

Abstract
How phosphorylation of the epithelial sodium channel (ENaC) contributes to its regulation is incompletely understood. Previously, we demonstrated that in outside-out patches ENaC activation by serum- and glucocorticoid-inducible kinase isoform 1 (SGK1) was abolished by mutating a serine residue in a putative SGK1 consensus motif RXRXX(S/T) in the channel’s α-subunit (S621 in rat). Interestingly, this serine residue is followed by a highly conserved proline residue rather than by a hydrophobic amino acid thought to be required for a functional SGK1 consensus motif according to in vitro data. This suggests that this serine residue is a potential phosphorylation site for the dual-specificity tyrosine phosphorylated and regulated kinase 2 (DYRK2), a prototypical proline-directed kinase. Its phosphorylation may prime a highly conserved preceding serine residue (S617 in rat) to be phosphorylated by glycogen synthase kinase 3 β (GSK3β). Therefore, we investigated the effect of DYRK2 on ENaC activity in outside-out patches of Xenopus laevis oocytes heterologously expressing rat ENaC. DYRK2 included in the pipette solution significantly increased ENaC activity. In contrast, GSK3β had an inhibitory effect. Replacing S621 in αENaC with alanine (S621A) abolished the effects of both kinases. A S617A mutation reduced the inhibitory effect of GKS3β but did not prevent ENaC activation by DYRK2. Our findings suggest that phosphorylation of S621 activates ENaC and primes S617 for subsequent phosphorylation by GSK3β resulting in channel inhibition. In proof-of-concept experiments, we demonstrated that DYRK2 can also stimulate ENaC currents in microdissected mouse distal nephron, whereas GSK3β inhibits the currents.
Keywords: Epithelial sodium channel (ENaC), Serum- and glucocorticoid-induced kinase isoform 1 (SGK1), Dual-specificity tyrosine phosphorylated and regulated kinase 2 (DYRK2), Glycogen synthase kinase 3 beta (GSK3β), Xenopus laevis oocytes, Microdissected mouse distal nephron, Patch clamp
Introduction
The epithelial sodium channel (ENaC) is the rate-limiting step for Na+ absorption in a variety of epithelia and plays a critical role in maintaining Na+ balance and controlling long-term blood pressure in mammals [28, 34, 63]. ENaC is probably a heterotrimer consisting of three homologous subunits (α, β, and γ) [60]. Each subunit has two transmembrane domains, a large extracellular loop and cytosolic N- and C-termini (Fig. 1a). In different epithelial tissues, ENaC activity is under the tight control by a range of hormones and local mediators [37, 49, 65].
Fig. 1.
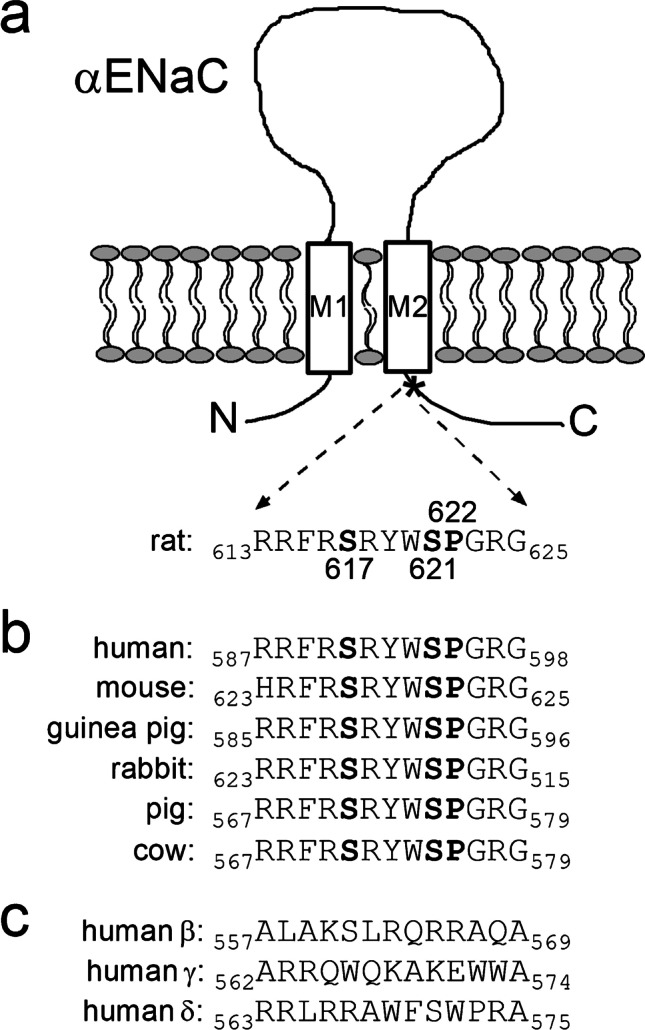
Two serine residues and one proline residue are highly conserved in a C-terminal region of αENaC close to the second transmembrane domain. a Schematic representation of αENaC illustrating the extracellular loop, two transmembrane domains (M1 and M2), and intracellular N- and C-termini. The amino acid sequence of rat αENaC (residues 613–625) corresponds to the C-terminal region indicated by a star (*) and contains the serine residues 617 (S617) and 621 (S621) and the proline residue 622 (P622) highlighted in bold. b Amino acid sequence alignment of this highly conserved C-terminal region from several mammalian αENaC subunits. The residues homologous to S617, S621, and P622 in rat αENaC are highlighted in bold. c Amino acid sequence alignment of homologous C-terminal regions from human β-, γ-, and δENaC subunits
At the cellular level, ENaC regulation involves a complex interplay of extracellular factors and intracellular signal transduction pathways including a number of protein kinases [3, 37, 65]. Kinases are important regulators of a wide range of cellular processes and consequently may modify ENaC function by acting at many different levels. Effects of kinases on ENaC include the phosphorylation and modification of regulatory proteins associated with ENaC or the phosphorylation of the channel itself with modulatory effects on its interaction with regulatory proteins. A prominent example is the ubiquitin ligase Nedd4-2 [65, 72, 73] which promotes endocytic retrieval and subsequent proteasomal degradation of ENaC. Phosphorylation of Nedd4-2 at specific sites, e.g., by serum- and glucocorticoid-induced kinase 1 (SGK1) [18, 25, 42], reduces its ability to bind to PY-motifs in the cytosolic C-termini of the channel resulting in reduced channel retrieval. This mechanism is thought to contribute to the increase of ENaC expression at the cell surface elicited by aldosterone because the latter has a strong stimulatory effect on SGK1 [12]. On the other hand, phosphorylation of specific sites in the C-termini of β- and γENaC has been reported to modify the channel’s interaction with Nedd4-2 thereby facilitating or impeding Nedd4-2 mediated channel retrieval [22, 41, 68].
In Xenopus laevis oocytes, the stimulatory effect of coexpressed SGK1 on ENaC whole-cell currents can be attributed mainly to an increased channel abundance at the cell surface [2, 18, 21, 62]. There is good evidence that this is due to inhibition of Nedd4-2-mediated channel retrieval, but a stimulation of channel forward trafficking may contribute to the effect [2, 45, 46]. This latter concept is also supported by the observation that in the oocyte system, a co-expressed neuronal isoform of SGK increased cell surface expression of homomeric human δENaC known to lack PY-motifs [82]. In addition, we have previously shown that recombinant active SGK1 included in the pipette solution can rapidly activate ENaC in excised outside-out patches from oocytes most probably by increasing the open probability of channels present in the plasma membrane [20]. This direct stimulatory effect on ENaC activity was also observed with recombinant protein kinase B alpha (PKBα) in the pipette solution [21]. Importantly, the stimulatory effect of both kinases was critically dependent on the serine residue S621 in rat αENaC [20, 21]. Originally, the putative phosphorylation site S621 was identified by searching for a conserved SGK/PKB consensus motif RXRXX(S/T) [38, 39] in the cytosolic termini of ENaC. The identified site is located in the carboxyl terminal region of αENaC close to the second transmembrane domain and is highly conserved in mammals (Fig. 1b). A corresponding site is absent in the β-, γ-, and δ-subunits of ENaC (Fig. 1c).
Presently, it remains an open question whether SGK1 and PKBα directly phosphorylate the channel at the identified phosphorylation site S621. Indeed, it is conceivable that the stimulatory effect of the recombinant kinases is mediated by modifying the activity of endogenous kinases or phosphatases present in the patch, thereby indirectly favoring channel phosphorylation at this site. Thus, SGK1 and PKBα may not phosphorylate the channel directly but may mediate their effect by activating or inhibiting an endogenous kinase or phosphatase, respectively. This hypothesis is supported by evidence from in vitro studies that the RXRXX(S/T) motif has to be followed by a bulky hydrophobic residue to become a preferred SGK/PKB consensus motif [1, 38]. In contrast, in αENaC (Fig. 1b), the RXRXX(S/T) motif is followed by a highly conserved proline residue (P622 in rat). Interestingly, this proline residue makes the preceding serine residue (S621 in rat) a rather poor phosphorylation site for SGK/PKB [1, 85] but a potential phosphorylation site for proline-directed kinases.
