

Abstract
Risk stratification in stage II and III colorectal cancer (CRC) patients is critical, as it allows patient selection for adjuvant chemotherapy. In view of the inadequacy of current clinicopathological features for risk-stratification, we undertook a systematic and comprehensive biomarker discovery effort to develop a risk-assessment signature in CRC patients. The biomarker discovery phase examined 853 CRC patients, and identified a gene signature for predicting recurrence-free survival (RFS). This signature was validated in a meta-analysis of 1212 patients from nine independent datasets, and its performance was compared against established prognostic signatures and consensus molecular subtypes (CMS). In addition, a risk-prediction model was trained (N=142), and subsequently validated in an independent clinical cohort (N=286). As a result, this mesenchymal-associated transcriptomic signature (MATS) identified high-risk CRC patients with poor RFS in the discovery (hazard ratio (HR) :1.79), and nine validation cohorts (HR:1.86). In multivariate analysis, MATS was the most significant predictor of RFS compared to established prognostic signatures and CMS subtypes. Intriguingly, MATS robustly identified CMS4-subtype in multiple CRC cohorts (AUC=0.92–0.99). In the two clinical cohorts, MATS stratified low and high-risk groups with a 5-year RFS in the training (HR:4.11) and validation cohorts (HR:2.55), as well as predicted response to adjuvant therapy in stage II and III CRC patients. We report a novel prognostic and predictive biomarker signature in CRC, which is superior to currently used approaches and have the potential for clinical translation in near future.
Keywords: Colorectal cancer, recurrence, mesenchymal, adjuvant therapy, prognosis
Introduction
Clinical decision making for adjuvant chemotherapy, which includes selection of most appropriate patient subgroups, as well as optimal treatment regimens, remains the most pressing challenge in the management of stage II and III colorectal cancer (CRC) patients1. This is an even greater concern in stage II CRC patients, where even though various risk factors are routinely considered for guiding adjuvant chemotherapy decisions2–4, there is a no consensus biomarkers available that allow selection of patients who might benefit the most from adjuvant treatments and spare the rest from their toxicity and expense5. To address this important unmet clinical need for the identification of high-risk stage II CRC patients, several multigene expression signatures have been reported6–9. However, in spite of best intentions (e.g. Oncotype DX), thus far, none of these gene signatures have been successfully translated into routine clinical practice.
In stage III CRC patients, six months of oxaliplatin-containing adjuvant chemotherapy is the standard treatment regimen following radical surgery5; however, oxaliplatin often causes sustained, severe neuropathy in many patients10. Recently, the International Duration Evaluation of Adjuvant Chemotherapy (IDEA) trial examined the efficacy of a shorter duration of oxaliplatin-containing adjuvant chemotherapy to minimize or prevent such severe neuropathy. Interestingly, patients at low risk for tumor recurrence (T1–3, N1) treated with a shorter duration of an oxaliplatin-containing regimen, exhibited no significant differences in disease-free survival (DFS) but reported markedly reduced neurotoxicity compared to the standard six-month treatment regimen in stage III CRC patients. These findings highlight the importance of selecting low-risk stage III CRC patients who can potentially be managed by regimes with as less toxic as possible. Collectively, there is the imperative need of robust prognostic and predictive biomarkers that can facilitate optimal risk stratification for low and high-risk stage II and III CRC patients. Availability of such markers will lead to more appropriate treatment modalities and potentially result in improved survival outcomes in CRC patients.
Recently, the colorectal cancer subtyping consortium (CRCSC) encompassing of six large international groups proposed consensus molecular subtypes (CMS), which segregate all CRCs into four distinct molecular categories11. Among these, the CMS4 subtype is characterized by increased expression of epithelial-to-mesenchymal transition (EMT) genes and correlates with poor overall survival (OS) and relapse-free survival (RFS). Although such a CMS-based classification has been a significant step forward in recognizing unique subgroups of CRCs, the translation of this approach into the clinic for patient prognostication has been hampered as it requires analysis of hundreds and thousands of genes. Furthermore, the current CMS classification method is challenging for interrogating individual patient response prediction; hence, simpler approaches in delineating the CRC subtypes is urgently needed to aid in clinical decision making12. In view of the increased recognition for the participation of EMT pathway and its association with the CMS4 subtype with poor outcomes in CRC patients, herein, we utilized a unique, first of its kind approach, by focusing our biomarker discovery efforts on developing a mesenchymal-associated transcriptomic signature (MATS) for patient prognosis and predicting response to adjuvant chemotherapy. To this end, we performed whole genome transcriptomic profiling using laser capture microdissected (LCM) colorectal cancer specimens and specifically identified genes associated with EMT. We subsequently evaluated and validated the performance of MATS in multiple, large cohorts of CRC patients, as well as demonstrated its superiority against various commercial platforms (e.g. OncotypeDX, CRCassigner, ColoGuidePro) for determining patient survival, as well as its ability to identify patients who could benefit from 5FU-based adjuvant chemotherapy.
Materials and Methods
Biomarker discovery
To identify a transcriptomic signature associated with the mesenchymal subtype in CRC, we analyzed multiple datasets which included analysis of our own gene expression profiling data, as well as large datasets available in the public domain. First, we performed gene expression profiling in RNA derived from 152 CRC tissue specimens that were carefully microdissected using laser capture microscopy (LCM), for tumor cell enrichment, and the dataset is available at GSE7122213. In order to identify genes associated with an EMT phenotype, we first selected the genes that were positively correlated with vimentin (VIM). We prioritized candidate genes whose correlation coefficient with VIM were greater than 0.6. Thereafter, the candidate genes were prioritized in the GSE41258 dataset comprising of 186 CRC and 54 normal colon mucosa specimens14 by selecting genes that were upregulated in CRC (adjusted P value < 0.05). This led to the identification of 34 candidate genes, which were subsequently evaluated in another large dataset of 461 stage II and III CRC patients (GSE39582), for evaluating their prognostic potential by using the least absolute shrinkage and selection operator (LASSO) Cox regression analysis15. This finally, resulted in an eight-gene panel, which was finally defined as a Mesenchymal Associated Transcriptomic Signature (MATS). The flow chart of the study design is shown in Supplementary Figure S1.
Determination of association between MATS and CMS4 in CRC
To determine the association between the MATS genes and CMS4 subtype, we analyzed six publicly available datasets, which included GSE39582 (N=566), GSE17536 (N=177), GSE33113 (N=90), TCGA microarray (N=209), TCGA RNA seq (N=323) and GSE104645 (N=193). CMS status of GSE39582, GSE17536, GSE33113 and TCGA datasets were obtained from the CRCSC11, while the CMS status of GSE104645 dataset was obtained from the corresponding publication16. We plotted receiver operator characteristic (ROC) curves to evaluate the accuracy of MATS in the identification of CMS4 CRC patients.
