times. Surprisingly, this system exhibited buildup consistent with both short-range and long-range correlations (Fig. 4b). In particular, maximal N-Cα and N-CO buildup is observed with a mixing time of 1.3 ms, followed by transfer to the sidechains at 5.6 ms, similar to the uniformly labeled sample. As these mixing times are too short to capture the expected long-range correlations, we suspected that the buildup reflects transfer to the 1% natural abundance 13C atoms on H3. This was confirmed by a control sample that only contained 15N-labeled H3 with natural abundance 13C for all histones and DNA (Fig. S9). This sample exhibited a similar TEDOR buildup profile (Fig. 4c).
While the short-range transfer appears to involve the dilute 13C natural abundance network, both mixed 15N H3/13C H4 and control 15N H3 samples also have noticeable buildup at long mixing times, including transfer to the unique deoxyribose natural abundance 13C atoms of the nucleosomal DNA. Since both samples have very similar buildup profiles, it appears that long-range 1D 15N-13C TEDOR transfer in chromatin systems is also dominated by the 13C natural abundance network and selective isotopic enrichment of specific histones does not offer much advantage. This can be rationalized by the number of 15N-13C contacts within 5 Å for H3 and H4. This number is relatively small, while the natural abundance network that includes all H3 atoms, nearby H2A, H2B and DNA is much larger and may contribute significantly even if only ~ 1% of the carbon atoms are enriched. This situation is very different from measurements in mixed 15N/13C amyloid fibrils where the monomers adopt a parallel, in-register β-sheet arrangement [56, 67, 68]. In such a geometry, every backbone 15N amide atom in one monomer is typically within 5 Å from an enriched 13C atom on a neighboring monomer ensuring that relevant intermolecular distances can be captured selectively. In principle, enriched 15N-13C contacts between H3 and H4 can be observed in multidimensional experiments with much higher selectivity. However, even with the enhancement afforded with DNP, we were not able to obtain a 2D ZF TEDOR experiment at long mixing times with sufficient sensitivity to detect such contacts.
4. Conclusions
Nucleosome arrays present a significant challenge for structural investigations by MAS NMR spectroscopy due to the presence of a large number of components, the inherent dynamics in the histone tails, and the complexity of the involved protein-protein interfaces [13]. Here, we have taken a critical look at how DNP can contribute to such investigations by providing much needed sensitivity. Despite the expected line-broadening at low temperatures, 13C,15N-labeled H4 nucleosome array samples present well resolved 13C-13C spectra where many correlations can be identified from their room temperature assignments. This allowed us to evaluate chemical shift changes due to the freezing process and to obtain de novo assignments for aromatic correlations that are otherwise invisible in conventional MAS NMR experiments. Low temperature chemical shift changes are typically less than 1 ppm with more significant changes observed for residues that are positioned at histone-histone and histone-DNA interfaces. This indicates that there are potentially some slight differences between nucleosome structures obtained at ambient and low temperatures that might also be of relevance to other structural methodologies that routinely investigate structures at low temperatures such as X-ray crystallography and cryo-EM [4].
We took advantage of the well-resolved nucleosome array spectra to show that 13C-13C correlations can be used to detect meaningful structural changes upon removal of DNA. Such experiments may prove useful in the structural investigations of chromatin interacting proteins that perturb DNA, including transcription factors and chromatin remodelers. The situation is much more complicated if the detection of histone-histone interactions is desired. The complex nature of histone interactions and the significant natural abundance 13C network present a challenge for the measurement of intermolecular correlations through uniformly labeled 15N/13C mixed histone samples. There are, however, other possibilities. For example, 15N/13C or 15N/31P correlations can be measured between 15N-labeled histones and the unique deoxyribose 13C or phosphate 31P atoms of the DNA. Alternatively, 19F-based correlations might offer the unique ability to capture interactions on a much longer length-scale (10 – 20 Å) [69–71] such as those between individual nucleosomes. When combined with DNP [58], such strategies may allow the structural investigation of precious amounts of post-translationally modified nucleosome array samples or the characterization of protein-protein interactions in multicomponent chromatin assemblies where the concentration of each individual component may be severely limiting. Current and future developments in polarization agents, polarization transfer mechanisms, low temperature probes and high-field microwave technologies will no doubt continue to push the sensitivity and resolution capabilities of DNP to help capture elusive interactions in this complex biological system [72–74].
