

Abstract
Transposable elements (TEs) have the ability to alter individual genomic landscapes and shape the course of evolution for species in which they reside. Such profound changes can be understood by studying the biology of the organism and the interplay of the TEs it hosts. Characterizing and curating TEs across a wide range of species is a fundamental first step in this endeavor. This protocol employs techniques honed while developing TE libraries for a wide range of organisms and specifically addresses: (1) the extension of truncated de novo results into full length TE families, (2) the iterative refinement of TE multiple sequence alignments, (3) the use of alignment visualization to assess model completeness and subfamily structure.
Keywords: curation, hidden Markov Model, RepeatModeler, RepeatMasker, alignment, transposable elements
INTRODUCTION:
Transposable elements (TEs) are discrete pieces of DNA that have the ability to mobilize within a genome, causing rearrangement and/or disruption of the structure of the host genome (Gray, 2000). As a result, TE activity alters epigenetic marks, gene expression and consequently protein interactions, potentially altering regulatory pathways (Chuong et al., 2017). Negative effects of regulatory pathway disruption are seen via disease phenotypes, such as cancer (Kazazian & Moran, 2017; Richardson et al., 2014). The high mutational pressure has led host genomes to build elaborate defense mechanisms, some of which are well-known; for example, both microRNA regulation and DNA methylation may have originated to suppress TEs. Conversely, exaptation of TEs can occur to form novel functions that are of benefit to the host (Chuong et al., 2017). For example, the origin of the adaptive immune system in jawed vertebrates lies in the expansion of a TE transposase, producing RAG1 and RAG2 proteins (Kapitonov & Jurka, 2005). These proteins are essential proteins for generating variability of antibodies and T-cell receptors. Though still largely underappreciated, the impact of TEs on genome and organismal evolution has thus been significant. This impact depends on the character and abundance of the TEs to which a genome has been exposed at any one time. Therefore, the first step in studying TE influence involves identifying and accurately characterizing these elements in the genome.
De novo tools for TE identification provide an unbiased view of the TE content and diversity present in a genome. Such tools are particularly important for analyzing understudied genomes that lack a well-defined TE library, as each genome contains a unique repertoire of TE insertions. In general, de novo repeat finders identify transposable elements either by taking advantage of the repetitive nature of TE copies or by identifying conserved sequence structures of known TE classes. Several TE discovery pipelines combine these two strategies to take advantage of the strengths and mitigate the weaknesses of each, including: RepeatModeler (Flynn et al., 2020), REPET (Flutre et al., 2011), and EDTA (Ou et al., 2019). By combining multiple TE discovery strategies designed to detect specific aspects of TEs, a greater likelihood of discovering all of the TE families in a genome can be reached (Arensburger et al., 2016; Arkhipova, 2017).
Each package differs in the combination of de novo programs and as such, will produce overlapping, yet potentially differing results. Of the packages described, all generate TE consensi and utilize alignments, but the preservation of seed alignments as output is unique to RepeatModeler alone. Seed alignments are multiple sequence alignments (MSAs) of representative family members (Wheeler et al., 2013). Despite the advantages, many researchers keep only the TE consensi and discard the seed alignments, ignoring the wealth of information contained within.
Maintaining the seed alignment for each model provides not only the provenance of the model, but also guidance for a TE curator. The seed alignment provides information regarding TE subfamily composition which may be observed as multiple divergence or deletion patterns (Hubley et al., 2016; Vijayabaskar, 2017; Wheeler et al., 2013). The seed alignment is also essential for generating a more sensitive profile hidden Markov model (HMM) for each TE family. By searching the genome for repetitive sequences using HMMs, the likelihood of detecting older, highly diverged copies of TEs is increased.
Generating a complete and accurate alignment of a TE family is perhaps the most important aspect of curation. Commonly used alignment methods include MAFFT (Katoh et al., 2002), MUSCLE (Edgar, 2004), ClustalW (J. D. Thompson et al., 1994), ProbCons (Do et al., 2005), DIALIGN (Morgenstern, 2004), Kalign (Lassmann & Sonnhammer, 2005; Morgenstern, 2004) and T-Coffee (Magis et al., 2014). However, many of these programs were optimized for protein alignments (Julie D. Thompson et al., 2011) and underperform when used with highly fragmented and diverged DNA sequences. Protein alignments are often guided by domain and structural information, the sequences of which are under evolutionary pressure and exhibit higher conservation than the nucleic acid counterparts. This is in contrast to non-coding DNA, and in particular transposable elements, the majority of which lack structural information to guide alignments and are neutrally-evolving inhabitants in the genome. These conditions make TE alignments challenging.
In this protocol, careful consideration has been paid to the methodology and parameters involved in the alignment of TE derived DNA. For example, we derived the log odds substitution matrices and gap initiation and extension penalties by studying the predominantly neutrally accumulated mutations in copies of ancient DNA transposons in different GC backgrounds in the human genome. We calculated four sets of matrices and associated gap penalties representing a 14, 18, 20 and 25% substitution level. These matrices and parameters have been used since 1998 in our program RepeatMasker (Smit et al., 2013) and have been adopted by external tools such as Censor (Kohany et al., 2006). Consensus sequences are not simply a majority call, but represent for each site the highest scoring nucleotide, using a derivative substitution matrix and a method to predict CpG sites (which, due to methylation, quickly change to CA or TG sites).
The alignment method used herein is a variation on iterative transitive search, whereby a MSA is generated transitively from the individual pairwise alignments of each sequence to a reference sequence. A consensus sequence obtained from the MSA may then be used as the reference in an iterative fashion, improving the quality of the consensus and the MSA. When used with a starting reference sequence lacking significant indels, this method has proven to be capable of reconstructing extremely ancient (highly diverged, and fragmented) TE families; for example, over the years we’ve recreated dozens of TEs that predate our speciation from reptiles and birds over 300 million years ago, like OldHat and AmnSINE1 (Nishihara et al., 2006).
While powerful, the output generated by de novo TE finders, even when the seed alignment is provided, is not perfect and requires manual curation to polish the TE models they produce. For instance, the models that these programs produce are often truncated and do not accurately represent the full-length TE. As such, the boundaries of the TE are not reached. In addition, the output generated may contain redundant TE consensi and require additional filtering. Therefore, detailed curation necessarily requires knowledge of TE biology.
Broadly divided by their movement intermediate into Class I, retrotransposons, and Class II, DNA transposons (Finnegan, 1989), TEs can be further divided into families and subfamilies (Wicker et al. 2007; Piegu et al. 2015) which are separated by structural features and phylogenetic analyses, respectively (Arkhipova, 2017; Piégu et al., 2015; Wicker et al., 2007). Examples of structural features are the terminal inverted repeats (TIRs) of some DNA transposable elements and the fixed length of target site duplications (TSDs) for LTR/ERV families. These structural features inform the movement mechanism of the TE and are the underlying framework by which some de novo programs identify both autonomous and non-autonomous subfamilies of TEs.
This protocol aims to elucidate the process of TE curation by leveraging the information provided by the seed alignment of a TE model in combination with knowledge of TE biology to provide researchers with detailed knowledge behind the decisions made during the TE curation process. The Basic Protocol guides the user through the processes of seed alignment extension through subfamily analysis. Extension and consensus refinement is achieved by using the alignAndCallConsensus.pl program by applying specialized scoring matrices. Following a short discussion of TE termini sequence structure, the protocol leads the user through two different strategies to analyze subfamily structure, COSEG and a cd-hit-based pipeline. Lastly, example usage of AutoRunBlocker.pl, a program to resolve gaps with the alignment, is provided. This program takes a region in the consensus within a smaller sliding window and determines if a user-defined minimum number of copies agree to have a different length than the current consensus, and provides an alternate consensus sequence with the suggested length changes.
To get the user comfortable with the protocol, example files are provided with the output detailed for each step (Table 1).
Table 1.
Example files for the Basic Protocol.
| TE type | Consensus | Elements | Species | Analysis |
|---|---|---|---|---|
| soloLTR | example1_con.fa | example1_elements.fa | Aotus nancymaae | Extension; cd-hit subfamily |
| SINE | example2_con.fa | example2_elements.fa | Mus musculus | COSEG subfamily |
| DNA | example3_con.fa | example3_elements.fa | Drosophila melanogaster | cd-hit subfamily |
BASIC PROTOCOL
EXTENSION AND EDGE POLISHING OF CONSENSI AND SEED ALIGNMENTS DERIVED FROM DE NOVO REPEAT FINDERS
The core of this protocol describes and provides example usage of a script which performs an alignment of a set of instances of a TE family to a representative copy or a putative consensus. The alignments are then used to generate a transitively induced MSA, initially for the purpose of consensus calling, but which eventually will become the refined seed alignment. If changes were made to the consensus, the copies can be realigned to the new consensus. By iteratively applying this method, a type of hill climbing optimization, the consensus will improve and stabilize. The protocol also describes how this process may be used to incorporate flanking sequences and extend the consensus to reach the full extent of the TE family. Finally, the resulting seed alignment may be analyzed for apparent subfamily structure using additional tools.
Necessary Resources
Hardware
A computer with a Linux OS, or appropriate virtualization technology e.g. Docker for Mac or VirtualBox, 100 GB of free hard drive space, and 32 GB of RAM
Software
RepeatModeler 2.0.2 (or higher)
To use the scripts in this protocol RepeatModeler must also be configured with a path for the UCSC toolkit, in particular faToTwoBit, twoBitInfo, and twoBitToFa.
RepeatModeler 2.0.2 is available at http://www.repeatmasker.org/RepeatModeler/. For an alternative installation method, the Dfam TE Tools docker/singularity container and instructions for use can be found at https://github.com/Dfam-consortium/TETools/.
RepeatMasker
COSEG
Files
Reference genome in FASTA and 2bit format
You can convert FASTA to 2bit with `faToTwoBit [-long] genome.fa genome.2bit` (You will need the `-long` option if the genome is larger than 4GB)
You can convert 2bit to FASTA with `twoBitToFa genome.2bit genome.fà
Interspersed repeats in FASTA format, a.k.a., the sequences to be aligned Consensi associated with the aforementioned interspersed repeats in FASTA format Example files (Table 1)
Protocol steps and annotations
TE model extension and consensus refinement
Note: In this protocol we will use the alignAndCallConsensus.pl script, which is located in the RepeatModeler/util/ directory. The two main inputs to this program are a FASTA file containing the consensus sequence(s), and a FASTA file containing the “elements” or “instances” to be aligned to the consensus. The instances can include some flanking sequence both before and after the region that aligns to the consensus (example1; example2) or contain only sequence that matches to the consensus with no flanking sequence (example3). For easier analysis, group the consensi and element files into the same directory. To avoid confusion or conflicts between output files belonging to distinct input files, we additionally recommend working on each example (pair of consensus + elements) in its own directory.
