

Abstract
Single-molecule imaging in living cells enables the investigation of molecular dynamics and interaction underlying the physiology of a cell. We recently developed a method to visualize translation events at the single mRNA resolution in living cells. Here we describe how we apply this method to visualize mRNA interactions with stress granules in the context of translational activity during cell stress.
Keywords: single-molecule imaging, stress granules, translation
1. Introduction:
When exposed to environmental stress, cells conserve resources by shutting down the translation of the majority of mRNA molecules. Stressed cells also form stress granules (SGs) and a fraction of mRNAs translocate into SGs1,2. Because these processes occur at the same time, it was not clear precisely when, where, and how mRNAs stop translation and interact with SGs. To address these questions, we recently developed technology to track individual mRNA molecules and simultaneously monitor their translational status and dynamic interactions with SGs on the seconds timescale in living cells3–5.
At the core of our technology is the MS2 stem-loop system developed by several members of the Singer lab over the past twenty years to track individual mRNA molecules live6,7. This system works as follows: The operator region of the MS2 bacteriophage genome forms an RNA stem-loop that is bound by an MS2 coat protein (MCP). By repeating the stem-loop sequence (this sequence is generally referred to as MS2) in a reporter gene (typically 24 repeats placed in the 3’-UTR) and introducing this reporter gene into cells expressing fluorescently labeled MCP, single mRNA molecules can be visualized. This is possible because of the signal amplification that occurs when multiple MCPs bind multiple stem loops in a single reporter mRNA. MS2 stem-loops are bound by MCP as soon as they are transcribed, so the MS2 system makes it possible to track single mRNA molecules throughout their full lifecycles, from transcription to degradation7–10. In addition to MS2, several other similar yet orthogonal systems have been developed to tag and track single RNA molecules in multiple colors, including the PP7 and lamdaN systems11–13.
Although MS2 makes it possible to track individual mRNA, it is not possible to visualize their translation using MS2 alone. One workaround technique, coined TRICK (translating RNA imaging by coat protein knock-off), was developed by the Chao lab14. Briefly, PP7 stem-loops are placed in the coding region of an MS2-tagged reporter mRNA. With this arrangement, fluorescent PP7 coat proteins bound to the PP7 stem loops are knocked off the mRNA reporter by the first ribosome that translates it. While this is a powerful way to distinguish translated from untranslated mRNA, the TRICK assay cannot be used to continuously monitor mRNA translation. To achieve this, our lab and others created a new type of protein tag that fluoresces brightly within seconds of its translation3,15–18. In essence, this new type of tag is a protein-analog of the MS2 tag. Just like MS2 stem loops in the 3’ UTR of a reporter mRNA, we fuse repeats of a short peptide epitope sequence onto the N-terminus of a reporter protein. When the reporter protein is translated in cells that co-express fluorescent antibody-based probes against the epitope (either Fab, scFv, or nanobodies, the analogs of MCP), the nascent peptide chain can be visualized as it is being translated (analogous to the visualization of transcription using MS2). Here, the signal is amplified not only by the number of repeats of the epitope tag, but also by the number of ribosomes translating the mRNA in polysomes. By combining this new protein tag with an MS2 mRNA tag, we are able to track individual mRNA molecules in living cells and continuously monitor their translational status over multiple rounds of translation19.
In this protocol, we describe in detail how we used this new technology in cells expressing a fluorescently labeled SG marker to quantify the dynamics of translation shut off of single mRNAs and the interaction between mRNAs and SGs after stress (Figure 1). We first describe how to set up the experiments, including a method to introduce all of the necessary components into living cells at optimal levels as well as suitable imaging conditions. We then describe the basics of image analysis to elucidate the dynamic parameters of mRNAs during cellular stress.
