

Summary
Neuroinflammation exacerbates the progression of SOD1-driven amyotrophic lateral sclerosis (ALS), although the underlying mechanisms remain largely unknown. Herein, we demonstrate that misfolded SOD1 (SOD1Mut)-causing ALS results in mitochondrial damage, thus triggering the release of mtDNA and an RNA:DNA hybrid into the cytosol in an mPTP-independent manner to activate IRF3- and IFNAR-dependent type I interferon (IFN-I) and interferon-stimulating genes. The neuronal hyper-IFN-I and pro-inflammatory responses triggered in ALS-SOD1Mut were sufficiently robust to cause a strong physiological outcome in vitro and in vivo. cGAS/DDX41-STING-signaling is amplified in bystander cells through inter-neuronal gap junctions. Our results highlight the importance of a common DNA-sensing pathway between SOD1 and TDP-43 in influencing the progression of ALS.
Subject areas: Pathophysiology, Neuroscience, Immunology
Graphical abstract
Highlights
-
•
Constitutive basal activation of IFN-I was found in the SOD1-ALS animal model
-
•
SOD1-ALS damaged mitochondria to release mtDNA and RNA:DNA to activate the STING-pathway
-
•
Blocking cGAS and STING diminishes neurodegeneration in vivo in the SOD1-ALS model
-
•
Connexin and pannexin channels are required to propagate neuroinflammation in SOD1-ALS
Pathophysiology; Neuroscience; Immunology
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating, rapidly progressing paralytic neurodegenerative disease (ND) characterized by severe upper and lower motor neuronal (MN) loss in the brainstem and spinal cord of humans. ALS has a prevalence of 5.4 per 100,000 individuals (Chiò et al., 2013). In certain regions, especially Guam, the Kii Peninsula in Japan, and Guinea, the prevalence is estimated to be ∼50–100 times higher than the rest of the world (Pablo et al., 2009). Typically, older adults and men represent a high-risk group for being diagnosed with ALS (Wijesekera and Leigh, 2009). Patients will succumb to the disease within 2–5 years of clinical onset because of the development of severe muscular atrophy and paralysis. The hallmark of the disease is the loss of muscle control that occurs when signal transduction between the brain and muscles lacks coordination leading to eventual loss of voluntary movements, muscle fasciculation, and skeletal muscle atrophy. Subsequently, paralysis of voluntary muscles occurs slowly culminating in death owing to respiratory failure. Unlike for other diseases, there is a paucity of universal consensus on the parameters that trigger the onset of ALS in humans or in preclinical investigations employing ALS mice models. Furthermore, specific stimuli always lead to the same endpoint in this disease. Several genes and pathophysiological pathways play a paramount role in ALS disease pathophysiology. At the same time, there is an ongoing debate about whether the pathological, clinical, and phenotypic heterogeneity observed in ALS should be treated as a single entity or if it requires categorization based on specific molecular signals that are only partially responsible because of their complexity (Turner and Swash, 2015). ALS has become a multisystemic condition with diverse clinical manifestations and multiple etiologies responsible for its pathogenesis. Conversely, specific etiological factors such as viral infections, autoimmune responses, oxidative stress, glutamate excitotoxicity, defective axonal transport, glial cell impairment, impaired DNA and RNA metabolism, metabolic impairment, heavy metal toxicity, apoptosis, proteinopathies, and proteome homeostasis have been implicated in the degenerative processes (Hardiman et al., 2017; Saberi et al., 2015; Taylor et al., 2016). Available evidence suggests that ALS is a multi-step disease with different phases that vary across patient groups, although in individuals with underlying genetic abnormalities the number of stages in the disease course are fewer than those without (Chiò et al., 2018; Vucic et al., 2019).
In general, over 95% of all ALS patients are classified as sporadic, defined as having no known familial history of the disease. The remaining 5% of the patients are classified as familial and typically appear to have inherited the trait in an autosomal dominant fashion. Mutations in the SOD1 gene, encoding superoxide dismutase 1, were the first to be identified as genetic factors associated with aggressive ALS progression (Bruijn et al., 1997, 1998; Rosen et al., 1993). SOD1 mutations represent the second most common genetic cause of ALS to date, with approximately 20% of familial cases, and with more than 100 different reported gene mutations (Chen et al., 2013; Mejzini et al., 2019). Elegant studies using transgenic rodent models carrying mutant forms of SOD1 have shown that non-neuronal cells also play a crucial role in the development of ALS, contributing to MN death by non-cell-autonomous mechanisms. Similarly, in in vitro studies murine SOD1 mutant astrocytes and microglia, as well as human astrocytes from sporadic and familial ALS patients can induce MN death. This suggests that ALS is a deadly convergence of insults developed across multiple cell types and stages that culminated in neuromuscular failure. However, the precise mechanisms through which each neuronal cell type contributes to ALS disease progression remains ambiguous.
One of the most striking hallmarks of misfolded SOD1 (SOD1 mutants, SOD1Mut)-driven ALS shared by familial and sporadic patients, as well as by rodent models, is neuroinflammation, characterized by microglia and astrocyte activation, T lymphocyte infiltration, and excessive production of pro-inflammatory cytokines and chemokines (Endo et al., 2016; Ringer et al., 2017; Yoshihara et al., 2002). These phenomena have been demonstrated in association with MN degeneration in both animal and human tissues, even during the pre-symptomatic phase of SOD1Mut-ALS. Accumulating evidence from preclinical work has suggested that immune cells either exert deleterious or protective effects on MN survival depending on the state of activation and disease stage; however, the underlying mechanisms are far from being fully elucidated.
Microglia are the most prominent immune cells within the central nervous system (CNS) and represent a specialized subset of macrophages accounting for ∼10–15% of the cells found in the brain. These cells survey the surrounding environment and respond to infections, abnormal or misfolded proteins, antigen complexes, and “danger signals”—more commonly known as alarmins or “danger-associated molecular patterns” produced by apoptotic tissues within the CNS. In response to these stimuli, “resting” microglia will become “activated” and produce reactive oxygen species, cytokines, and other toxic molecules, leading to the acceleration of neuronal dysfunction and death (Hanisch and Kettenmann, 2007; Liao et al., 2012). Previous studies have shown that activated microglia are widely present in the CNS of ALS patients, with a significant correlation between the intensity of microglia activation in the brain cortex region and the severity of clinico-neurological impairment (Turner et al., 2004). Studies using SOD1Mut transgenic mice further revealed that the replacement of SOD1Mut microglia with wild-type (WT) microglia through transplantation leads to neuroprotection by delaying neuronal cell death and improving the median lifespan of neurons (Beers et al., 2006; Boillée et al., 2006). Moreover, neutralizing microglia-derived superoxide also significantly increases the lifespan of SOD1Mut mice (Harraz et al., 2008). SOD1Mut astrocytes, a sub-type of glial cells, also exhibit increased expression of many pro-inflammatory and chemokine genes, accelerating MN death through necroptosis by enhancing microglia activation, supporting the likely role of astrocytes in regulating microglia activation (Yamanaka et al., 2008). Similarly, transplantation of WT to replace SOD1Mut astrocytes appears to attenuate astrocyte-mediated toxicity and neuronal loss (Papadeas et al., 2011). Hence, the role of astrocytes should not be undervalued. Despite all these efforts, genetic approaches to eliminate specific inflammatory genes in ALS murine models have largely remained unsuccessful, highlighting the complexity of neuroinflammation in ALS. For these reasons, a successful therapeutic approach is likely to derive from a better understanding of the intricate molecular mechanisms driving the inflammatory response throughout the course of the disease.
Currently, riluzole and edaravone are the only approved drugs available to treat ALS, even though the drugs only confer modest benefits to patients. Because the mechanism of neuroinflammation in SOD1-causing ALS remains unclear, we sought to revisit this topic and investigate the underlying rationale of different neuronal and non-neuronal cells that mediate MN death in ALS, with a primary focus on microglia. In the present study, we investigated two extensively characterized SOD1 mutations, i.e., G93A and G85R; the former inducing impaired in zinc-binding loops and associated with a more aggressive disease progression; whereas the latter is characterized by a normal zinc-binding loop, but greater susceptibility to disulfide and metal chelating agents, associated with a slower disease progression in mice (Tiwari et al., 2005; Yamashita and Ando, 2015). Here, we demonstrated that SOD1Mut leads to constitutive basal activation of type I interferon (IFN-I) and interferon-stimulating genes (ISGs), which in turn amplify other pro-inflammatory signaling cascades to promote neuroinflammation and neuronal death. These processes are achieved by damaging mitochondria and the release of mitochondria DNA (mtDNA) and mt(RNA:DNA) hybrid structures into the cytoplasm, to activate cGAS/DDX41-STING pathway for IFN-I induction. Furthermore, we found that neuronal connexin 36 and pannexin 1 were critical for extending the immune responses to bystander cells. Together, these data indicate that activation of the cGAS/DDX41-STING pathway in microglia is a novel molecular mechanism underlying MN death in SOD1-driven ALS. Moreover, our findings also identified a new therapeutic regimen suggesting a strategy for early intervention against the rapid progression of ALS and possibly other NDs in which stimulator of interferon genes (STING) activation could also play a likely role.
Results
Prevalence of IFN-I, chemokine, and pro-inflammatory cytokine activation in ALS-SOD1 models
To better characterize the neuroinflammatory state in ALS-SOD1-relevant models, we pursued an unbiased approach to identify a transcriptomic signature associated with SOD1-mutations. We first isolated the whole brain and performed genome-wide transcriptomic profiling on tissue samples obtained during the disease progression in a murine model that carried the G85R mutation. In the absence of any treatment, the unsupervised transcriptomic analysis revealed that, of the 770 annotated Refseq genes, ∼73 genes exhibited markedly (mean FC≥2-fold) increased expression, and ∼697 genes remained unchanged in SOD1-G85R whole brain samples when compared to the WT counterpart (Figure 1A). We subsequently conducted a systematic unbiased analysis to characterize the functional protein-protein correlation network. To this end, we performed gene ontology annotation of the modules with the STRING package with the addition of terms from the Kyoto Encyclopedia of Genes and Genomes database and the Geneset Enrichment Analysis databases evaluated using the Fisher exact test (Benjamini–Hochberg false discovery rate [FDR] < 0.05). Using this high-confidence network as baseline, we identified significantly enriched in genes encoding products associated with Toll-like receptors, NF-κB-associated signaling, and host defense responses, which showed the highest enrichment among the upregulated genes (Figures 1B and S1A), with ∼22% (16 of 73) genes corresponding to ISGs, including Irf1, Irf7, Rsad2, Ifih1, Ddx58, Gbp2, Oas1, Stat1, and Ifits. Further, we also detected significantly elevated expression of the pro-inflammatory cytokine gene, Tnf, as well as a panel of well-known chemokines genes, with some of them, also specifically induced by IFN-I, including Ccl2, Ccl5, Ccl7, Cxcl9, and Cxcl10 (Figure 1C). Furthermore, we also observed similar results after re-performing an unbiased analysis using the public transcriptome sequencing dataset of microglia isolated from another well-known ALS murine model SOD1-G93A (Krasemann et al., 2017).
Figure 1.

Basal activation of pro-inflammatory cytokines, chemokines, and IFN-I response in the SOD1-G85R in vivo model
(A) Whole brain tissues were isolated from WT and G85R mice at day-10 after birth. Purified whole RNA was subjected to NanoString multiplex gene analysis using the murine neuroinflammation panel that consists of 770 genes. Heat-map representation of differentially expressed genes with upregulated related genes (fold-change >2.0 as the cut-off threshold) by G85R expressed as fold changes relative to WT controls.
(B) The upregulated genes from (A) were subjected to unbiased protein network and enrichment analysis via the STRING-CytoScape.
(C) Representative heatmap of upregulated immune mediator profile (fold-change ≥2.0) in G85R mice after clustering into their respective families.
(D) Spinal cord from WT or G85R mouse models were isolated and dissected into three sections as depicted (left). RNA from each section were harvested and subjected to analysis of the indicated mRNA (right).
(E) Von Kossa and immunohistochemistry staining of brain tissue from WT or G85R mice with the indicated antibodies. Quantification of staining was performed via optical density determination.
(F) Kaplan-Meier survival plots of age-matched G93A mice challenged either with vehicle or MCP-1 neutralizing antibody (500 μg/week). Number of mice that survived at the endpoint are indicated.
(G) Primary cells were isolated from the brain of WT or G85R mice at day-10 after birth through immunopanning procedures. RNA was purified and subjected to RT-qPCR analysis.
(H) Newborn WT or G85R mice were intracranially injected with vehicle (DMEM), 500, 10,000, or 50,000 plaque-forming units (pfus) of CVB3 at day-8. The brain and the entire blood was harvested at two time-points (Day 10, and Week 60), respectively. Vp1 and 2A mRNA were quantified via RT-qPCR.
(I) C20 cells were first pre-treated with serum harvested from WT or G85R mice, followed by infection with CVB3 (MOI = 1.0). Vp1, Rsad2, and Isg15 mRNA were measured via RT-qPCR.
(J) Serum collected from endpoint WT or G85R mice (week-60) were subjected to ELISA analysis for IFN-β protein. All RT-qPCR assays are normalized to β-actin. Data represent the mean of at least three independent experiments with n = 3 biological replicates (mean ± SEM). Number of dots = number of mice/biological replicates. ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (D, E, and G–J: unpaired Student’s t test; F: Log rank Mantel-Cox test). A.U. = arbitrary units. See also Figure S1.
Next, we sought to determine whether the hyper-expression of inflammation-associated genes can be recapitulated in other organs in vivo. We isolated the spinal cord, a region crucial for the transmission of nerve signals from the motor cortex to the whole body, and further dissected it into three sections: namely cervical spine (CS), thoracic spine (TS), and lumbar spine (LS). We then examined the basal mRNA expression of Ifnb1, Tnf, Il6, Il1b, and Cxcl10 for each section by RT-qPCR, and found a significantly increased expression of all genes (Figures 1D and S1B). We also purified the RNA extracted from the spleen, liver, and lungs of both WT and SOD1-G85R mice and measured the gene expression. Our analysis identified elevated expression of the genes in all examined organs, including Ccl5, and Rsad2, a putative chemokine and ISGs (Figures S1C–S1E). Von Kossa staining showed a marked increase of calcium deposition within the brain, which is a sign of persistent inflammation. Immunohistochemical examination further revealed a significant increase in signal intensity of GFAP by ∼4-fold (p< 0.01), infiltration of immune cells in the brain tissues of SOD1-G85R, with a modest but statistically significant number of microglia (Iba1+), NK cells (NK1.1+), T-lymphocytes (CD8+), and B-lymphocytes (CD19+) (Figure 1E), indicating that neuronal astrogliosis and microgliosis are increased in G85R mice compared to age-matched controls. Chemokines genes were shown to play crucial role in the development of several neuroinflammatory diseases, including but not limited to tau pathology, and brain abscesses (Kielian et al., 2001; Semple et al., 2010). Overall, CCL/CXCL-family genes accounted for the largest mean fold-change in our NanoString profiling study; thus, we sought to investigate whether the constitutive activation of basal chemokines could contribute to SOD1-related pathologies. SOD1-G93A mice, another well-characterized transgenic rodent of ALS (Van Den Bosch, 2011),] received either vehicle, or anti-MCP-1 neutralizing antibody to abrogate the function of the critical chemoattractant molecule: MCP-1 (encoded by the Ccl2 gene). By comparing the viability of the anti-MCP-1 with vehicle-treated mutant mice, the abrogated functions of MCP-1 offered significant protection and prolonged survival (Figure 1F). Collectively, our results indicated that the IFN-I pathway is activated concurrently with the ongoing neuroinflammation to promote the pathogenesis in the ALS-SOD1-mutant model.