-
1
To begin refining your alignment and consensus sequence, run the following at a terminal:
$ alignAndCallConsensus.pl -c example1_con.fa -e example1_elements.fa -int
The terminal output includes the alignment engine, matrix, and average Kimura divergence of the alignment in question. For example1, the alignment engine used is RMBlast (Figure 1A, orange box), the matrix used is based on a 25% divergence (25p41g) with 41% CG background (25p41g) (Figure 1A, blue box), and the Kimura divergence for the alignment is 15% (Figure 1A, green box). In this case, the more appropriate 14% divergence substitution matrix (and its accompanying gap penalties) should be used (-ma 14) for refinement instead of the default 25% matrix (Options available are: 14, 18, 20 or 25). Using high divergence parameters, i.e. smaller penalties for mismatches and gaps, for low divergence copies may result in alignment of non-related DNA flanking the end of a fragment to the consensus and/or inclusion of short foreign insertions or inversions. These false alignments tend to have a much higher divergence than the true alignment. These sequences were obtained from a mammalian species, so a 41% CG background is appropriate (Options available are: 37–53; odd numbers only).
The “-int” option chosen will allow you to choose what changes you would like to be made to the consensus sequence. The purpose of this step is NOT to change the sequence, but rather to see the divergence of this alignment for appropriate matrix selection (Figure 1A, green box). Press “d” and then enter to terminate the program.
-
2
Re-run the alignment with the 14% matrix, by adding “-ma 14” to the command line.
$ alignAndCallConsensus.pl -c example1_con.fa -e example1_elements.fa -ma 14 -hp 7 -int
To see if the alignment can be extended beyond the termini of the current consensus, one can add to the consensus termini a string of letters that in our matrices score positive (~⅓ of a nucleotide match) to all nucleotides. We chose the letter H as it is part of the IUPAC code (A, C or T) and is accepted by all search engines, but is not found in consensus sequences or sequence assemblies. The option “-hp” sets the length of this string. Such an addition results in an ungapped extension of all copies that approached the end. If the extended sequences have a common origin, a consensus can be called. If not, a majority of positions will be called “N”. When working with low divergence sequences, longer H-pads can be added to speed up the extension process. When copies are highly diverged, accumulated deletions and insertions result in misalignments the further the ungapped alignment is attempted and the many Ns called will cause the extension to stop prematurely.
After running the program again with a different substitution matrix the Kimura divergence has changed (14.7% to 12.8%) as well as the alignment settings (Figure 1B). The parameters for the 25% scoring matrix were too permissive of substitutions for the true age of the sequences, leading to misalignment. Improved accuracy of the alignment using an appropriate matrix corresponds with a reduced kimura divergence.
In addition, several output files are generated: a MSA file (example1_con.ali), an RMBlast pairwise alignment output file (example1_con.out), and a CG modified pairwise alignment file (example1_con.out.CGmodified). These file names are based on the FASTA sequence name(s) contained within the consensus file, which are not necessarily the same as the name of the consensus file itself.
-
3
Extend and/or improve the consensus sequence
You will be prompted to either skip making changes to the consensus sequence or opt to improve the sequence in Figure 1B. In order to allow for extension, seven “H” characters have been added to the 5’ and 3’ edges of the consensus sequence (-hp 7). This parameter only needs to be used at the beginning of the protocol for extension, and does not need to be used in subsequent steps, as the H-pad will have been added directly to the termini of the sequences within the consensus file. In this case, there are several suggested nucleotide improvements (Figure 1B - transversions, transitions and resolution of ambiguous bases). In addition, extension on both the 5’ and 3’ ends of the consensus sequence is recommended due to the alignment of the H-pad to ambiguous stretches of nucleotides on both the 5’ and 3’ edges of the alignment (Figure 1B, red boxes). For this example, select the “x” option, allowing for the consensus sequence to improve as suggested while also extending both edges of the consensus sequence for re-alignment. “x” is not always the appropriate choice; other options include extending to the 5’ end only (5 or b(egin)), the 3’ end only (3 or e(nd)), only accepting the changes in between the optional H-pad (c), skipping the suggested changes to the current consensus sequence (s), or exiting the program without making any further changes (d).
-
4
Repeat consensus refinement (i.e., continue running alignAndCallConsensus.pl with the parameters for this model) until no additional changes are suggested.
After choosing an appropriate matrix, running alignAndCallConsensus.pl one iteration at a time can be tedious. Using the interactive “-int” option allows for multiple iterations to be completed in a single run of the program.
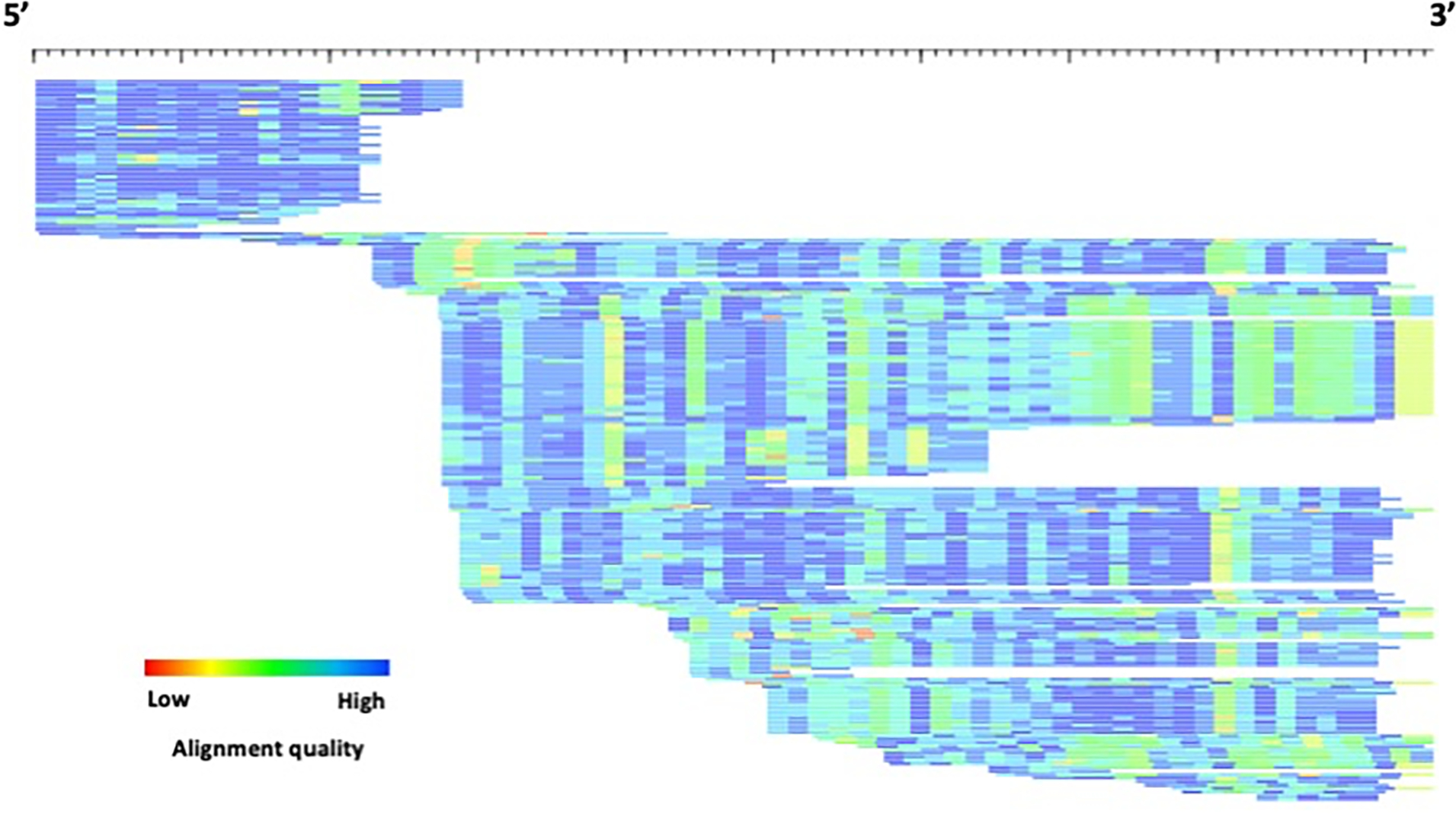
The process of iterative alignment and consequent consensus refinement should be repeated until no additional changes are suggested (the consensus sequence stabilizes), or the program is stopped by entering “d” at the interactive prompt. In this example, the 5’ end of the consensus continues extending (Figure 2), but extending the 3’ end would start including ambiguous sequence.
Note: The example used here is an examination of the long terminal repeat (LTR) of an endogenous retrovirus (ERV). The element file contains both solitary LTRs (the internal region is usually deleted through homologous recombination) as well as LTR sequences flanking either the 5’ or 3’ end of the internal sequence of the ERV. Continuing to extend into the internal coding region will result in a consensus sequence appearing to require continuous expansion on one edge, but not the other (Figure 2). In this case, the consensus sequence may never “stabilize”, as the 5’ edge will continue extending. For extensive model extension, it is recommended to occasionally check the alignment (.ali) file associated with the consensus in question.
-
5
Visually assess the edges of the model:
$ less example1_con.ali
As mentioned above, the sequences contributing to a solo LTR model can be a mixture of solo LTR sequences as well as LTRs connected to internal sequences, leading to extension of the consensus into the internal coding region (Figure 3; Minor Edge Polishing). It is important to note two things: first, the position within each of the TE instances where they start aligning to the consensus sequences (Figure 3, blue box). In this example several instances stop aligning to the consensus near their 5’ end, as indicated by the numbers less than ~20. Second, that a large number of sequences (the solo LTR instances) start aligning at roughly the same position within the consensus (Figure 3, red line).
In order to only assess the LTR portion of the alignment, it is useful to remove the sequences containing the internal region. After removing these sequences, iterative refinement can be continued until the consensus sequence stabilizes. This can be accomplished by pruning the consensus sequence on either side to the known LTR edge (5’ TG, 3’ CA) by limiting the alignment to include sequences that do not extend past the LTR edge. Here, the number of sequences that do not extend past the LTR edge is 34. Note that this number does not include the two header sequences (top sequence - “consensus”; second sequence - “ref:example1_con” Figure 3).
-
6
Limit the number of sequences that align by using the “-p” option and continue to improve the consensus without extending until the sequence stabilizes, a.k.a., no additional sequence changes are suggested.
$ alignAndCallConsensus.pl -c example1_con.fa -e example1_elements.fa -int -ma 14 -p 34
After pruning, the 5’ edge of the sequence has improved (Figure 4A). The 3’ edge might appear as though it needs additional polishing. However, the red arrows suggest two different edges as the result of at least two different subfamilies. Two distinct blocks of sequences end at a CA dinucleotide (Figure 4B, blue box). The ambiguous bases at the 3’ edge of the consensus sequence (polyN) indicate the edge of the alignment that comprises all possible subfamilies. This is supported by the non-conserved sequence underlined in black (Figure 4B). This apparent difference between the 5’ and 3’ edges can be observed by taking advantage of the alignAndCallConsensus.pl “-html” option (Figure 5).
-
7
Visualize the alignment
To visualize the alignment, run the alignAndCallConsensus.pl with the “-html” option, and enter “s” to skip any suggested changes, as this consensus sequence has already been improved:
$ alignAndCallConsensus.pl -c example1_con.fa -e example1_elements.fa -int -ma 14 -p 34 -html
By viewing the alignment in this manner, a possible subfamily structure is visible. This is evident in the alignment patterns grouping sequence as well as the “truncated” appearance of blocks of sequence that indicate either a recombination product or deletion event (Figure 5).