Supplementary Material
Acknowledgments
We thank Debelouchina lab members for helpful discussions. This work was supported by NIH R35 GM138382 and NSF MRI CHE 2019066 grants to G.T.D., and T32 GM008326 fellowships to N.E and B.E.A.
References
- [1].Kornberg RD, Chromatin structure: a repeating unit of histones and DNA, Science, 184 (1974) 868–871. [DOI] [PubMed] [Google Scholar]
- [2].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 A resolution, Nature, 389 (1997) 251–260. [DOI] [PubMed] [Google Scholar]
- [3].McGinty RK, Tan S, Recognition of the nucleosome by chromatin factors and enzymes, Curr Opin Struct Biol, 37 (2016) 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ, Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution, J Mol Biol, 319 (2002) 1097–1113. [DOI] [PubMed] [Google Scholar]
- [5].Allis CD, Jenuwein T, Reinberg D, Epigenetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 2007. [Google Scholar]
- [6].McGinty RK, Tan S, Principles of nucleosome recognition by chromatin factors and enzymes, Curr Opin Struct Biol, 71 (2021) 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G, Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units, Science, 344 (2014) 376–380. [DOI] [PubMed] [Google Scholar]
- [8].Machida S, Takizawa Y, Ishimaru M, Sugita Y, Sekine S, Nakayama JI, Wolf M, Kurumizaka H, Structural Basis of Heterochromatin Formation by Human HP1, Mol Cell, 69 (2018) 385–397 e388. [DOI] [PubMed] [Google Scholar]
- [9].Morrison EA, Bowerman S, Sylvers KL, Wereszczynski J, Musselman CA, The conformation of the histone H3 tail inhibits association of the BPTF PHD finger with the nucleosome, Elife, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kato H, van Ingen H, Zhou BR, Feng H, Bustin M, Kay LE, Bai Y, Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR, Proc Natl Acad Sci U S A, 108 (2011) 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rabdano SO, Shannon MD, Izmailov SA, Gonzalez Salguero N, Zandian M, Purusottam RN, Poirier MG, Skrynnikov NR, Jaroniec CP, Histone H4 Tails in Nucleosomes: a Fuzzy Interaction with DNA, Angew Chem Int Ed Engl, 60 (2021) 6480–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ, HP1 reshapes nucleosome core to promote phase separation of heterochromatin, Nature, 575 (2019) 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ackermann BE, Debelouchina GT, Emerging Contributions of Solid-State NMR Spectroscopy to Chromatin Structural Biology, Front Mol Biosci, 8 (2021) 741581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao M, Nadaud PS, Bernier MW, North JA, Hammel PC, Poirier MG, Jaroniec CP, Histone H3 and H4 N-terminal tails in nucleosome arrays at cellular concentrations probed by magic angle spinning NMR spectroscopy, J Am Chem Soc, 135 (2013) 15278–15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiang S, le Paige UB, Horn V, Houben K, Baldus M, van Ingen H, Site-Specific Studies of Nucleosome Interactions by Solid-State NMR Spectroscopy, Angew Chem Int Ed Engl, 57 (2018) 4571–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shi X, Prasanna C, Nagashima T, Yamazaki T, Pervushin K, Nordenskiold L, Structure and Dynamics in the Nucleosome Revealed by Solid-State NMR, Angew Chem Int Ed Engl, 57 (2018) 9734–9738. [DOI] [PubMed] [Google Scholar]
- [17].Shi X, Prasanna C, Pervushin K, Nordenskiold L, Solid-state NMR (13)C, (15)N assignments of human histone H3 in the nucleosome core particle, Biomol NMR Assign, (2020). [DOI] [PubMed] [Google Scholar]