IFN-I plays a crucial role in host immune responses as a pleiotropic modulator critical for determining the cellular susceptibility to viral infections, cellular growth, and immune cells activation. In the quiescent state, IFN-I is constitutively produced at low levels and exerts profound physiological effects on homeostasis through coordinated tonic signaling. Accumulating evidence suggests that exaggerated production of IFN-I could have a profound negative impact on the host (Stetson et al., 2008; Stetson and Medzhitov, 2006). Furthermore, Ccl2 has previously been reported as an interferon-α/β Receptor (IFNAR)-dependent gene. Limited Ccl2 expression, and thus, reduced production of MCP-1 protein was observed in IFNAR-deficient macrophages and tissue homogenates in vivo as measured using enzyme-linked immunosorbent assay (ELISA) (Lehmann et al., 2016; Palomino-Segura et al., 2019). These led us to hypothesize that IFN-I mis-regulation is the key to promoting the neuroinflammatory phenotype in the ALS-SOD1-G85R model. To test hypothesis, we isolated and cultured several key primary neuronal and non-neuronal cells from SOD1-G85R mice through immunopanning procedures. The identity of each respective cell type was verified via the mRNA expression of well-known markers (Figure S2). As shown in Figure 1G, both Ifnb1, and Ifna4 mRNA expression were markedly induced at baseline in all tested primary cells, including murine microglia, motor neurons, oligodendrocytes, and astrocytes. Comparable results were obtained for other chemokines and pro-inflammatory genes, including Cxcl10, Tnf, and Il6 (Figure S1F). Interestingly, we also detected a significant upregulation of microglia-specific ISGs, including Axls, B2m, and Aif1 in SOD1-G85R primary microglia (Figure S1G), which defined the microglial neurodegenerative phenotype (MGnD) gene signature. These observations prompted us to confirm the presence of IFN-I within the animal bloodstream using two bioassays. First, we assayed viral replication in vivo by infecting mice with coxsackievirus-B3 (CVB3), a neurotropic RNA virus via bilateral intracerebroventricular stereotaxic injection. The results indicated that there was a substantially decreased level of CVB3 replication (as indicated by Vp1 and 2A mRNA) within the whole brain and blood from day 10 and at week 60 when SOD1-G85R mice were compared to WT mice (Figure 1H). Second, we assayed viral replication and gene expression when serum-treated cells were infected with CVB3. We observed that SIM-A9, a murine brain-resident macrophage pre-treated with SOD1-G85R serum, displayed remarkably increased resistance to CVB3 infection. RT-qPCR analysis also indicated that there was a significant decrease in CVB3 replication (Vp1), followed by a substantial increase in the expression of ISGs (Rsad2 and Isg15) in human microglia (C20) that were pre-conditioned with serum from SOD1-G85R but not from serum from WT animals (Figure 1I). Quantitative ELISA further confirmed that the basal proteins levels of IFN-β were significantly elevated in the blood serum of SOD1-G85R-mutated model animals (Figure 1J). Aberrant expression of IFN-I is the cause of several autoimmune disorders (Crowl et al., 2017; Lee-Kirsch, 2017). In our assays, both ALS-SOD1Mut-mice models further displayed a significant increase in total serum immunoglobulin or in anti-nuclear antibody levels, two diagnostic markers of autoimmune diseases (Figure S1H). Taken together, these results indicate that the ALS-SOD1Mut-in vivo model produces substantially elevated levels of biologically active IFN-I, which may contribute to the development of an autoimmune-like phenotype in ALS.
Cell-type specific involvement in ALS-SOD1Mut-mediated IFN-I response
The basal gene expression in the in vivo model (Figure 1) is primed and subject to numerous feedforward and feedback loops; thus, we sought to address the potential cell-type specificity of hyper-responsive IFN-I associated gene expression in SOD1-driven ALS pathogenesis. We analyzed the activation of the IFN-I signaling pathway and its associated changes in gene expression upon SOD1Mut expression in various cell types. Both HeLa and HEK293T cells, the two most widely used human somatic cell lines in biological research, were co-transfected with either (1) p125-luc, a reporter plasmid containing the full-length IFN-β promoter; (2) p55C1B-luc, a reporter plasmid containing repetitive activated IRF3-binding sites of the IFN-β gene promoter; or (3) p55A2-luc, an NF-κB driven reporter plasmid comprising three repeats of the PRDII domain of the IFN-β promoter, and each of the SOD1Mut plasmids. Approximately 36 h later, the cells were assayed for luciferase activity. The results indicated that the relative luciferase activity from all three reporter plasmids in both SOD1Mut-transfected cell lysates were robustly induced (≥2-fold) as compared to both empty vector (EV) and SOD1-WT cells (Figure 2A). Further analysis indicated that the SOD1Mut activated all three promoter activities in a dose-dependent manner (Figure S3A). We also found that both SOD1Mut upregulated the expression of Ifnb1 mRNA at 12 h post-transfection, with its expression remaining persistently elevated and continued to peak and plateau over a 24-h period but started to decrease after 48 h (Figure S3B). Strikingly, none of the above observations were reproducible in HEK293T cells (Figure S3C).
Figure 2.

Overexpression of ALS SOD1-mutants activate IFN-I response in vitro and confer resistance to viral infection
(A and B) HeLa WT or murine RAW264.7 cells expressing indicated reporter were co-transfected with plasmids encoding EGFP-, human SOD1-WT,-G85R, or-G93A mutants. 36 h after transfection, cell lysates were collected and analyzed for luciferase activity.
(C) THP-1-derived MΦ cells were transfected with 0.01 μg of indicated plasmids for 36 h, indicated mRNA levels were assessed via RT-qPCR.
(D) Similar conditions in panel (C) were performed in an indicated neuronal cell lineage (H: Human; M: Murine).
(E) C20 cells were transfected with indicated plasmids, followed by CVB3 (MOI = 1.0) infection at indicated time-points. CVB3 Vp1 mRNA was measured via RT-qPCR.
(F) Supernatant from panel E were collected and subjected to the plaque assay to measure the release of viral particles.
(G) Newly seeded C20 cells were pre-treated with supernatant collected from C20 cells expressing the indicated plasmids for 24 h. Cells were then infected with HCoV-OC43 (MOI = 1.0) for 72 h. In a parallel experiment, C20 cells were treated with 100 ng/mL of IFN-β proteins for 24 h as positive control. HCoV-OC43 nucleocapsid RNA, Isg15, and Rsad2 mRNAs were measured using RT-qPCR.
(H and I) C20 cells transfected with the indicated plasmid were infected with the indicated MOI of CVB3 and assessed 24 h later for cell viability with crystal violet staining and the LDH released assay.
(J) NanoString analysis for gene expression of indicated family of transcription factors comparing whole brain of WT and SOD1-G85R mice (n = 3 for each genotype).
(K) Immunoblot analysis of phosphorylated or total TBK1 and IRF3 or β-actin in C20 cells after transfection with the indicated plasmids.
(L) (1) TBK1, or IRF3-deficient C20 cells using CRISPR/Cas9 system expressing the indicated plasmids for 36 h. Ifnb1 and Rsad2 mRNA measured via RT-qPCR. Transfected poly(I:C) [p(I:C), 0.1 μg] served as positive control. (2) Knockout efficiency of the positive clones was confirmed by western blotting. Clone#1 of two lineage were selected for subsequent studies. Data represent the mean of three independent experiments with n = 3 biological replicates (mean ± SEM). ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (A–I, and L: two-tailed, unpaired Student’s t test). See also Figure S3.
Next, we pursued the same question in a more physiologically relevant cellular context. The brain contains two sets of monocyte-macrophage cells—blood-borne monocytes-derived macrophages (BBMΦ), and brain-resident macrophages (microglia). BBMΦ, primarily located within the perivascular spaces, the leptomeninges, and the choroid plexus, serve as the main antigen-presenting cells of the CNS and have recently emerged as an essential component of the disease-associated microenvironment in the brain and have been described as playing critical roles in the progression of ND. We tested whether the observation-mentioned for HeLa cells could be recapitulated in BBMΦ by co-transfecting each reporter with indicated SOD1Mut constructs into RAW264.7, a murine BBMΦ-lineage cell line. As shown in Figure 2B, all three reporters were robustly activated in RAW264.7 expressing either of the SOD1Mut proteins. Moreover, ectopic expression of G85R, G93A, MAVS, or IRF3 5D (a constitutively active phosphomimetic IRF3 mutant) also resulted in robust induction of Ifnb1 mRNA not only in phorbol-12-myristate-13-acetate (PMA)-differentiated THP-1 macrophages (a human monocytic leukemia cell line) (Figure 2C), but also in several other popular in vitro neuronal cell lines. These include SIM-A9 and C20 (murine and human microglia) (Garcia-Mesa et al., 2017), M03.13 (human oligodendrocytes) (McLaurin et al., 1995), HASTR/ci35 (human astrocytes) (Furihata et al., 2016; Kitamura et al., 2018), Neuro2a and SH-SY5Y (murine and human neuroblastoma), and NSC-34 (murine motor neuron-like cells) (Figures 2D and S3D). These results collectively indicate that ALS-SOD1Mut could robustly activate the IFN-I pathway in HeLa cells and in most of the key neuronal populations within CNS in a cell-type-specific manner.
To evaluate whether the hyper-production of cytokines by ALS-SOD1Mut is mutation specific, we further characterized seven other ALS-associated SOD1 mutations (i.e., V190G, A4V, G37R, H46R, V148I, E100G, and G127X) in both HEK293T cells and microglia. The results indicated that exogenous expression of all seven SOD1Mut also robustly activated the expression of Ifnb1, Tnf, Il6, and Ccl5 in microglia as compared to both EV and WT cells. No induction in gene expression was observed when using the HEK293T cell line as the model, even when they were transfected with dosage-increasing levels of SOD1Mut (Figures S3E–S3G). These results further confirmed that the observed neuronal hyper-inflammatory response is a highly conserved feature across most, if not all ALS-associated SOD1Mut.
Albeit statistically significant, the basal induction of IFN-I by SOD1Mut was small (<10-fold), we questioned whether these observations could truly induce any physiological consequences to the host. To evaluate this, microglia were infected with CVB3, and the intracellular viral replication was measured through RT-qPCR. Ectopic expression of indicated SOD1Mut significantly suppressed CVB3 replication across all three post-infection time-points (Figure 2E). Quantification of virus particles in the supernatant collected at the 16 h post-infection time-point from Figure 2E also produced similar outcomes (Figure 2F). To demonstrate that the viral resistance in SOD1Mut expressing cells was due to IFN-I, supernatants from microglia expressing EV, WT, or SOD1Mut were transferred to cultures of newly seeded microglia. After 24 h, the supernatant was removed and cells were infected with human beta-coronavirus strain OC43 (HCoV-OC43), another neurotropic RNA virus that is associated with meningitis and acute disseminated encephalomyelitis (Cheng et al., 2020; Jacomy et al., 2006; Kasereka and Hawkes, 2021; Morfopoulou et al., 2016; Nilsson et al., 2020). The conditioned media from SOD1Mut but not EV and WT expressing cells strongly induced the expression of ISGs, and conferred resistance to HCoV-OC43 infection (Figure 2G). Our results demonstrated that the ISG-inducing capacity and the resistance to HCoV-OC43 are mediated by the elevated production of biologically active IFN-I proteins in the supernatant of SOD1Mut expressing cells. To further substantiate this, EV, WT, or specific SOD1Mut microglia were infected with CVB3 at various multiplicities of infection (MOIs). By evaluating cell death [percentage of lactate dehydrohenase (LDH) released and crystal violet staining), we observed that SOD1Mut overexpressing cells significantly reduced the susceptibility of cells to CVB3 infection in all MOIs tested (Figures 2H and 2I). Taken together, these results demonstrate that neuronal hyper-IFN-I and pro-inflammatory responses triggered by ALS-SOD1Mut are sufficiently robust to cause strong physiological implications, for instance, broad-spectrum resistance to infection by RNA viruses in vitro.
The transcription of IFN-I and pro-inflammatory genes depends on kinase TBK1 and converges on latent transcription factors (TF) such as NF-κB, IRF3, and AP-1 subunits. We, therefore, analyzed the expression and activation of these components upon SOD1Mut expression to determine the mechanistic impact on the pathway. Consistent with previous data, we found only the expression of Irf1 and Irf7, both ISGs, were highly upregulated, whereas the expression of other IRFs, NF-κB, and AP1 family members remained comparable in the SOD1-G85R brain compared to WT brain samples (Figure 2J). Furthermore, the ectopic expression of SOD1Mut in microglia resulted in phosphorylation of TBK1 and IRF3 (Figure 2K). Transcriptional induction of Ifnb1 and Rsad2 by SOD1Mut was completely abrogated in both TBK1 and IRF3 CRISPR-Cas9-targeted microglia (Figure 2L). In summary, these results suggest that, although, SOD1Mut does not target the expression of TF, the activation of the IFN-I pathway promotes neuroinflammation in ALS.
ALS-SOD1Mut triggered IFN-I responses through cGAS-STING pathway
Because the induction of IFN-I-associated genes by SODMut are blocked in TBK1-IRF3-deficient cells, we speculated that ALS-SOD1Mut interfered directly or acted upstream of the TBK1-IRF3-axis. During pathogen recognition receptor signaling, coordination of upstream sensors with their respective adaptor molecules is critical for activating the IRF3-NF-κB-AP1 enhanceosome complex to initiate the IFN-I response. Therefore, we next examined whether these components were necessary for engaging SOD1Mut-dependent IFN-I induction by generating a series of CRISPR-Cas9 knockout microglia (Figure 3A). As shown in Figure 3B, SOD1Mut expression in microglia-deficient in RIG-I (encoded by Ddx58), MDA5 (encoded by Ifih1), and MAVS continued to produce comparable levels of Ifnb1 mRNA. This observation was not cell-type specific, as it was also confirmed using MAVS-deficient human astrocytes, HASTR/ci35 cells (Figure 3C). TIR-domain-containing adapter-inducing IFN-β (TRIF, encoded by Ticam-1)-deficient microglia also fully responded to SOD1Mut expression (Figure 3D). These results indicate that ALS-SOD1Mut is able activate the IFN-I response independently of both RIG-I-like receptors (RLR) and Toll-like receptors pathways.
Figure 3.

Activation of IFN-I response by ALS SOD1-mutants is dependent on the cGAS-STING signaling pathway
(A) Knockout efficiency of Ddx58 (RIG-I), Ifih1 (MDA5), Mavs, or Ticam-1 (TRIF) gene in C20 cells examined via western blotting.
(B) WT, DDX58, IFIH1, or MAVS-CRISPR/Cas9-deficient C20 cells either transfected with 0.01 μg Flag-SOD1 WT, G85R, G93A, or MAVS plasmids for 36 h, or 0.5 μg poly(I:C) for 8 h. Ifnb1 mRNA measured using RT-qPCR.
(C) Similar conditions as in panel B performed using WT or MAVS-CRISPR/Cas9-deficient HASTR/ci35 cells, except human MAVS plasmid was replaced with human IRF3 5D plasmid. Ifnb1 mRNA assessed via RT-qPCR.
(D–F) WT, TICAM, cGAS or STING-CRISPR/Cas9-deficient C20 cells either transfected with 0.01 μg Flag-SOD1 WT, G85R, G93A, or MAVS plasmids for 36 h, treated with LPS (100 ng/mL) for 4 h or transfected with HT-DNA (5.0 μg/mL) for 8 h. Ifnb1 mRNA measured via RT-qPCR. The knockout efficiency of cGAS, or STING assessed via western blot and functional assays.
(G) WT C20 cells either treated with RU.521 (5.0 μg/mL) or H.151 (0.5 μg/mL) for 24 h. Cells were then transfected with 0.01 μg of Flag-SOD1 WT, G85R, G93A, or MAVS plasmids, or HT-DNA (1.0 μg/mL). Ifnb1 mRNA was measured via RT-qPCR.
(H) Schematic diagram of Flag-tagged cGAS-WT and mutants (location indicated by asterisk), STING-WT and deletion mutants.
(I) HEK293T cells stably expressing low amounts of Flag-STING-WT clones were first generated. The clones were further co-reconstituted with low expression of either Flag-cGAS-WT or point mutated plasmids. After selection, the double-positive clones cells were then transfected with 0.01 μg of Flag-SOD1 WT, G85R, or G93A plasmids. Ifnb1 mRNA was assessed via RT-qPCR. Protein expression for cGAS-WT and mutants from the 1.0 μg transfected samples were examined. Expression efficiency for each plasmid was evaluated via western blotting.