Figure 1.
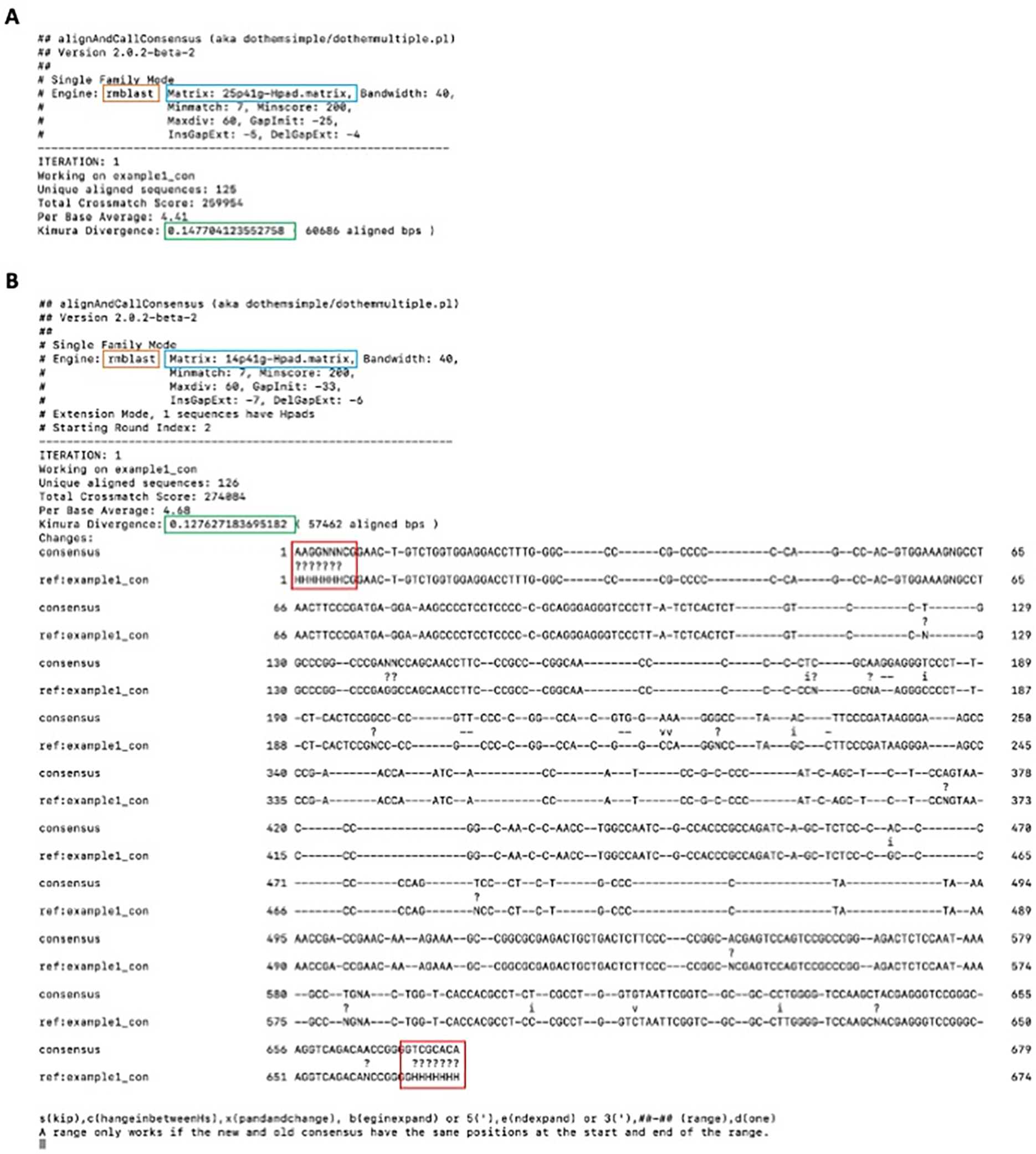
Terminal output of alignAndCallConsensus.pl performed with A) the default 25% substitution matrix and B) a 14% substitution matrix. The orange box indicates the search engine utilized, while the blue box indicates the substitution matrix used for this alignment. 25p41g indicates that a 25% substitution matrix with a 41% CG background was used. The green box highlights the average Kimura-model substitution level of all the aligned copies compared to the consensus. The presented sequences are the newly calculated consensus (“consensus”) and the previous consensus or reference sequence (“ref:example1_com”) as they appear in the complete MSA (gaps in both sequences indicate that copies exist with an insertion ther). “v” in between the sequences indicates a transversion, while an “i” indicates a transition. “?” indicates a nucleotide aligned to the ambiguous base (like “N” or “H”). The red boxes highlight the consensus of the bases aligned to the terminal “H-pads”.
Figure 2.

Continued 5’ extension of the example1 consensus sequence. The red box highlights the option selected to extend the 5’ edge of the alignment while the green box indicates the changing kimura divergence value as the number of aligned bp changes. The extension was terminated after an initial “x” selection with subsequent “5” extensions. Note that only the last 3 iterations of the program are shown.
Figure 3.

example1_con.ali alignment of the 5’ edge to the consensus sequence. The blue box indicates the position at which that sequence starts to align to the consensus sequence while the red line indicates a block of sequences that have a common start position.
Figure 4.

Example1_con.ali in the terminal after pruning. A) The 5’ alignment edge after pruning. The black bracket indicates the 5’ sequence past the conserved sequence. C) 3’ alignment edge. The blue box indicates the start position of the instance compared to the consensus sequence. The red arrows indicate two different 3’ edges. The black box highlights the lack of conservation past the consensus sequence.
Figure 5.
HTML visualization of example1_con.html. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. The length of the consensus sequence is 690 bp, including the H-pad.
In example1, a soloLTR, the edges of the model were clearly discernible. However, depending upon the TE model assessed, different termini sequence structures will be presented, and in the case of novel TEs, no known terminal sequence structure will be available as a visual guide to determine the consensi edges.
Note: For all examples your output may differ slightly from what is depicted in this protocol. This is dependent on the choices made during the alignAndCallConsensus.pl program.
Minor edge polishing
Regardless of the method chosen to identify TE families, it is important to note that polishing the edges of your TE consensi and seed alignments after extensive model extension is a crucial step in producing a high-quality library. TE terminal sequence structures for a wide range of classes have been well-studied and form the basis of many structure-based de novo discovery programs. As such, there are many excellent reviews providing details on these motifs (Arkhipova, 2017; Piégu et al., 2015; Wicker et al., 2007). This method is particularly useful for elements that have a tendency to have internal deletion (e.g., non-autonomous DNA transposons) or recombinant products that may disrupt the coding region and obscure element classification. In this section, we will discuss what to look for in the seed alignment sequences to indicate the true edge of the element has been reached in some common examples.
Many classes of DNA transposable elements are flanked by TIRs such as hAT, CACTA, Maverick and Merlin superfamilies, while others that lack TIRs still have somewhat conserved 5’ and 3’ termini such as the Helitron and Crypton. Autonomous class II elements encoding transposases recognize specific TIRs resulting in the split of TEs into different families. In addition, many of the class II superfamilies have target site duplications (TSDs) of a specific length or sequence composition (Wicker et al. 2007).
Retrotransposable element families can be split into LTR and non-LTR groups. Endogenous retroviruses (ERVs) are flanked by matching 5’ and 3’ LTRs as well as TSDs of a fixed length. In addition, the LTRs themselves have a recognizable 5’ TG and 3’ CA to distinguish them from the internal sequence (Figures 3 & 4).
Non-LTR elements, such as the autonomous long interspersed elements (LINEs) and their non-autonomous short interspersed element (SINE) counterparts are characterized by a 5’ GC-rich sequence and a 3’ polyA tail, A-rich region, or simple repeat. The 3’ end of most SINE elements also corresponds with the 3’ end of their autonomous LINE counterpart. SINE elements also contain an internal Pol III promoter close to the 5’ end of the element. Both LINE and SINE elements have TSDs as a result of their movement mechanism, but the length and sequence composition is unique to each insertion. Taken together, these characteristics are indicative of movement via target primed reverse transcription (TPRT).
In lieu of the presence of well-known termini sequence structures, the seed alignment and border of conserved sequence should guide the consensus sequence boundaries. If your TE instances require more flanking sequence to extend into, follow step 5 in the Support Protocol.
Subfamily analysis
Seed alignments have many strengths, including preserving the provenance of a TE model, the ability to generate more sophisticated models such as pHMMs for more sensitive TE genomic homology-based searches, and re-alignment during extension in order to improve the consensus sequence. Another strength is straightforward visualization of subfamily structure.
Once it has been determined that a subfamily analysis is necessary, there are several characteristics to check to select the most appropriate program for subfamily determination. The first is the length of the contributing interspersed insertions to the model and consequently, the consensus (Figure 6). Full-length insertions compared to the consensus as well as the presence of several distinct groups of divergence patterns indicate that COSEG is the most appropriate program to analyze potential subfamilies. COSEG groups sequences with co-segregating mutations which can separate diverged blocks of sequences to form subfamilies. Use of COSEG involves running four programs in the following order: 1) alignAndCallConsensus.pl, 2) bestwindow.pl, 3) preprocessAlignments.pl and 4) runcoseg.pl. If subfamily assessment is not required for a TE model, skip to the last step regarding gap resolution.
Figure 6.