- [18].Shi X, Prasanna C, Soman A, Pervushin K, Nordenskiold L, Dynamic networks observed in the nucleosome core particles couple the histone globular domains with DNA, Commun Biol, 3 (2020) 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Clark ET, Sievers EE, Debelouchina GT, A Chemical Biology Primer for NMR Spectroscopists, Journal of Magnetic Resonance Open, (2022) 100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kaplan M, Narasimhan S, de Heus C, Mance D, van Doorn S, Houben K, Popov-Celeketic D, Damman R, Katrukha EA, Jain P, Geerts WJC, Heck AJR, Folkers GE, Kapitein LC, Lemeer S, van PMP Bergen En Henegouwen, M. Baldus, EGFR Dynamics Change during Activation in Native Membranes as Revealed by NMR, Cell, 167 (2016) 1241–1251 e1211. [DOI] [PubMed] [Google Scholar]
- [21].Gupta R, Lu M, Hou G, Caporini MA, Rosay M, Maas W, Struppe J, Suiter C, Ahn J, Byeon IJ, Franks WT, Orwick-Rydmark M, Bertarello A, Oschkinat H, Lesage A, Pintacuda G, Gronenborn AM, Polenova T, Dynamic Nuclear Polarization Enhanced MAS NMR Spectroscopy for Structural Analysis of HIV-1 Protein Assemblies, J Phys Chem B, 120 (2016) 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Uluca B, Viennet T, Petrovic D, Shaykhalishahi H, Weirich F, Gonulalan A, Strodel B, Etzkorn M, Hoyer W, Heise H, DNP-Enhanced MAS NMR: A Tool to Snapshot Conformational Ensembles of alpha-Synuclein in Different States, Biophys J, 114 (2018) 1614–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Narasimhan S, Scherpe S, Lucini Paioni A, van der Zwan J, Folkers GE, Ovaa H, Baldus M, DNP-Supported Solid-State NMR Spectroscopy of Proteins Inside Mammalian Cells, Angew Chem Int Ed Engl, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Goldberga I, Li R, Chow WY, Reid DG, Bashtanova U, Rajan R, Puszkarska A, Oschkinat H, Duer MJ, Detection of nucleic acids and other low abundance components in native bone and osteosarcoma extracellular matrix by isotope enrichment and DNP-enhanced NMR, Rsc Adv, 9 (2019) 26686–26690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frederick KK, Michaelis VK, Corzilius B, Ong TC, Jacavone AC, Griffin RG, Lindquist S, Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus, Cell, 163 (2015) 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smith AN, Marker K, Piretra T, Boatz JC, Matlahov I, Kodali R, Hediger S, van der Wel PCA, De Paepe G, Structural Fingerprinting of Protein Aggregates by Dynamic Nuclear Polarization-Enhanced Solid-State NMR at Natural Isotopic Abundance, J Am Chem Soc, 140 (2018) 14576–14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sergeyev IV, Itin B, Rogawski R, Day LA, McDermott AE, Efficient assignment and NMR analysis of an intact virus using sequential side-chain correlations and DNP sensitization, Proc Natl Acad Sci U S A, 114 (2017) 5171–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hall DA, Maus DC, Gerfen GJ, Inati SJ, Becerra LR, Dahlquist FW, Griffin RG, Polarization-enhanced NMR spectroscopy of biomolecules in frozen solution, Science, 276 (1997) 930–932. [DOI] [PubMed] [Google Scholar]
- [29].Maly T, Debelouchina GT, Bajaj VS, Hu KN, Joo CG, Mak-Jurkauskas ML, Sirigiri JR, van der Wel PC, Herzfeld J, Temkin RJ, Griffin RG, Dynamic nuclear polarization at high magnetic fields, J Chem Phys, 128 (2008) 052211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Corzilius B, High-Field Dynamic Nuclear Polarization, Annu Rev Phys Chem, 71 (2020) 143–170. [DOI] [PubMed] [Google Scholar]