The dual-specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2) is a prototypical proline-directed kinase and a member of a family comprising at least seven mammalian isoforms [5]. DYRK2 has multiple functions and is expressed in a broad range of tissues [16, 75, 84]. These include colonic [81] and renal tubular epithelial cells [10, 78], where DYRK2 may be co-expressed with ENaC. Kinases of the DYRK family (DYRKs) autophosphorylate a critical tyrosine residue in their own activating loop during the translational process at the ribosome [48]. After complete translation and release from the ribosome, tyrosine-kinase-activity is lost and DYRKs phosphorylate their substrates on serine or threonine residues [47]. DYRKs are called proline-directed kinases due to their strong preference for a proline residue at the P + 1 position in combination with an arginine residue at P − 3 position. Thus, the typical phosphorylation recognition sequence of DYRK2 is RXX(S/T)P [8, 16, 70]. This suggests that S621 in the carboxyl terminus of rat αENaC located in the sequence 618RYWSP622 (Fig. 1b) is a good target for DYRK2-mediated phosphorylation.
Interestingly, DYRK2 phosphorylates several substrates which are subsequently recognized and further phosphorylated by the glycogen synthase kinase 3 β (GSK3β) [16]. Among those are eukaryotic initiation factor 2B (eIFB) [83], glycogen synthase [69], collapsin response mediator protein 4 (CRMP4) [15], transcription factors c-Myc, and c-Jun [74]. GSK3β phosphorylation targets are determined by other so-called priming kinases, because GSK3β preferentially phosphorylates its substrates when another phosphoserine (PS) or phosphothreonine residue (PT) is present four residues C-terminal to the site of GSK3β phosphorylation. Thus, a typical GSK3β recognition motif has the following sequence: SXXX(PS/PT) [23, 24]. This suggests that phosphorylation of S621 in the α-subunit of rat ENaC may prime the preceding serine residue 617 (S617) for phosphorylation by GSK3β (Fig. 1b).
Therefore, the aim of the present study was to investigate whether DYRK2 and GSK3β can affect ENaC function and whether this involves the two adjacent serine residues S621 and S617 located in the α-subunit of rat ENaC. For this purpose, we tested the effects of recombinant DYRK2 and GSK3β on rat ENaC heterologously expressed in Xenopus laevis oocytes using the outside-out patch clamp technique and site-directed mutagenesis. To confirm the oocyte findings in proof-of-concept experiments in native renal tissue, we also studied the effects of recombinant DYRK2 and GSK3β on ENaC currents in patch-clamp recordings from microdissected mouse distal nephron.
Materials and methods
cDNA clones
Full-length cDNAs for rat wild-type α-, β-, and ΓENaC [9] and for αS621AENaC [20], αS617AENaC, and αP622FENaC mutants were in pGEM-HE vector. Extension overlap PCR for site directed mutagenesis was used to generate αS617AENaC and αP622FENaC mutants. For the S617A mutation, a mutagenic forward primer with the sequence 5′-CTACGCCGGTTCCGGGCCCGGTACTGGTCTCCA-3′ and a reverse primer with the sequence 5′-TGGAGACCAGTACCGGGCCCGGAACCGGCGTAG-3′ were used to introduce a triplet mutation from CCA at nucleotides 1848–1851 to TTT. To generate αP622FENaC, a mutagenic forward primer with the sequence 5′-GCCGGTACTGGTCTTTTGGACGAGGGGCCAG-3′ and a reverse primer with the sequence 5′-CTGGCCCCTCGTCCAAAAGACCAGTACCGGC-3′ were used to introduce a triplet mutation from GGC at nucleotides 1864–1867 to AGC. Mutations were confirmed by sequence analysis. Linearized plasmids were used as templates for cRNA synthesis using T7 RNA polymerase (mMessage mMachine, Ambion, Austin, TX, USA).
Isolation of Xenopus laevis oocytes and injection of cRNA
Isolation of oocytes was performed essentially as described previously [20, 21, 30, 32, 62]. Oocytes were injected with cRNA using 0.1–0.2 ng of cRNA per ENaC subunit per oocyte. To prevent Na+ overloading [33], injected oocytes were incubated in low-sodium modified Barth’s saline (in mM, 1 NaCl, 40 KCl, 60 NMDG-Cl, 0.4 CaCl2, 0.3 Ca(NO3)2, 0.8 MgSO4, and 10 HEPES adjusted to pH = 7.4 with HCl) supplemented with 100 U/ml sodium penicillin and 100 µg/ml streptomycin sulfate. Ocytes were studied 48–72 h after injection.
Recordings in outside-out macropatches excised from Xenopus laevis oocytes
Current recordings from outside-out membrane patches were performed essentially as described previously [20, 21, 31, 40, 44] using conventional patch-clamp technique. Patch pipettes were pulled from borosilicate glass capillaries and had a tip diameter of about 5–7 μm after fire polishing. Pipettes were filled with K-gluconate pipette solution (in mM, 90 K-gluconate, 5 NaCl, 2 Mg-ATP, 2 EGTA, and 10 mM HEPES adjusted to pH = 7.28 with Tris). Seals were routinely formed in a low-sodium NMDG-Cl bath solution (in mM, 95 NMDG (N-methyl-D-glucamine)-Cl, 1 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, and 10 HEPES adjusted to pH 7.4 with Tris). In this bath solution, the pipette resistance averaged about 3 MΩ. In NaCl bath solution, NMDG-Cl was replaced by 95 mM NaCl. For continuous current recordings, the holding potential was set to − 70 mV using an EPC9 amplifier (HEKA Elektronik, Lambrecht, Germany). Using a 3 M KCl flowing boundary electrode, the liquid junction (LJ) potential occurring at the pipette/NaCl bath junction was measured to be 12 mV (bath positive) [44]. Thus, at a holding potential of − 70 mV, the effective trans-patch potential was − 82 mV. This value is close to the calculated equilibrium potential of Cl− (ECl = − 77.4 mV) and K+ (EK = − 79.4 mV) under our experimental conditions. Experiments were performed at room temperature. To change from one bath solution to another, a conventional gravity-fed system controlled by a magnetic valve system (ALA BPS-8) was used in combination with a TIB14 interface (HEKA Elektronik, Lambrecht, Germany). Pulse 8.78 software (HEKA Elektronik, Lambrecht, Germany) was used for data acquisition. Amiloride-sensitive current (∆IAmi) in outside-out membrane patches was determined by subtracting the current value recorded in the presence of amiloride (2 μM) from the corresponding value recorded prior to its addition. The current traces were filtered at 200 Hz and sampled at 800 Hz.
Preparation of mouse renal tubules
For this study, we used 29 male mice (C57BL/6 J originally acquired from Charles River Laboratories, Sulzfeld, Germany) aged 6–8 weeks. Mice were bred and maintained in the animal facility of Friedrich-Alexander-Universität Erlangen Nürnberg (FAU). Mice received a standard diet (Na+ content 3.2 g/kg, Cat-No 1310 from Spezialfutter GmbH & Co. KG, Lage, Germany) with free access to tap water. Isolation of renal tubules was essentially performed as described previously [57, 58]. Renal tubules were separated manually using fine forceps. We identified and isolated tubular segments with characteristic branching indicative of the transition from connecting tubules (CNT) to cortical collecting ducts (CCD). Under the dissecting microscope, there are no clear-cut boundaries between CNT and initial CCD. In particular, after transfer of the tubular segments to the perfusion chamber and after opening the tubules, it is difficult to distinguish between CNT and initial CCD. Therefore, we have to assume that our recordings include recordings from CNT as well as from CCD. We did not attempt to distinguish these recordings and pooled the data. The microdissected tubular segments were attached to small pieces of glass coverslips coated with Cell-Tak (Collaborative Research, Bedford, MA, USA) and were transferred to a temperature controlled perfusion chamber (37 °C) mounted on an inverted microscope (Leica DM IRB) to perform patch clamp recordings.