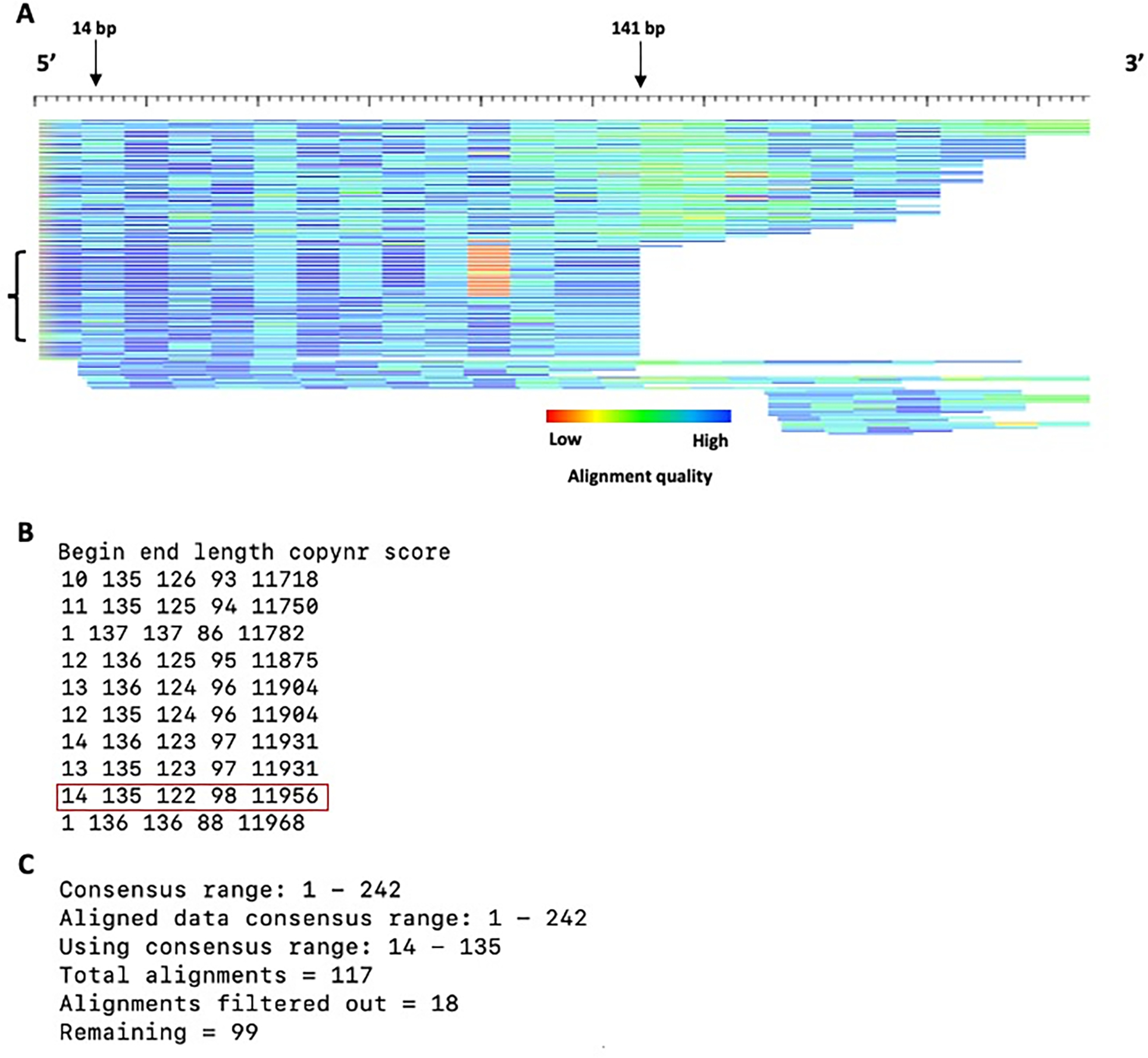
Alignment of example2 in HTML format. A) HTML alignment format. The bracket indicates a possible subfamily as observed by the divergence pattern differences for sequences. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. The length of the HTML alignment is 242 bp, including the H-pad. B) Terminal output of bestwindow.pl. The highest scoring 10 sequences are shown. C) Terminal output of preprocessAlignments.pl. Note that the consensus range length may differ slightly from your terminal output.
-
8
To generate pairwise alignments to group sequences into subfamilies, run the following in terminal:
$ alignAndCallConsensus.pl -c example2_con.fa -e example2_elements.fa -ma 14 -int -hp 7
After improving the consensus sequence if necessary (see steps 1–7), several output files are generated. For the next step in the coseg pipeline, you will need the example2_con.out file.
-
9
Coseg only takes into consideration those sequences that are aligned at least over the region of the consensus the user tells it to examine. To determine the part of the consensus / window within the alignment that captures the maximum amount of information we use the bestwindow.pl script, located in the RepeatModeler/util/ directory.
$ bestwindow.pl example2_con.out 115
The inputs are the pairwise alignments generated in the previous step (example2_con.out) and the minimum length of the windows to examine in the alignment. For example2, the consensus sequence is 246 bp. However, not all of the instances in the alignment are full length. To include the most sequence data for subfamily determination, bestwindow.pl recommends a window length 122 (Figure 6A, arrows). Use a conservatively small minimum window size to capture the optimum length to obtain the bulk of the sequences in the MSA (Figure 6A, arrows).
The output is printed directly to the terminal (Figure 6B). The combination of the beginning and end consensus positions are listed from lowest to highest scoring. The score is simply the multiplication of the copy number and the window length (not the total aligned bases). The window size that captures the most information with a high score for this model is from 14 bp to 135 bp, with a window size of 122, capturing 98 out 117 alignments, representing 100 TE instances (Figure 6B). Note that there are more alignments than TE instances as some sequences align to the consensus sequence multiple times.
When one or both termini of the TE have already been reached, do not include the positions corresponding to the H-pad at those ends in the window, as these are flanking DNA unrelated to the TE and introduce noise that may reduce the significance of observed co-segregating sites.
-
10
Filter the output of the RMBlast output for subfamily generation. Note that preprocessAlignments.pl (and runcoseg.pl used in the following step) are provided with coseg, not in RepeatModeler/util/.
$ preprocessAlignments.pl -consensus example2_con.fa -alignments example2_con.out -minConsRange 14 -maxConsRange 135
Here, the consensus file is the same consensus file that was utilized in step 1 earlier. The alignment file is example2_con.out. The purpose of this program is to filter the alignment output based on the start and stop parameters derived from the bestwindow.pl program.
Summary information from this step is printed on the terminal (Figure 6C). This output indicates the number of starting alignments (Figure 6C, “total alignments”) based upon the input parameters to the preprocessAlignment.pl program (consensus range), and how many sequences were filtered based on the aforementioned parameters.
-
11
Parse the processed alignments from the previous step into subfamilies based on co-segregating substitutions:
$ runcoseg.pl -filePrefix example2_con.out -m 10 -t
For this example, the filePrefix is example2_con.out (not to be confused with the alignment file of the same name), which encompasses the common name shared by the files required to run coseg: example2_con.out.seqs, example2_con.out.outliers, example2_con.out.ins, example2_con.out.fasta, and example2_con.out.cons. The minimum parameter (-m) is set slightly lower than the approximate number of the smallest putative subfamily (Figure 6A). The default value is 50.
With the “-t” option, runcoseg.pl checks for significant co-segregation of mutations at three sites, before analyzing pairs of sites. This option occasionally finds significant clusters in an alignment of ancient, highly diverged copies that falls under the radar of the default method which is based only on pairs.
Another option to runcoseg.pl is “-u”, which sets the minimum distance between diagnostic sites that coseg will consider. The default is 10. For short elements, like some SINEs, a smaller minimum distance may be chosen. The minimum is 1, for adjacent bases, but this should be avoided, since pairs of bases may mutate simultaneously. Most commonly, aligned TG and CA dinucleotides sequences are the result of a single differential decay of a CpG site and do not constitute co-segregating individual substitutions.
The product of this pipeline are several files, one of which, example2_con.out.seqs.subfamilies.seq, contains the two subfamily consensi as predicted by COSEG. To improve upon the consensi based on the TE instances for the model, now models, run alignAndCallConsensus.pl as described in the extension steps above. Not only does this program analyze individual consensus sequences, but can also analyze multiple consensi simultaneously.
-
12
Improve upon the COSEG output by aligning your TE instances to the new consensi and extending.
$ alignAndCallConsensus.pl -c example2_con.out.seqs.subfamilies.seq -e example2_elements.fa -ma 14 -int -hp 7
For the matrix parameter, the same matrix value that was utilized for model extension, if performed, should be used here. If previously unextended, you should use step 1 to determine the matrix required for the model of interest. In this example the consensus was trimmed to base pairs 14 through 135 in the previous steps, so you will need to extend on both the 5’ and 3’ sides again to reach the ends.
Note that this model is a SINE element. Therefore, continue improving and extending the consensus until the 3’ polyA sequence is reached as well as the 5’ GC-rich region (See “Minor Edge Polishing”). Depending upon when extension is halted, due to some subjectivity for stopping extension in the polyA tail, the HTML visualization may appear slightly different than depicted in this protocol (Figure 6).
Visualization of the alignments (“-html” option) of the TE instances to each family indicates a subfamily with a higher divergence (Figure 7A) and a subfamily with a lower divergence (Figure 7B), highlighting the utility of COSEG to separate higher (older) and lower (younger) divergence subfamilies.
Figure 7.
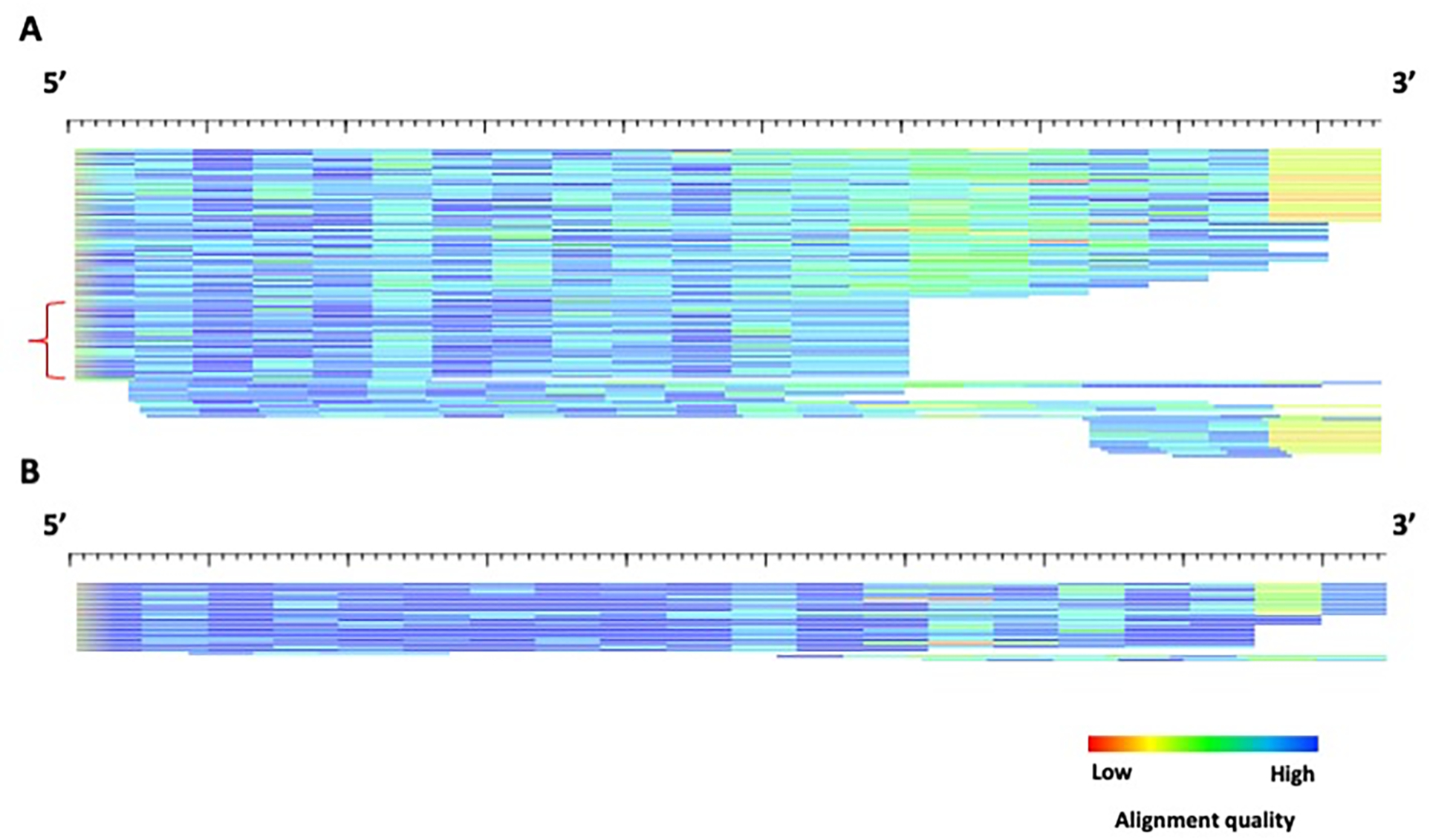
HTML format of the example2 TE subfamilies produced by COSEG. A) subfamily0 alignment. The red bracket indicates a possible subfamily that may have been missed by COSEG. B) subfamily1 alignment. Alignment of TE instances to 5’ edge of the example2 subfamily consensi generated by COSEG. A) subfamily0. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. The lengths for the alignments for subfamily0 and subfamily1 are 220 and 201 bp, respectively.