Meta-analysis of MATS for predicting recurrence-free survival in CRC patients
To validate the prognostic potential of MATS, we performed a meta-analysis of this signature in nine publicly-available gene expression datasets that we have complied and published previously17. In brief, patients were identified, and corresponding gene expression data was downloaded from the GEO repository (https://www.ncbi.nlm.nih.gov/geo/). Following quality control evaluation, each case was MAS5 normalized, and the mean expression of each case was centered to 1000. The MATS signature was established by using the mean expression of the eight selected genes. For each gene, JetSet was used to select the most reliable probe set representing that given gene18. The only exception was BCAT1, because this gene was not present in the HGU133A arrays, and JetSet selection is based on the ranking in HGU133A. Survival analysis was performed by computing Cox proportional hazards regression. Survival differences were visualized using a Kaplan-Meier plot. Survival analysis was performed using the MATS signature and relapse-free survival (RFS) using the survival R package and GraphPad Prism Ver. 6.0 (GraphPad Software, San Diego, CA). In the multivariate analysis, the continuous MATS signature was converted to 0/1 (high/low) using the median as a cut-off. Subsequently, the MATS signature was compared with various features including the MSI status, gender, stage, tumor location, and mutational status of the TP53, KRAS, and BRAF genes. In addition, we also compared the performance of MATS with previously established genes signatures including the OncotypeDX, CRCassigner, ColoGuidePro, Budinska, DeSousa, and Marisa, as described previously17, in a multivariate analysis. When comparing the expression levels in different cohorts, Kruskal-Wallis H-test was used. Statistical significance was set at P<0.05, and the false discovery rate was (FDR) computed to account for multiple testing correction.
Patient cohorts
To validate the results from the biomarker discovery efforts, performance of MATS was evaluated in 428 stage II and III CRC patients, enrolled at two independent institutions. Initially, for biomarker training, we analyzed fresh frozen tissues specimens from 142 CRC patients enrolled at the National Cancer Center Hospital, Tokyo, Japan, between 2004 and 2006 (the training cohort). For the validation of MATS, we analyzed another independent cohort of formalin-fixed paraffin-embedded (FFPE) tissues specimens from a cohort of 286 CRC patients enrolled at the Tokyo Medical and Dental University Hospital, Tokyo, Japan, between 2007 and 2011 (the validation cohort). The clinicopathological characteristics of the training and validation cohorts are shown in Supplementary Table S1. Patients who received radiotherapy or chemotherapy prior to surgery were excluded. All patients in an adjuvant setting were treated with fluoropyrimidine-based drugs (5FU+leucovorin, capecitabine, S-1), and none of patients received oxaliplatin-containing adjuvant treatment in either of our in-house clinical cohorts. A written informed consent was obtained from all patients for their willingness to participate in this study, and the study was approved by the institutional review boards of all participating institutions. The RFS time periods were calculated from the date of surgery, as defined previously11. The Tumor Node Metastasis (TNM) staging was performed according to the American Joint Committee on Cancer (AJCC) established criteria.
MATS for predicting palliative chemotherapy response in CRC patients
We also explored the predictive significance of MATS in a palliative setting where metastatic CRC patients received either FOLFOX or an anti- epidermal growth factor receptor (EGFR) therapy, by analyzing GSE28702 and GSE5851 datasets, where RECIST outcomes were available for determining patient response. In GSE28702, metastatic CRC patients were treated by FOLFOX as a first line chemotherapy. In GSE5851, patients already received at least one prior chemotherapeutic regimen for their metastatic lesion. Then they were treated by cetuximab monotherapy. The detail of patients’ characteristics were described previously19 20.
RNA extraction and qRT-PCR
Total RNA extraction from fresh frozen and FFPE specimens was performed using the RNeasy Mini kit and Allprep FFPE kits (QIAGEN, Hilden, Germany) respectively, according to the manufacturer’s instructions. Thereafter, cDNA was synthesized from 2 μg of total RNA by using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA). The qRT-PCR assays were performed using QuantStudio 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). We used 5 ng of cDNA in each well and the SensiFast Low-rox probe Master Mix (Bioline, London, UK). The following PCR cycling conditions were used: 2 min at 95°C for enzyme activation, 50 cycles of 95°C for 10 s and 60°C for 50 s for denaturation, annealing and extension. The primer sequences used in this study are shown in Supplementary Table S2.
Microsatellite instability (MSI) analysis
Microsatellite instability (MSI) analysis was conducted using five mononucleotide repeat microsatellite markers (BAT-25, BAT-26, NR-21, NR-24, and NR-27) in a pentaplex PCR system, as described earlier21.
Statistical analysis
The expression level of target genes was normalized against ACTB using the 2−Δct method. Association between the gene signature and various clinicopathological factors were assessed by the χ2 test. All P values were 2-sided, and those less than 0.05 were considered statistically significant. We calculated the Hazard Ratios (HRs) for RFS using univariate cox-regression analysis for individual genes in MATS. To build the multi-gene MATS, we performed multivariate cox-regression analysis by including all individual genes. Subsequently, for Kaplan-Meier (KM) analysis, we stratified patients based on Youden’s index derived cut-off thresholds22. The qRT-PCR–based MATS risk scores were calculated using the formula derived from the multivariate Cox model. The coefficients of each of the genes are listed in Supplementary Table S3. In these analyses, we classified patients with a risk score of 0.57 or higher as high-risk of disease recurrence (high-risk group), and those with a risk-score lower than 0.57 as low-risk of disease recurrence (low-risk group). The same coefficients and cut-off thresholds derived from the training cohort were applied to the independent validation cohort. KM-analysis and log-rank test were conducted for estimating and comparing the survival rates in CRC patients with low and high-risk MATS scores. Univariate and multivariate Cox proportional hazards analysis were performed to compare the performance of MATS with other clinicopathological features, in both the clinical training and validation cohorts. The predictive accuracy of MATS for determining therapeutic benefit from fluoropyrimidine-based adjuvant chemotherapy was undertaken by comparing the RFS rates in patients with and without chemotherapy. Considering that a majority of stage II CRC patients in our in-house clinical cohorts (93% in the training cohort and 89% in the validation cohort) did not receive adjuvant chemotherapy, the predictive biomarker potential of MATS was limited to the stage III patients only. We performed statistical analyses using the GraphPad Prism Version 6.0 (San Diego, CA), Medcalc version 16.1 (Ostend, Belgium) and the R-software version 3.3.1.
Results
Identification of a mesenchymal-associated transcriptomic signature (MATS) for predicting tumor recurrence in CRC patients
As illustrated in the Supplementary Figure S1, we first performed a careful laser capture microdissection of tissue specimens (to enrich for tumor cells) in a cohort of 152 CRC patients, which was followed by gene expression profiling analysis. Subsequently, we identified a panel of 87 candidate genes that significantly correlated with vimentin, which is a key EMT-associated gene (Supplementary Table S4). Thereafter, we further narrowed down this list of candidate genes by validating them in another cohort of CRC and matched normal tissues, which led us to identify a panel of 34 genes that were consistently and significantly associated with a mesenchymal phenotype and were upregulated in CRC patients (Supplementary Table S5). We next performed Lasso Cox regression analysis to establish a clinically-actionable, gene classifier for predicting recurrence-free survival (RFS), by analyzing another large cohort of 461 CRC patients with a stage II and III disease. This effort resulted in the identification of an eight-gene panel (COL1A2, COL3A1, FN1, POSTN, FSTL1, BCAT1, DKK3 and PRR16), which we referred to as a mesenchymal-associated transcriptomic signature (MATS). In this initial exploratory cohort, MATS demonstrated a promising prognostic potential, as evident from its ability to robustly stratify all CRC patients into low and high-risk groups, which exhibited five-year RFS rates of 69% and 52%, respectively (HR: 1.79 [1.32–2.44], P<0.001; Figure 1a).