(J) Similar conditions as in I were used but replaced with Flag-cGAS-WT plasmid co-transfected with either STING-WT or deletion mutant (Δ368). Ifnb1 mRNA was measured using RT-qPCR.
(K) Protein lysates collected from C20 cells expressing the indicated plasmids were subjected to immunoblotting with the indicated antibodies.
(L) Immunofluorescence staining of endogenous cGAS in primary astrocytes cells derived from day-10 WT, or G85R transgenic mice.
(M) Both supernatant and lysates from C20 cells overexpressed with the indicated plasmids were collected and subjected to ELISA analysis of cGAMPs.
(N) Sera was prepared from whole blood derived from SOD1 WT or G85R mice, followed by ELISA analysis of cGAMPs (day-10). All RT-qPCR of RNA samples were normalized to β-actin. Data represent the mean of three independent experiments (mean ± SEM). ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (B–G, M, and N: unpaired Student’s t test). See also Figure S4.
HEK293T cells have been shown to be deficit of the cyclic GMP-AMP synthase (cGAS)-STING DNA-sensing pathway, rendering them very easy to transfect with DNA plasmids (Langereis et al., 2015). Because HEK293T cells do not respond to SOD1Mut, we hypothesized that the cGAS-STING-axis was essential. Remarkably, we found that microglia lacking either cGAS or STING failed to respond to both Herring-testes DNA (HT-DNA, a positive control) and SOD1Mutwhile retaining a full response to MAVS (Figures 3E and 3F). Similar observations were found when the SOD1Mut vectors were expressed in microglia treated with either RU.521 (a cGAS inhibitor) or H-151 (an STING inhibitor) (Figure 3G). We then tested several cGAS and STING mutants to further elucidate this mechanism (Figure 3H). As expected, the reconstitution of cGAS catalytic domain deletions (E225A-D227A), the DNA-binding (C396/397A), phosphorylation (Y215E), and oligomerization (K394E) domain mutants in HEK293T cells failed to respond to SOD1Mut compared with cGAS-WT (Figure 3I). Similarly, STING-WT robustly upregulated Ifnb1 expression when co-expressed with cGAS-WT in HEK293T but not with the deletion mutant STING-Δ368, which lost its ability to bind TBK1 (Figure 3J). Importantly, SOD1Mut overexpression resulted in phosphorylation of cGAS at residue Tyr-215 (Figure 3K). In agreement with this data, condensation of cGAS to form cytoplasmic puncta as previously described (Du and Chen, 2018) also occurred in primary astrocytes derived from SOD1-G85R, but not in WT mice (Figure 3L). Besides, we also detected the presence of secondary messenger GMP-AMP (cGAMP) in both lysate and supernatant from SOD1Mut stably expressing microglia (Figure 3M). This further prompted us to examine the level of cGAMPs in vivo. As shown in Figure 3N, we observed a significant increase in total serum levels of cGAMPs in the G85R model. Collectively, these data showed that the ALS-SOD1Mut initiated an innate immune response through the cGAS-STING pathway to promote neuroinflammation and neuronal cell death in the SOD1Mut-ALS model.
IFNAR signaling is required for ALS-SOD1Mut to promote neuronal inflammation
Secreted IFN-β proteins, after binding to transmembrane IFNAR, will induce the translocation of the interferon-stimulating-gene factor-3 (ISGF3) complex (comprising IRF9-STAT1-STAT2) into the nucleus to initiate the transcription of ISGs. We then investigated the contribution of IFNAR signaling to the inflammatory response in the context of SOD1-induced ALS pathology. As expected, the induction of ISGs mRNA were completely abrogated in STAT1-deficient microglia upon SOD1Mut expression. Because the cGAS-TBK1-IRF3-axis remained unaffected, the lack of a positive amplification feedback loop only resulted in a partial loss of Ifnb1 mRNA expression (Figures S4A and S4B). Furthermore, the SOD1Mut-induced transcription of chemokines and NF-κB-dependent genes (Cxcl10, Ccl3, Il6, Nos2) were also significantly downregulated in STAT1-deficient cells (Figures S4C and S4D). Consistent with this, pre-treatment of WT microglia with IFN-β proteins further enhanced the levels of ISG and inflammatory gene expression (Figure S4E). To test the translational potential of these findings, we treated SOD1-G93A mice with vehicle, or IFNAR-neutralizing antibody weekly starting at the age of 8-weeks-old. Antibody treatment abrogated ISG expression, improved motor function, and survival (Figures S4F–S4H), demonstrating that transient inhibition of the IFN response can improve the functional outcome of ALS. Collectively, these results indicated that IFNAR signaling is the key SOD1Mut-controlled mechanism for activating the neuroinflammatory responses.
Therapeutic benefits of cGAS and STING inhibitors in a murine model of ALS
Consistent with our earlier findings (Figure 3), we observed that cGAS inhibition with pharmacological inhibitors, RU.521, potently diminished G93A-induced IFN-β reporter activities, with a 50% effective inhibitory concentration (EC50) of 0.621 μM (Figure 4A), whereas two of the STING inhibitors tested—H-151 (EC50 of 0.7319 μM) and C-176 (EC50 of 0.007 μM) on IFN-β reporter activities (Figure 4B, red curve) and a EC50 of 0.6439 μM by H-151 on NF-κB reporter activities (Figure 4B, black curve). To this end, we hypothesized that STING inhibition would attenuate disease in SOD1-G93A mice. To evaluate this relationship, we generated genetically modified animals by crossing SOD1-G93A with Sting−/− deficient mice to obtain G93A/Sting−/− double-positive mice. Remarkably, STING-deletion, and thus cGAS/IFN-I inactivation, resulted in a pronounced 107-day extension in median survival [G93A, 145.0 ± 2.311 days; G93A/Sting−/−, 252.0 ± 1.42 days (logrank Mantel p = 0.0034)] (Figure 4C), although disease onset did not differ between both groups (Figure 4D).
Figure 4.

cGAS and STING pharmacological inhibitors ameliorates ALS neuropathology in the SOD1-G93A model
(A and B) SIM-A9 cells co-transfected with luciferase reporters and 0.01 μg of the indicated SOD1 mutant plasmids in the presence of RU.521, H-151, or C-176. Estimated EC50 values from dose-response curves of the indicated inhibitors were measured.
(C–E) Kaplan-Meier plot of disease-onset days, survival rate, or motor performance of indicated time-point for G93A, Sting−/−, or G93A/Sting−/−-double-positive mice. Schematic diagram indicates how the gait performance (stride-length, blue line) of each mouse was measured.
(F) In vivo ChAT and LDH levels from brain and spinal cord homogenates isolated at the indicated time-points in G93A, Sting−/−, or G93A/Sting−/−-double-positive mice.
(G) Expression of the indicated mRNA in vivo in brain isolated from week-21 of G93A, Sting−/−, or G93A/Sting−/−-double-positive mice.
(H–J) Body weight of vehicle and inhibitor-treated groups were measured (H). Kaplan-Meier plot of disease-onset days, or survival time (I and J) of vehicle and inhibitor-treated groups. Number of mice used in each group as indicated.
(K) Grip strength, hindlimb grasping, or gait performance test of G93A mice after challenge with vehicle, RU.521 (500 μg), or C-176 (500 μg).
(L and M) Basal gene expression of indicated mRNA in vivo in brain and spinal cord isolates from G93A mice challenged with vehicle, RU.521 (500 μg, i.p.), or C-176 (500 μg, i.p.) at Week 21 analyzed by RT-qPCR. Number of dots = number of mice (biological replicates) used in each condition. All RT-qPCR of RNA samples was normalized to β-actin. Unless indicated, data represent the mean of at least three independent experiments (mean ± SEM). ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (E–H and K–M: unpaired Student’s t test; C, D, I, and J: Log rank Mantel-Cox test).
We then evaluated motor functions through gait performance, measuring motor limb strength and coordination in ALS models using stride analyses. Stride is defined as the distance between each successive mice paw print on a single side of a sheet of paper. As the disease progress, mice are unable to move their hindlimbs as much with each step, and therefore, the stride distance decreases. This measure also reflects the complication of front limb weakness, such that mice were unable to extend their front limbs normally, as a result, the back-to-front stride distance also declined. This was evident on weeks 18 and 21, whereas all vehicle treatment groups, continued to decline at a steady rate. We observed a drastic improvement of stride length (p< 0.05) in G93A/Sting−/−-double-positive mice (Figure 4E, left). Analysis of hindlimb grasping score, a second method used to assess motor functions, indicated that the STING-depletion group also consistently displayed remarkable recovery in feet-clasping capability (Figure 4E, right). We further examined motor neuron cell survival by measuring the level of choline acetyltransferase (ChAT), an enzyme that synthesizes the neurotransmitter acetylcholine, a typical marker for healthy spinal cord motor neurons (Casas et al., 2013). As shown in Figure 4F, the number of healthy spinal cord motor neurons steadily declined between weeks 15–21; conversely, STING-deletion provided drastic protection (p< 0.01) and effectively blocked spinal motor neurons cell death in SOD1-G93A mice on weeks 18 and 21. We also observed a significant reduction in LDH release from the brain and spinal cord homogenates collected from G93A/Sting−/−-double-positive mice as compared to the vehicle group (Figure 4F, p< 0.05). RT-qPCR quantification of brain mRNA confirmed the reduction of cytokine genes (Figure 4G, p< 0.05). These results indicated that the genetic ablation of the STING pathway achieved significantly greater neuro-protective efficacy and helped improve motor performance in the SOD1-G93A mutant ALS model.
Based on the promising results from G93A/Sting−/− mice, we next examined whether in vivo administration of cGAS, and STING inhibitors could provide therapeutic benefit on muscle performance and disease progression. To test this, we continued to monitor the motor function, weight loss, and atrophy using the rapidly progressive ALS mouse model SOD1-G93A injected either with vehicle, RU.521, or C-176, weekly starting at 8-week of age. Beginning at 8-week of age, all SOD1-G93A mice received intraperitoneal injection of either 500 μg of C-176, RU.521, or an equivalent amount of vehicle, twice weekly for the first four weeks to achieve a maximal cumulative dose, then once per week thereafter. These treatments were given until the experimental endpoint. As shown in Figure 4H, both groups showed the same trend of initial weight gain followed by a gradually weight loss until the endpoint. However, compared to aged-matched controls, animals treated with RU.521 and C-176 maintained and/or gained weight (p≤ 0.05) up to week-18 and week-21, beyond the expected endpoint of disease progression. Consistently, the onset of the disease in SOD1-G93A mice also showed a marginal improvement after treatment with RU.521, and C-176; although this difference did not reach statistical significance (logrank test for survival plot: p = 0.426 for C-176; p = 0.0728 for RU.521) (Figure 4I). Conversely, the mean survival period displayed a significant increase from 50 to 78 days in treated mice (Vehicle, 117.5 ± 1.528 days; RU.521, 168.0 ± 2.363 days [log rank p = 0.0001]; C-176, 188.0 ± 3.672 days [log rank p = 0.0045]) (Figure 4J). We next evaluated whether both RU.521 and C-176 could improve the functional performance of SOD1-G93A mice, as assessed by stride length, hindlimb grip strength, and the clasping test at several age-points. Compared to aged-matched controls, animals treated with RU.521 and C-176 displayed improved motor function based on grip strength from weeks 19–27 and the differences at weeks 19 and 21 were statistically significant (One-way ANOVA, Week 19, p = 0.0069; Week 20, p = 0.0009; Week 21, p = 0.0006) (Figure 4K, left). In addition, most control-treated mice showed a complete paralysis of the hindlimbs, and hence did not manifest a feet-clasping phenotype upon tail suspension, whereas both the RU.521 and C-176-treated groups showed a marked improvement in the feet-clasping phenotype and rearing activity from weeks 14–21 (Figure 4K, middle). In line with this result, gait performance by measuring the footprint stride length further indicated that mice receiving either RU.521 or C-176 showed significantly better maintenance of the motor phenotype of the hindlimbs at both 18 and 21-weeks of age, compared with vehicle group, which showed gait abnormalities (i.e., shorter stride length, p< 0.05) (Figure 4K, right). This suggests that blocking cGAS and/or STING conferred protection to neuronal damaged in vivo. Furthermore, we also observed a significant reduction in mRNA levels of IFN-I, ISGs, pro-inflammatory, and chemokines genes in the end-stage brain and spinal cord from inhibitor-treated mice (Figure 4L). H-151, another potent, irreversible small molecule antagonist of STING (Haag et al., 2018; Vincent et al., 2017), also behaved similarly in reducing Ifnb1, Rsad2, and Il6 mRNA expression within the spinal cord of G93A mice, albeit less robust than C-176 (Figure 4M). Nonetheless, as the disease progressed and SOD1-G93A mice reached the end stage (i.e., the length of time between disease onset and the disease endpoint), mice became weak and immobile, whereas inhibitor-treated mice showed much better health conditions. They groomed and explored their cages, illustrating marked beneficial effects of RU.521 and C-176 on muscle performance as the disease progressed. Collectively, these results demonstrated that administration of cGAS or STING inhibitors in SOD1-ALS in vivo model ameliorated the ongoing inflammation found in the CNS, which further contributed to improve motor function and lifespan.
ALS-SOD1Mut damaged mitochondria and the released mtDNA activated the cGAS-STING pathway
cGAS is a well-known innate immune sensor that surveys the cytosol for the presence of self- and non-self-DNA (Gao et al., 2013; Härtlova et al., 2015; Sun et al., 2013). We next questioned the source of DNA that activated cGAS in SOD1Mut-ALS. Pathological changes including mitochondrial abnormalities in the SOD1 model have been elucidated previously (Jaarsma et al., 2000, 2001; Pedrini et al., 2010), whereas other studies have shown that the released mtDNA from apoptotic cells could trigger an immune response (West et al., 2015), the relationship between these has not been described in SOD1Mut-ALS. Thus, we examined whether SOD1Mut expression could trigger the release of mtDNA to the cytoplasm. SOD1Mut transfected microglia were subjected to subcellular fractionation. After confirming the purity of each fraction (Figure 5A), we found that the expression of SOD1Mut in microglia and astrocytes showed a significant two- to 5-fold enrichment of mtDNA (dloop, Mt-atp6) in the cytosolic fraction compared to EV and WT (Figure 5B). Similarly, fractionation of the extracts derived from the whole brain, spinal cord, primary microglia, and astrocytes of the G85R transgenic mice also showed a significant enrichment of other mtDNA genes, including Mt-co1 and Mt-nd1 (Figures 5C–5E). Confocal microscopic analysis confirmed that SOD1Mut-induced elevated levels of dsDNA in the cytosol. We also found that the mitochondria were more intact and were localized perinuclearly in EV or WT compared to a more fractionated and stretchy morphology in SOD1Mut-expressing microglia (Figure 5F), which coincided with the dissipation of mitochondrial membrane potential in primary microglia and astrocytes of the G85R-mutated mice (Figure 5G), confirming the dysfunction and mitochondrial damage observed in SOD1-ALS pathology.
Figure 5.

ALS SOD1-mutants damage mitochondria and release mtDNA to activate the IFN-I response
(A and B) WT C20 and HASTR/ci35 cells were either treated with 1.0 μg/mL of tunicamycin for 8 h or transfected with indicated plasmids and 36 h after transfection, cells were subjected to fractionation and blotted with indicated antibodies (A). Total DNA extracted from cytosolic and nuclear fractions as treated in A. Cytosolic mtDNA genes was assessed via RT-qPCR (B).
(C and D) Total DNA was harvested from the cytosolic and nuclear fractions as in A from whole brain and spinal cord lysates of WT or G85R mice at Week 60. Cytosolic mtDNA genes was assessed.
(E) Primary microglia and astrocytes derived from WT or G85R mice at day-10 after birth were subjected to subcellular fractionation as in A. Cytosolic mtDNA genes were assessed.
(F) Immunofluorescence staining of dsDNA in C20 cells transfected with plasmids encoding EGFP-SOD1 WT,-G85R, or-G93A.
(G) Primary microglia and astrocytes derived from age-matched WT or G85R mice (day-10) were harvested and subjected to MMP assay.