Example2 is a prolific SINE element obtained from a model generated by RepeatModeler2 on the mouse genome (mm39). As such, the model is generated by a representative set of sequences. Although COSEG nicely separates the starting material into two subfamilies, subfamily0 is still a mixture of instances of divergences with no obvious pattern, which most likely represent a mixture of several consensi, but not enough instances of each to group them into discernible models. In this case, it is recommended that more TE instances are collected from the genome using rmblast.pl, located in the RepeatModeler/util/directory, using the consensus as the query and the genome as a target. Then follow the Support Protocol starting from step 3 with the resulting “.out” file as the input, and repeat the Basic Protocol.
In the event that the subfamily structure for a model presents with a “truncated” appearance (Figure 5; Figure 8), a different strategy should be utilized. COSEG is not appropriate because depending upon the number of sequences that appear “truncated”, a large section of sequences will be missed. Specifically, a section of sequences will all begin or end at the exact same point in the alignment, indicative of a deletion or recombination product. A suggested program to group subfamilies with different lengths and/or recombination products is cd-hit T (Li & Godzik, 2006) combined with a post-processing script ‘ClusterPartialMatchingSubs.pl’ which processes the cd-hit output for use as input for the alignAndCallConsensus.pl program.
Figure 8.
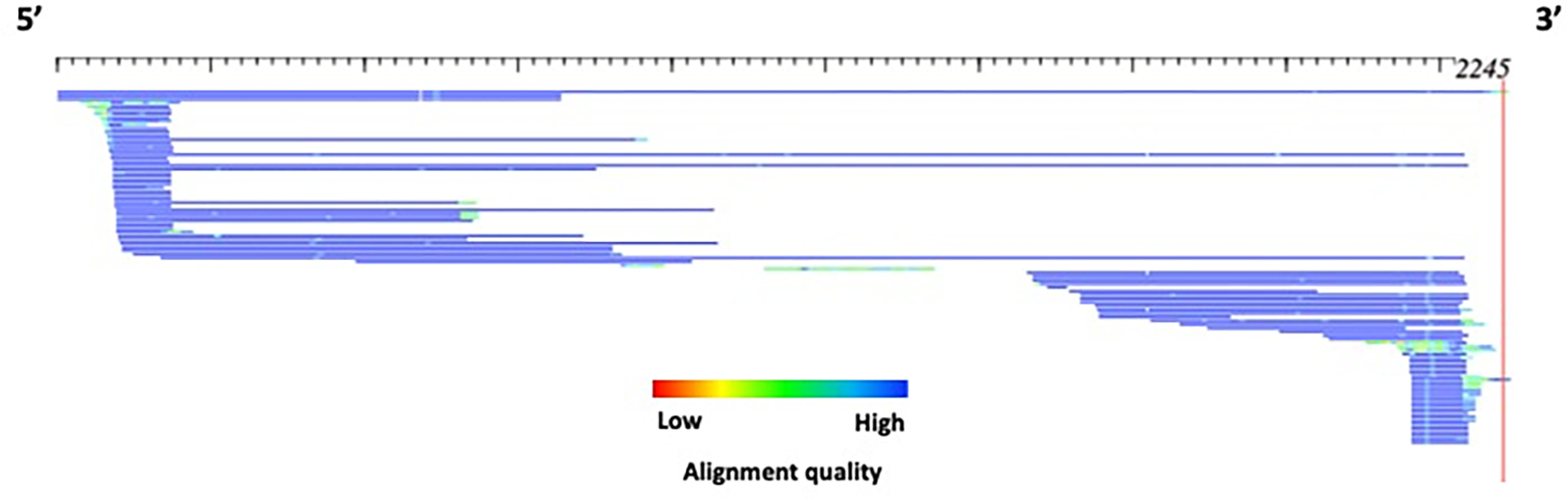
HTML alignment of example3. The alignment shows possible deletion products which may represent a subfamily structure. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. The total length of this alignment is 2284 bp.
The cd-hit program clusters sequences by length and subsequently using a sequence identity threshold to group the sequences. In this manner, it is more appropriate to use this program than COSEG for the analysis of TE models that comprise a subfamily structure that includes deletion and recombination products differing in length, such as example1 and example3 (Figure 5; Figure 8).
With the default settings, cd-hit analyzes the global sequence identity rather than the local sequence identity. The default settings also include a sequence identity threshold of 0.9, or 90%, calculated as the number of identical nucleotide sequences in the alignment. The two output files of cd-hit are a FASTA file of representative sequence and a text file of lists of clusters.
The suggested application for the example1 soloLTR (steps #.a) and example3 DNA element (steps #.b) is as follows:
-
13
Cluster TE instances by running the following in your terminal:
-
$ ClusterPartialMatchingSubs.pl example1_elements.fa -n 3 -lmax 600 -lmin 300 -a
The 133 sequences in the elements file contains small fragments of LTRs up to full-length ERVs, which variety in length derails cd-hit’s attempts to cluster subfamilies. The options “-lmax 600” and “-lmin 300” filter sequences longer than 600 bp and shorter than 300 bp from the input file, providing cd-hit a set of 101 mostly full-length solo LTRs.
By default, the script performs two cd-hit runs in succession. The first finds clusters of copies >= 90% (“-maxid” default value) identical to a chosen reference copy, the second finds clusters >80% (“-minid” default value) similar to the reference, taking the previously detected clusters into account. Such a hierarchical clustering prevents very similar sequences joining different clusters.
The choice of “minid” should be based on the estimated age (divergence levels) of the TE copies. For copies < 10% diverged from their original sequence, 80% is a good cutoff, as individual copies will on average be <80% diverged from each other. Older TEs require a lower minid. The number of runs can be changed by increasing or decreasing the distance between successive cutoffs, using the option -step (default 10%).
The results are combined and a directory is created for each cluster of 3 or more sequences. This minimum cluster size can be increased with the “-n” option. For example1, eight clusters are generated, with corresponding directories for each cluster. In addition, a file containing only the consensi is located within a file called clusterconsensi.
Within each directory, the sequences that contributed to a cluster are printed to the file repseq in the appropriate directory. For each cluster, the consensus is printed to the file rep, is the reference sequence used by cd-hit to initiate the cluster. The script then runs alignAndCallConsensus.pl up to three times. Default parameters are 14% divergence with a 41% background, but these can be changed by altering the “-di(vergence)” and “-g(clevel)” options. As the reference sequence tends to be longer than the TE copy, the alignments are automatically pruned. By default, the MSA will be pruned at both ends until the higher of 3 or the number of sequences in the cluster divided by three sequences are aligned at a position. A prune value of “-p 4” will add 4 to the number of required aligned sequences, while “-p −1” will subtract 1 sequence. However, this value cannot be less than 2.
By default, the script AutoRunBlocker.pl is run, which often fixes some indels in the consensus. Invoking the “-a” option skips this step, which is the slowest part of the procedure. In this example “-a” is invoked so that AutoRunBlocker can be run manually to illustrate its use (see step 15).
The consensus sequences for each cluster are concatenated in the file “clusterconsensi”.
To assess if the identified clusters created significantly different consensus sequences and how the relate to each other, perform a quick rmblast comparison:
$ rmblast.pl clusterconsensi clusterconsensi -bw 100 -mm 20 -ms 2000 -masklevel 101
Use a high bandwidth (-bw) to overcome large gaps, a large seed length/minimum match (-mm) to only align very similar regions, and high minimum score (-ms) to only see the better matches between subfamilies. “-masklevel 101” returns all matches over the cutoff score, and not only the best.
The rmblast.pl output shows that all but the Cluster2 consensus match each other from the beginning to the end (Table 2). Cluster2 appears ca 30 bp short of the 5’ end. Clusters 7 and 8 have one and three extra bases preceding the initial TG, which can be removed with an editor.
-
ClusterPartialMatchingSubs.pl example3_elements.fa -n 4 -a
The “-lmax” and “-lmin” parameters are not used here as the products of this alignment are all internal deletion products of a DNA transposable element. In addition, the default parameters for percent identity clustering are sufficient as the TE instances are almost identical (Figure 8). The use of “-n 4” was used here, as a value of 3 returned an additional two subfamilies that were weakly supported (data not shown).
The output of this program produces three low divergence consensi separated by length (Figure 11; Figure12). This subfamily analysis pipeline works particularly well to separate the TE instances exclusively by length because the copies are nearly identical (Figure 8), as opposed to an MSA with a wide range of divergences that cause a problem for clustering TE instances into subfamilies (Figure 7A).
-
Table 2.
Rmblast.pl output of the eight subfamilies produced by ClusterPartialMatchingSubs.pl analysis of example1.