Figure 1:
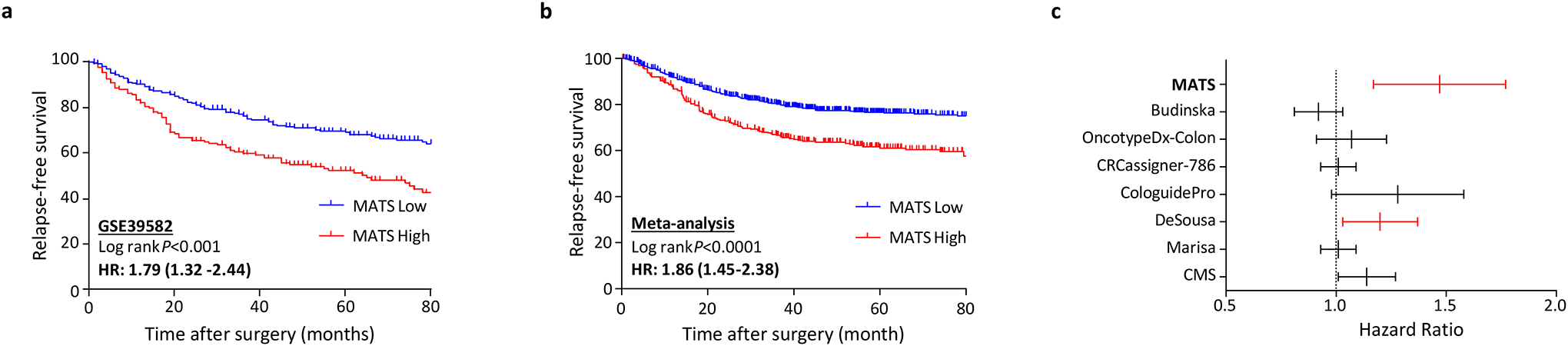
Mesenchymal associated transcriptomic signature (MATS) and its association with recurrence free survival in comparison to earlier published gene signatures and its ability to predict chemotherapy response (a) Kaplan-Meier survival curve derived from the LASSO cox regression model of MATS in the GSE39582 exploratory cohort. (b) Meta-analysis of the MATS classifier in predicting recurrence free survival using 9-independent microarray gene expression cohorts (c) Multivariate analysis of MATS in comparison to the earlier published gene expression signatures and subtypes
MATS is highly accurate in identifying CMS4-positive CRC patients
As our initial hypothesis stemmed from identifying a robust epithelial mesenchymal gene signature, we were enthused to understand the relationship between MATS and the CMS4 subtype – considering that this specific CRC subtype associates with a mesenchymal phenotype and poor prognosis. It was encouraging to observe that our biomarker discovery process was indeed quite robust, because each of the eight MATS-associated genes were significantly upregulated in the CMS4 positive CRC patients; a finding that was validated in six, large, independent CRC datasets (Supplementary Figures S2–7). When we performed a binary logistic regression analysis in these six datasets to distinguish CMS4 vs. CMS1–3 patients, the ROC curve analysis and corresponding area under the curve (AUC) values for MATS in each of these cohorts were remarkably high: 0.94 (GSE39582), 0.92 (GSE17536), 0.99 (GSE33113), 0.95 (TCGA-microarray), 0.97 (TCGA RNA seq) and 0.92 (GSE104645). Not only the cumulative AUC value of MATS was high, it was very reassuring to observe that even each of the individual genes exhibited remarkably robust AUC values across all the datasets (Supplementary Table S6) for the identification of CMS4 subtypes, ranging from: 0.86–0.93 (COL1A2), 0.80–0.94 (COL3A1), 0.81–0.89 (FN1), 0.77–0.91 (POSTN), 0.76–0.97 (FSTL1), 0.48–0.92 (BCAT1), 0.76–0.92 (DKK3), 0.62–0.89 (PRR16). These data highlight the robustness of MATS in identifying CMS4 associated CRC patients; as supported by its promising prognostic and predictive potential in this malignancy.
The recurrence prediction potential of MATS was successfully validated in multiple, large, CRC datasets
Next, to further validate the prognostic potential of MATS, we performed a meta-analysis by combining gene expression profiling data from nine publicly-available datasets (GSE41258, GSE14333, GSE37892, GSE39582, GSE33113, GSE17538, GSE12945 and GSE38832). Interestingly, in line with our initial discovery and exploratory phase analysis, MATS-derived risk scores significantly discriminated low vs. high-risk patients in terms of 5-year RFS rates (HR=1.86 [1.45–2.38]; Figure 1b). To rule out any potential confounding effects for the use of adjuvant chemotherapy, we analyzed treated and untreated patients separately; where once again, MATS was a significant predictor of poor RFS in high-risk patients, independent of adjuvant chemotherapy (Figure 2a). In addition, MATS significantly associated with CMS4 (Figure 2b) as well as other known clinicopathological risk factors such as a higher TNM stage (Figure 2c) and proximal tumor location (Figure 2d), while no significant associations were observed with regards to the MSI status (Figure 2e), TP53, KRAS and BRAF gene mutations (Supplementary Figure S8).