Whole-cell and outside-out patch clamp recordings from microdissected mouse distal nephron
A computer-controlled EPC-9 patch clamp amplifier (HEKA Elektronik, Lambrecht, Germany) was used to perform conventional whole-cell and outside-out patch clamp recordings as previously described [55–59]. To gain access with the patch pipette to the apical cell membrane, tubules were cut open with a broken glass pipette attached to a micromanipulator. Principal cells expressing ENaC were identified according to their characteristic shape and responsiveness to amiloride. Pipettes were pulled from borosilicate glass capillaries and had a tip diameter of about 1.5 μm after fire polishing. Pipettes were filled with a pipette solution containing the following (in mM): 85 K gluconate, 40 CsOH, 5 Na gluconate, 2 Mg ATP, 2 EGTA Na, 2 MgCl2, 20 TEA-OH, and 10 HEPES. Its pH was adjusted to 7.2 with gluconic acid. The bath solution had the following composition (in mM): 145 Na gluconate, 5 K gluconate, 2 CaCl2, 5 barium acetate, 1 MgCl2, 3 glucose, and 5 HEPES; pH was adjusted to 7.4 with Tris. Pipette resistance measured in the bath solution was about 4–6 MΩ. Seals were formed at the apical surface of principal cells by using gentle suction. Seal resistance ranged from 3 to 10 GΩ. Series resistance was in the order of 10 to 30 MΩ and was not compensated. For continuous whole-cell as well as outside-out current recordings, the holding potential (Vhold) was set at − 60 mV. In each experiment, ∆IAmi was initially measured in the whole-cell configuration and subsequently in the outside-out configuration provided that membrane patch excision was successful. In both configurations, ∆IAmi was determined by subtracting the current measured in the presence of amiloride (2 μM) from that measured in its absence. The current traces were filtered at 250 Hz and sampled at a rate of 2 kHz. For further analysis, they were digitally re-filtered at 70 Hz. Data were analyzed using the program “Patch for Windows” written by Dr. Bernd Letz (HEKA Elektronik, Lambrecht, Germany).
Chemicals
Recombinant constitutively active human SGK1 (∆1–60, S422D) and recombinant active full length human DYRK2 were purchased from Biomol GmbH (Hamburg, Germany) as 2 μg (SGK1) and 5 μg (DYRK2) vials in 50 μl stock solution both containing as main components 50 mM Tris–HCl, 0.1 mM EGTA, 0.1% 2-mercaptoethanol, 0.15 mM NaCl, and 270 mM sucrose. SGK1 and DYRK2 pipette solutions were freshly prepared on the day of the experiment by adding stock solutions to 1 ml of the corresponding pipette solutions giving a final SGK1 and DYRK2 concentration of 80 U/ml (for experiments in oocytes) and 20 U/ml (for experiments in microdissected renal tubules). Recombinant GSK3β and a selective GSK3β inhibitor (CHIR99021) were kindly provided by Prof. Philip Cohen (Dundee, UK). GSK3β in a concentration of 1.54 mg/ml was stored in a stock solution containing as main components 50 mM Tris–HCl, 0.1 mM EGTA, 0.1% 2-mercaptoethanol, 0.15 mM NaCl, and 50% glycerol. On the day of the experiment, the GSK3β pipette solution was freshly prepared by adding stock solution to pipette solution giving a final GSK3β concentration of 16 U/ml (for experiments in oocytes) and 8 U/ml (for experiments in microdissected renal tubules). CHIR99021 was stored in DMSO as stock solution with a concentration of 10 mM. On the day of the experiment, stock solution was added to pipette solution giving a final CHIR99021 concentration of 2 μM [54]. To preserve SGK1, DYRK2, and GSK3β activity, the pipette solutions were supplemented with dithiothreitol (Sigma-Aldrich, Taufkirchen, Germany) in a concentration of 0.1 mM in experiments with oocytes and 0.05 mM in experiments with microdissected renal tubules. Control experiments were performed using identical pipette solutions with SGK1, DYRK2, or GSK3β after heat inactivating the solutions at 68° C for 45 min. Moreover, in additional control experiments, we confirmed that dithiothreitol per se had no detectable effect on ENaC activity in the concentrations used (data not shown). Amiloride hydrochloride was purchased from Sigma-Aldrich (Taufkirchen, Germany) and was added from an aqueous 10 mM stock solution.
Statistics
Data are presented as mean values ± SEM; n indicates the number of individual recordings. In oocyte experiments, N indicates the number of different batches of oocytes used. In animal experiments, N is number of mice used in a set of recordings. Data from different oocyte batches and from different animals were pooled. Statistical analysis of nested experiments using N and n was not performed due to the low number of recordings obtained per animal and oocyte batch. Normal distribution of data was assessed using D’Agostino–Pearson omnibus test. Statistical significance was assessed by an appropriate parametric test: paired or unpaired Student’s t-test and Student’s ratio t-test. Significance was accepted for p < 0.05. Statistical analysis was performed using Graph Pad Prism 5.04.
Results
Recombinant DYRK2 stimulates ENaC currents in outside-out patches from Xenopus laevis oocytes
To investigate whether DYRK2 can modify ENaC activity, we performed patch-clamp recordings using Xenopus laevis oocytes expressing α-, β-, and γ-subunits of rat ENaC (αβγENaC). Recordings from outside-out macropatches were started about 4 min after patch excision. To minimize spontaneous channel rundown known to occur in the presence of a high extracellular Na+ concentration [80], patches were maintained for most of the time in NMDG-Cl solution containing only 1 mM Na+. In this solution, only a negligible inward current was detectable at a holding potential of − 70 mV. In contrast, periodic changes to NaCl bath solution revealed sizeable inward currents consistent with a current component carried by Na+ influx via ENaC. This current component was largely inhibited by the application of 2 μM amiloride, a concentration known to specifically inhibit ENaC. Using this protocol, the amiloride-sensitive sodium current (∆IAmi) was repeatedly determined to monitor ENaC activity over time. Figure 2a (left panel) shows a representative control recording with heat inactivated DYRK2 (inactive DYRK2) in the pipette solution. Data from similar experiments are summarized in the right panel of Fig. 2a and demonstrate that under control conditions ENaC currents recorded in outside-out patches remain pretty stable for about half an hour. This is consistent with previous control recordings using vehicle control in the pipette solution or other heat-inactivated kinases [20, 21]. In contrast, when catalytically active DYRK2 was included in the pipette solution, ∆IAmi significantly increased by about twofold within ~ 25 min (Fig. 2b). Using a similar experimental approach, we have previously shown that the serine residue S621 in the α-subunit of ENaC is essential for mediating channel activation by SGK1, PKBα, or the phosphatase inhibitor ocadaic acid included in the pipette solution [20, 21]. To test whether this residue is also important for mediating the stimulatory effect of DYRK2, we performed recordings in outside-out patches obtained from oocytes expressing mutant αS621AβγENaC in which S621 of the α-subunit was replaced by alanine. No stimulatory effect of DYRK2 was observed on mutant αS621AβγENaC (Fig. 2c). Similarly, no stimulatory effect of DYRK2 was detected in outside-out patches obtained from oocytes expressing mutant αP622FβγENaC in which P622 of the α-subunit was replaced by phenylalanine (Fig. 2d). These findings indicate that ENaC activation by DYRK2 requires the phosphorylation site S621 and in addition the proline residue P622 most likely as recognition site for the proline-directed kinase which enables it to phosphorylate the preceding serine residue. Phenylalanine was chosen as replacement for P622 to test the functional role of this proline residue and to create an optimal SGK1 consensus site (see next paragraph).
Fig. 2.

Recombinant DYRK2 stimulates ENaC currents in outside-out patches from Xenopus laevis oocytes. a, b, c, and d, Left panels, representative current traces recorded in outside-out patches of αβγENaC, αS621AβγENaC, and αP622FβγENaC expressing oocytes at a holding potential (Vhold) of − 70 mV. As indicated by the bars, bath solution was changed from a low Na+ (NMDG-Cl; [Na+] = 1 mM) to a normal Na+ containing solution (NaCl; [Na+] = 96 mM) without or with amiloride (Ami, 2 μM). Heat-inactivated DYRK2 (inactive DYRK2) or active recombinant DYRK2 (80 U/ml) were included in the pipette solutions as indicated under the traces. Right panels, summary of normalized ∆IAmi values obtained from similar experiments as shown in the representative traces (left panels). Each grey line corresponds to an individual outside-out patch clamp recording and connects ∆IAmi values obtained at different time points. The black lines in each graph connect average ∆IAmi values (mean ± SEM; αβγENaC/inactive DYRK2, n = 8; αβγENaC/DYRK2, n = 13; αS621AβγENaC/DYRK2, n = 7; αP622FβγENaC/DYRK2, n = 6). ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 4 min using paired Student’s ratio t-test. ** p < 0.01; *** p < 0.001
Recombinant SGK1 fails to stimulate mutant αP622FβγENaC
As stated in the introduction, in vitro studies indicate that an RXRXX(S/T) motif has to be followed by a large hydrophobic residue to become a functional SGK1 consensus motif [1, 38]. Thus, the P622F mutation in the channel’s α-subunit (αP622FENaC) theoretically converts the original motif (616RSRYWS621P622, Fig. 1) into an optimal SGK1 consensus site (616RSRYWS621F622). Therefore, we asked the question whether SGK1 can activate mutant αP622FβγENaC under our experimental conditions. To address this, ENaC activity was assessed by measuring ∆IAmi in outside-out macropatches excised from oocytes expressing αβγENaC or αP622FβγENaC. In these recordings, we confirmed our previous findings [20, 21] that ∆IAmi remained stable over time with heat inactivated SGK1 (inactive SGK1) included in the pipette solutions (Fig. 3a) but significantly increased by about threefold within ~ 20 min with catalytically active SGK1 in the pipette solution (Fig. 3b). Importantly, SGK1 failed to stimulate αP622FβγENaC (Fig. 3c) despite the predicted optimal SGK1 consensus site of the mutant channel. We cannot exclude the possibility that introducing a hydrophobic residue in the C-terminal domain may cause a structural rearrangement of this flexible loop, thereby preventing SGK1 from accessing its target. However, the well-preserved channel function of αP622FENaC argues against this possibility. Thus, our finding that SGK1 failed to stimulate αP622FβγENaC supports the hypothesis that S621 is not directly phosphorylated by SGK1 but that the stimulatory effect of SGK1 is mediated by an indirect effect resulting in the phosphorylation of S621.