Figure 1:
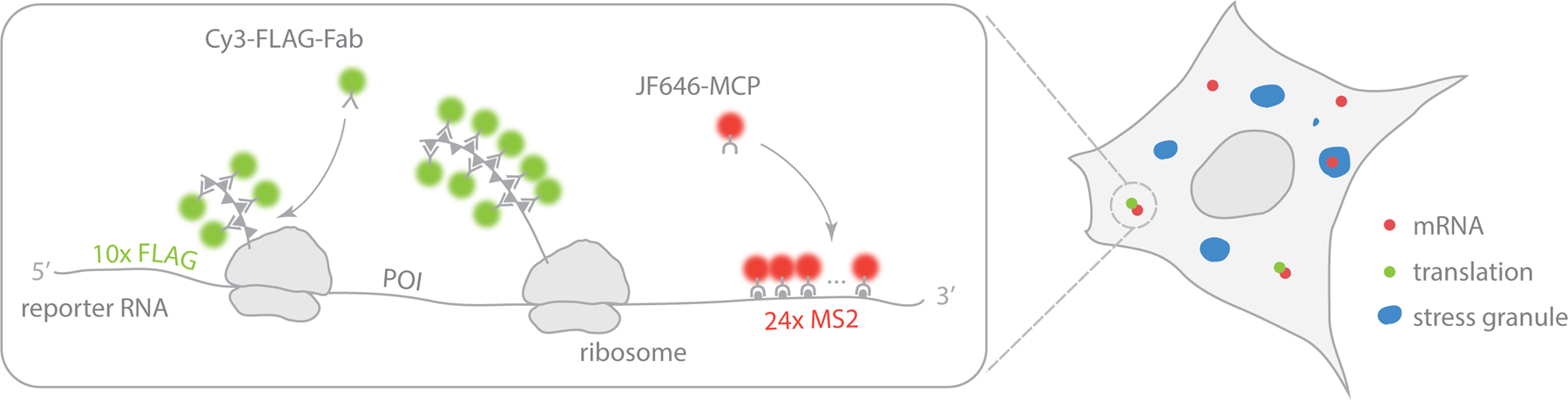
Schematic illustrating a single-molecule translation reporter system in a SG marker cell.
2. Materials:
Prepare all solutions in sterile conditions. Sodium Arsenite is a carcinogen and a teratogen. Handle with extreme caution and follow waste disposal regulations.
-
Sample prep
Detailed in our recent protocol paper (Koch et al. Bio Protoc., under review)
-
Reporter plasmid
Reporter plasmids can be acquired from Addgene (ID81082–81085). Replace promoter, 5’, 3’- UTR, and/or the protein-coding sequence of interest by subcloning (see Note a).
-
Cy3 anti-FLAG Fab
Prepare anti-FLAG Fab from anti-FLAG antibody with the Fab preparation kit. Label anti-FLAG Fab with Cy3 NHS ester and then purify Cy3 anti-FLAG Fab with ultrafiltration. The working stock should be ~1 mg/mL.
-
Purified Halo-MCP
Standard protein expression and purification from E.coli should be performed. The expression vector is available upon request. Alternatively, the genetically-encoded plasmid can be transfected (see Note b).
-
-
Tissue culture
-
U-2 OS cells stably expressing the SG marker GFP-G3BP1
Isolate GFP-G3BP1 U-2 OS cells from GFP-G3BP1/mRFP-DCP1a U-2 OS cells20(see Note c).
Phosphate buffered saline (PBS)
Trypsin
-
DMEM (+)
Add 50 mL fetal bovine serum, 5 mL Penicillin-Streptomycin (10,000 U/mL), and 5 mL L-Glutamine (200 mM) to 500 mL Dulbecco’s Modified Eagle Medium (DMEM, high glucose, no glutamine).
-
-
Imaging
Glass bottom dishes (see Note d)
-
106 um glass beads
Sterilize glass beads in 2 M NaOH for 2 hours. Wash the beads with Milli-Q water until the pH is neutral. Then rinse the beads with 100% ethanol. Keep the beads dry (see Note e).