(H) C20 cells transfected either with HT-DNA (5.0 μg/mL), or Flag-MAVS/IRF3 5D plasmids (0.1 μg each) were either Sham, ddC, or EtBr treated. Ifnb1 mRNA was measured via RT-qPCR (left). Protein expression was analyzed through western blotting (right).[Note: MAVS in standard form (∼52 kDa) are marked by asterisk (∗∗∗); splicing variants, truncated forms, midi and mini-MAVS (<52 kDa) are marked by vertical blue line (Brubaker et al., 2014; Qi et al., 2017); MAVS in higher molecular weight (>52 kDa) are marked by vertical red line (Hou et al., 2011)].
(I) C20 cells transfected with indicated plasmids were either sham or EtBr (150 ng/mL) treated for 4 days. Samples were subjected to fractionation and DNA extraction, followed by RT-qPCR analysis of mtDNA genes and the indicated mRNA. Statistical analysis corresponding to the same condition in the Sham treated group.
(J) C20 cells transfected with the indicated plasmids were either sham or ddC (50 μg/mL) for 6-day. Samples were subjected to fractionation and DNA extraction, followed by RT-qPCR analysis of mtDNA gene and the indicated mRNA. Statistical analysis corresponds to the same conditions as the Sham treated group.
(K) Knockdown efficiency of the Ppid gene in HASTR/ci35 cells was validated (left). Experimental design of HASTR/ci35 cells co-transfected with indicated siRNA (10 nM) and 0.01 μg plasmids (middle). 48 h later, cells were subjected to fractionation described in A and quantification of mtDNA genes.
(L) Similar experimental design as in panel K using C20 cells. mtDNA genes and Ifnb1 mRNA was quantified.
(M) C20 cells transfected with indicated plasmids were either vehicle (DMSO) or CsA (20 μM) treated. Samples were subjected to fractionation and DNA extraction, followed by measuring of Mt-co1. RT-qPCR of all mtDNA genes was normalized to genomic DNA, TERT. RT-qPCR of RNA samples was normalized to β-actin. Data represent the mean of four independent experiments (mean ± SEM). Number of dots = number of mice. ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (B–E, G, and I–L: unpaired Student’s t test). (RFU = relative fluorescence unit; A.U. = arbitrary unit). See also Figure S5.
Next, we examined whether the released of mtDNA was the ligand that activated IFN-I through the cGAS-STING pathway. To this end, we depleted mtDNA by treating cells with ethidium bromide (EtBr) or dideoxycytidine (ddC) to inhibit mtDNA replication (Kai et al., 2006; Kao et al., 2012; Nelson et al., 1997; Yu et al., 2007). We confirmed that the protein expression induced by the transfected plasmid and the signaling pathway remained unaffected (Figure 5H), although these treatments resulted in a significant reduction in mtDNA and a reduction in the expression of Ifnb1 and Cxcl10 mRNA in SOD1Mut-transfected microglia (Figures 5I and 5J). Next, we generated TFAM-deficient microglia (Figure S5A), as we speculated that depletion of this mtDNA-associated protein may enhance the accessibility of mtDNA to activate the innate immune response. Notably, TFAM-depleted microglia enhanced detectable levels of cytosolic mtDNA upon tunicamycin treatment (a positive control for triggering mtDNA accumulation) (Sun et al., 2017; Yuzefovych et al., 2013) or transfection with SOD1Mut plasmids (Figures S5B and S5C). Ifnb1, Cxcl10, and ISG expression were also enhanced in SOD1Mut-transfected TFAM-depleted cells (Figure S5D). We further substantiated this by transfecting the pure cytosolic fraction of SOD1Mut cells treated with proteinase K (Figure S5E). As shown in Figure S5F, the proteinase K treated purified cytosolic fraction showed a complete absence of TFAM proteins and increased detectable levels of cytosolic mtDNA upon SOD1Mut expression (Figure S5G). Consequently, we observed a significant upregulation of Ifnb1 expression compared with the untreated control fraction of SOD1Mut-transfected cells, and such an enhancement was notably abrogated in cGAS- or STING-depleted cells (Figures S5H and S5I). Finally, while this study was in preparation, the group of Seth Masters reported a similar observation in the Transactive response DNA-binding protein-43 (TDP-43 mutants, TDP-43Mut)-driven ALS (Yu et al., 2020). Other authors have reported that TDP-43 triggers the release of mtDNA through the mitochondrial permeability transition pore (mPTP) and activates the cGAS-STING pathway (Yu et al., 2020). Hence, we questioned whether SOD1Mut could also trigger the released of mtDNA through a similar mechanism. We employed the siRNA approach to deplete the expression of peptidyl-prolyl isomerase D (PPID, encoded by the Ppid gene), an integral constituent of the mPTP complex. After confirming the knockdown efficiency (Figure 5K, left), we then co-transfected both siRNA and plasmids expressing either SOD1Mut or TDP-43Mut into the human astrocyte HASTR/ci35 cell lines and subjected them to subcellular fractionation (Figure 5K, middle). Our results revealed that the release of mtDNA in SOD1Mut expressing cells remained unaffected in the cytoplasmic fraction of Ppid-depleted cells when compared to controls (Figure 5K, right). Conversely, and to our surprise, we found no significant differences in the abundance of mtDNA triggered by TDP-43 A315T in Ppid-depleted cells versus controls. This prompted us to question whether the observation was cell-type specific. We then performed the similar experiments using another neuronal cell lineage, C20 cells. Again, we obtained a similar result, with concurrent robust Ifnb1 mRNA expression in both controls and Ppid-depleted cells (Figure 5L). We further substantiated these findings by treating cells with cyclosporin A (CsA), a potent inhibitor of cyclophilin D and a well-established pharmacological inhibitor of the mPTP pore. Our results indicated that the abundance of the cytoplasmic fraction of mtDNA remained comparable in either the SOD1Mut or TDP-43Mut expressing C20 cells in the absence or presence of CsA treatment (Figure 5M), suggesting that the release of mtDNA in SOD1Mut-expressing neuronal cells was not dependent on mPTP. Collectively, our data suggest that ALS-SOD1Mut damaged mitochondria, and the released of mtDNA further activated neuronal innate immune responses through the cGAS-STING axis.
ALS-SOD1Mut triggered the released of mt(RNA:DNA) hybrid to activate the DDX41-STING pathway
We observed a complete abrogation of IFN-I levels in STING-depleted cells, while retaining residual signals in cGAS-depleted cells (Figure 3F), which led us to speculate that another sensor functioning upstream of STING likely contributed to the SOD1Mut-dependent IFN-β response. As mtDNA contains RNA hybridized to DNA, damaged mitochondria may leak these structures into the cytoplasm. Remarkably, we could detect the presence of RNA:DNA hybrid intermediates in the cytosol of primary astrocytes and motor neurons extracted from transgenic G85R but not in extracts from WT mice (Figures 6A and 6B). These prompted us to validate the S9.6 staining by subjecting the fixed G85R primary astrocytes samples to mock or enzymatic treatments before immunostaining. Notably, pre-treatment with RNase H (RNase H), an enzyme which specifically degrades the RNA strand of the RNA:DNA hybrid, led to a significant decrease of total cellular S9.6 staining, with a reduction of approximately 4-fold (Figure S6A) (p< 0.001). In comparison, treatment with RNase T1, an endoribonuclease that specifically targets single-stranded RNA, did not have an effect on total cellular S9.6 staining when compared to mock treated samples (Figure S6A), indicating that the majority of the S9.6 signals derived from immunostaining of fixed G85R primary astrocytes stemmed from the specific binding of S9.6 to mt(RNA:DNA) hybrids. Next, we assessed whether the mt(RNA:DNA) hybrid could contribute to the SOD1Mut-induced IFN-I response. We treated the purified cytosolic fraction with RNase H and re-transfected them into WT microglia (Figure 6C). As shown in Figure 6D, the RNase H-treated cytosolic fraction from SOD1Mut-transfected cells showed a significant reduction in Ifnb1, Isg15, and Cxcl10 mRNA expression, these inductions were abrogated in microglia lacking cGAS or STING (Figures 6E and 6F). However, buffer-treated cytosolic fraction from SOD1Mut-expressing cells continued to produce a residual expression in cGASKO cells (≈4-fold) but not in STINGKO microglia (Figures 6E and 6F; Red versus blue arrow), indicating that mt(RNA:DNA) hybrid contributed to SOD1Mut-induced IFN-I response independent of cGAS but still required STING.
Figure 6.

ALS SOD1-mutants trigger mt(RNA:DNA) hybrid release and activate IFN-I responses through the DDX41-STING axis
(A and B) Primary neurons and astrocytes extracted from WT or G85R-mice at day-10 after birth were fixed and immunostained with indicated antibodies and DAPI (blue).
(C) Schematic diagram of DNase I/RNase H treatment experiments. Pure cytosolic fraction from C20 cells transfected with 0.01 μg of Flag-SOD1 WT or SOD1-mutant vectors for 36 h were incubated with either buffer, DNase I or RNase H, followed by DNA extraction. These samples were re-transfected into cells and subjected to gene quantification.
(D) RT-qPCR analysis of the indicated mRNA from samples derived from experimental design described in panel C.
(E) WT C20 cells treated with RU.521 (5.0 μg/mL) or H-151 (0.5 μg/mL) for 24 h. Cells were then transfected with DNA extracted from EV and G85R-transfected samples as described in C. RNA was purified and Ifnb1 mRNA was measured using RT-qPCR.
(F) WT, cGAS, or STING-deficient C20 cells were transfected with DNA extracted as described in panel C, Ifnb1 mRNA was measured via RT-qPCR.
(G) CRISPR/Cas9 DDX41-deficient C20 cells validated by western blotting.
(H) WT or DDX41-deficient C20 cells were transfected with 0.01 μg of indicated plasmids, and the indicated mRNA was measured using RT-qPCR.
(I) WT C20 cells transfected with the indicated plasmids and treated with DMSO or LFM-A13 (100 μM). RNA was purified and Ifnb1 mRNA was measured via RT-qPCR.
(J) DNA extracted from G85R or G93A-transfected samples and prepared as described in panel C were re-transfected into WT, cGAS, or DDX41-deficient cells for 6 h. Ifnb1 mRNA expression was measured.
(K) DNA extracted from G85R-transfected samples prepared as described in C were re-transfected into HEK293T cells expressing either EV, Flag-tagged STING-WT, or STING Δ368. RNA was purified and Ifnb1 mRNA was measured using RT-qPCR. All RT-qPCR of RNA samples was normalized to β-actin. Data represent the mean of three independent experiments (mean ± SEM). White scale bars correspond to 10 μm ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (D–F and H–K: unpaired Student’s t test). See also Figure S6.
Both DDX41 and cGAS have been shown to recognize RNA:DNA intermediates (Mankan et al., 2014; Stavrou et al., 2018). Our study has excluded the latter. Hence, we asked whether DDX41 was critical for SOD1Mut-induced IFN-I and thus, generated DDX41-deficient microglia (DDX41KO, Figure 6G). Although the mtDNA levels are comparable with WT (Figure S6C), Ifnb1 and Cxcl10 mRNA expression was significantly downregulated in DDX41KO microglia upon expression of ALS-SOD1Mut (Figure 6H). To further substantiate this observation, mt(RNA:DNA) derived from the fractionated cytoplasmic fraction of SOD1Mut-expressing microglia were immunoprecipitated with the S9.6 antibody. Small aliquots of the pulled-down lysates were re-transfected into HeLa cells for validation through immunostaining. As shown in Figure S6B, cells transfected with the S9.6 pulled-down lysate showed cytoplasmic puncta staining but this was not observed in cells transfected with IgG-control pulled-down lysate, confirming the predominant constituent of the S9.6 pull-down product was mt(RNA:DNA). We then re-introduced the remaining lysates into WT, cGASKO, or DDX41KO microglia, followed by RT-qPCR analysis. We observed that the mt(RNA:DNA)-induced Ifnb1 mRNA expression was significantly impaired (≈10-fold, p< 0.001) in DDX41KO cells but not in WT and cGASKO microglia (Figure S6B, right). Further, cells treated with LFM-A13, a molecular inhibitor of Bruton’s tyrosine kinase (BTK), which phosphorylates DDX41 to induce the IFN-I response (Lee et al., 2015), also significantly downregulated the SOD1Mut-induced Ifnb1, Cxcl10, and Irf7 expression in microglia, oligodendrocytes, or motor neuron cells (Figures 6I and S6D–S6F). Furthermore, transfection of DDX41-but not in cGAS-deficient microglia with the RNase H-treated cytosolic fraction from SOD1Mut cells produced a comparable amount of Ifnb1 mRNA compared to buffer-treated controls (Figure 6J), suggesting the importance of DDX41 in sensing cytosolic mt(RNA:DNA). To determine whether cGAS or DDX41 is the primary sensor for mtDNA or mt(RNA:DNA), respectively, we treated microglia with siRNA targeting IFI16 and ZBP1, other known critical DNA sensors in non-neuronal cells (Almine et al., 2017; Takaoka et al., 2007; Unterholzner et al., 2010). Knockdown of both proteins did not influence Ifnb1 expression derived from the buffer-treated SOD1Mut-cytosolic fraction (Figures S6G and S6H). Finally, we accessed whether STING functioned downstream of DDX41 on sensing mt(RNA:DNA). Given that HEK293T still expresses DDX41, but not STING. Reconstitution of STING-WT restored Ifnb1 expression in HEK293T cells transfected with buffer-treated but not with the RNase H-treated cytosolic fraction. No induction was observed in STING-Δ368 co-transfected cells (Figure 6K). Collectively, these findings indicate that mtDNA or mt(RNA:DNA) released from damaged mitochondria primarily activate the cGAS- or DDX41-STING pathway, respectively, to induce the IFN-I response in the SOD1Mut-ALS model.
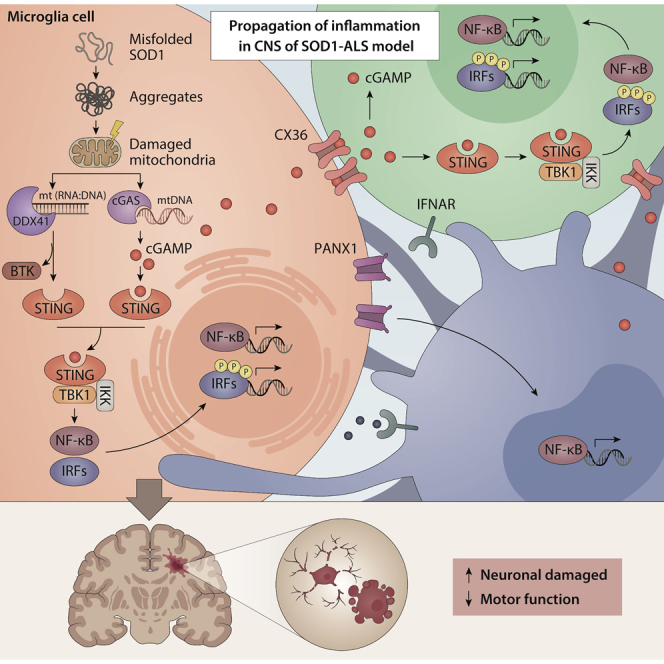
Cell-cell contact was essential for the propagation of ALS-SOD1Mut-dependent IFN-I responses
Activation of IRF3 and NF-κB is the hallmark to trigger robust IFN-I and pro-inflammatory responses. To better characterize the molecular mechanisms induced by ALS-SOD1Mut, we used human microglia and transduced them with an amino-terminally EGFP-tagged SOD1-G85R construct and analyzed both IRF3 and NF-κB activation by immunofluorescence microscopy. As expected, transient expression of EGFP-SOD1-G85R in the microglia led to the activation and re-localization of IRF3 and NF-κB to nuclear complexes when compared to non-SOD1Mutexpressing cells (Figure S7A, asterisk). Strikingly, we also observed the nuclear localization of IRF3 and NF-κB in microglia lacking pEGFP-SOD1-G85R expression, which were located adjacent to pEGFP-SOD1G85R-expressing cells (Figure S7A, white arrows). Notably, this phenomenon of bystander cell activation was not observed when the microglia were transfected with polyinosinic:polycytidylic acid [poly(I:C), p(I:C)], a synthetic analog of double-stranded RNA (dsRNA) acting as a molecular mimic associated with viral dsRNA produced during viral replication (Figure S7A, right lower panel). Because polyI:C activates the cytosolic RNA sensor RLR pathway, these results indicate that both IRF3 and NF-κB activation of surrounding cells occurs via an event that is linked to cGAS activity.