Fig. 3.

Recombinant SGK1 fails to stimulate αP622FβγENaC. Left panels, representative current traces recorded in outside-out patches of αβγENaC (a, b) or αP622FβγENaC (C) expressing oocytes as described in Fig. 2. Heat-inactivated SGK1 (inactive SGK1) or constitutively active recombinant SGK1 (80 U/ml) were included in the pipette solutions as indicated under the traces. Right panels, summary of normalized ∆IAmi values obtained from similar experiments as shown in the representative traces (left panels) using the same symbols as in Fig. 2. αβγENaC/inactive SGK1, n = 5; αβγENaC/SGK1, n = 7; αP622FβγENaC/SGK1, n = 7. ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 4 min using paired Student’s ratio t-test. * p < 0.05; ** p < 0.01
Recombinant GSK3β inhibits ENaC currents
As noted in the introduction, phosphorylation of S621 may prime the preceding S617 for phosphorylation by GSK3β, because the latter preferentially phosphorylates its substrates four residues N-terminal to a phosphoserine (PS) or phosphothreonine residue (PT) [23, 24]. Therefore, we tested the effect of GSK3β on ENaC activity using the same approach as described above for DYRK2 and SGK1. In control experiments, we detected no effect of heat-inactivated GSK3β (inactive GSK3β) on ENaC currents (Fig. 4a). However, catalytically active GSK3β significantly reduced ∆IAmi to less than 50% of its original value (Fig. 4b). In contrast, the inhibitory effect of GSK3β was not observed, when the specific GSK3β inhibitor CHIR99021 [54] was included in the pipette solution in addition to GSK3β. This supports the conclusion that the inhibitory effect GSK3β on ENaC is specific and due to its kinase activity. Importantly, GSK3β failed to inhibit ENaC currents in outside-out patches from oocytes expressing αS617AβγENaC (Fig. 4d) or αS621AβγENaC (Fig. 4e). Thus, the inhibitory effect of GSK3β depended on the presence of S617 and also on the presence of S621. This suggests that phosphorylation of S621 is required as a priming site for GSK3β to phosphorylate S617.
Fig. 4.

Recombinant GSK3β inhibits ENaC currents. Left panels, representative current traces recorded in outside-out patches of αβγENaC (a, b, c), αS617AβγENaC (d), or αS621AβγENaC (e) expressing oocytes as described in Fig. 2. Heat-inactivated GSK3β (inactive GSK3β) or active recombinant GSK3β (16 U/ml) were included in the pipette solutions as indicated under the traces. In c, the GSK3β inhibitor CHIR99021 (2 µM) was included in the pipette solutions together with GSK3β. Right panels, summary of normalized ∆IAmi values obtained from similar experiments as shown in the representative traces (left panels) using the same symbols as in Fig. 2. αβγENaC/inactive GSK3β, n = 9; αβγENaC/GSK3β, n = 20; αβγENaC/GSK3β + CHIR99021, n = 8; αS617AβγENaC/GSK3β, n = 7; αS621AβγENaC/GSK3β, n = 6. ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 4 min using paired Student’s ratio t-test. *** p < 0.001
Recombinant DYRK2 stimulates αS617AβγENaC
Since mutating S617 or S621 in the channel’s α-subunit abolished the inhibitory effect of GSK3β, we wondered whether the stimulatory effect of DYRK2 was preserved in outside-out patches from oocytes expressing αS617AβγENaC. In control recordings, we demonstrated that inactive DYRK2 had no significant stimulatory effect on αS617AβγENaC (Fig. 5a). Importantly, the stimulatory effect of catalytically active DYRK2 on αS617AβγENaC was fully preserved with a ~ 2.5-fold increase of ∆IAmi within ~ 25 min (Fig. 5b). Taken together with the findings shown in Fig. 2, this indicates that the stimulatory effect of DYRK2 is dependent on S621 and P622, but independent of S617.
Fig. 5.

Recombinant DYRK2 activates αS617AβγENaC. a and b, left panels, representative current traces recorded in outside-out patches of αS617AβγENaC expressing oocytes as described in Fig. 2. Heat-inactivated DYRK2 (inactive DYRK2) or active recombinant DYRK2 (80 U/ml) were included in the pipette solutions as indicated under the traces. Right panels, summary of normalized ∆IAmi values obtained from similar experiments as shown in the representative traces (left panels) using the same symbols as in Fig. 2. αS617AβγENaC/inactive DYRK2, n = 3; αS617AβγENaC/DYRK2, n = 6. ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 4 min using paired Student’s ratio t-test. ** p < 0.01
DYRK2 can stimulate ENaC in outside-out patches from principal cells of microdissected mouse distal nephron
Our oocyte data indicate that DYRK2 and GSK3β can stimulate and inhibit ENaC activity, respectively, and that these effects are mediated by specific phosphorylation sites in the C-terminus of the channel’s α-subunit. To explore a possible regulatory role of these kinases in native renal tissue, we tested the effects of DYRK2 and GSK3β on ENaC currents in microdissected mouse distal nephron using an established experimental technique [55–59]. Tubular epithelial cells were approached from the apical membrane, and amiloride-sensitive ENaC currents (ΔIAmi) were recorded in the whole-cell and outside-out configuration of the patch-clamp technique. Microdissected and split open tubular fragments of the connecting tubule (CNT) and cortical collecting duct (CCD) were used which are known to express ENaC in principal cells. For the purpose of this study, we did not distinguish between recordings from CNT and CCD and pooled the data from all successful experiments (see Experimental Procedures).
At the beginning of the experiments, whole-cell currents were recorded at a holding potential of − 60 mV (Fig. 6a and 6b, left panels). Recordings were started in the presence of amiloride (2 μM). Washout of amiloride revealed an ENaC-mediated inward current component which was rapidly inhibited upon reapplication of amiloride. A detectable amiloride-sensitive whole-cell current was taken as evidence that the cell under investigation was a principal cell expressing ENaC. After determining ∆IAmi in the whole-cell configuration, we routinely tried to excise an outside-out patch and to record from the same cell ∆IAmi also in the outside-out configuration. In successful attempts, we monitored ∆IAmi over time by repeated application and washout of amiloride (Fig. 6a and 6b, right panels). Representative current traces from experiments with active recombinant DYRK2 or heat-inactivated DYRK2 in the pipette solution are shown in Fig. 6a and 6b, respectively. Data from similar recordings are summarized in Fig. 6c, d, and e. Interestingly, the amiloride-sensitive whole-cell currents were not significantly different in the two groups with ∆IAmi averaging 253 ± 54 pA (n = 14; N = 11) in control experiments and 193 ± 57 pA (n = 9; N = 8) in experiments with active DYRK2 included in the pipette solution (Fig. 6c). A possible explanation for this is that active DYRK2 included in the pipette solution does not reach ENaC in the plasma membrane in a sufficient concentration. Interestingly, recordings performed in the outside-out configuration revealed a significant difference between the two groups. In the control group with inactive DYRK2, the initial ∆IAmi measured in the outside-out configuration averaged 1.54 ± 0.63% (n = 14) of the corresponding ∆IAmi determined in the whole-cell mode (Fig. 6d). This percentage roughly reflects the ratio of the area of the pipette tip to the area of the entire apical cell membrane. Importantly, in experiments with active DYRK2 in the pipette solution, the initial ΔIAmi in outside-out patches was increased to 8.96 ± 3.08% of ΔIAmi determined in the corresponding whole-cell recordings. This value was significantly higher than that observed in control recordings with heat-inactivated DYRK2 (p < 0.01) (Fig. 6d). Moreover, in outside-out patches with active DYRK2 in the pipette solution ΔIAmi further increased over time (Fig. 6a, right panel) reaching on average ~ 160% of its initial value within ~ 10 min (Fig. 6e). In contrast, in control recordings in outside-out patches with inactive DYRK2 in the pipette solution, ΔIAmi remained relatively stable (Fig. 6b, right panel; Fig. 6e). These findings indicate that DYRK2 can stimulate ENaC currents in outside-out patches from native renal tubules. Apparently, the main stimulatory effect occurs within the initial 2–3 min needed to establish the outside-out configuration with some further stimulation after patch excision.