Opti-MEM
-
White DMEM (+)
Phenol-free DMEM (+)
-
Janelia Flour 646 HaloTag Ligands21
Dissolve Janelia Flour 646 HaloTag Ligands (JF646-Halo) to 10 mM in DMSO for the stock solution. Dilute the stock solution to 0.2 mM in DMSO for the working stock solution. Store at −20 C (see Note f).
-
Puromycin
Dissolve puromycin dihydrochloride to 50 mg/mL in sterile H2O.
-
Sodium arsenite
Dissolve sodium arsenite to 50 mM in sterile H2O.
3. Methods
-
Imaging
Seed cells on glass-bottom dishes at 75% confluency the night before imaging (see Note g).
Two hours before bead-loading, wash cells two times with PBS and exchange media to Opti-MEM supplemented with 10% FBS (v/v) (see Note h).
Prepare sample for bead-loading by mixing the following: 750 ng of reporter plasmid, ~0.5 ug of Cy3 anti-FLAG Fab (~1 uL of the working stock), ~0.13 ug of purified Halo-MCP (~1 uL of the working stock), PBS to a total volume of 4 uL (see Note i). Do not vortex.
Remove media from the glass-bottom dish, and set aside in a conical tube (see Note j). Immediately after removing the media, add the 4 uL sample mix to the glass area of the dish, then sprinkle glass beads onto the cells as a monolayer. Tap the dish ~10 times against the hood bench. Decant the media that was set aside back into the dish22.
3 – 4 hours after bead-loading, wash cells three times with PBS. Next, replace the media with white DMEM(+) supplemented with 200 nM JF646-Halo. DMSO concentration should not exceed 0.1%. Incubate the cells at 37C for 30 minutes. Wash the cells three times with PBS, and replace the media with white DMEM(+). There can be some glass beads remaining in the dish, but this should not affect the imaging provided they do not cover the area being imaged. Keep cells in an incubator until ready to image.
Place cells onto a stage top incubator at 37C and 5% CO2 on a fluorescence microscope (see Note k). Take a fraction of medium into a 1.5 mL microcentrifuge tube, and keep it at 37C. Place the lid of the dish loosely on such that the lid can be removed without perturbing the dish when drugs are added. A microinjection system can be employed for this purpose.
Search for cells containing ~10 to ~100 mRNAs in the cytoplasm. mRNA should appear as distinct puncta excited by ~637 nm laser (red dots in Figure 2). Some mRNAs should be co-localized with distinct puncta excited by ~561 nm laser (green dots in Figure 2). GFP-G3BP1 should be uniformly diffused throughout the cell (blue haze) (see Note l).
Set up imaging conditions depending on your experiments. The entire cell volume should be imaged such that mRNAs do not go out of focus. It is highly recommended to use a TTL-triggered piezo stage to go through the entire cell volume as quickly as possible. To study the global trend of change in mRNA dynamics before and after stress, we introduced a 3 minute interval between each volume capture. To investigate the interaction between mRNA and SGs, we introduced a 2 sec interval between each volume capture.
Acquire pre-stress images.
Dilute sodium arsenite in the medium that was set aside in step a-vi, add it back to the dish, and pipette several times to mix the medium between each volume capture. The final concentration of sodium arsenite should be 0.5 mM (see Note m). Acquire post-stress images. SGs should start forming ~10 to 20 minutes after adding sodium arsenite (blue blobs in Figure 2).
To investigate translation resumption after stress relief, wash the cells ten times with white DMEM(+)
Image TetraSpeck Fluorescent Microspheres mounted on a slide for camera alignment if multiple cameras are used for imaging.
-
Image analysis
-
Pre-processing movies
Create a maximum-intensity projection through z.
Create a moving-average projection through time (using a few frames) if the signal-to-noise ratio is low.
Define a mask that contains an entire cell to analyze in order to avoid including any molecules from adjacent cells.
Apply a bandpass filter to individual images from movies to accentuate mRNAs, translation signals, and SGs of a given size range.