| SW | divergence | % del | % ins | Query | Query start | Query end | A_left | Target | Target start | Target end | T_left |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4920 | 0.88 | 0 | 0 | Cluster0 | 1 | 571 | 0 | Cluster0 | 1 | 571 | 0 |
| 2704 | 2.1 | 0.35 | 32.33 | Cluster0 | 1 | 571 | 0 | Cluster12 | 1 | 433 | 0 |
| 2009 | 11.88 | 2.21 | 3.06 | Cluster0 | 210 | 571 | 0 | Cluster8 | 132 | 490 | 0 |
| 4051 | 0.22 | 0 | 0 | Cluster10 | 1 | 465 | 0 | Cluster10 | 1 | 465 | 0 |
| 2941 | 10.54 | 7.1 | 0.2 | Cluster10 | 1 | 465 | 0 | Cluster7 | 2 | 498 | 0 |
| 2725 | 8.6 | 13.76 | 1.34 | Cluster10 | 1 | 465 | 0 | Cluster6 | 1 | 522 | 0 |
| 3822 | 0.23 | 0 | 0 | Cluster12 | 1 | 433 | 0 | Cluster12 | 1 | 433 | 0 |
| 2735 | 2.77 | 32.33 | 0.35 | Cluster12 | 1 | 433 | 0 | Cluster0 | 1 | 571 | 0 |
| 4579 | 0.19 | 0 | 0 | Cluster2 | 1 | 522 | 0 | Cluster2 | 1 | 522 | 0 |
| 3488 | 1.72 | 0 | 14.73 | Cluster2 | 1 | 522 | 0 | Cluster5 | 32 | 486 | 0 |
| 3076 | 4.62 | 1.93 | 14.5 | Cluster2 | 4 | 522 | 0 | Cluster8 | 29 | 490 | 0 |
| 4268 | 0.21 | 0 | 0 | Cluster5 | 1 | 486 | 0 | Cluster5 | 1 | 486 | 0 |
| 3503 | 5.56 | 2.06 | 1.85 | Cluster5 | 1 | 486 | 0 | Cluster8 | 4 | 490 | 0 |
| 3452 | 1.98 | 14.73 | 0 | Cluster5 | 32 | 486 | 0 | Cluster2 | 1 | 522 | 0 |
| 4562 | 0 | 0 | 0 | Cluster6 | 1 | 522 | 0 | Cluster6 | 1 | 522 | 0 |
| 2787 | 11.3 | 1.53 | 6.64 | Cluster6 | 1 | 522 | 0 | Cluster7 | 2 | 498 | 0 |
| 2734 | 7.66 | 1.34 | 13.76 | Cluster6 | 1 | 522 | 0 | Cluster10 | 1 | 465 | 0 |
| 4354 | 0 | 0 | 0 | Cluster7 | 1 | 498 | 0 | Cluster7 | 1 | 498 | 0 |
| 2941 | 9.86 | 0.2 | 7.1 | Cluster7 | 2 | 498 | 0 | Cluster10 | 1 | 465 | 0 |
| 2806 | 11.87 | 6.64 | 1.53 | Cluster7 | 2 | 498 | 0 | Cluster6 | 1 | 522 | 0 |
| 4346 | 0.41 | 0 | 0 | Cluster8 | 1 | 490 | 0 | Cluster8 | 1 | 490 | 0 |
| 3524 | 5.54 | 1.85 | 2.06 | Cluster8 | 4 | 490 | 0 | Cluster5 | 1 | 486 | 0 |
| 3045 | 5.19 | 14.5 | 1.93 | Cluster8 | 29 | 490 | 0 | Cluster2 | 4 | 522 | 0 |
| 2053 | 11.98 | 3.06 | 2.21 | Cluster8 | 132 | 490 | 0 | Cluster0 | 210 | 571 | 0 |
The bolded rows highlight the comparisons of the cluster2 consensus with other subfamily consensi. The red numbers highlight the differences between the 5′ edges between the bolded consensi in the row.
SW: Smith-Waterman score Divergence: percent divergence
% del: percent of deletions in the query sequence
% ins: percent of insertions in the query sequence Query: name of query sequence
Query start: starting position match in the query sequence Query end: ending position of match in the query sequence
Q_left: number of bases in query sequence past the ending position of match Target: name of target sequence
Target start: starting position match in the target sequence Target end: ending position of match in the target sequence
T_left: number of bases in target sequence past the ending position of match
Figure 11.
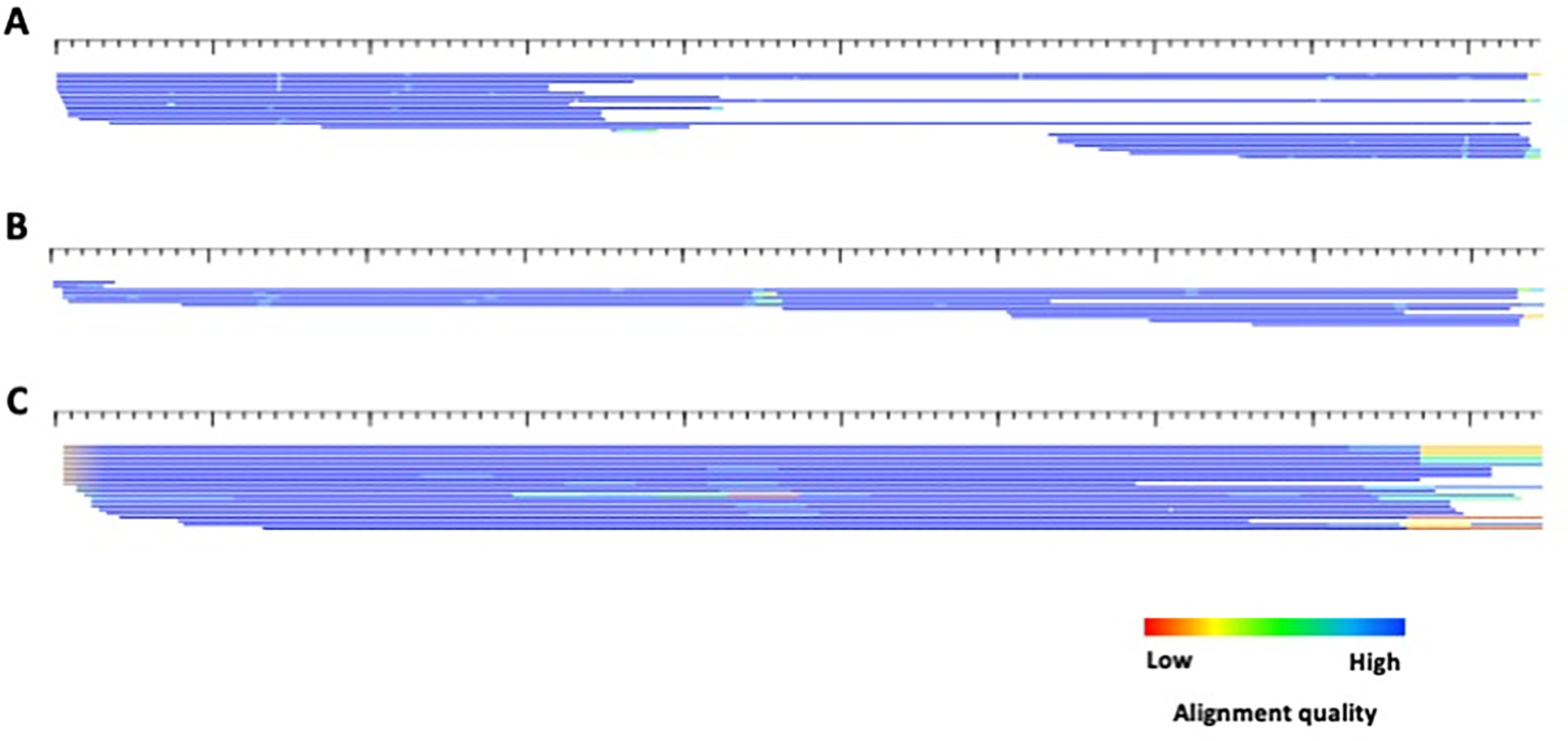
HTML alignments of the 3 consensi produced for example3 by ClusterPartialMatchingSubs.pl. A) cluster0; B) cluster1; C) cluster6.. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. All sequences are in the 5’ to 3’ orientation. The consensi lengths for cluster0, cluster1 and cluster6 are 2124, 1154, and 192, respectively.
Figure 12.

Length comparison of the example3 consensi to the original example3_con sequence. This image was generated using alignAndCallConsensus.pl and overlaying thicker and colored lines to highlight the difference length of the derived consensi and the original consensus sequence. The purple line is cluster0, green is cluster1 and orange is cluster6. The colors do not correspond to alignment quality.
Steps 13a and 13b highlight the differences between class I and class II subfamily relationships. Class II subfamilies are internal deletion products of full-length DNA elements as a result of the movement mechanism (Figure 8). Class I elements are more prone to recombination and present with a different alignment pattern (Figure 5).
-
14
Refine consensus and visualize subfamily alignments
-
Of the 133 instances in the original elements file for example 1, only 94 are present in the individual cluster alignments. Moreover, some copies may have ended up in the wrong cluster. By running alignAndCallConsensus.pl again using the original elements file and clusterconsensi as the consensus file all 133 sequences will be aligned to the best matching among the eight consensus sequences.
Since 7 of the 8 consensus sequences are already full length, the quickest way to stabilize them is to add with an editor an H-pad only to the 5’end of the Cluster2 consensus (in this example we add 7 Hs) and run the command
$ alignAndCallConsensus.pl -c clusterconsensi -e example1_elements.fa -ma 14 -re 4
The “-re(fine) 4” option iterates the command 4 times or stops earlier when none of the consensus sequences has changed in the last round. After 4 rounds the Cluster2 consensus should have expanded 4×7 = 28bp 5’, which is approximately how much it seemed to be truncated. After 2 cycles the other consensus sequences stabilize, and, by comparison of the resulting consensus file with itself as above, we see that after 4 cycles Cluster2 consensus is 4 bp overextended. After removing the H-pad and first 4 bases of the Cluster2 consensus with an editor, run alignAndCallConsensus one last time..
$ alignAndCallConsensus.pl -c clusterconsensi -e example1_elements.fa -ma 14 -hp 8 -f b -html
The “-f(inishedext) b” option tells the script that the consensus sequences are full length and do not need to be extended, although (because of -hp 8) they are flanked by H-pads. “-f 5” would prevent it from extending 5’ and “-f 3” likewise 3’. The H-pads still perform a function: 1) for more diverged copies they act as attractors so that copies with a few mutations near the end are still fully aligned. This improves the seed alignment (which will have the columns under the Hs removed), especially when HMM profiles are prepared from them. 2) Full ERVs and solo LTRs are flanked by a constant length target site duplication (TSD) as a result of their integration mechanism. For different groups of ERVs these are 4, 5 or 6 bp. Observing these TSDs confirms that the consensus sequences are really full length, while identifying their lengths sometimes helps to classify the LTR. The script TSD.pl returns the summed alignment score of all 5’ and 3’ flanking 1 to 8-mers to each other (random sequence would score negative or near 0), lists the flanking sequences of the highest scoring length and calculates a possible preferential target site.
To confirm the edges of all subfamilies has been reached following consensus refinement, run the following program on each subfamily (example for cluster7):
$ TSD.pl Cluster7.ali
TSD.pl on any of the other .ali files returns a clear 5 bp TSD (Figure 9), indicating the edges of the soloLTR have been reached. Unfortunately, in this case the length is misleading, as a 5 bp target is usually indicative of a class III ERV, while the internal sequences of these LTRs contain class I ERV protein coding regions which classically generates a 4bp TSD.
Figure 10 shows the final subfamily alignments in HTML format. Notice the lack of fragmentation and truncation and decreased divergence compared to when all TE instances were aligned to one consensus sequence (Figure 5).
-
alignAndCallConsensus.pl -c clusterconsensi -e example3_elements.fa -ma 14 -f b -html
Because class II subfamilies are internal deletion products, the edges are clearly defined. Therefore, the H-pad (-hp) option is not required, nor the interactive option (“-re” or “-int”) to iteratively and/or interactively refine the consensus as the TE instances are nearly identical (Figure 8). Figure 11 shows the separation of TE instances by length, while Figure 12 shows the subfamily relationships to the original consensus sequence (example3_con.fa; Table 1), a clear example of class II TE subfamily relationships.
-
Figure 9.

Terminal output of TSD.pl for cluster7.ali. The red box highlights the highest-scoring TSD length out of possible lengths 1–8 bp.
Figure 10.