Fig. 6.

DYRK2 stimulates ENaC currents in outside-out patches excised from the apical membrane of principal cells in split open microdissected mouse renal tubules. a and b Representative current traces from whole-cell (left panels) and subsequent outside-out (right panels) patch-clamp recordings at a continuous Vhold of − 60 mV. Black bars indicate the presence of amiloride (Ami, 2 μM) in the bath solution. Active recombinant DYRK2 (40 U/ml; a) or heat-inactivated DYRK2 (inactive DYRK2; b) were included in the pipette solutions as indicated under the traces. c–e Summary of data from similar experiments as shown in the representative traces (a and b, left panels) with DYRK2 (n = 9) or inactive DYRK2 (n = 14) in the pipette solution. In c and d, black dots correspond to measurements from individual patches, and open columns with error bars represent mean values ± SEM. ∆IAmi values shown in c were determined in the whole-cell configuration. Data shown in d represent ∆IAmi values from outside-out patches (∆IAmi(o/out)) expressed as percentage of the corresponding whole-cell ∆IAmi values (% of ∆IAmi (w/c)). DYRK2, n = 9; inactive DYRK2, n = 14. In c and d, statistical significance was assessed by unpaired Student’s t-test and unpaired Student’s ratio t-test, respectively. e Averaged normalized ΔIAmi recorded in outside-out patches as illustrated in a (right panel) at different times after patch excision with DYRK2 (solid triangles, n = 9) or inactive DYRK2 (open circles, n = 14) in the pipette solution. In e, ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 2 min using paired Student’s ratio t-test. * p < 0.05; ** p < 0.01; n.s. not significant
GSK3 has an inhibitory effect on ENaC in outside-out patches from principal cells of microdissected mouse distal nephron.
In similar experiments, we also tested the effect of GSK3β on ENaC in microdissected mouse distal nephron. Constitutively, active recombinant GSK3β or heat-inactivated GSK3β were added to the pipette solution. Representative traces from these experiments are depicted in Fig. 7a and 7b, respectively, and the results are summarized in Fig. 7c, d, and e. Similar to the experiments with DYRK2, ΔIAmi measured in the whole-cell configuration was not different in the group with active GSK3β compared to ΔIAmi in the control group with inactive GSK3β (Fig. 7a and 7b, left traces; Fig. 7c). The initial ΔIAmi determined in the outside-out configuration appeared to be slightly reduced in the group with active GSK3β. The ratio between the initial ΔIAmi in outside-out patches and the corresponding ΔIAmi in the whole-cell configuration averaged 1.24 ± 0.49% (n = 7; N = 6) in experiments with GSK3β in the pipette solution and 2.43 ± 0.45% (n = 10; N = 7) in control experiments with inactive GSK3β (Fig. 7d). This difference did not reach statistical significance (p = 0.096). However, ΔIAmi declined more rapidly in outside-out patches with active GSK3β in the pipette solution than in patches with inactive GSK3β (Fig. 7a and 7b, right panels). Within 4 min after patch excision, relative ΔIAmi was significantly lower in outside-out patches with active GSK3β in the pipette solution than relative ΔIAmi in outside-out patches from the control group with inactive GSK3β (Fig. 7e). After 8 min, this difference was even more pronounced, and ΔIAmi had declined to 46 ± 1.1% (n = 7; N = 6) of its initial value in the experiments with active GSK3β but only to 78 ± 0.9% (n = 10; N = 7) in control experiments with inactive GSK3β (p = 0.02). These findings indicate that GSK3β can inhibit ENaC in native renal tubules.
Fig. 7.

GSK3β inhibits ENaC currents in outside-out patches excised from the apical membrane of principal cells in split open microdissected mouse renal tubules. a and b Representative current traces from whole-cell (left panels) and subsequent outside-out (right panels) patch-clamp recordings from experiments similar to those shown in Fig. 6. Active recombinant GSK3β (8 U/ml; a) or heat-inactivated GSK3β (inactive GSK3β; b) were included in the pipette solutions as indicated under the traces. c–e Summary of data from similar experiments as shown in the representative traces (a and b, left panels) with GSK3β (n = 7) or inactive GSK3β (n = 10) in the pipette solution. In c and d, black dots correspond to measurements from individual patches, and open columns with error bars represent mean values ± SEM. ∆IAmi values shown in c were determined in the whole-cell configuration. Data shown in d represent ∆IAmi values from outside-out patches (∆IAmi(o/out)) expressed as percentage of the corresponding whole-cell ∆IAmi values (% of ∆IAmi (w/c)). GSK3β, n = 7; inactive GSK3β, n = 10. In c and d, statistical significance was assessed by unpaired Student’s t-test and unpaired Student’s ratio t-test, respectively. e Averaged normalized ΔIAmi recorded in outside-out patches as illustrated in a (right panel) at different times after patch excision with GSK3β (solid triangles, n = 7) or inactive GSK3β (open circles, n = 10) in the pipette solution. In e, ∆IAmi values determined at individual time points were compared with the corresponding initial ∆IAmi value at 2 min using paired Student’s ratio t-test. * p < 0.05; n.s. not significant
Discussion
The key findings of the present study are the following: (i) DYRK2 stimulated and GSK3β inhibited ENaC activity in outside-out patches from oocytes; (ii) the stimulatory effect of DYRK2 depended on the amino-acid residues S621 and P622 in the C-terminus of rat αENaC which supports the hypothesis that S621 is a phosphorylation site for a proline directed kinase; (iii) the inhibitory effect of GSK3β depended on both S617 and S621, consistent with the idea that phosphorylation of S621 primes S617 to be phosphorylated by GSK3β; (iv) the S617 phosphorylation site is necessary for channel inhibition by GKS3β but not required for channel activation by DYRK2; (v) in proof-of-concept experiments we demonstrated a stimulatory effect of DYRK2 and an inhibitory effect of GSK3β on ENaC activity also in microdissected mouse distal nephron.
This study confirms our previous finding that in outside-out patch clamp recordings from oocytes an intact S621 phosphorylation site in the C-terminus of rat αENaC is critical for mediating acute channel activation by SGK1 included in the pipette solution [20]. So far, it remained unclear whether SGK1 directly phosphorylates S621 or whether a downstream kinase is needed to mediate the S621-dependent stimulatory effect of SGK1. The findings of the present study support the hypothesis that due to a proline residue at the P + 1 position, the serine residue S621 is a phosphorylation site for proline-directed kinases, e.g., DYRK2. Indeed, replacing P622 by phenylalanine prevented ENaC activation by DYRK2, highlighting the functional importance of this proline residue to make S621 a suitable phosphorylation site for this prototypical proline-directed kinase. Interestingly, the P622F mutation also inhibited the stimulatory effect of SGK1, despite converting the original motif into an optimal SGK1 consensus site (616RSRYWS621F622). Moreover, the proline residue following S621 makes the latter an unlikely SGK1 phosphorylation site according to in vitro studies [1, 38, 85]. Taken together, this argues against a direct phosphorylation of S621 by SGK1, although its stimulatory effect is lost when S621 is replaced by an alanine [20, 21].
We have previously shown that the stimulatory effect of SGK1 can be mimicked by including the broad range phosphatase inhibitor okadaic acid or protein phosphatase inhibitor type 2 (PIP2) in the pipette solution [20]. Importantly, the stimulatory effect of phosphatase inhibition on ENaC also critically depended on S621 and was abolished by mutating this site [21]. Moreover, ENaC activity in outside-out patches decreased to very low levels, when Mg2+ was omitted from the pipette solution. This is consistent with the concept that an endogenous Mg2+-dependent kinase activity is present in the patch and is involved in maintaining baseline ENaC activity [20]. Inclusion of phosphatase inhibitors probably shifts the balance between endogenous kinase and phosphatase activity in the patch to favor phosphorylation of the stimulatory S621 site. Similarly, inclusion of SGK1 or DYRK2 in the pipette solution is likely to favor phosphorylation of S621 resulting in ENaC activation above baseline level. In case of DYRK2, this is most likely a direct effect which can be inhibited by mutating the proline residue P622. The latter residue is critical for DYRK2 to recognize the S621 phosphorylation site. In contrast, SGK1 may indirectly lead to S621 phosphorylation by stimulating an endogenous oocyte kinase or by inhibiting an endogenous phosphatase. In both cases, the resulting ENaC activation is due to increased phosphorylation of S621 as indicated by the finding that mutating this residue abolished the stimulatory effect of both SGK1 and DYRK2. The identity of the endogenous oocyte kinase and phosphatase involved in the phosphorylation and dephosphorylation of S621, respectively, is presently unknown.