Figure 2:
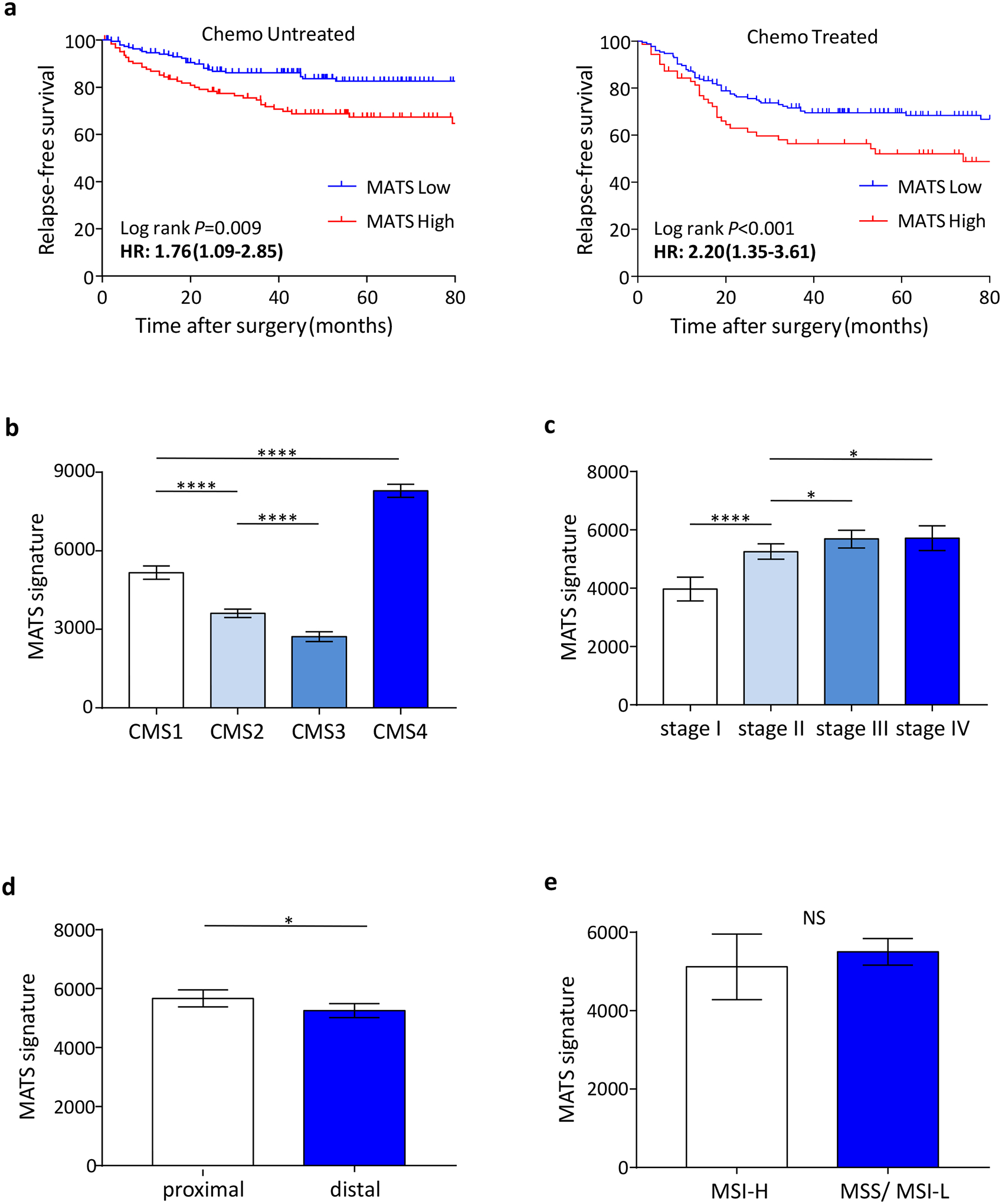
Association of MATS with adjuvant chemotherapy and other known molecular markers. (a) MATS was a significant predictor of poor RFS, independent of adjuvant chemotherapy. (b, c, d and e) MATS significantly associated with CMS4 as well as other known clinicopathological risk factors such as a higher TNM stage and proximal tumor location, while no significant associations were observed with regards to the MSI status. *P < 0.05; ****P<0.0001.
To further understand the clinical significance of MATS in the context of previously published gene signatures and CRC subtypes (DeSousa, Marisa, Budinska and CMS classifications), as well as several commercially marketed assays (OncotypeDX® colon, CRCassigner-786 and CologuidePro), we performed a multivariate analysis by including gene signatures from all these panels. Intriguingly, among all these signatures, only the MATS and Desousa classifications were found to be independent predictors of RFS in CRC patients, with MATS being significantly superior as reflected by significantly higher HRs (HR=1.47 [1.17–1.77)] for MATS vs. HR=1.20 [1.03–1.37] for Desousa; Figure 1c).
The recurrence prediction potential of MATS was independently validated in the in-house training and validation cohorts of CRC patients
To further ascertain and validate the prognostic significance of MATS in a clinical setting, we performed a qRT-PCR-based training and validation in two independent in-house clinical cohorts. One of the eight genes (PRR16) did not consistently amplify in these assays; hence, we reduced the MATS signature to the remaining seven genes. The Figure 3a depicts univariate analysis for the prediction of RFS for each of the seven genes individually, and cumulatively as MATS. The HRs of individual genes ranged from 1.95 to 3.11. Subsequently, we combined all seven genes in a Cox regression model to build a prognostic training classifier for the prediction of RFS in CRC patients. When we used this training classifier for risk prediction, we observed that patients with low-risk scores derived from MATS generally demonstrated better RFS vs. those with high-risk scores (HR=4.11, 95% CI 2.02–8.42; P<0.0001; Figure 3b).
Figure 3:
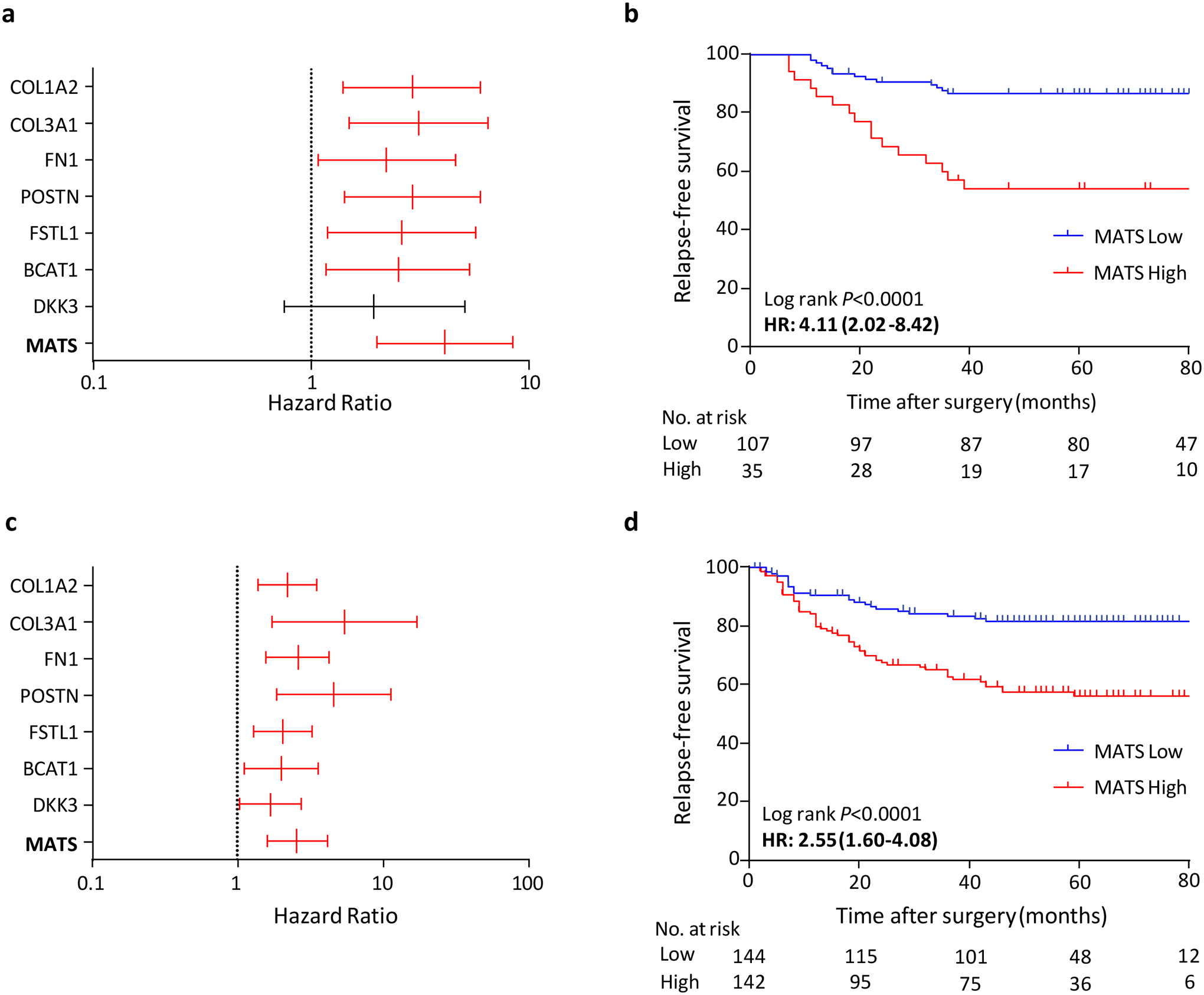
Constructing and validating mesenchymal associated transcriptomic signature (MATS) by qRT-PCR in independent in-house clinical cohorts. Univariate cox proportional hazard model derived hazard ratios (HR) and 95% CIs for individual MATS genes in predicting relapse-free survival; training cohort (a) and the validation cohort (c). Kaplan-Meier survival curve (b) of MATS for relapse-free survival in the training cohort. Kaplan-Meier survival curve (d) of MATS for relapse-free survival in the validation cohort.