Binarize the bandpass-filtered images to make masks for each channel (one for mRNAs, one for translation signals, and one for SGs, Figure 2). This is achieved by choosing an appropriate threshold value and setting all pixels above that value to one and all pixels below that value to zero. The threshold value should be chosen such that the vast majority of pixels within mRNAs (translation signals, or SGs) are above the threshold, whereas the vast majority of pixels outside mRNAs (translation signals, or SGs) are below the threshold.
- Fit all detected mRNAs and translation signals to a 2D Gaussians of the following form to determine the precise coordinate of spots:
where IBG is the background fluorescence, I the spot intensity, (σx, σy) spreads of the spot, and (x0, y0) the spot location with super-resolution. Apply a correction factor on either the determined centroids (for step b-ii, b-iii, and b-v) or mask images (for step b-iv) to adjust for potential camera misalignment if multiple cameras are used for imaging (see Note n).
-
Global translation shut off dynamics
Calculate the distance between mRNAs and their closest translation signals. Categorize mRNAs into translating mRNAs if the distance from the mRNA to the closest translation signal is less than 3 pixels (= 390 nm).
Create a mask image of mRNAs by drawing disks with a radius of 2 pixels (260 nm) centered at the super resolved locations (determined in step b-i-6 or b-i-7). Multiply this mask image with the mask image of SGs to find mRNAs in SGs.
Count the number of non-translating mRNAs in the cytosol or SGs, and the number of translating mRNAs in the cytosol or SGs. Divide those numbers by the total number of mRNAs at each time point to determine the fraction of each population. Plot the fraction of each population as a function of time for each cell.
- Normalize the number of SGs to the fraction of the maximum number of SGs detected in the cell to plot the SG formation curves for each cell. Fit the SG formation curve to the following equation such that all the curves plotted above can be aligned for the cell-to-cell variability in the timing of the beginning of SG formation:
where NSG is the number of SGs, A, B, and C are normalization factors, and t is the time. Shift all the single cells curves such that their values at 10% of their maxima aligns from the fitted results.
-
Single mRNA molecule translation shut off dynamics
-
Track mRNA molecules with the nearest-neighbor tracking algorithm. Any single-molecule tracking package can be used, such as TrackMate23.
Hand-curate tracks to merge tracks that get split accidentally because of dimmer signals for a few frames and/or to omit tracks that are indistinguishable from other mRNAs due to dense environments.
Measure the intensity of each translation signal and background signal by using a disk and a ring mask, respectively (Figure 3). While 2D Gaussian fitting can serve this purpose, we find it sometimes fails to detect near-zero intensities of translation signals. The disk-ring mask method ensures that the intensity of the translation signal will be sensible, even when low.
Plot the location of mRNA with the intensity of the translation signal along with the location of SGs.
-
-
mRNA-SG interaction dynamics
Use the Mathematica command “ComponentMeasurements” (or an equivalent command in Python or Matlab) to measure the centroid position, area, total intensity, and convex vertices of each masked object determined in step b-i-5 or b-i-7 (i.e., each region of connected ones in the binarized images).
Track the masked SGs (i.e., perform particle tracking, as described above in step b-iii-1, but now on the masked images of SGs).
Create a sequence of 2-colored binarized sub-images from each tracked SG. This is achieved by extracting pixels within the smallest box that contains the convex vertices of each SG at each time point, as determined in step b-iv-1 above. In each sub-image, one channel should correspond to the mRNA and the other to the SG.
For each tracked SG, analyze the corresponding sequence of binarized sub-images as follows: (1) Starting from the beginning of the image sequence, check if an mRNA is overlapping with the SG. In this case, there will be pixels with ones in both channels in the binarized sub-image. If so, count how many consecutive time points this is true. Set this length of time to be the mRNA-SG interaction time. Repeat until the end of the SG track is reached (see Note o).
Repeat for all tracked SG to produce a distribution of mRNA-SG interaction times. If an individual interaction persists until the final frame, ignore the interaction as its precise length of time cannot be determined (see Note p). If an individual interaction lasts for less than two consecutive frames, ignore the interaction as it may simply reflect spurious imaging noise.