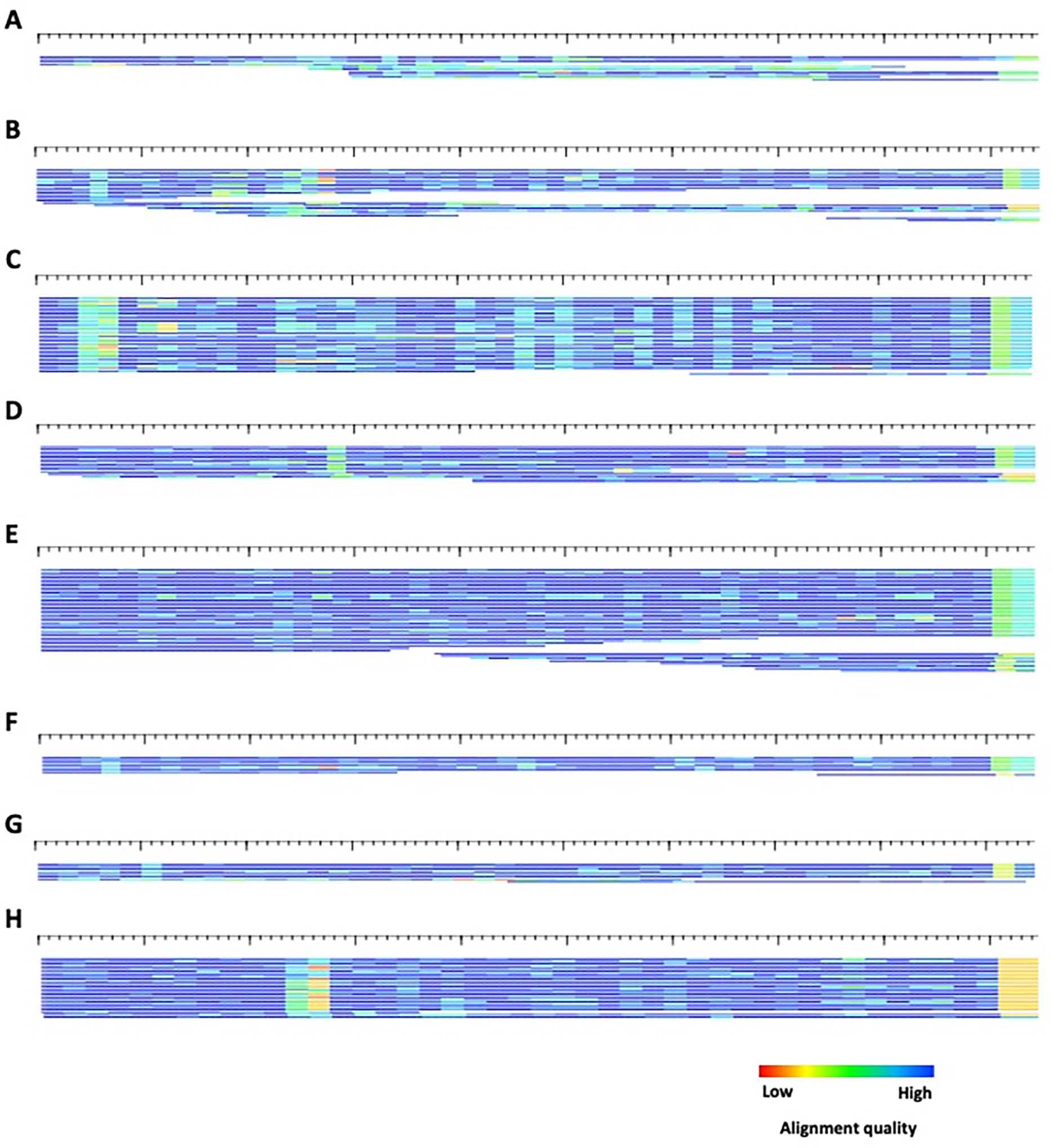
HTML alignments of the 8 consensi produced for example1 by ClusterPartialMatchingSubs.pl. A) cluster0 – 586 bp ; B) cluster2 – 572 bp; C) cluster5 – 502 bp; D) cluster6 – 522 bp ; D) cluster7 - 513 bp; E) cluster8 – 503 bp; G) cluster10 – 481 bp; H) cluster12 – 449 bp. Each sequence is represented by a single row (sorted by start position) where the color gradient indicates alignment quality (red=low; blue=high) over 10bp non-overlapping windows. All sequences are in the 5’ to 3’ orientation.
Often, gaps within the alignment and resulting consensus sequence will resolve themselves during the iterative refinement process, but not always. To aid in the proper calling of gaps, an additional program, AutoRunBlocker.pl, can be utilized to properly assign gaps based on a user-defined majority. This program can also be useful in separating models that may differ by length. Figure 10H indicates an additional subfamily structure within cluster12.
-
15
Resolve gaps within the alignment
$ AutoRunBlocker.pl -l Cluster12.ali -windowSize 12 -mc 3 -mr 1 -p
AutoRunBlocker.pl looks at a sliding window of a size -w(indowSize) over the MSA to determine if a plurality of copies have a different length in it than the consensus (= the window size). When the plurality includes “-mc” (“minCopyAgreement”) or more copies and is at least “-mr” (“minRatioAgreement”) more common than the consensus length, a new consensus is derived from the ungapped alignment of all copies with the plurality length. In the current version of the script, two or more overlapping windows that meet the conditions are merged and presented as a larger window in order not to present the same conflict multiple times. In the -p(prompt) mode, the user is asked if the new consensus should replace the old consensus with the new one. Without the -p option, all locations where the conditions are met are replaced. When finished, the number of changes made are indicated and a new full consensus is printed to the screen.
We integrate AutoRunBlocker.pl in a number of automated pipelines, as it generally improves the consensus. This is especially apparent when reconstructing TEs with open reading frames. We recommend using shorter window sizes for more diverged repeats, though given an observed problem area, larger windows may have to be tried to improve the MSA.
With the above command, AutoRunBlocker.pl suggests to replace one portion of the cluster12 consensus with a shorter sequence starting at position 118 within the alignment (Figure 13B). Notice that due to the merging of windows, the final consensus window over this region was 33 bp. To re-analyze the subfamilies, one could replace the Cluster12 consensus in the clusterconsensi file with the new consensus and re-run alignAndCallConsensus.pl.
However, it is clear from Figure 13B that the Cluster12 MSA still represents at least two different clusters of copies, one 15 bp longer than the other. A ClusterPartialMatchingSubs.pl run on the elements file “repseq” in the Cluster12 directory shows that this indel is basically the only difference between the two and we would probably decide to represent them with one model and seed alignment.
Figure 13.

AutoRunBlocker.pl analysis of the cluster12 subfamily derived from example1. A) Terminal output of AutoRunBlocker.pl. B) Cluster12.ali visualization. The red box highlights the sequence for which AutoRunBlocker.pl suggests an alternate length.
SUPPORT PROTOCOL
GENERATING SEED ALIGNMENTS USING A LIBRARY OF CONSENSI AND A GENOME ASSEMBLY
Many TE libraries may be maintained only with consensi. As such, the provenance of the consensus is not maintained. If the library was obtained via a de novo TE finder, it is likely that extension and further polishing is required. The main protocol starts from both TE consensi and instances; this protocol aims to help the researcher generate MSAs including instances in order to build the most accurate TE library possible.
Necessary Resources
Hardware
A computer with a Linux OS, or appropriate virtualization technology e.g. Docker for Mac or VirtualBox, 100 GB of free hard drive space, and 64 GB of RAM
Software
RepeatModeler 2.0.2 (or higher)
RepeatMasker
Files
Consensus library, in FASTA format
Reference genome in both FASTA and 2bit format
You can convert FASTA to 2bit with `faToTwoBit [-long] genome.fa genome.2bit` (You will need the `-long` option if the genome is larger than 4GB)
You can convert 2bit to FASTA with `twoBitToFa genome.2bit genome.fà
-
Remove any sequences that are mostly composed of tandem repeats (TRs) or simple repeats. These sequences will attract many redundant hits and there is little utility in building good models for them, with the possible exception of specific Satellite sequences which might be of particular interest. In many workflows TRs will be masked by methods specifically designed to handle tandem and simple repeats.
To filter the consensi for tandem repeats, run the following command on your terminal. The fasta-trf-filter.pl program can be found in the RepeatModeler/util/ directory.
$ fasta-trf-filter.pl consensi.fa >consensi.filtered.fa
Or do the filtering manually. We use these criteria:
TRF parameters: 2 7 7 80 10 50 and finally a max period of 20
If a sequence is less than 80% masked OR has at least one 100bp or longer contig, then keep that consensus.
-
Run RepeatMasker with the consensi as the search library to collect interspersed repeats. Searching for homologous sequences with RepeatMasker recapitulates seed alignments corresponding to the input consensi, and competes consensi against each other when determining which (sub)family model they should be assigned to in the case of redundancy or subfamily structure.
$ RepeatMasker -a -e rmblast -lib consensi.filtered.fa genome.fa
The “-a” option produces the “*.align” or “*.align.gz” file used for the next step to generate the seed alignments. The “-lib” option specifies to use the custom consensus library instead of any configured/installed search libraries. In this example, we used rmblast (“-e rmblast”), but cross_match or abblast could also be used.
-
Generate seed alignments for your input TE consensi. Make a new directory to contain your alignments, and run the generateSeedAlignments.pl command inside that directory. generateSeedAlignments.pl can be found in the RepeatModeler/util/ directory.
$ mkdir stks
$ cd stks
$ generateSeedAlignments.pl -taxon ‘Genus species’ -assemblyFile ../genome.2bit ../genome.fa.align >generateSeedAlignments.log 2>&1
The generateSeedAlignments.pl program creates one file in the Stockholm MSA format (.stk) for each newly generated seed alignment, and produces detailed log output.
generateSeedAlignments.pl can also be used with a “.out” file produced by rmblast.pl instead of a .align file.
-
Split the stk files into consensus and elements files. This can be done with the Linup program, also in the RepeatModeler/util/ directory:
$ Linup family1.stk -consensus >family1_con.fa
$ Linup family1.stk -fasta >family1_elements.fa
You may also find it convenient to convert all the files at once ahead of time, depending on your own workflow preferences. For instance:
$ for infile in *.stk; do Linup “$infile” -consensus >“$infile”_con.fa; Linup “$infile” -fasta >“$infile”_elements.fa; echo “done: $infile”; done
-
The extension method used in alignAndCallConsensus.pl requires the elements file to include flanking sequences, which are not included in the output from generateSeedAlignments.pl. To add flanking sequences to an existing set of instances, first run alignAndCallConsensus.pl once with an appropriate matrix to generate a .out file. Then, run extendFlankingSeqs.pl (also in the RepeatModeler/util/ directory) to append additional sequence from the original genome assembly onto both sides of each sequence in the elements file:
$ alignAndCallConsensus.pl -c family1_con.fa -e family1_elements.fa -int -ma <matrix>
$ extendFlankingSeqs.pl -i family1_con.out -o family1_elements.fa -d genome.2bit -flank 200
COMMENTARY
CRITICAL PARAMETERS & TROUBLESHOOTING:
RepeatModeler, RepeatMasker, and tools used in this protocol make use of other programs (“dependencies”) which must also be installed and configured appropriately for your computing environment. Our software attempts to detect some of the most common problems with installation, but it is impossible to automatically detect all possible issues. At this time we generally recommend installing these tools manually, or the use of preinstalled Singularity or Docker environments provided by the Dfam TE Tools project (https://github.com/Dfam-consortium/TETools) to obtain the correct versions and configuration.