Next, we hypothesized that SOD1Mut-expressing cells would be capable of propagating IFN-I to bystander cells. We employed a trans-well culture system that could separate donor and recipient cells. This experimental set-up could effectively block the bystander cell activation. With this aim, we established four different experimental groups involving both co-cultures or trans-well system: Group 1 comprised only NSC-34 motor neurons cells; Group 2 comprised WT HEK293T and NSC-34 cells in co-culture; Group 3 comprised HEK293T cells expressing STING co-incubated with NSC-34 expressing SOD1Mut in the trans-well system; and Group 4 comprised HEK293T expressing FLAG-STING cells co-cultured together with SOD1Mut expressing NSC-34 cells (Figure S7B). We observed robust transactivation of the IFN-β reporter and the induction of IFN-I associated genes in Group 4 but not in Groups 1, 2, and 3 conditions (Figures S7C and S7D), indicating that a cell-to-cell contact-dependent propagation mechanism was responsible for relaying IFN-I signals.
Next, we investigated whether a similar mechanism was responsible for the release of NF-κB-dependent pro-inflammatory cytokines. We first performed experiments based on the original set-up described in Figure S7D, with slight modifications, whereby STING-stably expressing HEK293T cells in both Groups 3 and 4 were replaced with WT HEK293T cells (Figure S7E). These conditions were originally hypothesized to act as a negative control in parallel with the experimental conditions described in Figure S7D. Unexpectedly, we still observed the induction of Tnf or Tnfaip3 (a TNFα-inducible gene) in Group 4. Furthermore, this bystander pro-inflammatory gene activation was also observed when WT HEK293T cells were co-incubated with SOD1Mut-expressing NSC-34 cells in the Trans-well system (Group 3, Figure S7F), suggesting that the contact free bystander propagation of pro-inflammatory responses may also occur through a cGAS-STING-independent pathway. To provide additional supporting evidence, we used similar experimental conditions as those described in Figure S7E but replaced the WT HEK293T cellswith HEK293T cells expressing NF-κB luciferase reporters (Figure S7G). Switching the recipient cells also produced comparable results: NF-κB promoter transactivation was observed in both Groups 3 and 4 but not in Groups 1 and 2 (Figure S7H). To validate that this cGAS-STING-independent NF-κB activation in trans was not because of the migration of cells through the semipermeable membrane of the trans-well system, we replaced NSC-34 cells with microglia (Figure S7I). Consistently, we could still detect Tnf, but not Ifnb1 expression in Group 3 (Figure S7J, blue arrow). Furthermore, RT-qPCR analysis also failed to detect the expression of either Iba1 or Cd11b genes (microglial cell markers) in the wells containing HEK293T-cells (Figure S7J, black bars), which excluded that the Tnf-producing cells as observed in Group 3 were from migrated microglia. We then questioned whether bystander activation of NF-κB could occur via a paracrine loop. Immunofluorescence microscopy analysis of microglia treated with tumor necrosis factor receptor 1 (TNFR1) blocker indicated that nuclear NF-κB translocation could still be observed in bystander cells upon SOD1Mut expression (Figure S7K, arrows), suggesting that paracrine signaling was not the mediator of bystander activation. To investigate the role of protein secretion more broadly, we investigated whether these phenomena could be impaired when the cellular secretory pathway was abolished by the protein transport inhibitor brefeldin A (BFA). To determine whether drug treatment blocked cytokine secretion (Hong et al., 2019), using poly(I:C) as a negative control, BFA exerted no effect on bystander activation upon SOD1Mut expression in microglia (Figure S7L, arrows), suggesting that cell-cell propagation of pro-inflammatory signals was not mediated by the extracellular secretory pathway. Overall, these results collectively conveyed two messages: (1) ALS-SOD1Mut propagates and amplifies IFN-I signaling to bystander cells through intact physical cell-to-cell contact; and (2) Cell contact free bystander propagation of SOD1Mut-induced pro-inflammatory cytokines also occurs via a route independent of the cGAS-STING pathway.
CX36 propagated cGAS-STING-dependent IFN-I signals to bystander cells in SOD1-ALS
To directly assess the functional role of cGAS-STING-axis in bystander activation, we next generated monoclonal HEK293T cGAS cells with low constitutive expression of cGAS (cGASlow). This monoclonal line would require the extrinsic DNA stimulation to activate STING and that of the downstream IRF3. As shown in Figure 7A, titrating the number of HEK293T cGASlow cells over STING competent cells in conjunction with G85R transfection revealed a dose-dependent increase in IFN-β promoter transactivation [Group (i) versus (ii)]. This bystander STING activation phenomenon was also observed when HEK293T STING cells co-cultured with G85R-expressing NSC-34 cells that are inherently competent for cGAS (Figure 7B). We then further investigated this mechanism using a genetic approach. Amino-terminal EGFP-tagged SOD1Mut (G85R or G93A) expressing WT human microglia were seeded and co-cultured either together with WT (Group i), or STINGKO human microglia (Group ii). RT-qPCR analysis revealed that the mRNA expression of antiviral genes Ifnb1, and ISGs (Rsad2, Irf7) was significantly downregulated when SOD1Mut-expressing cells were co-cultured with STINGKO as compared to WT (Figure 7C). Moreover, switching SOD1Mut-expressing WT microglia to SOD1Mut-expressing cGASKO microglia completely abrogated Ifnb1 mRNA expression while retaining a residual of Tnf gene (Figure 7D). Taken together, these results provide direct functional evidence for cGAS-dependent STING activation of IFN-I in trans, in parallel with the cGAS-STING independent bystander propagation of pro-inflammatory responses.
Figure 7.

Connexin-36 mediate cGAMP(2′ 5′) intercellular transfer in human microglia
(A and B) HEK293T and HEK293T-STING cells were co-cultured with HEK293T cGASlow or NSC-34 cells (ratio ranging from 1:0.5 to 1:0.125), followed by transfection with p125-Gluc (IFN-β), whereas transactivation of the reporter was assessed after 24 h.
(C and D) WT (C, brown), or cGAS-deficient C20 cells (D, green) expressing either EV, SOD1-WT, or mutants were co-cultured with WT [marked by asterisk (∗), blue] or STING-deficient C20 cells (orange). Relative induction of indicated mRNA from each group (i, ii, iii, or iv) was measured via RT-qPCR.
(E) C20 cells expressing EV, WT, or mutants SOD1 in the absence or presence of CBX (150 μM) were subjected to RT-qPCR of Ifnb1 mRNA (upper). The protein level of indicated SOD1 mutants in C20 cells after treatment with CBX was assessed via western blotting using specific antibodies (lower).
(F) CX36-deficient C20 cells generated through CRISPR/Cas9 system are validated by western blotting.
(G) Schematic diagram of experimental design. WT or CX36-deficient C20 cells (white) were co-cultured with increasing ratios of STING-deficient C20 cells (ratios ranging from 1:0.5 to 1:0.125) containing either SOD1-G85R mutants or HT-DNA (0.1 μg/mL), followed by transfection with p125-/NF-κB-pLuc. Luciferase activity was measured after 24 h.
(H) STING-deficient C20 cells expressing SOD1-G85R/93A mutants [marked with asterisk (∗)] were co-cultured with CX36-deficient cells alone or reconstituted with CX36 WT or W277A mutant plasmids. Relative expression (RE) of Ifnb1 mRNA from each designated group (i, ii, or iii) was measured with RT-qPCR.
(I) STING-deficient C20 cells expressing EV, SOD1-G85R/93A mutants [marked with asterisk (∗)] were co-cultured with either WT or CX36-deficient C20 cells. After 36 h, relative expression of indicated mRNA from designated group (i or ii) were measured via RT-qPCR. All RT-qPCR of RNA samples were normalized to β-actin. Data represent the mean of at least three independent experiments (mean ± SEM). ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (C and D: unpaired Student’s t test). See also Figure S7.
The detection of DNA by cGAS leads to the production of secondary messenger cGAMP, which can pass through cellular gap junctions to activate the STING-dependent IFN-I pathway in bystander cells (Ablasser et al., 2013). We, therefore, evaluated the impact of carbenoxolone (CBX), an inhibitor of cellular gap junctions. WT human microglia treated with increasing doses of CBX produced significantly less Ifnb1 mRNA upon SOD1Mut expression without affecting the protein levels (Figure 7E), indicating the involvement of cellular gap junctions in bystander propagation. Gap junctions formed by the connexins family represent a well-established route of cell-cell contact-dependent intercellular communication. Consequently, to investigate the mechanisms of intercellular cGAMP transfer in the ALS-SOD1Mut model, we simultaneously targeted CX36 (CX36KO) or CX43/45 (CX43-CX45DKO-double knockout) in human microglia using the CRISPR-Cas9 system (Figures 7F and S8A). CX36KO, or CX43-CX45DKO cells showed normal responsiveness in Ifnb1 mRNA expression toward LPS, poly(I:C), and IRF3 5D, indicating that IFN-I responses were intact in these cells (Figure S8B). However, co-cultured of CX43-CX45DKO cells with an increased number of STINGKO microglia did not result in substantial differences of IFN-β reporter activities, as well as Ifnb1, Tnf, or Il6 mRNA expression upon both SOD1Mut expression and HT-DNA transfection (Figures S8C and S8D). Conversely, CX36KO cells significantly reduced the levels of SOD1Mut-dependent IFN-β reporter activity, while retaining NF-κB signals (Figure 7G, red arrows), as supported by FACS-single-cell analysis, in which co-culturing of fluorescence-labeled-cGAMPs pre-loaded STINGKO cells (serving as the baseline) either with WT or CX43-CX45DKO cells led to a significant increase in the total number of cells positive for cGAMP-labeled fluorescence (p< 0.01). However, co-culturing with CX36KO had a minimal effect with the average number of cells that were cGAMP fluorescence-positive remained comparable to those found at baseline (Figure S8E, middle). These observations were congruent with the induction of Ifnb1 mRNA expression (Figure S8E, right), suggesting that CX36 was the critical gap junction in the CNS that responsible for transferring cGAMPs to bystander cells for STING/IFN-I activation. Moreover, the reconstitution with the CX36 WT construct succeeded in restoring the SOD1Mut-induced Ifnb1 mRNA induction, but not of the W277A defective mutant (Siu et al., 2016) (Figure 7H). Consistently, only a fraction of pro-inflammatory genes was reduced (Figure 7I). Collectively, CX36 was essential for the inter-neuronal propagation of IFN-I responses in the ALS-SOD1 model.
CX36 and PANX1 channels cooperatively potentiated the neuronal inflammatory reaction
The activation of the NF-κB signaling pathway is required to mount an effective inflammatory response. To further delineate the molecular mechanisms that allow ALS-causing SOD1Mut to propagate pro-inflammatory cytokine responses to neighboring cells as observed in Figures S7E–S7H, we first asked whether these phenomena were NF-κB-dependent. To this end, we employed an siRNA approach using four experimental groups: Group (i) WT human microglia, C20, were transfected with siRNA-targeting RelA (p65, a key member of the NF-κB family) and were co-incubated with cGASKO C20 cells expressing G85R in the trans-well system; Group (ii) Similar cell types and conditions as in Group (i) but without the trans-well system; Group (iii) RelA-depleted C20 cells alone, and Group (iv) RelA-depleted C20 cells co-cultured with cGASKO C20 cells (Figure 8A). We failed to detect the induction of Ifnb1 mRNA in the four experimental groups, including Groups (i) and (ii), these results were expected as the cells lacked cGAS protein. Consistently, we still observed a mild induction of Tnf expression in the presence of the trans-well system, and a more robust induction when these were co-cultured together. However, such induction was completely abrogated in the presence of RelA-siRNA depleted C20 cells (Figure 8B). Co-culturing of RelA-depleted C20 cells on top of cGASKO C20 cells expressing G85R also revealed a dosage-reduced induction of Tnf mRNA expression in control cells, while drastically reduced in RelA-depleted cells (Figure 8C). Furthermore, co-incubation of NSC-34 expressing SOD1Mut with an increasing number of HEK293T cells expressing the NF-κB reporter construct further showed a dosage-increment of luciferase activities (Figure 8D). Subsequently, we performed immunofluorescence staining analysis by co-culturing cGASKO C20 cells expressing EGFP-SOD1-G85R with WT C20 cells. We observed the nuclear translocation of NF-κB in bystander cells (Figure 8E). Collectively, these results suggest that ALS-associated SOD1Mut cells are capable of propagating inflammatory responses to bystander cells independently of cGAS-STING and cell-cell contact, and that the expression of these inflammatory genes in bystander cells was dependent on NF-κB.
Figure 8.

Pannexin-1 and connexin-36 cooperatively propagate NF-κB dependent cytokines to bystander cells
(A and B) Schematic diagram showing the experimental design. cGAS-deficient C20 cells expressing G85R plasmid [marked with asterisk (∗)] were co-cultured with WT C20 cells transfected with 20 nM of indicated siRNAs [marked with epsilon (ψ)] in the presence or absence of the trans-well system. The expression of indicated mRNA were measured via RT-qPCR. Knockdown efficiency of RelA gene is shown (B, right).
(C) cGAS-deficient C20 cells expressing G85R plasmid [marked with asterisk (∗)] co-cultured with titrated number of WT C20 cells transfected with 20 nM of the indicated siRNAs [marked with epsilon (ψ)] in increasing ratios of (1:0.5/0.25/0.125). Expression of Ifnb1, or Tnf quantified via RT-qPCR.
(D) HEK293T cells expressing NF-κB reporter plasmid and NSC-34 cells expressing SOD1 mutants were culture alone or co-cultured before the luciferase assay.
(E) Confocal analysis of pSOD1-G85R expressing cGAS-deficient cells co-cultured with WT C20 cells. EGFP-G85R stably expressing cells are marked with asterisk (∗). Nuclear translocation of NF-κB in bystander cells is indicated by white arrows. Nuclear-membrane is marked by white-dotted lines.
(F) Knockout efficiency of PANX1 using CRISPR-Cas9 verified using immunoblotting. Amount of ATP within cultured supernatant were measured using an ATP-assay. Treatment with 1.0 μM oligomycin A served as the positive control.
(G) cGAS-deficient C20 cells expressing G85R or G93A plasmid (indicated as asterisk) were co-incubated with WT or PANX1-deficient cells in the trans-well system. Statistical analysis shown in Group (ii) was performed comparing the same condition in Group (i).
(H) Similar experimental design as in (G) was used but the cell was replaced with WT C20 cells treated with vehicle (water) or 100 μM probenecid in the trans-well system. Statistical analysis shown in Group (ii) was performed comparing the same condition in Group (i).
(I) WT, PANX1-deficient (yellow), or PANX1-CX36-double deficient (light green) C20 cells were co-cultured with STING-deficient C20 cells expressing SOD1-mutants [marked by asterisk (∗), orange]. Relative induction of the indicated mRNA from designated groups (i, ii, or iii) were quantified via RT-qPCR. Fold-reduction is indicated.
(J) PANX1-CX36-double deficient (light green) C20 cells were reconstituted with WT PANX1 or CX36 plasmids before co-culturing with STING-deficient C20 cells expressing G85R for 36 h. Relative induction of Ifnb1, Tnf, or Il1b mRNA was measured. All RT-qPCR data were normalized to β-actin. Data represent the mean of at least three independent experiments (mean ± SEM). ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001; (B, C, and F–H: unpaired Student’s t test).