Next, using the same model coefficients and cutoff scores derived from the training cohort, we applied these in an independent validation cohort (N=286). As illustrated in Figure 3c, the univariate analysis for each of the seven genes in the validation cohort yielded HRs ranging from 1.68 to 5.44. Consistent with the results from the training cohort, patients with lower risk-scores demonstrated better survival than those with higher risk-scores (5-year RFS rates were 56% for the high-risk group, and 82% for the low-risk group; HR=2.55, 95% CI 1.60–4.08, P<0.0001; Figure 3d). The detailed associations between MATS-derived risk scores and various clinicopathological factors in the training and validation cohorts are shown in Table 1. In univariate analysis, MATS, T4, lymphatic invasion and lymph node metastasis were significant predictors of RFS in both patient cohorts. In multivariable analysis, only MATS and T4 emerged as independent predictors of RFS in both cohorts (Table 2). These results further highlights the predictive potential of MATS, a signature which was independently validated across multiple CRC cohorts including a qRT-PCR based internal validation cohort, highlighting its potential applicability in routine clinical settings. Furthermore, we evaluated the RFS using this model in patients with stage II and stage III CRC separately in the validation cohort. This model efficiently distinguished RFS in patients with both stage II and stage III CRC as depicted in the Kaplan-Meier curve analysis (HR: 2.70, P =0.006 and HR: 2.04, P < 0.02, respectively, Supplementary Figure 9A, B).
Table 1:
Association between MATS risk score and clinicopathological factors
| Training Cohort | Validation Cohort | ||||||
|---|---|---|---|---|---|---|---|
| MATS risk score | MATS risk score | ||||||
| Variables | Low | High | P value | Low | High | P value | |
| (N=107) | (N=35) | (N=144) | (N=142) | ||||
| Gender | |||||||
| Male | 58 | 26 | 0.03 | 73 | 89 | 0.04 | |
| Female | 49 | 9 | 71 | 53 | |||
| Age | |||||||
| <65 | 71 | 24 | 0.80 | 42 | 54 | 0.11 | |
| ≥65 | 36 | 11 | 102 | 88 | |||
| Location | |||||||
| Colon | 57 | 14 | 0.17 | 104 | 80 | 0.005 | |
| Rectum | 50 | 21 | 40 | 62 | |||
| Histology | |||||||
| Differentiated | 99 | 30 | 0.22 | 135 | 126 | 0.13 | |
| Undifferentiated | 8 | 5 | 9 | 16 | |||
| Tumor size | |||||||
| ≤45mm (median) | 58 | 13 | 0.07 | 69 | 43 | 0.002 | |
| >45mm | 49 | 22 | 70 | 94 | |||
| not available | 5 | 5 | |||||
| T stage | |||||||
| T1–3 | 79 | 22 | 0.21 | 108 | 90 | 0.03 | |
| T4 | 28 | 13 | 36 | 52 | |||
| Lymphatic invasion | |||||||
| Absent | 73 | 19 | 0.13 | 70 | 61 | 0.36 | |
| Present | 34 | 16 | 74 | 80 | |||
| not available | 0 | 0 | 0 | 1 | |||
| venous invasion | |||||||
| Absent | 43 | 10 | 0.21 | 18 | 10 | 0.12 | |
| Present | 64 | 25 | 126 | 131 | |||
| not available | 0 | 0 | 0 | 1 | |||
| Lymphnode Metastasis | |||||||
| Absent | 40 | 14 | 0.78 | 95 | 65 | <0.001 | |
| Present | 67 | 21 | 49 | 77 | |||
| Preoperative CEA | |||||||
| <5 | 75 | 21 | 0.26 | 85 | 77 | 0.53 | |
| 5≤ | 32 | 14 | 58 | 61 | |||
| not available | 0 | 0 | 1 | 4 | |||
Table 2:
Univariate and multivariate analyses of MATS and clinicopathological parameters in the in-house training and validation cohorts
| Training Cohort (N=142) | Validation Cohort (N=286) | |||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| Age (≥65) | 1.39 (0.67–2.87) | 0.37 | 1.22 (0.77–1.94) | 0.39 | ||||
| Gender (M vs. F) | 2.41 (1.04–5.60) | 0.04 | 1.81 (0.74–4.39) | 0.19 | 1.00 (0.65–1.54) | 1 | ||
| Location (Rectum) | 1.64 (0.79–3.40) | 0.18 | 1.93 (1.25–2.97) | 0.002 | 1.66 (1.04–2.65) | 0.03 | ||
| Differentiation | 1.16 (0.35–3.80) | 0.81 | 0.95 (0.44–2.06) | 0.91 | ||||
| T4 vs. T2&3 | 2.91 (1.43–5.95) | 0.003 | 2.10 (1.00–4.40) | 0.04 | 1.90 (1.23–2.94) | 0.004 | 1.71 (1.07–2.75) | 0.02 |
| Venous Invasion | 2.10 (0.91–4.88) | 0.08 | 2.31 (0.85–6.27) | 0.1 | ||||
| Lymphatic Invasion | 2.31 (1.13–4.73) | 0.02 | 1.68 (0.80–3.52) | 0.17 | 1.90 (1.20–3.01) | 0.006 | 1.27 (0.75–2.16) | 0.37 |
| Lymphnode metastasis | 2.37 (1.02–5.52) | 0.04 | 2.25 (0.94–5.40) | 0.06 | 2.49 (1.59–3.90) | 0.0001 | 1.64 (0.99–2.73) | 0.05 |
| Examined LN Number (<12) | 1.09 (0.34–3.60) | 0.88 | 1.28 (0.74–2.20) | 0.38 | ||||
| CEA (≥5) | 0.92 (0.42–2.01) | 0.84 | 1.82 (1.17–2.81) | 0.007 | 1.79 (1.14–2.81) | 0.01 | ||
| MSI | 4.20 (1.04–16.9) | 0.04 | 3.16 (0.76–13.9) | 0.11 | ||||
| MATS | 4.11 (2.02–8.42) | 0.0001 | 3.75 (1.78–7.89) | 0.0005 | 2.10 (1.28–3.44) | 0.0001 | 2.10 (1.28–3.44) | 0.003 |
MATS is also a robust predictor of response to adjuvant and palliative chemotherapy in CRC patients
To evaluate the benefit from 5FU-based adjuvant chemotherapy, we first analyzed 88 stage III patients who were stratified into low- and high-risk groups based on MATS within the training cohort. We noticed that the 5FU-based adjuvant chemotherapy associated with a higher RFS rates in stage III, MATS low-risk patients (5 year survival rates were 89% with chemotherapy vs. 69% with no chemotherapy, HR: 2.96; P=0.05, Figure 4a); while no differences were observed in MATS high-risk patients, HR: 1.41; P=0.65 (Figure 4b).