From the remaining SG-mRNA interaction times, calculate the interaction time survival probability curve as follows: (1) count the fraction of interactions that last longer than N seconds, with N ranging from the shortest interactions of interest (in our case 6 s) to the longest interactions of interest (in our case, 300 s); (2) Fit the fraction of tracks lasting longer than N seconds versus time to a double exponential curve of the form A exp(−t/t1) + B exp(−t/t2); (3) Renormalize the fraction of interactions lasting longer than N seconds according to the fit so that the fraction is 100% at t = 0 s (that is A + B = 100).
From the fitted interaction time survival probability curve, t1 and t2 give the timescales of rapid and slow binding, respectively (assuming t1 < t2 in fit). The fraction of fast and slow interactions can be calculated as A/(A + B) and B/(A + B), respectively.
-
Diffusion of mRNA
Calculate the mean squared displacement (MSD) from the super-resolved locations of tracked mRNA in step b-iii-1.
Calculate the MSD of SGs using the intensity-centroid of SGs.
To calculate the MSD of mRNA inside an SG, the diffusive movement of the SG must be corrected. One way to easily do this is to use the position of another mRNA within the SG as a reference position. Thus, for each SG with more than one mRNA in it, do the following: (1) for each pair of mRNA in the SG choose one to be the reference mRNA; (2) normalize the coordinates of the other mRNA in the pair to the reference mRNA (i.e., the position of the reference mRNA defines the (0,0) position); (3) calculate the MSD from the normalized coordinates; (4) divide the obtained normalized MSD by a factor of two to remove the impact of the movement of the reference mRNA.
-
Figure 2:
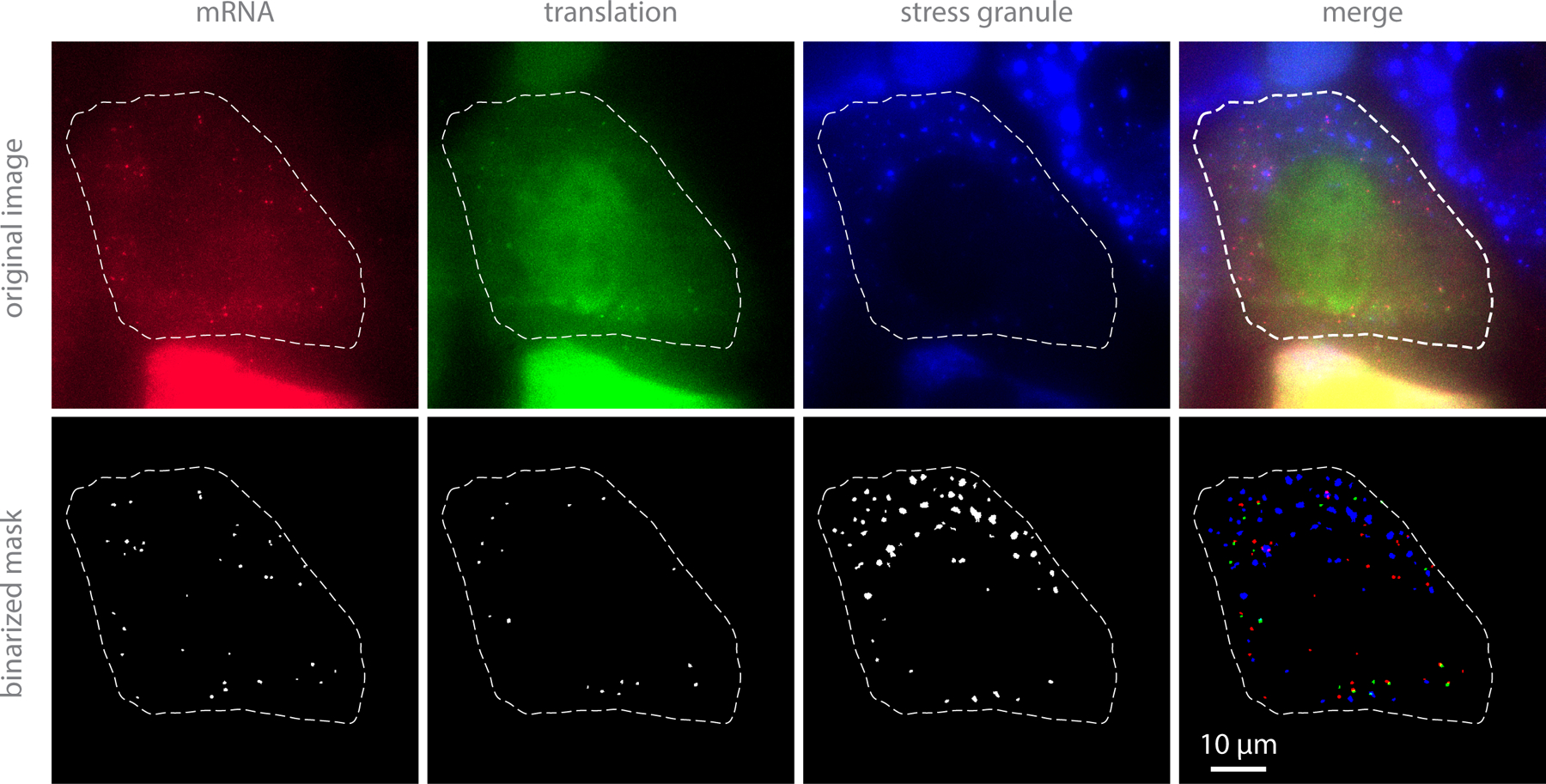
Example images of cells under stress conditions in each channel and corresponding mask images.
FIgure 3:
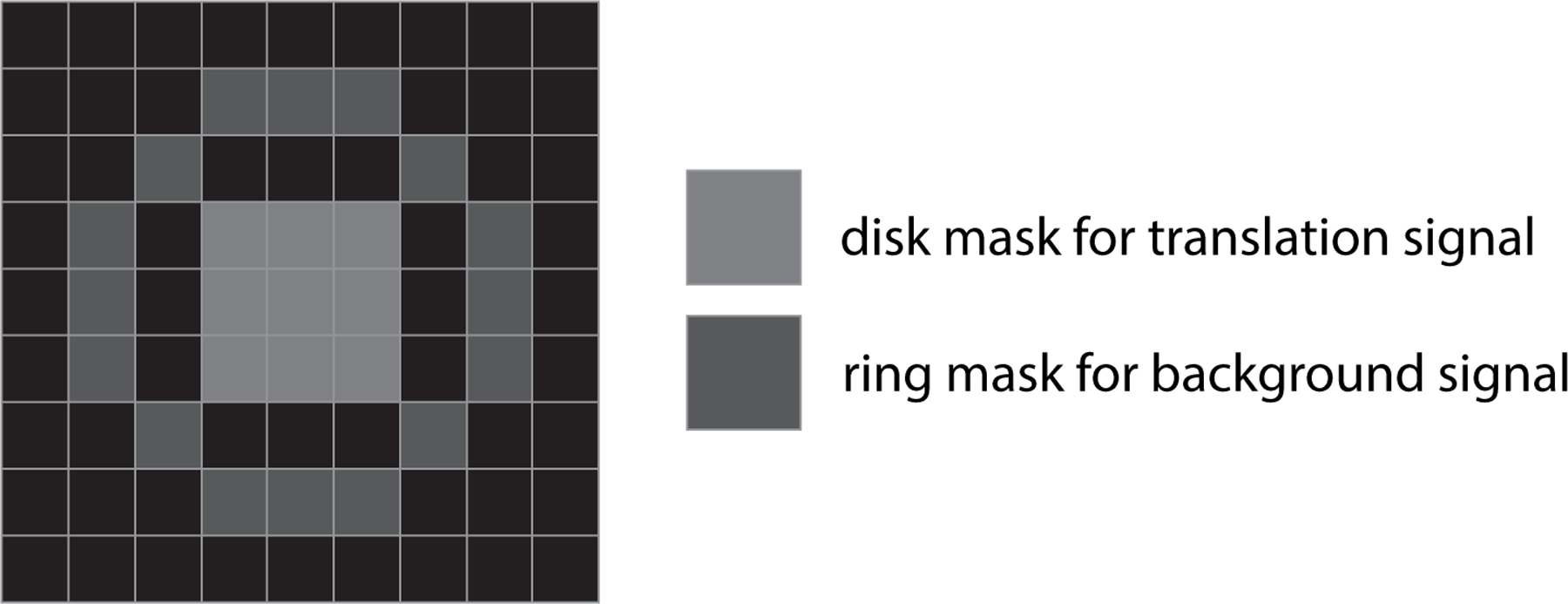
Disk and ring masks to estimate translation signal and background signal, respectively, for mRNAs whose translation activity drops down to near-zero.