Pannexin-1 (Panx1) channels are capable of propagating signals through intercellular space. There has been growing evidence demonstrating that both Panx1 channels work cooperatively with connexin under pathological conditions, including neurodegeneration, inflammation, and epilepsy, allowing direct intercellular communication, bidirectional flow of ions, and signaling molecules like ATPs and glutamate to greatly facilitate the transmission of signals (Lohman et al., 2015; Makarenkova et al., 2018; Scemes et al., 2007; Zhou et al., 2019). To establish whether PANX1 is responsible for these phenomena, we generated PANX1-deficient microglia using the CRISPR-Cas9 system. According to protein expression detected by western blot, PANX1 sgRNA targeted pools displayed significant lower expression of PANX1 compared to control sgRNA targeted pool. Accordingly, functional analyses verified that the extracellular concentration of ATP was also drastically reduced across all clones (Figure 8F). Clone#1 cells were used in the subsequent analysis. Two experimental groups using trans-well system were designed as follows: Group (i) cGASKO C20 expressing SOD1Mut were co-incubated with WT C20 cells and or Group (ii) cGASKO C20 expressing SOD1Mut cells were co-incubated with PANX1KO C20 cells. We observed that CRISPR-mediated deletion of PANX1 significantly mitigated NF-κB-dependent gene expression in the trans-well culture conditions (Figure 8G). Similar findings were observed when WT C20 cells were treated with probenecid, an inhibitor of the PANX1 channel (Figure 8H), suggesting that the PANX1 channel plays a crucial role in propagating long-distance intercellular NF-κB-dependent inflammatory responses. We then attempted to establish whether PANX1 and CX36 could synergistically promote neuroinflammation in response to aberrant SOD1. To this end, three independent co-culture systems were established, whereby STINGKO C20 cells expressing either of SOD1Mut were co-incubated with: Group (i) WT C20 cells, or Group (ii) PANX1KO C20 cells, or Group (iii) PANX1-CX36-double deficient C20 cells (PANX1-CX36DKO). We found that co-cultures with PANX1KO cells showed a reduction in the overall inflammatory IFN-I and NF-κB signaling, while PANX1-CX36DKO cells drastically diminished both (Figure 8I). Specifically, the fold-reduction in NF-κB signaling was higher than for IFN-I when PANX1 alone was depleted (Figure 8I, 5-fold vs 2-fold; Group i vs ii), indicating PANX1 possessed a stronger role over CX36 in regulating NF-κB gene expression. To provide further evidence, we reconstituted the expression of PANX1 and/or CX36 in PANX1-CX36DKO C20 cells, which was followed by co-incubation with STINGKO cells expressing aberrant SOD1. Consistently, SOD1Mut alone did not lead to the induction of any tested genes, while co-expressing SOD1Mut with CX36 led to a significantly higher induction of Ifnb1 mRNA overTnf, and Il1b (Figure 8J, blue arrow). Conversely, restoring PANX1 expression with SOD1Mut resulted in higher expression of NF-κB-dependent genes as compared to IFN-I (Figure 8J, red arrow). In summary, the in vitro data document a mechanism by which PANX1 and CX36 channels cooperatively enhance the long-distance intra- and intercellular propagation of IFN-I and NF-κB-dependent inflammatory response to bystander cells, with CX36 for IFN-I, and PANX1 for NF-κB signaling.
Discussion
The role of neuroinflammation in worsening the progression of different ND is gaining increased recognition. These phenomena are dependent on a broader transcriptional program induced in neuronal-related cells, as well as other related cell types. Significant progress has been made in delineating how this transcriptional program is induced and regulated and a plethora of factors acting at the level of signaling pathways and TFs collectively contribute to the outcome of these responses. The mammalian genome encodes various regulatory factors that directly or indirectly controls immune gene expression in the CNS. Although the consequences of inflammation in ALS are well-established, the underlying etiology remains unclear. IFN-I signaling has been shown to play a key role in the pathogenesis of Parkinson’s (Sliter et al., 2018), Alzheimer’s (Roy et al., 2020), and ALS (Wang et al., 2011). Although the mechanisms remain ambiguous, gene expression and immunohistochemical analyses of tissues derived from SOD1-G93A animals and sporadic ALS patients revealed elevated levels of ISGs (Wang et al., 2011). Only very recently, the cGAS/STING signaling pathway was shown to be associated with ALS resulting from a genetic aberration in TDP-43 (Yu et al., 2020) and chromosome nine open reading frame 72 (C9orf72) (McCauley et al., 2020); the former triggering mtDNA release and activating IFN-I via the cGAS-STING pathway, and the latter, harnessing the expansion of hexanucleotide repeat (GGGGCC) in the C9orf72 gene leading to sub-optimal control of inflammation induced by STING-mediated IFN-I responses. Nevertheless, the mechanism underlying how STING is activated, whether via cGAS or via other sensors (if any) are involved in the activation, and if so, how this activation occurs, largely remain unexplored.
In this study, we demonstrated that cGAS/DDX41-STING-driven activation of IFN-I signaling exacerbates and contributes to progression of neurodegeneration in SOD1-driven ALS. Furthermore, we demonstrate that aberrant SOD1 damages cellular mitochondria and enhances the cytosolic release of mtDNA and RNA:DNA hybrids to initiate IFN-I activation. Using both gain-of-function and loss-of-function approaches, we provide critical insights into the biological implications of ALS-causing SOD1Mut both at the cellular and organism level. Unbiased transcriptome profiling of SOD1Mut-derived brain tissues has demonstrated that the presence of SOD1Mut can selectively regulate the expression of a large number of immune-associated genes, all of which playing key roles in host defense against pathogens. Importantly, we have also demonstrated that the ectopic expression of SOD1Mut in various human neuronal, and non-neuronal cell lineages, including macrophages, induces IRF3 activation, followed by the expression of IFN-I associated genes, and NF-κB-dependent pro-inflammatory genes. Our study also demonstrated that STING-driven activation of IFN-I signaling is also elevated in vivo as was evident from our experiments showing that the ALS mouse model (G85R) produced elevated levels of IFN-I associated cytokine proteins. Pharmacological blockade of cGAS, STING, IFNAR, or IFNAR-dependent gene products, CCL2/MCP-1 in the rapidly progressive model (G93A) significantly ameliorated disease progression and prolonged the survival of these animals as compared to their control counterparts. However, our results showed that both cGAS and STING inhibition did not significantly delay the median time of disease onset. These observations are congruent with Yu et al., (2020), who stated that STINGKO-TDP43 double-positive mice did not demonstrate any impact on disease onset. Although inflammation is strongly associated with ALS, it is widely accepted that inflammation does not solely determine the onset of ALS. The exact mechanisms of how SOD1 and other mutated proteins trigger ALS remains unclear. Although we cannot be certain that all these effects are due only to the presence of ALS-causing SOD1Mut, these results indeed provide compelling evidence for the in vivo physiological implications of SOD1Mut in the IFN-I signaling pathway.
In addition to identifying the physiologic functions of SOD1Mut both in isolated cells and in vivo, we further provide mechanistic insights into how SOD1Mut induces IFN-I gene expression. Although cGAS was required to maximize IFN-β gene expression, cGAS deficiency did not completely abrogated the Ifnb1 mRNA abundance in SOD1Mut-expressing microglia. However, the STING-dependent pathway was crucial for a complete abrogation. One explanation for this result is that cGAS and other DNA sensor(s) may induce redundant signaling pathways. We found that expression of ALS-linked SOD1Mut enhanced mitochondrial damage and stimulated cytosolic mtDNA and RNA:DNA release to activate cGAS-, or DDX41-dependent IFN-β gene expression in primary neuronal cells. As for the latter, sensing of mt(RNA:DNA) further recruited BTK to phosphorylate DDX41, and facilitated the activation of STING independent of any secondary messengers for IFN-I signaling. This is well supported by various genetic-knockout models, biochemical fractionation, and functional compensation studies using inhibitors, and enzyme treatments. Moreover, immunofluorescence staining of G85R-derived primary motor neurons and astrocytes clearly revealed the presence of these ligands. Moreover, evidence from our membrane potential (ΔΨ) and confocal experiments collectively revealed the disorganized and fragmentation of mitochondrial structure, lending support to the idea that SOD1Mut may act directly on this organelle, rather than indirectly via other major regulators within the cell.
TBK1 is an important signaling hub active downstream of cGAS. Recruitment of TBK1 to STING is essential to mediate IRF3 phosphorylation and nuclearization, leading to IFN-I production. Heterozygous loss-of-functions mutations in Tbk1 gene have been implicated in familial ALS (Cirulli et al., 2015; Freischmidt et al., 2015), whereas heterozygous deletion of the Tbk1 gene in vivo significantly alleviated end-stage neuroinflammation and delayed disease progression in the SOD1 model (Brenner et al., 2019). Conversely, the basal induction of IFN-I remained unaffected, our experimental model suggested that TBK1 signaling remained intact. In the physiological setting, our observations were concordant with those of a previous study whereby a two-stage meta-analysis involving >4000 ALS patients concluded that the probability of TBK1 mutations was extremely low and was not commonly found in ALS patients (∼1.0–1.8%), despite the fact that their presence, indeed, increased the risk of developing ALS (Cui et al., 2018). Notably, it was reported that TDP-43 triggers the release of mtDNA through mPTP (Yu et al., 2020). However, we confirmed that ALS-causing SOD1Mut do not share such a mechanism. Unlike TDP-43, which invades mitochondria, SOD1 is ubiquitously expressed throughout the cytoplasm, nucleus, endoplasmic reticulum (ER), as well as mitochondria. Evidence also indicates that SOD1Mut are recruited into stress granules (Gal et al., 2016; Lee et al., 2020) and disruption of stress granules have been shown to reduce IFN-I signaling (Ng et al., 2013; Onomoto et al., 2012). Delineating these crosstalk pathways may advance our understanding of the underlying mechanisms of the IFN-I response in SOD1-ALS. Surprisingly, in our system, we observed that the depletion of mPTP does not affect the released of mtDNA by TDP-43 A315T. The underlying mechanism(s) for this discrepancy remains unclear. A potential factor could be the cell-line system used by the different studies. For example, we utilized two neuronal-related cell lines (an immortalized human microglia and astrocytes) in our system, whereas Yu et al., 2020 mainly utilized HEK293T and murine embryonic fibroblasts. Nevertheless, it has been well-characterized that HEK293T cells can serve as a natural “knockout” cell line as they do not express detectable cGAS or STING protein, and therefore, fail to activate IFN-I expression in response to DNA (Burdette et al., 2011; Sun et al., 2013). Nevertheless, it remains unclear how expression of the ALS-linked TDP-43 mutants (i.e., A315T and Q331K) in WT HEK293T cells could still activate the expression of Ifnb1 mRNA as shown in other study (Yu et al., 2020). Future work is required to understand these discrepancies and to demonstrate how SOD1Mut damages the mitochondria.
Our mechanistic studies also indicate that SOD1Mut triggers a unique in trans immune signaling mechanism in microglia, where the cGAMP(2′-5′) produced by cGAS is delivered to bystander cells specifically through the gap junction CX36, which cooperatively amplifies the overall inflammatory reactions and PANX1. The mechanism whereby PANX1 propagates NF-κB-specific genes independently of cell-cell contact and cGAS/STING remains to be fully defined. At least two possibilities exist; first, PANX1 was previously shown to amplify the downstream effect after chronic exposure to key mediators of inflammation, including IL-1, and/or TNF-α through multiple feed-forward and feedback loops by promoting IL-1β production via MyD88-NF-κB activation and extracellular Ca2+ uptake (Yang et al., 2020). Alternatively, PANX1 has been shown to regulate ATP efflux that could serve as a paracrine and autocrine signal to activate P2X purinoceptor 7 (P2X7) in neighboring cells for the inflammasome signalosome (Albalawi et al., 2017; Parzych et al., 2017; Wang et al., 2013). Occasionally, we did observe some SOD1Mut expressing cells with weaker IRF3/NF-κB nuclearization than in bystander cells without SOD1Mut, although the underlying phenomenon remains ambiguous. One hypothesis we developed is that proper localization of STING to the mitochondria-ER-associated membrane is necessary to achieve robust IFN-I signaling (Barber, 2015). SOD1Mut damages mitochondria and disruption of STING localization weakens the signaling cascade, resulting in a weaker nuclearization of IRF3/NF-κB. As for bystander cells, activation of the IRF3/NF-kB pathway is simply based on the transfer of cGAMPs from neighboring cells. The mitochondrial-ER structure remains intact and STING localization is not affected, which leads to more robust IRF3/NF-κB nuclearization. Nonetheless, further clarification is warranted in the future.
In summary, the present study is the first demonstration that loss or inhibition of cGAS/STING signaling significantly extends lifespan and improves the functional performance of a mutant SOD1 disease model. The findings also suggest that cGAS/DDX41-STING signaling contributes to motor neuron death and eventual ALS disease progression. Hence, reducing the downstream consequences of STING activation through specific inhibitors is a promising approach for therapeutic intervention to decelerate ALS disease progression. The insights obtained from this investigation shall further advance our understanding of the physiological roles of SOD1Mut in STING-mediated IFN-I signaling and will contribute to expand the significance of these pathways in NDs.