Figure 4:
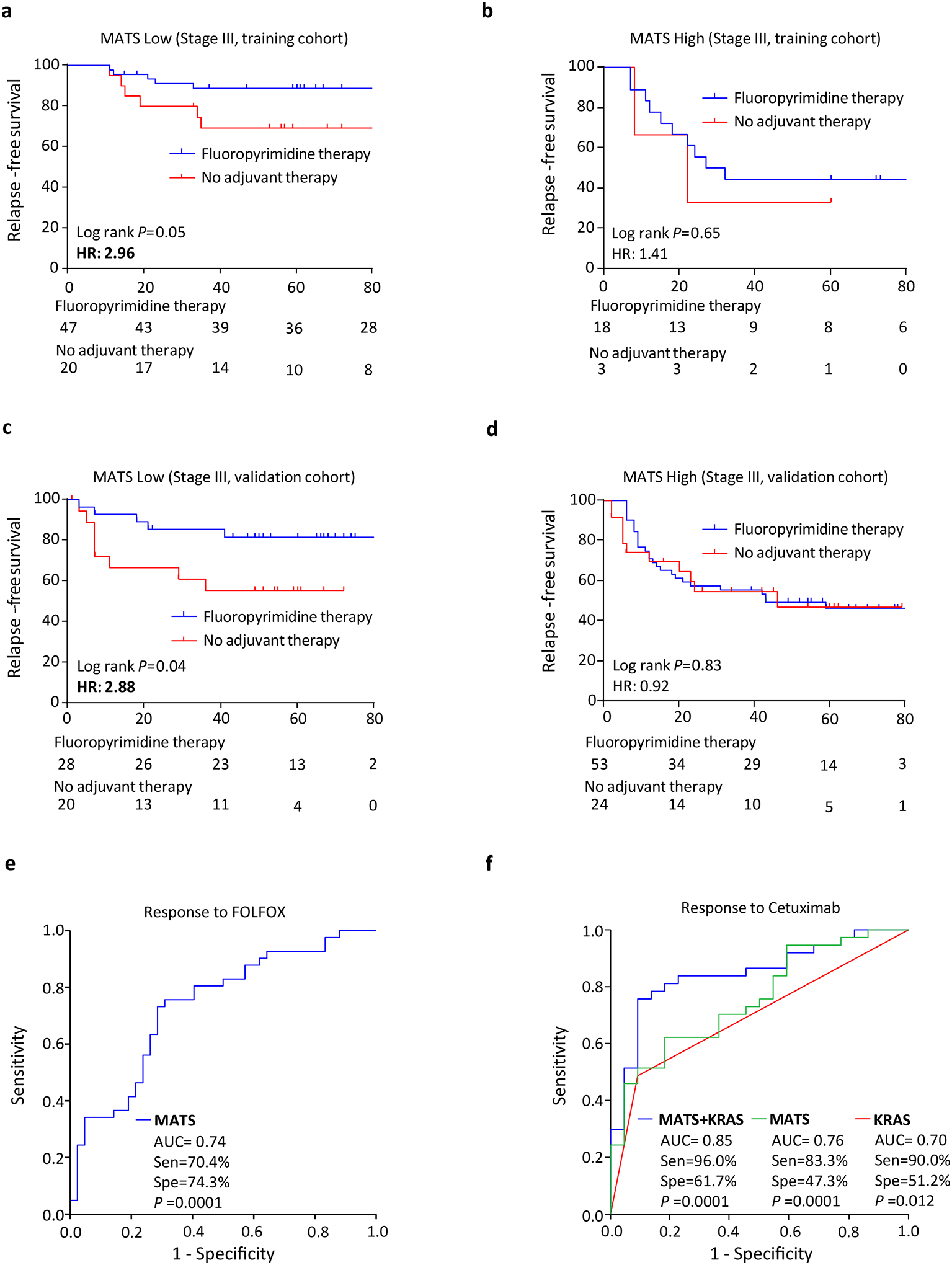
Chemotherapy predictive ability of MATS in both adjuvant and palliative setting (a) Kaplan-Meier survival curve for stage III patients in MATS low group, which were stratified by the receipt of fluoropyrimidine based chemotherapy alone in the in-house training cohort (b) Kaplan-Meier survival curves for stage III patients in MATS high group in the in-house training cohort (c) Kaplan-Meier survival curves for stage III patients in MATS low group in the in-house validation cohort (d) Kaplan-Meier survival curves for stage III patients in MATS high group in the in-house validation cohort (e) ROC curve of MATS in predicting FOLFOX response in mCRC patients analyzed using GSE28702 external validation cohort (f) ROC curve of MATS, KRAS as well as MATS+KRAS in predicting cetuximab response in mCRC patients analyzed using GSE5851 external validation cohort. Sen: Sensitivity, Spe: Specificity
Likewise, in the validation cohort of 125 stage III patients, the 5FU-based adjuvant chemotherapy correlated with a higher rates of RFS in the MATS low-risk group (5 year survival rates were 82% with chemotherapy vs. 56% with no chemotherapy, HR: 2.88; P=0.04; Figure 4c), while no significant differences were noted in the MATS high-risk group, HR: 0.92; P=0.83 (Figure 4d).