5. Acknowledgments:
We thank Nancy Kdersha for providing GFP-G3BP1/mRFP-DCP1a U-2 OS cells and Esther Braselmann and Theresa Nahreini for isolating GFP-G3BP1 U-2 OS cells at the BioFrontiers Institute Flow Cytometry Core facility. We thank Dr. Luke Lavis for kindly providing JF646 labeled HaloTag ligand. We thank all members of the Stasevich lab for their support and helpful discussions. TJS and TM were supported by the NIH (grant no. R35GM119728).
Footnotes
The epitope tag sequences are recommended to be placed at the N-terminal end of the protein to maximize the fluorescence intensity at the translation site. If placed at the C-terminal end, the ribosome will leave soon after translating the epitope tag sequence, resulting in an immediate loss of fluorescence. On the other hand, if the epitope tags is placed at N-terminus of the protein, a ribosome remains for a while after translating the epitope tags, resulting in a sustained fluorescence signal. This also allows the researcher to estimate the translation elongation rate. Complementary epitope tags can be introduced to study non-canonical translation or dissect the translation dynamics of the relative location of two distinct translation sites, including stalling and termination rates24,25.
The mammalian expression vectors for MCP is available from Addgene (ID64540). We prefer to bead-load the purified MCP because we can control the concentration of MCP in the cells in a more precise manner. The proper concentration of MCP (or anti-FLAG Fab) is critical to get the best signal-to-noise ratio due to the nature of the system relying on amplification.
U-2 OS cells are suitable to visualize translation from single RNA molecules because of their relatively thinner cytoplasm.
We use MatTek glass bottom dishes with a 14 mm glass microwell. This microwell serves to retain 4 uL of sample during bead-loading.
106 um glass beads can be harmful to the lungs, so do not inhale them.
Make aliquots of 10 uL to avoid freeze-thaw cycles.
Patches of cells can be detached while bead-loading if the confluency is close to or over 100%. Some cells (for example HEK293 cells) are easy to detach while bead-loading even though the confluency is not high.
This step is optional. We have empirically found that cells in Opti-MEM with FBS attach to the glass surface better and stay healthier after bead-loading.
This amount is sufficient for a 14 mm glass microwell. The amount of components and total volume need to be scaled accordingly.
Make sure to remove all media especially from the glass area so that the remaining media does not dilute the sample to bead load. The bead loading procedure should be performed promptly so the cells do not dry out.
Because of the nature of the system which relies on signal amplification, it is important to have thin sectioning when imaging. We therefore recommend either a HiLO microscope26, a light-sheet microscope, or a confocal microscope.
In order to make sure translation signals (green dots) are bona fide translation sites, the translation elongation inhibitor puromycin can be added. All translation signals should disappear within a minute of adding 50 – 100 ug/mL of puromycin, while mRNA signal (red dots) should persist.