Limitations of the study
This study also had limitations. First, all analyses were conducted using SOD1-G85R and G93A mice or were based on the SOD1-G85R and G93A genetic background, which have limitations in terms of modeling all forms of ALS, particularly TDP-43, FUS, C9orf72, and other major hallmarks of sporadic and familial ALS. Another major limitation is that we did not attempt to longitudinally investigate the kinetics of the immune response in correspondence to disease progression in the CNS. Immune activation is often considered a dynamic process and may change throughout the course of the illness; therefore, a longitudinal study would have been preferred for a cross-sectional investigation. Finally, the present data obtained using murine models may imperfectly predict efficacy in humans. Our conclusions could therefore be strengthened by repeating the analyses using actual patient samples from an ALS clinic. Larger sample sizes are preferred, as this would increase the statistical power of the analysis, and hence improve the capacity to detect smaller phenotypes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CX43 | Cell Signaling Technology | Cat#3512S; RRID: AB_2294590 |
| Rabbit monoclonal anti-VDAC (D73D12) | Cell Signaling Technology | Cat#4661; RRID: AB_10557420 |
| Rabbit monoclonal anti-RIG-I (D14G6) | Cell Signaling Technology | Cat#3743; RRID: AB_2269233 |
| Rabbit monoclonal anti-MDA5 (D74 × 104) | Cell Signaling Technology | Cat#5321; RRID: AB_10694490 |
| Rabbit anti-SOD1 | Cell Signaling Technology | Cat#2770; RRID: AB_2302392 |
| Rabbit monoclonal anti-MAVS (D5A9E, H) | Cell Signaling Technology | Cat#24930; RRID: AB_2798889 |
| Rodent specific anti-MAVS (Mouse/Rat) | Cell Signaling Technology | Cat#4983; RRID: AB_823566 |
| Rabbit monoclonal anti-COX IV (3 × 1011) | Cell Signaling Technology | Cat#4850; RRID: AB_2085424 |
| Rabbit monoclonal anti-STAT1 (D1K9Y) | Cell Signaling Technology | Cat#14994; RRID: AB_2737027 |
| Rabbit monoclonal anti-STING (D2P2F) | Cell Signaling Technology | Cat#13647; RRID: AB_2732796 |
| Rabbit monoclonal anti-TBK1/NAK (D1B4) | Cell Signaling Technology | Cat#3504; RRID: AB_2255663 |
| Rabbit monoclonal anti-phoshoTBK1/NAK1 (Ser172, D52C2) | Cell Signaling Technology | Cat#5483; RRID: AB_10693472 |
| Rabbit monoclonal anti-phospho-IRF3 (Ser396, 4D4G) | Cell Signaling Technology | Cat#4947; RRID: AB_823547 |
| Mouse monoclonal anti-dsDNA [35I9 DNA] | Abcam | Cat#ab27156; RRID: AB_470907 |
| Rabbit polyclonal anti-DDX41 | ABclonal Inc. | Cat#A6576; RRID: AB_2767170 |
| Rabbit polyclonal Lamin A/C [KO validated] | ABclonal Inc. | Cat#A0249; RRID: AB_2757062 |
| Rabbit polyclonal anti-HSP60/HSPD1 | ABclonal Inc. | Cat#A0969; RRID: AB_2757488 |
| Rabbit polyclonal anti-IRF3 [KO validated] | ABclonal Inc. | Cat#A11118; RRID: AB_2861504 |
| Rabbit polyclonal anti-PANX1 | ABclonal Inc. | Cat#A13587; RRID: AB_2760449 |
| Rabbit polyclonal anti-GJC1/CX45 | ABclonal Inc. | Cat#A7053; RRID: AB_2767608 |
| Rabbit polyclonal anti-cGAS | ABclonal Inc. | Cat#A8335; RRID: AB_2770305 |
| Rabbit polyclonal anti-phospho-cGAS Y215 | ABclonal Inc. | Cat#AP0946; RRID: N/A |
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Mouse monoclonal anti-O4 | Sigma-Aldrich | Cat#O7139; RRID: AB_477662 |
| Mouse monoclonal anti-CD45 (clone 30-F11) | BD Pharmingen | Cat#550539; RRID: AB_2174426 |
| Sheep polyclonal anti-ITGB5 | R&D Systems | Cat#AF3824; RRID: AB_1151977 |
| Mouse monoclonal anti-L1, clone 324 | Millipore Sigma | Cat#MAB5272; RRID: AB_2133200 |
| Mouse monoclonal anti-DNA:RNA hybrid, clone S9.6 | Millipore Sigma | Cat#MABE1095; RRID: AB_2861387 |
| Mouse monoclonal anti-NK1.1 | Thermo Fisher Scientific | Cat#MA1-70100; RRID: AB_2296673 |
| Mouse monoclonal anti-NF-κB p65 (F-6) | Santa Cruz Biotechnology | Cat#sc-8008; RRID: AB_628017 |
| Mouse monoclonal anti-mtTFA (F6) | Santa Cruz Biotechnology | Cat#sc-166965; RRID: AB_10610743 |
| Mouse monoclonal anti-Pol II (A10) | Santa Cruz Biotechnology | Cat#sc-17798; RRID: AB_677355 |
| Rabbit polyclonal anti-TOM20 (FL-145) | Santa Cruz Biotechnology | Cat#sc-11415; RRID: AB_2207533 |
| Mouse monoclonal anti-GFAP | Santa Cruz Biotechnology | Cat#sc-33673; RRID: AB_627673 |
| Mouse monoclonal anti-Iba1 | Santa Cruz Biotechnology | Cat#sc-32725; RRID: AB_667733 |
| Mouse monoclonal anti-CD4 | Santa Cruz Biotechnology | Cat#sc-19641; RRID: AB_2009035 |
| Mouse monoclonal anti-CD19 | Santa Cruz Biotechnology | Cat#sc-; RRID: AB_667733 |
| Mouse monoclonal anti-β-Actin (C4) | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_2714189 |
| Mouse monoclonal anti-β-tubulin (G8) | Santa Cruz Biotechnology | Cat#sc-55529; RRID: AB_2210962 |
| Mouse monoclonal anti-CD68 | Santa Cruz Biotechnology | Cat#sc-20060; RRID: AB_627158 |
| Mouse monoclonal anti-VP1 Clone 31A2 | Mediagnost | Cat#M47; RRID: N/A |
| InVivoMAb anti-mouse IFNAR-1 | Bio X Cell | Cat#BE0241; RRID: N/A |
| InVivoMAb anti-mouse CCL2 (MCP-1) | Bio X Cell | Cat#BE0185; RRID: N/A |
| Bacterial and virus strains | ||
| CVB3 | ATCC | Cat#VR-300 |
| HCoV-OC43 | ATCC | Cat#VR-1558 |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI Medium 1640 (1X) | Gibco | Cat#11875-093 |
| RPMI Medium 1640 D-Glucose (−) | Gibco | Cat#11879-020 |
| Fetal Bovine serum (FBS) | Gibco | Cat#12483-020 |
| HEPES (1M) pH 7.5 | Gibco | Cat#15630-080 |
| 2-mercaptoethanol (1000X) | Gibco | Cat#21985-023 |
| N2-supplement (100X) | Thermo Fisher | Cat#17502-048 |
| B-27 supplement (50X), serum free | Thermo Fisher | Cat#17504044 |
| Cyclosporin A (CsA) | Sigma-Aldrich | Cat#SML1018 |
| Heparin-Binding EGF-Like Growth Factor | Sigma-Aldrich | Cat#E4643 |
| LFM-A13 | Sigma-Aldrich | Cat#435300 |
| 2′,3′-Dideoxycytidine (ddC) | Sigma-Aldrich | Cat#D5782 |
| Corn oil | Sigma-Aldrich | Cat#C8267 |
| Tween-80 | Sigma-Aldrich | Cat#P4780 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat#D8418 |
| Dexamethasone | Sigma-Aldrich | Cat#D4902 |
| Carbenoxolone disodium salt | Sigma-Aldrich | Cat#C4790 |
| Brefeldin A | Sigma-Aldrich | Cat#B5936 |
| Probenecid | Sigma-Aldrich | Cat#P3000000 |
| DMEM | Sigma-Aldrich | Cat#D6429 |
| DMEM Ham’s:F12 Mixture | Sigma-Aldrich | Cat#51445C |
| Dulbecco’s PBS | Sigma-Aldrich | Cat#D8662 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat#P4333 |
| Sodium pyruvate solution | Sigma-Aldrich | Cat#S8636 |
| Hematoxylin solution Gill No.2 | Sigma-Aldrich | Cat#GHS232 |
| Herring testes (HT)-DNA | Sigma-Aldrich | Cat#D6898 |
| Digitonin | Sigma-Aldrich | Cat#D141 |
| Tunicamycin | Abcam | Cat#ab120296 |
| RNase H | NEB | Cat#0297S |
| DNase I | NEB | Cat#M0303S |
| IFN-β-human recombinant proteins | Bio Basic Canada | Cat#RC217-16 |
| Image-iT FX Signal Enhancer | Invitrogen | Cat#I36933 |
| UltraPure™ Ethidium Bromide | Invitrogen | Cat#15585011 |
| Lipofectamine 2000 | Invitrogen | Cat#11668027 |
| Lipofectamine RNAiMAX | Invitrogen | Cat#13778075 |
| Normocin™ | Invivogen | Cat#ant-nr-1 |
| High molecular weight poly(I:C) | Invivogen | Cat#tlrl-pic |
| Protease inhibitor cocktail tablets | Roche | Cat#05892791001 |
| DNAfectin™ Plus | ABM Canada | Cat#G2500 |
| TransIT-X2 Dynamic Delivery System | Mirus Bio | Cat#MIR 6004 |
| TNFR1 Blocking Peptide | Fitzgerald | Cat#33R-10641 |
| RU.521 | Cayman Chemical | Cat#31765 |
| C-176 | Cayman Chemical | Cat#25859 |
| H-151 | Cayman Chemical | Cat#25857 |
| Fluo-cGAMP | BIOLOG Life Science Institute | Cat#C195-001 |
| Protein G-SepharoseTM 4 Fast Flow | GE Healthcare Biosciences | Cat#17-0618 |
| Critical commercial assays | ||
| CyQuant™ LDH Cytotoxicity kit | Thermo Fisher | Cat#C20301 |
| POWER SYBR® Green RNA-to-CTTM kit | Thermo Fisher | Cat#4389986 |
| ATP assay kit | Sigma-Aldrich | Cat#MAK190 |
| MMP-assay kit | Sigma-Aldrich | Cat# MAK147-1KT |
| Universal Mycoplasma detection kit | ATCC | Cat#30-1012K |
| QIAGEN Plasmid Plus kit | QIAGEN | Cat#12941 |
| RNeasy Mini-extraction kit | QIAGEN | Cat#74104 |
| QIAmp DNA Mini Kit | QIAGEN | Cat#51304 |
| Mouse ChAT ELISA Kit | Antibodies.com | Cat#A77879 |
| QuicKey Mouse IgG ELISA Kit | Elabscience | Cat#E-TSEL-M0003 |
| Mouse IgM ELISA Kit | Elabscience | Cat#E-EL-M3036 |
| Mouse ANA (Anti-Nuclear Antibody) ELISA | MyBioSource | Cat#MBS261480 |
| 2′3′-cGAMP ELISA Kit | Cayman Chemical | Cat#501700 |
| VeriKine mouse IFN Beta ELISA Kit, High Sensitivity | PBL Assay Science | Cat#42410-1 |
| Deposited data | ||
| RAW and analyzed data | This paper | GEO: GSE167974 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | Cat#CCL-2.2; RRID: CVCL_0030 |
| U2-OS | ATCC | Cat#HTB-96; RRID: CVCL_0042 |
| HEK293T | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| THP-1 | ATCC | Cat#TIB-202; RRID: CVCL_0006 |
| RAW264.7 | ATCC | Cat#TIB-71; RRID: CVCL_0493 |
| SH-SY5Y | ATCC | Cat#CRL-2266; RRID: CVCL_0019 |
| Neuro2a | ATCC | Cat#CCL-131; RRID: CVCL_0470 |
| SIM-A9 | ATCC | Cat#CRL-3265; RRID: CVCL_5I31 |
| HASTR/ci35 | From Dr. Tomomi Furihata (TUPLS, Japan) | Cat#N/A; RRID: CVCL_4U69; (Furihata et al., 2016; Kitamura et al., 2018) |
| C20 | From Dr. David Alvarez-Carbonell (CWRU, USA) | Cat#N/A; RRID: N/A; (Garcia-Mesa et al., 2017) |
| NSC-34 | From Dr. Neil R. Cashman (UBC, Canada) | Cat#N/A; RRID: CVCL_D356; (Cashman et al., 1992) |
| MO3.13 | From Dr. Neil R. Cashman (UBC, Canada) | Cat#N/A; RRID: CVCL_D357; (McLaurin et al., 1995) |
| Experimental models: Organisms/strains | ||
| Wild-type C57BL/6J background mice | The Jackson Laboratory | Cat#000664 |
| B6.Cg-Tg (SOD1∗G85R)148Dwc/J mice | The Jackson Laboratory | Cat#008248 |
| B6.Cg-Tg(SOD1∗G93A)1Gur/J mice | The Jackson Laboratory | Cat#004435 |
| B6.Cg-Tmem173tm1.2Camb/J mice | The Jackson Laboratory | Cat#025805 |
| Oligonucleotides | ||
| HCoV-OC43 Nucleocapsid (N): F: CGATG AGGCTATTCCGACTAGGT R: CCTTCCTGAGCCTTCAATATAGTAACC |
Integrated DNA Technologies (IDT) | (Hammitt et al., 2011) |
| CVB3 2A gene: F: GCTTTGCAGACATCCGTGATC R: CAAGCTGTGTTCCACATAGTCCTTCA |
Integrated DNA Technologies (IDT) | N/A |
| CVB3 VP1 gene: F: ACATGGTGCGAAGAGTCTATTGAG R: TGCTCCGCAGTTAGGATTAGC |
Integrated DNA Technologies (IDT) | N/A |
| Murine IFNA4 gene: F: CTGCTACTTGGAATGCAACTC R: CAGTCTTGCCAGCAAGTTGG |
Integrated DNA Technologies (IDT) | (Mattei et al., 2013) |
| Murine IFNB1 gene: F: GCCTTTGCCATCCAAGAGATGC R: ACACTGTCTGCTGGTGGAGTTC Human IFNB1 gene: F: CAACTTGCTTGGATTCCTACAAAG R: TATTCAAGCC TCCCATTCAATTG |
Integrated DNA Technologies (IDT) | N/A |
| Murine RSAD2 gene: F: TGGGGATGCTGGTGCCCACT R: ACCCCGGACCTGTGGCTGTT Human RSAD2 gene: F: TTGGACATTCTCGCTATCTCCT R: AGTGCTTTGAT CTGTTCCGTC |
Integrated DNA Technologies (IDT) | N/A |
| Murine ISG15 gene: F: CAAGCAGCCAGAAGCAGACT R: CCCAGCATCTTCACCTTTAGG Human ISG15 gene: F: CGCAGATCACCCAGAAGATCG R: TTCGTCGCAT TTGTCCACCA |
Integrated DNA Technologies (IDT) | N/A |
| Murine TNF gene: F: GTCCCCAAAGGGATGAGAAGTT R: GTTTGCTACGACGTGGGCTACA Human TNF gene: F: CCTCTCTCTAATCAGCCCTCTG R: GAGGACCTGG GAGTAGATGAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine CXCL10 gene: F: GCTGGGATTCACCTCAAGAA R: CTTGGGGACACCTTTTAGCA Human CXCL10 gene: F: GTGGCATTCAAGGAGTACCTC R: TGATGGCCTTC GATTCTGGATT |
Integrated DNA Technologies (IDT) | N/A |
| Human CCL3 gene: F: ATGCAGGTCTCCACTGCTGCCCTT R: GCACTCAGCTCCAGGTCGCTGACAT |
Integrated DNA Technologies (IDT) | (Chui and Dorovini-Zis, 2010) |
| Murine CCL5 gene: F: GCTGCTTTGCCTACCTCTCC R: TCGAGTGACAAACACGACTGC |
Integrated DNA Technologies (IDT) | N/A |
| Murine IL-6 gene: F: ACAACCACGGCCTTCCCTAC R:TCTCATTTCCACGATTTCCCAG Human IL-6 gene: F: ACTCACCTCTTCAGAACGAATTG R: CCATCTTT GGAAGGTTCAGGTTG |
Integrated DNA Technologies (IDT) | N/A |
| Murine IL-1B gene:F: GCAACTGTTCCTGAACTCAACT R: ATCTTTTGGGGTCCGTCAACT Human IL-1B gene: F: CCACAGACCTTCCAGGAGAATG R: GTGCAGTTC AGTGATCGTACAGG |
Integrated DNA Technologies (IDT) | N/A |
| Human NOS2 gene: F: AGGGACAAGCCTACCCCTC R: CTCATCTCCCGTCAGTTGGT |
Integrated DNA Technologies (IDT) | N/A |
| Human IRF7 gene: F: GCTGGACGTGACCATCATGTA R: GGGCCGTATAGGAACGTGC |
Integrated DNA Technologies (IDT) | N/A |
| Human TNFAIP3 gene: F: TTGTCCTCAGTTTCGGGAGAT R: ACTTCTCGACACCAGTTGAGTT |
Integrated DNA Technologies (IDT) | N/A |
| Murine AXL gene: F: ATGGCCGACATTGCCAGTG R: CGGTAGTAATCCCCGTTGTAGA |
Integrated DNA Technologies (IDT) | (Zou et al., 2020) |
| Murine B2M gene: F: ACAGTTCCACCCGCCTCACATT R: TAGAAAGACCAGTCCTTGCTGAAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine AIF1 gene: F: TCTGCCGTCCAAACTTGAAGCC R: CTCTTCAGCTCTAGGTGGGTCT |
Integrated DNA Technologies (IDT) | N/A |
| Human IBA1 gene: F: CCCTCCAAACTGGAAGGCTTCA R: CTTTAGCTCTAGGTGAGTCTTGG |
Integrated DNA Technologies (IDT) | N/A |
| Human CD11B gene: F: GGAACGCCATTGTCTGCTTTCG R: ATGCTGAGGTCATCCTGGCAGA |
Integrated DNA Technologies (IDT) | N/A |
| Murine SLC1A2 gene: F: ACAATATGCCCAAGCAGGTAGA R: CTTTGGCTCATCGGAGCTGA |
Integrated DNA Technologies (IDT) | N/A |
| Murine GFAP gene: F: CGGAGACGCATCACCTCTG R: AGGGAGTGGAGGAGTCATTCG |
Integrated DNA Technologies (IDT) | N/A |
| Murine FGFR3 gene: F: GGAGGACGTGGCTGAAGAC R: GGAGCTTGATGCCCCCAAT |
Integrated DNA Technologies (IDT) | N/A |
| Murine AQP4 gene: F: CTTTCTGGAAGGCAGTCTCAG R: CCACACCGAGCAAAACAAAGAT |
Integrated DNA Technologies (IDT) | N/A |
| Murine GJB6 gene: F: ACCAGCATAGGGAAGGTGTG R: TGCAGAGTGTTGCAGACAAAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine GJC2 gene: F: TCCACAATCATTCCACCTTCG R: CAGAAGCGCACATGAGACAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine SOX10 gene: F: ACACCTTGGGACACGGTTTTC R: TAGGTCTTGTTCCTCGGCCAT |
Integrated DNA Technologies (IDT) | N/A |
| Murine SIGLEC4A gene: F: CTGCCGCTGTTTTGGATAATGA R: CATCGGGGAAGTCGAAACGG |
Integrated DNA Technologies (IDT) | N/A |
| Murine MOG gene: F: `AGCTGCTTCCTCTCCCTTCTC R: ACTAAAGCCCGGATGGGATAC |
Integrated DNA Technologies (IDT) | N/A |