The concept of an indirect effect of SGK1 on S621 is also consistent with our previous observation that plasma membrane cholesterol removal abolishes the acute stimulatory effect of SGK1 on ENaC in outside-out patches [40]. Cholesterol removal is likely to compromise the function of cholesterol-rich lipid raft microdomains, which are thought to serve as signaling platforms for ENaC and may be important for the channel’s interaction with associated regulatory proteins. Thus, the finding that cholesterol depletion prevents ENaC stimulation by SGK1 suggests that additional regulatory proteins, e.g., an endogenous kinase or phosphatase, are required to mediate the stimulatory effect of SGK1 on the channel.
Our findings in microdissected tubules provide proof of principle that the prototypical proline-directed kinase DYRK2 can activate ENaC not only in the oocyte expression system but also in native renal tissue. DYRK2 is known to be expressed in the kidney [16] including in renal tubular epithelial cells co-expressing ENaC [10, 78]. Moreover, using RNA-seq analysis, we recently confirmed expression of DYRK2 in cultured mouse cortical collecting duct (CCD) cells from the mCCDcl1 cell line (unpublished observation). This cell line is known to express ENaC and is an established CCD model to study ENaC-mediated transepithelial sodium transport [26, 50–52]. Whether DYRK2 is the physiologically relevant kinase or whether other proline-directed kinases are involved in ENaC activation by targeting S621 remains to be investigated. Regulating mechanisms and stimuli able to modify the expression and activity of DYRK2 are still incompletely understood [16]. Thus, it is presently unclear how DYRK2 may be regulated to modify ENaC function according to physiological needs. Moreover, it is conceivable that in the presence of a suitable baseline kinase activity, the degree of S621 phosphorylation is determined by adjusting the activity of a phosphatase dephosphorylating this site rather than by modifying the kinase activity.
At present, it is unclear how phosphorylation at S621 mechanistically causes channel activation. Our previous findings indicate that in outside-out patches, the acute stimulatory effect of SGK1 on ENaC is not due to the insertion of additional channels into the plasma membrane. Instead, constitutively active SGK1 in the pipette solution seems to activate a population of near-silent channels present in excised outside-out patches from ENaC expressing oocytes [20]. Thus, phosphorylation at S621 probably affects ENaC gating and turns previously silent channels into channels with a rather high open probability. Interestingly, this is reminiscent of proteolytic ENaC activation observed in single-channel recordings from outside-out patches exposed to trypsin or chymotrypsin in the bath solution [19, 29]. Proteolytic channel activation is a unique feature of ENaC [64, 65], but the underlying mechanisms and the identity of the physiologically relevant proteases remain incompletely understood. There is good evidence that proteases stimulate ENaC by cleaving specific sites in the extracellular loops of its α- and γ-subunits [37]. This cleavage causes the release of inhibitory tracts which probably leads to a conformational change resulting in channel activation [35]. Recently published cryo-EM structural data of ENaC indicate that specific binding sites are present to allow a close interaction of the α- and γ-inhibitory tracts with their respective subunits [60, 61]. However, it is still unclear how the occupancy of these binding sites alters channel conformation. No cryo-EM structural information is presently available regarding the C-termini of ENaC including the S621 residue in α-ENaC. Thus, at present, it remains purely speculative to postulate that a phosphorylation at this site may result in a conformational change similar to that induced by proteolytic channel activation. It will be an interesting task for future studies to explore a possible link between proteases and kinases in acutely regulating ENaC activity at the level of the plasma membrane.
As stated in the introduction, DYRK2 is known to act as priming kinase for GSK3β [16]. Our findings support the hypothesis that phosphorylation of S621 primes S617 for phosphorylation by GSK3β. Interestingly, GSK3β inhibited ENaC expressed in X. laevis oocytes and in microdissected tubules. Moreover, in the oocyte expression system, its effect depended on both S617 and on S621. In contrast, the stimulatory effect of DYRK2 was fully preserved when S617 was mutated and S621 remained intact. Thus, phosphorylation of S617 by GSK3β may serve as feedback mechanism to limit ENaC activation induced by phosphorylation of S621. Our functional data provide indirect evidence that phosphorylation of these residues is relevant for ENaC regulation. It remains a challenge for future studies to demonstrate direct phosphorylation of these sites and to elucidate how the degree of phosphorylation varies according to different physiological conditions.
GSK3β was first discovered to phosphorylate glycogen synthase, a final enzyme in the glycogen synthesis pathway. GSK3β is expressed in many tissues [13, 14] including ENaC expressing renal epithelial cells [10, 11, 78]. Indeed, we recently confirmed co-expression of ENaC and GSK3β in mCCDcl1 cells using RNA-seq analysis (unpublished observation). Phosphorylation of glycogen synthase by GSK3β leads to its inactivation [13]. Inhibition of GSK3β mediates the effect of insulin on glycogen synthesis [17]. Protein kinase B alpha (PKBα, named also Act1), induced by insulin, phosphorylates GSK3β at serine residue S9, thereby inactivating it [66, 67]. Interestingly, insulin has been reported to activate ENaC [4, 6, 7, 53, 71, 77]. The stimulatory effect of insulin on ENaC may be mediated partially by PKBα, because it was shown that this kinase increases ENaC abundance in the plasma membrane [43] and acutely activates ENaC in outside-out patches [21]. The latter effect depends on S621. Since we have shown that ENaC is inhibited by GSK3β, it is tempting to speculate that inhibition of GSK3β by PKBα contributes to ENaC activation by insulin. Moreover, activation of ENaC by other kinases known to inactivate GSK3β may be partially attributed to their inhibitory effects on this kinase. For instance, SGK1 [66] and the protein kinase A (PKA) [76] can inactivate GSK3β through phosphorylation at S9. Thus, SGK1 and PKA, induced by aldosterone [27, 34] and vasopressin [36, 79], respectively, may activate ENaC at least in part by inactivation of GSK3β. Taken together, GSK3β may be involved in ENaC regulation as a tonic inhibitory factor and/or may serve as an inhibitory feedback mechanism after ENaC activation by phosphorylation at S621.
In summary, our study indicates that phosphorylation of S621 by a proline-directed kinase, e.g., DYRK2, stimulates ENaC activity and primes S617 to be phosphorylated subsequently by GSK3β which limits the stimulatory effect of the initial phosphorylation and results in channel inhibition. Moreover, our findings provide proof of principle that DYRK2 and GSK3ß can activate and inhibit ENaC also in microdissected renal tubules, respectively. This supports the concept that the opposing effects of the two adjacent phosphorylation sites in the C-terminus of the channel’s α-subunit play a role in acute regulation of ENaC activity in native tissue.
Acknowledgements
We thank Prof. Philip Cohen for his inspiring comment that S621 may be a suitable phosphorylation site for DYRK2 and for making recombinant GSK3β and a selective GSK3β inhibitor (CHIR99021) available to us.
Author contribution
Al.D. and C.K. conceived the study; Al.D., V.N., and C.K. designed the experiments and interpreted the data; Al.D., V.N., and An.D. performed the experiments and analyzed the data; Al.D. and C.K. wrote the paper. All authors edited and approved the final version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), SFB 1350 (project number 387509280; subproject A4).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval
Housing and care of the animals and all procedures were in accordance with the principles of German legislation, with approval by the animal welfare officer for FAU, and under the governance of the state veterinary health inspectorate (for mice: TS-1/ 03 ZellPhys and TS-10/2022 ZellPhys; for Xenopus laevis: 621–2531.32–05/02 and 55.2–2532-2–527).