- [31].Sauvee C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P, Highly efficient, water-soluble polarizing agents for dynamic nuclear polarization at high frequency, Angew Chem Int Ed Engl, 52 (2013) 10858–10861. [DOI] [PubMed] [Google Scholar]
- [32].Rosay M, Blank M, Engelke F, Instrumentation for solid-state dynamic nuclear polarization with magic angle spinning NMR, J Magn Reson, 264 (2016) 88–98. [DOI] [PubMed] [Google Scholar]
- [33].Lim BJ, Ackermann BE, Debelouchina GT, Targetable Tetrazine-Based Dynamic Nuclear Polarization Agents for Biological Systems, Chembiochem, 21 (2020) 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Linden AH, Franks WT, Akbey U, Lange S, van Rossum BJ, Oschkinat H, Cryogenic temperature effects and resolution upon slow cooling of protein preparations in solid state NMR, J Biomol NMR, 51 (2011) 283–292. [DOI] [PubMed] [Google Scholar]
- [35].Bauer T, Dotta C, Balacescu L, Gath J, Hunkeler A, Bockmann A, Meier BH, Line-Broadening in Low-Temperature Solid-State NMR Spectra of Fibrils, J Biomol NMR, 67 (2017) 51–61. [DOI] [PubMed] [Google Scholar]
- [36].van der Wel PC, Hu KN, Lewandowski J, Griffin RG, Dynamic nuclear polarization of amyloidogenic peptide nanocrystals: GNNQQNY, a core segment of the yeast prion protein Sup35p, J Am Chem Soc, 128 (2006) 10840–10846. [DOI] [PubMed] [Google Scholar]
- [37].Debelouchina GT, Bayro MJ, van der Wel PC, Caporini MA, Barnes AB, Rosay M, Maas WE, Griffin RG, Dynamic nuclear polarization-enhanced solid-state NMR spectroscopy of GNNQQNY nanocrystals and amyloid fibrils, Phys Chem Chem Phys, 12 (2010) 5911–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ni QZ, Markhasin E, Can TV, Corzilius B, Tan KO, Barnes AB, Daviso E, Su Y, Herzfeld J, Griffin RG, Peptide and Protein Dynamics and Low-Temperature/DNP Magic Angle Spinning NMR, J Phys Chem B, 121 (2017) 4997–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bajaj VS, van der Wel PC, Griffin RG, Observation of a low-temperature, dynamically driven structural transition in a polypeptide by solid-state NMR spectroscopy, J Am Chem Soc, 131 (2009) 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Marin-Montesinos I, Goyard D, Gillon E, Renaudet O, Imberty A, Hediger S, De Paepe G, Selective high-resolution DNP-enhanced NMR of biomolecular binding sites, Chem Sci, 10 (2019) 3366–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Takahashi H, Fernandez-de-Alba C, Lee D, Maurel V, Gambarelli S, Bardet M, Hediger S, Barra AL, De Paepe G, Optimization of an absolute sensitivity in a glassy matrix during DNP-enhanced multidimensional solid-state NMR experiments, J Magn Reson, 239 (2014) 91–99. [DOI] [PubMed] [Google Scholar]
- [42].Rogawski R, Sergeyev IV, Zhang Y, Tran TH, Li Y, Tong L, McDermott AE, NMR Signal Quenching from Bound Biradical Affinity Reagents in DNP Samples, J Phys Chem B, 121 (2017) 10770–10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Debelouchina GT, Gerecht K, Muir TW, Ubiquitin utilizes an acidic surface patch to alter chromatin structure, Nat Chem Biol, 13 (2017) 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lowary PT, Widom J, New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning, J Mol Biol, 276 (1998) 19–42. [DOI] [PubMed] [Google Scholar]
- [45].Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K, Reconstitution of nucleosome core particles from recombinant histones and DNA, Methods Enzymol, 375 (2004) 23–44. [DOI] [PubMed] [Google Scholar]