Many of the tools used in this protocol are part of the RepeatModeler 2.0.2 distribution and are located in the RepeatModeler/util directory. Others are part of coseg, cd-hit, or other software listed in the requirements section. It may be convenient to put some or all of these directories on your PATH instead of typing out full paths to the individual programs. The Dfam TE Tools container is configured so that all the tools used in this protocol and more are already on the PATH.
For the purpose of troubleshooting, it is important to keep logs, screen outputs, and temporary files. The shell redirection operators “>“ and “2>“ and output-recording utilities such as “tee” and “script” are useful for this purpose and available on most Linux systems. We also recommend running analyses on a local disk whenever possible, which is both faster and more reliable than network filesystems.
Comments, questions, concerns, and bug reports related to the RepeatMasker and RepeatModeler packages can be directed to the RepeatMasker and RepeatModeler GitHub pages (listed in the Internet Resources section) or by email to help@repeatmasker.org.
When subfamilies are not generated by COSEG, it is possible that the visual assessment of the alignment does not have co-segregating mutations detectable by the program. In these cases, it may be helpful to group the sequences by divergence level and generate consensi for the different divergent groups.
In the event that a conserved sequence edge has not been reached, but your TE instances do not contain additional flanking sequence, please see Support Protocol step 5 for instructions on how to extend the flanking sequence using the .out file and a 2bit genome.
UNDERSTANDING RESULTS:
The quality of the output data depends on the quality of the input. TE discovery in a genome is influenced by the sequencing method and the resulting genome assembly. Due to their repetitive nature, young and/or longer TE copies tend to be represented by strings of Ns in genome assemblies based exclusively on short reads, while this is hardly an issue with long-read sequencing technology (Peona et al., 2021). Low sequencing coverage will also compound the difficulty in TE discovery and placement. Such analyses limit the utility of TEs in comparative genomic studies.
Especially when planning a subfamily analysis, care should be taken not to collect multiple copies of the same TE insertion. These may have originated in silico from unassembled identical sequences in lower quality genome assemblies, or in vivo via processes other than the TE’s mechanism of transposition. The most problematic of the latter are TEs embedded in tandem sequence arrays or segmental duplication. A general characteristic of such duplicates is a much higher similarity to each other over a much longer region than the average between all copies. It is recommended to try to eliminate them before analysis.
An expectation of the nature of the TEs in a genome often helps to properly interpret the results. For example, mammalian genomes are rich in mostly old retrotransposable elements, specifically high-copy number SINEs and LINEs, and generally lack DNA transposons (Arensburger et al., 2016; Arkhipova, 2017). Many classes of transposons have not been found in mammals and the classification of a mammalian TE model as, say, a Zisupton or a Copia element should be considered dubious. On the other hand, many invertebrates have a wide variety of low-copy-number, young TE families and a MSA of many highly diverged copies probably indicates a mixture of TE families.
De novo methods can falsely identify high-copy host genes or gene families as a TE, such as rDNA or a zinc-finger domain. Consequently, it is important to assess your TE models by comparing the consensi to annotated genes in a database, such as Genbank. This search will also identify any highly-transcribed host genes reverse-transcribed and re-inserted into the genome as a processed pseudogene. These instances are by-products of TE mobilization and are not members of TE families. A genome may contain unclassified TE families that do not fall into any previously-defined TE category and have no terminal sequence similarity to known TEs. In that case, defining the ends of an element has to be guided by the conservation in the MSA and perhaps the detection of TSDs, rather than, for example, homology to a known TIR.
Another problematic issue is that de novo TE finders often produce redundant models for a TE or models representing fragments of a TE. With this in mind, you should analyze related groups of TE models concurrently, and be ready to combine redundant or related consensi, especially before endeavoring subfamily analysis. When substantially extending a consensus, you should regularly compare it to the library of models to check for new matches.
As pointed out in the LTR subfamily protocol above, subfamily analysis has an arbitrary stopping point. A 90/90 rule (90% identity over 90% sequence coverage) has been proposed and utilized to differentiate subfamilies (Wicker et al., 2018). However, the preferred level of subfamily splitting depends on many factors, including abundance of the repeat, phylogenetic relevance, age of the element (great interest exists in subtly different currently active TEs) and possible improvement of detection. As an example of the latter, two similar subfamilies of an ancient TE that differ significantly at one end may be split to allow full matching of members of both subfamilies. Subfamilies are often characterized and grouped based on phylogenetic analyses (Arkhipova, 2017; Wicker et al., 2007). Much work has been done in that regard on the abundant Alu SINEs in primates (Ray & Batzer, 2005). Alu subfamilies that differ by just a few substitutions, such as AluTa10 and AluTa15 in New World monkeys, can have a contrasting phylogenetic distribution. Here, ideally, a comparative genomic analysis should be done to confirm or reject the necessity of subfamily generation.
TIME CONSIDERATIONS:
The length of time it will take a TE curator to produce a curated model is highly dependent upon the researcher’s level of experience with TE biology and seed alignments. In addition, library fragmentation and required extension as well as TE composition, will increase the amount of time required per model. Therefore, the range of time required to complete a single model will vary. For example1, the approximate time to complete steps 1–7 (extension) is 15–45 minutes depending upon the level of expertise. Please note that for example1 minimal extension is required. COSEG analysis of example2 can be completed in 10–20 minutes. ClusterPartialMatchingSubs.pl analysis of examples 1 and 2 can be completed in 5–10 minutes. Resolving the gaps of example1 can be completed in 5–10 minutes. Example1 has gone through extension and consensus refinement, subfamily analysis and gap resolution. The total time for this process for example1 is 35–85 minutes.
For the support protocol, the RepeatMasker step will take the most time, potentially a few days depending on the amount of data and parallelism. The “-pa” option for RepeatMasker can be used to increase the amount of work done in parallel, reducing the time taken but increasing the required number of CPU cores and memory. generateSeedAlignments.pl takes much less time to run, but has a heavier memory footprint. We recommend at least 32GB of RAM.
ACKNOWLEDGEMENTS:
This work is funded by NHGRI grant # U24 HG010136, and NHGRI grant # RO1 HG002939.
Footnotes
INTERNET RESOURCES:
https://www.dfam.org/classification/dna-termini
Sequence repository containing the LOGOs, HMMs and consensi for the conserved termini of DNA transposons.
Instructions for downloading COSEG, RepeatMasker, RepeatModeler2, and their dependencies (e.g., TRF, rmblast)
https://github.com/rmhubley/RepeatMasker/
Source code and issue tracking for RepeatMasker
https://github.com/Dfam-consortium/RepeatModeler
Source code and issue tracking for RepeatModeler
http://weizhongli-lab.org/cd-hit/
Instructions for downloading cd-hit
http://www.bioinformatics.org/cd-hit/cd-hit-user-guide.pdf
Detailed user manual for additional cd-hit options
LITERATURE CITED:
- Arensburger P, Piégu B, & Bigot Y (2016). The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from the fables of Jean de la Fontaine? [Review of The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from the fables of Jean de la Fontaine?]. Mobile Genetic Elements, 6(6), e1256852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipova IR (2017). Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mobile DNA, 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, & Feschotte C (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nature Reviews. Genetics, 18(2), 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MSP, Brudno M, & Batzoglou S (2005). ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Research, 15(2), 330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan DJ (1989). Eukaryotic transposable elements and genome evolution. Trends in Genetics: TIG, 5(4), 103–107. [DOI] [PubMed] [Google Scholar]
- Flutre T, Duprat E, Feuillet C, & Quesneville H (2011). Considering transposable element diversification in de novo annotation approaches. PloS One, 6(1), e16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, & Smit AF (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America, 117(17), 9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray YH (2000). It takes two transposons to tango: transposable-element-mediated chromosomal rearrangements. Trends in Genetics: TIG, 16(10), 461–468. [DOI] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, Smit AFA, & Wheeler TJ (2016). The Dfam database of repetitive DNA families. Nucleic Acids Research, 44(D1), D81–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, & Jurka J (2005). RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biology, 3(6), e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, & Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr, & Moran JV (2017). Mobile DNA in Health and Disease. The New England Journal of Medicine, 377(4), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohany O, Gentles AJ, Hankus L, & Jurka J (2006). Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics, 7, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T, & Sonnhammer ELL (2005). Kalign--an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics, 6, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, & Godzik A (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics , 22(13), 1658–1659. [DOI] [PubMed] [Google Scholar]
- Magis C, Taly J-F, Bussotti G, Chang J-M, Di Tommaso P, Erb I, Espinosa-Carrasco J, & Notredame C (2014). T-Coffee: Tree-based consistency objective function for alignment evaluation. Methods in Molecular Biology , 1079, 117–129. [DOI] [PubMed] [Google Scholar]
- Morgenstern B (2004). DIALIGN: multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Research, 32(Web Server issue), W33–W36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Smit AFA, & Okada N (2006). Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Research, 16(7), 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, Lugo CSB, Elliott TA, Ware D, Peterson T, Jiang N, Hirsch CN, & Hufford MB (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology, 20(1), 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peona V, Blom MPK, Xu L, Burri R, Sullivan S, Bunikis I, Liachko I, Haryoko T, Jønsson KA, Zhou Q, Irestedt M, & Suh A (2021). Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. Molecular Ecology Resources, 21(1), 263–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piégu B, Bire S, Arensburger P, & Bigot Y (2015). A survey of transposable element classification systems--a call for a fundamental update to meet the challenge of their diversity and complexity. Molecular Phylogenetics and Evolution, 86, 90–109. [DOI] [PubMed] [Google Scholar]
- Ray DA, & Batzer MA (2005). Tracking Alu evolution in New World primates. BMC Evolutionary Biology, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit Arian F. A., Hubley Robert, and Green Philip. RepeatMasker Home Page, 2013. http://www.repeatmasker.org/
- Richardson SR, Morell S, & Faulkner GJ (2014). L1 retrotransposons and somatic mosaicism in the brain. Annual Review of Genetics, 48, 1–27. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, & Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22(22), 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Linard B, Lecompte O, & Poch O (2011). A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives. PloS One, 6(3), e18093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayabaskar MS (2017). Introduction to Hidden Markov Models and Its Applications in Biology. Methods in Molecular Biology , 1552, 1–12. [DOI] [PubMed] [Google Scholar]
- Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, Smit AFA, & Finn RD (2013). Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Research, 41(Database issue), D70–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Gundlach H, Spannagl M, Uauy C, Borrill P, Ramírez-González RH, De Oliveira R, International Wheat Genome Sequencing Consortium, Mayer KFX, Paux E, & Choulet F (2018). Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biology, 19(1), 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, & Schulman AH (2007). A unified classification system for eukaryotic transposable elements. Nature Reviews. Genetics, 8(12), 973–982. [DOI] [PubMed] [Google Scholar]