Avoid perturbing the dish as this could change your imaging field.
Binarize an image of TetraSpeck Fluorescent Microsphere to detect all spots for each channel. Fit all detected spots to a 2D Gaussians. Use the Mathematica command “FindGeometricTransform” (or an equivalent command in Python or Matlab) to calculate a geometric transformation function.
Using the strategy described above, if more than one mRNA mask overlaps with an SG mask, it is still counted as a single mRNA-SG interaction. While this slightly underestimates the number of long mRNA-SG interactions, it is recommended because it is difficult to precisely determine how many mRNA are in an individual SG. This difficulty arises because mRNAs within SGs often cluster and can be partially decayed, both of which make them difficult to precisely count.
An individual interaction that already exists at the beginning of the observation can be included.
6. References:
- 1.Buchan JR & Parker R Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 36, 932–941 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Protter DSW & Parker R Principles and Properties of Stress Granules. Trends Cell Biol 26, 668–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morisaki T et al. Real-time quantification of single RNA translation dynamics in living cells. Science 352, 1425–1429 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Moon SL et al. Multicolour single-molecule tracking of mRNA interactions with RNP granules. Nat. Cell Biol (2019) doi: 10.1038/s41556-018-0263-4. [DOI] [PMC free article] [PubMed]
- 5.Moon SL, Morisaki T, Stasevich TJ & Parker R Coupling of translation quality control and mRNA targeting to stress granules. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertrand E et al. Localization of ASH1 mRNA Particles in Living Yeast. Mol. Cell 2, 437–445 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Tutucci E et al. An improved MS2 system for accurate reporting of the mRNA life cycle. Nat. Methods 15, 81–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larson DR, Zenklusen D, Wu B, Chao JA & Singer RH Real-Time Observation of Transcription Initiation and Elongation on an Endogenous Yeast Gene. Science 332, 475–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulon A et al. Kinetic competition during the transcription cycle results in stochastic RNA processing. eLife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horvathova I et al. The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells. Mol. Cell 68, 615–625.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Hocine S, Raymond P, Zenklusen D, Chao JA & Singer RH Single-molecule analysis of gene expression using two-color RNA labeling in live yeast. Nat. Methods 10, 119–121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daigle N & Ellenberg J LambdaN-GFP: an RNA reporter system for live-cell imaging. Nat. Methods 4, 633–636 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Cawte AD, Unrau PJ & Rueda DS Live cell imaging of single RNA molecules with fluorogenic Mango II arrays. Nat. Commun 11, 1283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halstead JM et al. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347, 1367–1671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu B, Eliscovich C, Yoon YJ & Singer RH Translation dynamics of single mRNAs in live cells and neurons. Science 352, 1430–1435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan X, Hoek TA, Vale RD & Tanenbaum ME Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 165, 976–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Han B, Zhou R & Zhuang X Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell 165, 990–1001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichon X et al. Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J. Cell Biol 214, 769–781 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morisaki T & Stasevich TJ Quantifying Single mRNA Translation Kinetics in Living Cells. Cold Spring Harb. Perspect. Biol 10, a032078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kedersha N, Tisdale S, Hickman T & Anderson P Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods Enzymol 448, 521–552 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Grimm JB et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods (2015) doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed]
- 22.McNeil PL & Warder E Glass beads load macromolecules into living cells. J. Cell Sci 88 ( Pt 5), 669–678 (1987). [DOI] [PubMed] [Google Scholar]
- 23.Tinevez J-Y et al. TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Lyon K, Aguilera LU, Morisaki T, Munsky B & Stasevich TJ Live-Cell Single RNA Imaging Reveals Bursts of Translational Frameshifting. Mol. Cell 75, 172–183.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch A, Aguilera L, Morisaki T, Munsky B & Stasevich TJ Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nat. Struct. Mol. Biol 27, 1095–1104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tokunaga M, Imamoto N & Sakata-Sogawa K Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161 (2008). [DOI] [PubMed] [Google Scholar]