- [46].Morcombe CR, Zilm KW, Chemical shift referencing in MAS solid state NMR, J Magn Reson, 162 (2003) 479–486. [DOI] [PubMed] [Google Scholar]
- [47].Takegoshi K, Nakamura S, Terao T, 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR, Chem. Phys. Lett, 344 (2001) 631–637. [Google Scholar]
- [48].Jaroniec CP, Filip C, Griffin RG, 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly (13)C,(15)N-labeled solids, J Am Chem Soc, 124 (2002) 10728–10742. [DOI] [PubMed] [Google Scholar]
- [49].Lee W, Tonelli M, Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy, Bioinformatics, 31 (2015) 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Skinner SP, Fogh RH, Boucher W, Ragan TJ, Mureddu LG, Vuister GW, CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis, J Biomol NMR, 66 (2016) 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem, 25 (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
- [52].Stoll S, Schweiger A, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J Magn Reson, 178 (2006) 42–55. [DOI] [PubMed] [Google Scholar]
- [53].Liao SY, Lee M, Wang T, Sergeyev IV, Hong M, Efficient DNP NMR of membrane proteins: sample preparation protocols, sensitivity, and radical location, J Biomol NMR, 64 (2016) 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fricke P, Mance D, Chevelkov V, Giller K, Becker S, Baldus M, Lange A, High resolution observed in 800 MHz DNP spectra of extremely rigid type III secretion needles, J Biomol NMR, 65 (2016) 121–126. [DOI] [PubMed] [Google Scholar]
- [55].Thurber K, Tycko R, Low-temperature dynamic nuclear polarization with helium-cooled samples and nitrogen-driven magic-angle spinning, J Magn Reson, 264 (2016) 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, Griffin RG, Intermolecular structure determination of amyloid fibrils with magic-angle spinning and dynamic nuclear polarization NMR, J Am Chem Soc, 133 (2011) 13967–13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Frederick KK, Michaelis VK, Caporini MA, Andreas LB, Debelouchina GT, Griffin RG, Lindquist S, Combining DNP NMR with segmental and specific labeling to study a yeast prion protein strain that is not parallel in-register, Proc Natl Acad Sci U S A, 114 (2017) 3642–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lu M, Wang M, Sergeyev IV, Quinn CM, Struppe J, Rosay M, Maas W, Gronenborn AM, Polenova T, (19)F Dynamic Nuclear Polarization at Fast Magic Angle Spinning for NMR of HIV-1 Capsid Protein Assemblies, J Am Chem Soc, 141 (2019) 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Andreas LB, Barnes AB, Corzilius B, Chou JJ, Miller EA, Caporini M, Rosay M, Griffin RG, Dynamic nuclear polarization study of inhibitor binding to the M2(18–60) proton transporter from influenza A, Biochemistry, 52 (2013) 2774–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mak-Jurkauskas ML, Bajaj VS, Hornstein MK, Belenky M, Griffin RG, Herzfeld J, Energy transformations early in the bacteriorhodopsin photocycle revealed by DNP-enhanced solid-state NMR, Proc Natl Acad Sci U S A, 105 (2008) 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jakdetchai O, Eberhardt P, Asido M, Kaur J, Kriebel CN, Mao J, Leeder AJ, Brown LJ, Brown RCD, Becker-Baldus J, Bamann C, Wachtveitl J, Glaubitz C, Probing the photointermediates of light-driven sodium ion pump KR2 by DNP-enhanced solid-state NMR, Sci Adv, 7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jaudzems K, Bertarello A, Chaudhari SR, Pica A, Cala-De Paepe D, Barbet-Massin E, Pell AJ, Akopjana I, Kotelovica S, Gajan D, Ouari O, Tars K, Pintacuda G, Lesage A, Dynamic Nuclear Polarization-Enhanced Biomolecular NMR Spectroscopy at High Magnetic Field with Fast Magic-Angle Spinning, Angew Chem Int Ed Engl, 57 (2018) 7458–7462. [DOI] [PubMed] [Google Scholar]