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this article was revised: Originally, the article was published with an error. The affiliation of the author Christoph Korbmacher should be " Institut für Zelluläre und Molekulare Physiologie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Waldstr, 6, 91054 Erlangen, Germany
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
6/9/2022
A Correction to this paper has been published: 10.1007/s00424-022-02717-4
References
- 1.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999;274:37834–37839. doi: 10.1074/jbc.274.53.37834. [DOI] [PubMed] [Google Scholar]
- 3.Baines D. Kinases as targets for ENaC regulation. Curr Mol Pharmacol. 2013;6:50–64. doi: 10.2174/18744672112059990028. [DOI] [PubMed] [Google Scholar]
- 4.Baxendale-Cox LM, Duncan RL. Insulin increases sodium Na+ channel density in A6 epithelia: implications for expression of hypertension. Biol Res Nurs. 1999;1:20–29. doi: 10.1177/109980049900100104. [DOI] [PubMed] [Google Scholar]
- 5.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 6.Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI 3-kinase activity. Am J Physiol Cell Physiol. 2003;284:C1645–1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- 7.Blazer-Yost BL, Vahle JC, Byars JM, Bacallao RL. Real-time three-dimensional imaging of lipid signal transduction: apical membrane insertion of epithelial Na+ channels. Am J Physiol Cell Physiol. 2004;287:C1569–1576. doi: 10.1152/ajpcell.00226.2004. [DOI] [PubMed] [Google Scholar]
- 8.Campbell LE, Proud CG. Differing substrate specificities of members of the DYRK family of arginine-directed protein kinases. FEBS Lett. 2002;510:31–36. doi: 10.1016/s0014-5793(01)03221-5. [DOI] [PubMed] [Google Scholar]
- 9.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Chou CL, Knepper MA. A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol. 2021 doi: 10.1681/ASN.2020101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Paunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A. 2017;114:E9989–E9998. doi: 10.1073/pnas.1710964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A. 1999;96:2514–2519. doi: 10.1073/pnas.96.5.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 14.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 15.Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Correa-Saez A, Jimenez-Izquierdo R, Garrido-Rodriguez M, Morrugares R, Munoz E, Calzado MA. Updating dual-specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2): molecular basis, functions and role in diseases. Cell Mol Life Sci. 2020;77:4747–4763. doi: 10.1007/s00018-020-03556-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross DA, Watt PW, Shaw M, van der Kaay J, Downes CP, Holder JC, Cohen P. Insulin activates protein kinase B, inhibits glycogen synthase kinase-3 and activates glycogen synthase by rapamycin-insensitive pathways in skeletal muscle and adipose tissue. FEBS Lett. 1997;406:211–215. doi: 10.1016/s0014-5793(97)00240-8. [DOI] [PubMed] [Google Scholar]
- 18.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diakov A, Bera K, Mokrushina M, Krueger B, Korbmacher C. Cleavage in the γ-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J Physiol. 2008;586:4587–4608. doi: 10.1113/jphysiol.2008.154435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C-terminus of the channel’s alpha-subunit. J Biol Chem. 2004;279:38134–38142. doi: 10.1074/jbc.M403260200. [DOI] [PubMed] [Google Scholar]
- 21.Diakov A, Nesterov V, Mokrushina M, Rauh R, Korbmacher C. Protein kinase B alpha (PKBα) stimulates the epithelial sodium channel (ENaC) heterologously expressed in Xenopus laevis oocytes by two distinct mechanisms. Cell Physiol Biochem. 2010;26:913–924. doi: 10.1159/000324000. [DOI] [PubMed] [Google Scholar]
- 22.Dinudom A, Fotia AB, Lefkowitz RJ, Young JA, Kumar S, Cook DI. The kinase Grk2 regulates Nedd4/Nedd4-2-dependent control of epithelial Na+ channels. Proc Natl Acad Sci U S A. 2004;101:11886–11890. doi: 10.1073/pnas.0402178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem. 1987;262:14042–14048. doi: 10.1016/S0021-9258(18)47901-X. [DOI] [PubMed] [Google Scholar]
- 25.Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol. 2005;16:2279–2287. doi: 10.1681/ASN.2004100828. [DOI] [PubMed] [Google Scholar]
- 26.Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger JD, Rossier BC. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16:878–891. doi: 10.1681/ASN.2004121110. [DOI] [PubMed] [Google Scholar]
- 27.Garty H. Regulation of the epithelial Na+ channel by aldosterone: open questions and emerging answers. Kidney Int. 2000;57:1270–1276. doi: 10.1046/j.1523-1755.2000.00961.x. [DOI] [PubMed] [Google Scholar]
- 28.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 29.Haerteis S, Krappitz M, Diakov A, Krappitz A, Rauh R, Korbmacher C. Plasmin and chymotrypsin have distinct preferences for channel activating cleavage sites in the gamma subunit of the human epithelial sodium channel. J Gen Physiol. 2012;140:375–389. doi: 10.1085/jgp.201110763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber R, Krueger B, Diakov A, Korbmacher J, Haerteis S, Einsiedel J, Gmeiner P, Azad AK, Cuppens H, Cassiman JJ, Korbmacher C, Rauh R. Functional characterization of a partial loss-of-function mutation of the epithelial sodium channel (ENaC) associated with atypical cystic fibrosis. Cell Physiol Biochem. 2010;25:145–158. doi: 10.1159/000272059. [DOI] [PubMed] [Google Scholar]
- 31.Ilyaskin AV, Kirsch SA, Bockmann RA, Sticht H, Korbmacher C, Haerteis S, Diakov A. The degenerin region of the human bile acid-sensitive ion channel (BASIC) is involved in channel inhibition by calcium and activation by bile acids. Pflugers Arch. 2018;470:1087–1102. doi: 10.1007/s00424-018-2142-z. [DOI] [PubMed] [Google Scholar]
- 32.Ilyaskin AV, Korbmacher C, Diakov A. Inhibition of the epithelial sodium channel (ENaC) by connexin 30 involves stimulation of clathrin-mediated endocytosis. J Biol Chem. 2021;296:100404. doi: 10.1016/j.jbc.2021.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kellenberger S, Gautschi I, Rossier BC, Schild L. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest. 1998;101:2741–2750. doi: 10.1172/JCI2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 35.Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol. 2020;318:C150–C162. doi: 10.1152/ajpcell.00418.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleyman TR, Ernst SA, Coupaye-Gerard B. Arginine vasopressin and forskolin regulate apical cell surface expression of epithelial Na+ channels in A6 cells. Am J Physiol. 1994;266:F506–511. doi: 10.1152/ajprenal.1994.266.3.F506. [DOI] [PubMed] [Google Scholar]
- 37.Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu Rev Physiol. 2018;80:263–281. doi: 10.1146/annurev-physiol-021317-121143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339(Pt 2):319–328. doi: 10.1042/bj3390319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344(Pt 1):189–197. doi: 10.1042/bj3440189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krueger B, Haerteis S, Yang L, Hartner A, Rauh R, Korbmacher C, Diakov A. Cholesterol depletion of the plasma membrane prevents activation of the epithelial sodium channel (ENaC) by SGK1. Cell Physiol Biochem. 2009;24:605–618. doi: 10.1159/000257516. [DOI] [PubMed] [Google Scholar]
- 41.Krueger B, Yang L, Korbmacher C, Rauh R. The phosphorylation site T613 in the beta-subunit of rat epithelial Na+ channel (ENaC) modulates channel inhibition by Nedd4-2. Pflugers Arch. 2018;470:649–660. doi: 10.1007/s00424-018-2115-2. [DOI] [PubMed] [Google Scholar]
- 42.Lang F, Pearce D. Regulation of the epithelial Na+ channel by the mTORC2/SGK1 pathway. Nephrol Dial Transplant. 2016;31:200–205. doi: 10.1093/ndt/gfv270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J Biol Chem. 2007;282:29866–29873. doi: 10.1074/jbc.M701923200. [DOI] [PubMed] [Google Scholar]
- 44.Lefevre CM, Diakov A, Haerteis S, Korbmacher C, Grunder S, Wiemuth D. Pharmacological and electrophysiological characterization of the human bile acid-sensitive ion channel (hBASIC) Pflugers Arch. 2014;466:253–263. doi: 10.1007/s00424-013-1310-4. [DOI] [PubMed] [Google Scholar]
- 45.Liang X, Butterworth MB, Peters KW, Frizzell RA. AS160 modulates aldosterone-stimulated epithelial sodium channel forward trafficking. Mol Biol Cell. 2010;21:2024–2033. doi: 10.1091/mbc.E10-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lier N, Gresko N, Di Chiara M, Loffing-Cueni D, Loffing J. Immunofluorescent localization of the Rab-GAP protein TBC1D4 (AS160) in mouse kidney. Histochem Cell Biol. 2012;138:101–112. doi: 10.1007/s00418-012-0944-1. [DOI] [PubMed] [Google Scholar]