In view of the observed predictive potential of MATS in an adjuvant setting, we next questioned whether it can predict therapeutic response in patients treated in a palliative setting, by analyzing two cohorts of metastatic CRC patients. In the first cohort of 83 unresectable CRC patients with available RECIST scores19, MATS was able to achieve an AUC of 0.74 (sensitivity% 70.4%, specificity% 74.3%, P=0.0001, Figure 4e) in predicting response to FOLFOX as first line chemotherapy. Response rate was 29.5% in MATS high risk patients and 74.3% in MATS low risk patients, respectively. Interestingly, MATS also yielded an impressive AUC of 0.76 (sensitivity% 83.3%, specificity% 47.3%, P=0.001, Figure 4f) in predicting response to cetuximab in metastatic CRC patients in the Khambata-Ford20 cohort (N=68 with RECIST response status). While KRAS mutations alone yielded an AUC of 0.70 (sensitivity% 90.0%, specificity% 51.2%, P=0.012, Figure 4f), a combination of MATS together with KRAS mutations, further improved the AUC values to 0.85 (sensitivity% 96.0%, specificity% 61.7%, P=0.0001, Figure 4f) in predicting response to cetuximab treatment. The disease control rate was 16.6% in MATS high risk patients and 47.3% in MATS low risk patients, respectively. By combining MATS with KRAS mutation, the disease control rate could be predicted more precisely as 4.0% in high risk patients and 61.7% in low risk patients, respectively. Taken together, these results further emphasize the clinical utility of MATS, both in prognosis, as well as in predicting response to chemotherapy and anti-EGFR therapy in CRC patients.
Discussion
In this study, we primarily developed and validated a mesenchymal associated transcriptomic signature from laser capture microdissected (LCM) CRC samples to improve the current prognosis and adjuvant treatment prediction in stage II and III CRC patients using a comprehensive approach as well as utilizing multiple CRC patient cohorts. LCM was used to reliably identify the transcriptome of tumor epithelial cells which have undergone EMT. Our efforts have led to the identification of a clinically translatable mesenchymal transcriptomic signature that was quite robust in identifying poor CMS4 molecular subtype, and predicting RFS across multiple CRC cohorts. More importantly, MATS was independent and superior to earlier published prognostic signatures such as OncotypeDX, CRCassigner, ColoGuidePro, as well the molecular subtypes that are previously published along with the consensus molecular subtypes that were established in CRC recently. MATS was also superior and independent of the current clinical risk-factors put forward by National Comprehensive Cancer Network (NCCN) for identifying high-risk CRC patients.
Interestingly, in our in-house cohort stage III patients with MATS low-risk benefited significantly from 5FU-based adjuvant chemotherapy alone with an excellent prognosis. Conversely, those with MATS high-risk did not benefit from 5FU-based adjuvant chemotherapy alone. Recent NCCN guideline recommends oxaliplatin containing adjuvant chemotherapy for stage III patients. However, stage III patients are heterogeneous in survival patterns when classified by T and N categories23. Therefore, MATS low-risk stage III patients might be able to be treated by 5FU-based drug alone, sparing them from the potentially toxic and expensive oxaliplatin-based regimen. In addition, our exploratory analysis in metastatic CRC patients revealed two significant associations 1) MATS is an excellent predictive marker for FOLFOX therapy in first-line treatment of unresectable CRC patients, and 2)MATS is a better as well as an independent predictor of cetuximab response in metastatic CRC patients.
Taken together, MATS is not only beneficial to predict RFS but also helps in guiding treatment decisions. This is one of the major concerns of prognostic markers which are published thus far in CRC and one of the biggest strengths of the MATS classifier. In addition, MATS is probably one of the most robust and clinically translatable gene signature published so far that can identify CMS4 subtype patients with excellent accuracy. Early exploratory studies published recently show the value of CMS4 subtyping in predicting response to chemotherapy both in adjuvant as well as palliative settings16, 24–28. Therefore, MATS could play a huge role in clinical translation being able to identify the poor CMS4 subtype with utmost accuracy.
Recently, many researchers have studied blood-based biomarkers for CRC as it is minimally invasive and useful in several clinical purposes, from detecting early-stage cancer to monitoring tumor progression. As several researchers have recently reported mRNA blood markers29 30, we expect the stability and relative abundance of MATS genes in circulation. In future, MATS may eventually be translated into a blood-based marker for predicting outcome and treatment response, as well as surveillance.
The major limitation of this study is its retrospective nature. Therefore, our results should be further validated in a large prospective multicenter trials to evaluate the potential of MATS in recurrence prediction. In addition, the predictive ability of MATS in various clinical regimens that are administered in both adjuvant as well as palliative setting needs to be further validated in large retrospective as well as prospective multinational clinical cohorts.
In conclusion, our findings indicate that the MATS classifier can effectively assign patients with stage II and III CRC into low and high-risk groups, thereby adding complementary prognostic value to the traditional clinicopathological risk factors and mismatch repair status currently used to estimate the prognosis of these patients. Moreover, our study show that the MATS could help to identify low-risk stage III patients who can benefit from fluoropyrimidine-based adjuvant chemotherapy alone. MATS might facilitate reduction of unnecessary oxaliplatin-based adjuvant therapy currently being performed in patients with stage III CRC. Thus, MATS potentially offers clinical value in directing personalized medicine and tailored decision making in stage II and III CRC patients. Since we developed an RT-PCR based ‘risk prediction model’ using our gene signature, this score can be readily applied to independent, future prospective cohorts to evaluate the potential of this new classifier for decision making in CRC patients.
Supplementary Material
Novelty & Impact.
In view of the inadequacy of currently used clinicopathological features for risk-stratification in patients with colorectal cancer (CRC), we undertook a systematic and comprehensive biomarker discovery effort to develop a mesenchymal-associated gene signature, for risk-assessment in this disease. This signature was validated in a meta-analysis of 1212 patients with stage II and III CRCs from nine independent datasets, as well as two independent in-house patient cohorts. We report that our signature-derived low-risk patients with stage III disease significantly benefited from adjuvant chemotherapy with an excellent prognosis, whereas the high-risk patients did not. Furthermore, our exploratory analysis revealed that our signature was an excellent predictor for therapeutic response to FOLFOX and cetuximab, in metastatic settings. Taken together, our signature potentially offers clinical value in directing personalized treatment options for patients with colorectal cancer.
Acknowledgments
We thank Jacob Turner and Xuan Wang for their input on data analysis. We thank Y. Takagi and J. Inoue for their valuable contribution in FFPE sample collection and processing.
Funding
The present work was supported by the CA72851, CA181572, CA184792 and CA187956 grants from the National Cancer Institute, National Institute of Health; RP140784 from the Cancer Prevention Research Institute of Texas; grants from the Sammons Cancer Center and Baylor Foundation, as well as funds from the Baylor Research Institute, Dallas, TX, USA awarded to Ajay Goel. Balázs Győrffy was supported by the NVKP_16-1-2016-0037 and KH-129581 grants of the National Research, Development and Innovation Office, Hungary.