- [63].Michael AK, Grand RS, Isbel L, Cavadini S, Kozicka Z, Kempf G, Bunker RD, Schenk AD, Graff-Meyer A, Pathare GR, Weiss J, Matsumoto S, Burger L, Schubeler D, Thoma NH, Mechanisms of OCT4-SOX2 motif readout on nucleosomes, Science, 368 (2020) 1460–1465. [DOI] [PubMed] [Google Scholar]
- [64].Armache JP, Gamarra N, Johnson SL, Leonard JD, Wu S, Narlikar GJ, Cheng Y, Cryo-EM structures of remodeler-nucleosome intermediates suggest allosteric control through the nucleosome, Elife, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chen Q, Yang R, Korolev N, Liu CF, Nordenskiold L, Regulation of Nucleosome Stacking and Chromatin Compaction by the Histone H4 N-Terminal Tail-H2A Acidic Patch Interaction, J Mol Biol, 429 (2017) 2075–2092. [DOI] [PubMed] [Google Scholar]
- [66].Debelouchina GT, Bayro MJ, Fitzpatrick AW, Ladizhansky V, Colvin MT, Caporini MA, Jaroniec CP, Bajaj VS, Rosay M, Macphee CE, Vendruscolo M, Maas WE, Dobson CM, Griffin RG, Higher order amyloid fibril structure by MAS NMR and DNP spectroscopy, J Am Chem Soc, 135 (2013) 19237–19247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Helmus JJ, Surewicz K, Apostol MI, Surewicz WK, Jaroniec CP, Intermolecular alignment in Y145Stop human prion protein amyloid fibrils probed by solid-state NMR spectroscopy, J Am Chem Soc, 133 (2011) 13934–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Debelouchina GT, Platt GW, Bayro MJ, Radford SE, Griffin RG, Intermolecular alignment in beta2-microglobulin amyloid fibrils, J Am Chem Soc, 132 (2010) 17077–17079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Shcherbakov AA, Mandala VS, Hong M, High-Sensitivity Detection of Nanometer (1)H-(19)F Distances for Protein Structure Determination by (1)H-Detected Fast MAS NMR, J Phys Chem B, 123 (2019) 4387–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Roos M, Wang T, Shcherbakov AA, Hong M, Fast Magic-Angle-Spinning (19)F Spin Exchange NMR for Determining Nanometer (19)F-(19)F Distances in Proteins and Pharmaceutical Compounds, J Phys Chem B, 122 (2018) 2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shcherbakov AA, Roos M, Kwon B, Hong M, Two-dimensional (19)F-(13)C correlation NMR for (19)F resonance assignment of fluorinated proteins, J Biomol NMR, 74 (2020) 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Berruyer P, Bjorgvinsdottir S, Bertarello A, Stevanato G, Rao Y, Karthikeyan G, Casano G, Ouari O, Lelli M, Reiter C, Engelke F, Emsley L, Dynamic Nuclear Polarization Enhancement of 200 at 21.15 T Enabled by 65 kHz Magic Angle Spinning, J Phys Chem Lett, 11 (2020) 8386–8391. [DOI] [PubMed] [Google Scholar]
- [73].Mentink-Vigier F, Marin-Montesinos I, Jagtap AP, Halbritter T, van Tol J, Hediger S, Lee D, Sigurdsson ST, De Paepe G, Computationally Assisted Design of Polarizing Agents for Dynamic Nuclear Polarization Enhanced NMR: The AsymPol Family, J Am Chem Soc, 140 (2018) 11013–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mathies G, Caporini MA, Michaelis VK, Liu Y, Hu KN, Mance D, Zweier JL, Rosay M, Baldus M, Griffin RG, Efficient Dynamic Nuclear Polarization at 800 MHz/527 GHz with Trityl-Nitroxide Biradicals, Angew Chem Int Ed Engl, 54 (2015) 11770–11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.