- 47.Lochhead PA, Sibbet G, Kinstrie R, Cleghon T, Rylatt M, Morrison DK, Cleghon V. dDYRK2: a novel dual-specificity tyrosine-phosphorylation-regulated kinase in Drosophila. Biochem J. 2003;374:381–391. doi: 10.1042/BJ20030500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lochhead PA, Sibbet G, Morrice N, Cleghon V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell. 2005;121:925–936. doi: 10.1016/j.cell.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 49.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 50.Mansley MK, Korbmacher C, Bertog M. Inhibitors of the proteasome stimulate the epithelial sodium channel (ENaC) through SGK1 and mimic the effect of aldosterone. Pflugers Arch. 2018;470:295–304. doi: 10.1007/s00424-017-2060-5. [DOI] [PubMed] [Google Scholar]
- 51.Mansley MK, Neuhuber W, Korbmacher C, Bertog M. Norepinephrine stimulates the epithelial Na+ channel in cortical collecting duct cells via α2-adrenoceptors. Am J Physiol Renal Physiol. 2015;308:F450–458. doi: 10.1152/ajprenal.00548.2014. [DOI] [PubMed] [Google Scholar]
- 52.Mansley MK, Niklas C, Nacken R, Mandery K, Glaeser H, Fromm MF, Korbmacher C, Bertog M (2020)Prostaglandin E2 stimulates the epithelial sodium channel (ENaC) in cultured mouse cortical collecting duct cells in an autocrine manner.J Gen Physiol 15210.1085/jgp.201912525 [DOI] [PMC free article] [PubMed]
- 53.Marunaka Y, Hagiwara N, Tohda H. Insulin activates single amiloride-blockable Na channels in a distal nephron cell line (A6) Am J Physiol. 1992;263:F392–400. doi: 10.1152/ajprenal.1992.263.3.F392. [DOI] [PubMed] [Google Scholar]
- 54.Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Lang F, Wulff P, Kuhl D, Cohen P. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J. 2004;384:477–488. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nesterov V, Bertog M, Canonica J, Hummler E, Coleman R, Welling PA, Korbmacher C. Critical role of the mineralocorticoid receptor in aldosterone-dependent and aldosterone-independent regulation of ENaC in the distal nephron. Am J Physiol Renal Physiol. 2021;321:F257–F268. doi: 10.1152/ajprenal.00139.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nesterov V, Bertog M, Korbmacher C. High baseline ROMK activity in the mouse late distal convoluted and early connecting tubule probably contributes to aldosterone-independent K+ secretion. Am J Physiol Renal Physiol. 2022;322:F42–F54. doi: 10.1152/ajprenal.00252.2021. [DOI] [PubMed] [Google Scholar]
- 57.Nesterov V, Dahlmann A, Bertog M, Korbmacher C. Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am J Physiol Renal Physiol. 2008;295:F1052–1062. doi: 10.1152/ajprenal.00031.2008. [DOI] [PubMed] [Google Scholar]
- 58.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol. 2012;303:F1289–1299. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 59.Nesterov V, Krueger B, Bertog M, Dahlmann A, Palmisano R, Korbmacher C. In Liddle syndrome, epithelial sodium channel is hyperactive mainly in the early part of the aldosterone-sensitive distal nephron. Hypertension. 2016;67:1256–1262. doi: 10.1161/HYPERTENSIONAHA.115.07061. [DOI] [PubMed] [Google Scholar]
- 60.Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I (2018)Structure of the human epithelial sodium channel by cryo-electron microscopy.Elife 710.7554/eLife.39340 [DOI] [PMC free article] [PubMed]
- 61.Noreng S, Posert R, Bharadwaj A, Houser A, Baconguis I (2020)Molecular principles of assembly, activation, and inhibition in epithelial sodium channel.Elife 910.7554/eLife.59038 [DOI] [PMC free article] [PubMed]
- 62.Rauh R, Dinudom A, Fotia AB, Paulides M, Kumar S, Korbmacher C, Cook DI. Stimulation of the epithelial sodium channel (ENaC) by the serum- and glucocorticoid-inducible kinase (Sgk) involves the PY motifs of the channel but is independent of sodium feedback inhibition. Pflugers Arch. 2006;452:290–299. doi: 10.1007/s00424-005-0026-5. [DOI] [PubMed] [Google Scholar]
- 63.Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol. 2014;15:33–46. doi: 10.1016/j.coph.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 64.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 65.Rotin D, Staub O. Function and regulation of the epithelial Na+ channel ENaC. Compr Physiol. 2021;11:2017–2045. doi: 10.1002/cphy.c200012. [DOI] [PubMed] [Google Scholar]
- 66.Sakoda H, Gotoh Y, Katagiri H, Kurokawa M, Ono H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Fukushima Y, Abe M, Shojima N, Kikuchi M, Oka Y, Hirai H, Asano T. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J Biol Chem. 2003;278:25802–25807. doi: 10.1074/jbc.M301127200. [DOI] [PubMed] [Google Scholar]
- 67.Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416:307–311. doi: 10.1016/s0014-5793(97)01235-0. [DOI] [PubMed] [Google Scholar]
- 68.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, Garty H. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem. 2002;277:13539–13547. doi: 10.1074/jbc.M111717200. [DOI] [PubMed] [Google Scholar]
- 69.Skurat AV, Dietrich AD. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem. 2004;279:2490–2498. doi: 10.1074/jbc.M301769200. [DOI] [PubMed] [Google Scholar]
- 70.Soundararajan M, Roos AK, Savitsky P, Filippakopoulos P, Kettenbach AN, Olsen JV, Gerber SA, Eswaran J, Knapp S, Elkins JM. Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure. 2013;21:986–996. doi: 10.1016/j.str.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol. 2007;18:1652–1661. doi: 10.1681/ASN.2007010020. [DOI] [PubMed] [Google Scholar]
- 72.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996;15:2371–2380. doi: 10.1002/j.1460-2075.1996.tb00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taira N, Mimoto R, Kurata M, Yamaguchi T, Kitagawa M, Miki Y, Yoshida K. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J Clin Invest. 2012;122:859–872. doi: 10.1172/JCI60818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tandon V, de la Vega L, Banerjee S. Emerging roles of DYRK2 in cancer. J Biol Chem. 2021;296:100233. doi: 10.1074/jbc.REV120.015217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanji C, Yamamoto H, Yorioka N, Kohno N, Kikuchi K, Kikuchi A. A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3beta (GSK3β) and mediates protein kinase A-dependent inhibition of GSK-3beta. J Biol Chem. 2002;277:36955–36961. doi: 10.1074/jbc.M206210200. [DOI] [PubMed] [Google Scholar]
- 77.Tiwari S, Nordquist L, Halagappa VK, Ecelbarger CA. Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am J Physiol Renal Physiol. 2007;293:F178–185. doi: 10.1152/ajprenal.00447.2006. [DOI] [PubMed] [Google Scholar]
- 78.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F (2015) Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi:10.1126/science.1260419 [DOI] [PubMed]
- 79.Verrey F. Antidiuretic hormone action in A6 cells: effect on apical Cl- and Na+ conductances and synergism with aldosterone for NaCl reabsorption. J Membr Biol. 1994;138:65–76. doi: 10.1007/BF00211070. [DOI] [PubMed] [Google Scholar]
- 80.Volk T, Konstas AA, Bassalay P, Ehmke H, Korbmacher C. Extracellular Na+ removal attenuates rundown of the epithelial Na+-channel (ENaC) by reducing the rate of channel retrieval. Pflugers Arch. 2004;447:884–894. doi: 10.1007/s00424-003-1193-x. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y, Sun J, Wei X, Luan L, Zeng X, Wang C, Zhao W. Decrease of miR-622 expression suppresses migration and invasion by targeting regulation of DYRK2 in colorectal cancer cells. Onco Targets Ther. 2017;10:1091–1100. doi: 10.2147/OTT.S125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wesch D, Miranda P, Afonso-Oramas D, Althaus M, Castro-Hernandez J, Dominguez J, Morty RE, Clauss W, Gonzalez-Hernandez T, Alvarez de la Rosa D, Giraldez T. The neuronal-specific SGK1.1 kinase regulates δ-epithelial Na+ channel independently of PY motifs and couples it to phospholipase C signaling. Am J Physiol Cell Physiol. 2010;299:C779–790. doi: 10.1152/ajpcell.00184.2010. [DOI] [PubMed] [Google Scholar]
- 83.Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–615. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshida S, Yoshida K. Multiple functions of DYRK2 in cancer and tissue development. FEBS Lett. 2019;593:2953–2965. doi: 10.1002/1873-3468.13601. [DOI] [PubMed] [Google Scholar]
- 85.Zhu G, Fujii K, Belkina N, Liu Y, James M, Herrero J, Shaw S. Exceptional disfavor for proline at the P + 1 position among AGC and CAMK kinases establishes reciprocal specificity between them and the proline-directed kinases. J Biol Chem. 2005;280:10743–10748. doi: 10.1074/jbc.M413159200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.