Abbreviations:
- AJCC
American Joint Committee on Cancer
- AUC
area under the curve
- CMS
consensus molecular subtypes
- CRC
colorectal cancer
- CRCSC
colorectal cancer subtyping consortium
- DFS
disease-free survival
- EGFR
epidermal growth factor receptor
- EMT
epithelial-to-mesenchymal transition
- FDR
false discovery rate
- FFPE
formalin-fixed paraffin-embedded
- HR
hazard ratio
- IDEA
International Duration Evaluation of Adjuvant Chemotherapy
- KM
Kaplan-Meier
- LASSO
the least absolute shrinkage and selection operator
- LCM
laser capture microdissection
- MATS
mesenchymal-associated transcriptomic signature
- MSI
Microsatellite instability
- NCCN
National Comprehensive Cancer Network
- OS
overall survival
- RFS
recurrence-free survival
- ROC
receiver operator characteristic
- TNM
Tumor Node Metastasis
- VIM
vimentin
Footnotes
Disclosures
Yasuhide Yamada received honoraria from Taiho, Chugai, and Nipponkayaku Pharmaceutical. Marwan Fakih received honoraria from Amgen, Array, Bayer and Pfizer (Consulting/advisory relationship), Amgen and Guardant360 (Speaker’s Bureau), Amgen, AstraZeneca and Novartis (Research funding). The other authors have declared no conflict of interest
Data Accessibility
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Dienstmann R, Salazar R, Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J Clin Oncol 2015;33: 1787–96. [DOI] [PubMed] [Google Scholar]
- 2.Benson AB 3rd, Schrag D, Somerfield MR, Cohen AM, Figueredo AT, Flynn PJ, Krzyzanowska MK, Maroun J, McAllister P, Van Cutsem E, Brouwers M, Charette M, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol 2004;22: 3408–19. [DOI] [PubMed] [Google Scholar]
- 3.Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol 2012;23: 2479–516. [DOI] [PubMed] [Google Scholar]
- 4.Network. NCC. Colon Cancer (Version 1.2017)..
- 5.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F, de Gramont A. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27: 3109–16. [DOI] [PubMed] [Google Scholar]
- 6.O’Connell MJ, Lavery I, Yothers G, Paik S, Clark-Langone KM, Lopatin M, Watson D, Baehner FL, Shak S, Baker J, Cowens JW, Wolmark N. Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J Clin Oncol 2010;28: 3937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kopetz S, Tabernero J, Rosenberg R, Jiang ZQ, Moreno V, Bachleitner-Hofmann T, Lanza G, Stork-Sloots L, Maru D, Simon I, Capella G, Salazar R. Genomic classifier ColoPrint predicts recurrence in stage II colorectal cancer patients more accurately than clinical factors. Oncologist 2015;20: 127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agesen TH, Sveen A, Merok MA, Lind GE, Nesbakken A, Skotheim RI, Lothe RA. ColoGuideEx: a robust gene classifier specific for stage II colorectal cancer prognosis. Gut 2012;61: 1560–7. [DOI] [PubMed] [Google Scholar]
- 9.Gao S, Tibiche C, Zou J, Zaman N, Trifiro M, O’Connor-McCourt M, Wang E. Identification and Construction of Combinatory Cancer Hallmark-Based Gene Signature Sets to Predict Recurrence and Chemotherapy Benefit in Stage II Colorectal Cancer. JAMA Oncol 2016;2: 37–45. [DOI] [PubMed] [Google Scholar]
- 10.Beijers AJ, Mols F, Tjan-Heijnen VC, Faber CG, van de Poll-Franse LV, Vreugdenhil G. Peripheral neuropathy in colorectal cancer survivors: the influence of oxaliplatin administration. Results from the population-based PROFILES registry. Acta Oncol 2015;54: 463–9. [DOI] [PubMed] [Google Scholar]
- 11.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21: 1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, Goel A, Wang X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. Semin Cancer Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi H, Ishikawa T, Ishiguro M, Okazaki S, Mogushi K, Kobayashi H, Iida S, Mizushima H, Tanaka H, Uetake H, Sugihara K. Prognostic significance of Traf2- and Nck- interacting kinase (TNIK) in colorectal cancer. BMC Cancer 2015;15: 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A 2009;106: 7131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10: e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okita A, Takahashi S, Ouchi K, Inoue M, Watanabe M, Endo M, Honda H, Yamada Y, Ishioka C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018;9: 18698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sztupinszki Z, Gyorffy B. Colon cancer subtypes: concordance, effect on survival and selection of the most representative preclinical models. Sci Rep 2016;6: 37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 2011;12: 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsuji S, Midorikawa Y, Takahashi T, Yagi K, Takayama T, Yoshida K, Sugiyama Y, Aburatani H. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br J Cancer 2012;106: 126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25: 3230–7. [DOI] [PubMed] [Google Scholar]
- 21.Goel A, Nagasaka T, Hamelin R, Boland CR. An optimized pentaplex PCR for detecting DNA mismatch repair-deficient colorectal cancers. PLoS One 2010;5: e9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruopp MD, Perkins NJ, Whitcomb BW, Schisterman EF. Youden Index and optimal cut-point estimated from observations affected by a lower limit of detection. Biom J 2008;50: 419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkel S, Mansmann U, Papadopoulos T, Wittekind C, Hohenberger W, Hermanek P. The prognostic inhomogeneity of colorectal carcinomas Stage III: a proposal for subdivision of Stage III. Cancer 2001;92: 2754–9. [PubMed] [Google Scholar]
- 24.Mooi JK, Wirapati P, Asher R, Lee CK, Savas PS, Price TJ, Townsend A, Hardingham J, Buchanan D, Williams D, Tejpar S, Mariadason JM, et al. The prognostic impact of Consensus Molecular Subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: molecular analysis of the AGITG MAX clinical trial. Ann Oncol 2018. [DOI] [PubMed] [Google Scholar]
- 25.Linnekamp JF, Hooff SRV, Prasetyanti PR, Kandimalla R, Buikhuisen JY, Fessler E, Ramesh P, Lee K, Bochove GGW, de Jong JH, Cameron K, Leersum RV, et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ 2018;25: 616–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sveen A, Bruun J, Eide PW, Eilertsen IA, Ramirez L, Murumagi A, Arjama M, Danielsen SA, Kryeziu K, Elez E, Tabernero J, Guinney J, et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin Cancer Res 2018;24: 794–806. [DOI] [PubMed] [Google Scholar]
- 27.Thanki K, Nicholls ME, Gajjar A, Senagore AJ, Qiu S, Szabo C, Hellmich MR, Chao C. Consensus Molecular Subtypes of Colorectal Cancer and their Clinical Implications. Int Biol Biomed J 2017;3: 105–11. [PMC free article] [PubMed] [Google Scholar]
- 28.Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 2017;17: 268. [DOI] [PubMed] [Google Scholar]
- 29.Su C, Li H, Peng Z, Ke D, Fu H, Zheng X. Identification of plasma RGS18 and PPBP mRNAs as potential biomarkers for gastric cancer using transcriptome arrays. Oncol Lett 2019;17: 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue VW, Cheung MT, Chan PT, Luk LLY, Lee VH, Au TC, Yu AC, Cho WCS, Tsang HFA, Chan AK, Wong SCC. Non-invasive Potential Circulating mRNA Markers for Colorectal Adenoma Using Targeted Sequencing. Sci Rep 2019;9: 12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.