| Murine MBP gene: F: GGCGGTGACAGACTCCAAG R: GAAGCTCGTCGGACTCTGAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine HB9/MNX1 gene: F: CAGCACCTTCCAACTGGACCAG R: TTCGGCACTTCCCCAAGAGGTT |
Integrated DNA Technologies (IDT) | N/A |
| Murine ISL1 gene: F: GGTCTCTGGAACATCCCACATTGT R: CTGTTCCTACTCCCCATTCACT |
Integrated DNA Technologies (IDT) | N/A |
| Murine CHAT gene: F: GCTTGAATGGAGCGAATCGTTGG R: CACCAGGACGATGCCATCAAAAG |
Integrated DNA Technologies (IDT) | N/A |
| Murine ACTB gene: F: CATTGCTGACAGGATGCAGAAGG R: TGCTGGAAGGTGGACAGTGAGG Human ACTB gene: F: ACTGGAACGGTGAAGGTGAC R: GTGGACTTGGGA GAGGACTG |
Integrated DNA Technologies (IDT) | N/A |
| MT-DNA D-LOOP: F: AATCTACCATCCTCCGTGAAACC R: TCAGTTTAGCTACCCCCAAGTTTAA |
Integrated DNA Technologies (IDT) | N/A |
| MT-DNA MT-CO1: F: GACGTAGACACACGAGCATATTTCA R: AGGACATAGTGGAAGTGAGCTACAAC |
Integrated DNA Technologies (IDT) | N/A |
| MT-DNA MT-ND1: F: CCACCTCTAGCCTAGCCGTTTA R: GGGTCATGATGGCAGGAGTAAT |
Integrated DNA Technologies (IDT) | N/A |
| MT-DNA MT-ATP6: F: TAGCCATACACAACACTAAA GGACGA R: GGGCATTTTTAATCTTAGAGCGAAA |
Integrated DNA Technologies (IDT) | N/A |
| Nuclear TERT gene: F: CTAGCTCATGTGTCAAGACCC TCTT R: GCCAGCACGTTTCTCTCGTT |
Integrated DNA Technologies (IDT) | N/A |
| RIG-I/DDX58 sgRNA (Human): 5′-GGATTAT ATCCGGAAGACCC-3′ |
Applied Biological Materials Inc. | N/A |
| MDA5/IFIH1 sgRNA (Human): 5′-CGAATTCC CGAGTCCAACCA-3′ |
Applied Biological Materials Inc. | (Ng et al., 2020) |
| MAVS sgRNA (Human): 5′-ATTGCGGCAGAT ATACTTAT-3′ |
Applied Biological Materials Inc. | N/A |
| GJA1 (CX43) sgRNA (Human): 5′-CTTGTCAA GGAGTTTGCCTA-3′ |
Applied Biological Materials Inc. | N/A |
| GJC1 (CX45) sgRNA (Human): 5′-ATCGTCCTT ACAGCTGTAGG-3′ |
Applied Biological Materials Inc. | N/A |
| GJD2 (CX36) sgRNA (Human): 5′-AGCACTCCA CTATGATCGGG-3′ |
Applied Biological Materials Inc. | N/A |
| PANX1 sgRNA (Human): 5′-AATCGAGATCTC CTGCGCGA-3′ |
Applied Biological Materials Inc. | N/A |
| TBK1 sgRNA (Human): 5′-AGAGCACTTCTAA TCATCTG-3′ |
Applied Biological Materials Inc. | N/A |
| IRF3 sgRNA (Human): 5′-TTGGAAGCACGG CCTACGGC-3′ |
Applied Biological Materials Inc. | N/A |
| TICAM1/TRIF sgRNA (Human): 5′-GTAGGCC ACGTCCCGCAGCG-3′ |
Applied Biological Materials Inc. | N/A |
| cGAS/MAB21L3 sgRNA (Human): 5′-ATGGC CTTTCCGTGCCA-3′ |
Applied Biological Materials Inc. | N/A |
| STING/TMEM173 sgRNA (Human): 5′-ACGGG ATGGATGGATGC-3′ |
Applied Biological Materials Inc. | N/A |
| STAT1 sgRNA (Human): 5′-TCCCATTACAGG CTCAGTCG-3′ |
Applied Biological Materials Inc. | N/A |
| TFAM sgRNA (Human): 5′-GCGTTTCTCCG AAGCATGTG-3′ |
Applied Biological Materials Inc. | N/A |
| DDX41 sgRNA (Human): 5′-AAGAGCGACAT GAGCGCGTG-3′ |
Applied Biological Materials Inc. | N/A |
| siRNAs targeting human PPID gene: ID: s10913, s10914, and s10915 | Thermo Fisher | Cat#4390824 |
| siRNAs targeting human ZBP1 gene: ID: s37484, and s37485 | Thermo Fisher | Cat#4390420 |
| siRNAs targeting human IFI16 gene: ID: s7136, and s7137 | Thermo Fisher | Cat#4392420 |
| Recombinant DNA | ||
| p125-Luciferase reporter (IFN-β) | From Dr. Takashi Fujita (Kyoto University, Japan) | (Yoneyama et al., 1998) |
| p55C1B-Luciferase reporter (IRF3) | From Dr. Takashi Fujita (Kyoto University, Japan) | (Narita et al., 2014) |
| p55A2-Luciferase reporter (NF-κB) | From Dr. Takashi Fujita (Kyoto University, Japan) | (Narita et al., 2014) |
| pEF-Tak-Flag-MAVS | From Dr. Takashi Fujita (Kyoto University, Japan) | (Onoguchi et al., 2010) |
| pEF-Flag-IRF3 5D | From Dr. Takashi Fujita (Kyoto University, Japan) | (Mori et al., 2004) |
| pcDNA-Flag-cGAS WT, ΔcGAMP, ΔDNA, K394E, and Y215E. | From Dr. Nelson O. Gekara (Stockholm University, Sweden) | (Jiang et al., 2019) |
| pEGFP-SOD1-WT, G93A, and G85R | From Dr. Neil R. Cashman (UBC, Canada) | (Pokrishevsky et al., 2017) |
| pcDNA-Flag-SOD1-WT, A4V, G37R, H46R, E100G, G93A, G85R, V148G, V148I, and G127X | From Dr. Christine Vande Velde (Université deMontréal, Canada) | (Semmler et al., 2020) |
| pGL4.32 (luc2P/NF-κB-RE/Hygro) | Promega | Cat#E8491 |
| pRL-TK Renilla Luciferase Control reporter vector | Promega | Cat#E2231 |
| Software and algorithms | ||
| Prism version 8.0 | GraphPad | https://www.graphpad.com/ |
| Microsoft Office Home and Student 2019 | Microsoft Corporation | https://www.microsoft.com/microsoft-365 |
| Adobe Illustrator 2020 | Adobe Inc. | http://www.adobe.com |
Resource availability
Lead contact
Further information and reasonable requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chen Seng Ng (chenseng.ng@xmu.edu.my).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals and experimental procedures
WT C57BL/6J (#000664), transgenic SOD1-G85R, and transgenic SOD1-G93A mice expressing high copy numbers of mutant SOD1 in a C57BL6/J background were all obtained from The Jackson Laboratory. Sting−/− (also known asMPYS−/− or Tmem173−/−) and SOD1-G93A mice were crossed to generate Sting−/− G93A (G93A/Sting−/−)-double-positive mice. These mice were housed under a 12-h/12-h light-dark cycle under controlled-temperature conditions (∼20–22°C) and relative humidity (50%) with standard laboratory diet and water ad libitum unless otherwise indicated. Mice were bred and maintained in specific-pathogen free condition. Pregnant mice were individually housed in translucent plastic cages with corncob bedding. Animals were monitored weekly for changes in weight and behavior otherwise as further described in figure legends. The animals were euthanized with isoflurane at different time-points as indicated in the figure legends. Tissues including the brain, spinal cord, and other internal organs were harvested and either fixed with 4% paraformaldehyde or flash-frozen with liquid nitrogen then stored at −80°C. For the survival study, animals were inspected twice daily for health issues, and natural death was recorded for each animal. Moribund animals were euthanized, and every animal found dead was necropsied. For administration of neutralizing antibodies, and pharmacological inhibitors, WT, SOD1-G93A mice were injected with 500 μg of neutralizing antibodies (anti-IFNAR, and anti-MCP-1) or an equivalent amount of vehicle (PBS) by intraperitoneal injection two-times per week for the first 4-weeks, three-days apart for the second injection in each week, then once a week afterwards. As for inhibitors, 8-weeksold WT, and SOD1-G93A mice were injected with 500 μg of C-176, H-151, or RU.521 or an equivalent amount of vehicle (containing 90% corn oil+10% Tween80, v/v) intraperitoneally two-times per week for the first 4-weeks and three-days apart for the second injection in each week, then once a week thereafter. No adverse effects were detected in the mice during treatment. For all experiments, adequate measures were taken to minimize any discomfort or pain. The study was performed in compliance with the ethical standards of the IACC of UBC, as well as in accordance to Malaysian laws. Mice were sibling-matched between treatment groups, all WT and SOD1 (G85R and G93A) mutant mice used in experimentation were male mice, and 8-weeksold adult mice were used in most experimentation unless otherwise as indicated in respective figure legends.
Method details
Motor function test
Hindlimb-clasping test
In the hindlimb-clasping test, the mouse was gently lifted by the tail, grasped near its base, and the hindlimb position was observed for 10 s and scored as follows: if the hindlimbs were consistently splayed outward, away from the abdomen, displaying normal escape extension with no sign of limb clasping, it was assigned a score of 0; if one hindlimb was retracted toward the abdomen for 50% of the time suspended and showed a loss of mobility, it received a score of 1; if both hindlimbs were partially retracted toward the abdomen for 50% of the time suspended, and show loss of mobility, it received a score of 2; and if both hindlimbs were entirely retracted and touching the abdomen for 50% of the time while suspended, followed by loss of mobility, it received a score of 3. Finally, if both forelimbs and hindlimbs exhibited clasping and were crossed for 50% of the observation period, coupled with loss of mobility, it received a score of 4.
Grip-strength test
A digital grip strength meter (Columbus Instruments) was used to automatically measure maximal hindlimb muscle grip strength. Mice were held by their tail and lowered until their hindlimb grasped the standard pull-bar connected to the digital force gauge. The tail was lowered until the body was in line with the device, followed by gently pulling away the mice from the pull-bar with a smooth steady motion until both of their hindlimbs are released from the bar. The strength of the hindlimb grip was quantified in gram force. Each mouse was given five attempts and the average of the grip strength from these attempts are recorded.
Gait analysis
At the beginning of this test, the personnel responsible for testing and scoring were blinded to animal genotype or the experimental condition until gait analysis and scoring of papers has been completed for the entire study. The forepaws and hind paws were covered with two nontoxic, water-soluble color inks (green color for forepaws and red color for hind paws). Small amounts of sterilized sunflower seeds were placed in housing cages 2-days prior to testing to facilitate habituation. Mice were then allowed to walk along an 89.0-cm-long and 39.0-cm-wide runway with a goal chamber placed at the endpoint. On the day of testing, a small quantity of sunflower seeds was placed inside the goal chamber to encourage the mice to walk through without stopping. The runway was covered with sheets of white paper. After reaching the goal chamber, the mice were retrieved and gently the feet gently wiped with water-dampened Kimtech wipers. The footprints were analyzed manually at the indicated time-points after confirming the footprints were completely dry, and the stride length (defined as the distance between two sequential footprints created by the same foot) of the hindlimbs was quantified.
Cell lines, cell culture, and transfection conditions
Both HEK293T (ATCC® CRL-3216TM) and HeLa cells (ATCC® CCL-2TM) were maintained at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS, Gibco, Thermo Fisher), 1x Penicillin-Streptomycin (Pen-Strep, Sigma), and 100 μg/mL Normocin (Invivogen). THP-1 monocyte cells (ATCC® TIB-202TM) were maintained at 37°C and 5% CO2 in RPMI 1640 (Gibco) supplemented with 10% FBS, 1x Pen-Strep, 0.05 mM of β-mercaptoethanol, 10 mM HEPES, and 1 mM of sodium pyruvate. Immortalized human microglia (Clone#20, C20) were a kind gift from Dr. David Alvarez-Carbonell (Case Western Reserve University) (Garcia-Mesa et al., 2017). In brief, C20 were maintained at 37°C and 5% CO2 in DMEM/Nutrient Mixture F12 Ham with L-glutamine (F12:DMEM, Sigma) supplemented with 1x FBS, 1x Pen-Strep, 1.0 μM dexamethasone, and 100 μg/mL of normocin. Immortalized human astrocytes (HASTR/ci35) were cultured as previously described (Furihata et al., 2016). In brief, HASTR/ci35 were seeded in sterile culture dishes pre-coated with collagen I in the presence of DMEM media containing N2 supplement (1%), 10% FBS, penicillin-streptomycin, GlutaMax-I, and blasticidin (4.0 μg/mL) and incubated at 33°C with 5% CO2. DNA-related materials were transfected using TransIT-X2 transfection reagent according to manufacturer’s protocol. RNA-related materials were transfected using RNAi-MAX according to the manufacturer’s protocol. All tissue culture cells were tested for Mycoplasma and were confirmed to be negative using Mycoplasma detection kit. All the above experiments were performed in certified BSL-2 facilities.
Immunopanning of murine primary neuronal and non-neuronal cells and culture conditions
Microglia/macrophages, oligodendrocyte lineage cells, neurons and astrocytes were purified by immunopanning from the P10 mice cerebral cortex and cultured as previously described (Foo, 2013). In brief, mouse cortices were first enzymatically (papain) and then mechanically dissociated to generate a single-cell suspension that was incubated on multiple antigen-specific antibody plated immunopanning plates to isolate microglia (secondary antibody, Jackson ImmunoResearch and CD45 antibody), oligodendrocyte lineage cells (O4 antibody), neurons (L1 antibody), and astrocytes (ITGB5 antibody). Isolated microglia/macrophages were cultured in fresh DMEM/F1 media containing 10% FBS, 1% penicillin/streptomycin, 1% L-glutamine, and 1% sodium pyruvate (Sarkar et al., 2017). Isolated oligodendrocyte lineage cells were lysed for downstream analysis immediately after immunopanning. Isolated neurons were cultured in growth medium containing 50% DMEM and 50% neurobasal with 1 mM sodium pyruvate, 2 mM L-glutamine, 1% penicillin/streptomycin, 100 μg/mL human transferrin, 60 ng/mL progesterone, 16 μg/mL putrescine, 40 ng/mL sodium selenite, 40 ng/mL thyroxine, 30 ng/mL tri-idothyronine, and B27 Supplement (Dugas et al., 2008). Isolated astrocytes were cultured in growth medium containing 50% DMEM and 50% neurobasal with 1% penicillin/streptomycin, 1 mM sodium pyruvate, 292 μg/mL L-glutamine, 1x SATO and 5.0 μg/mL of N-acetyl cysteine. This medium was supplemented with the astrocyte-required survival factor HBEGF (Sigma-Aldrich) at 5 ng/mL (Foo, 2013).
Generation of CRISPR-Cas9-deficient cell lines
All CRISPR-Cas9 sgRNAs constructs used in this study were custom-made and synthesized through ABM. Constructs targeted isoform-2 of each respective protein in the human lineage. Sequences for sgRNAs are provided in the Key resources table. Cells of interest were seeded and house in 37°C with CO2 for overnight. Cells were then transfected with CRISPR-Cas9 expressing sgRNA targeting gene of interest for 48 h. Cells were then trypsinized and replated at a density of 10 cells/mL in a 96-well tissue culture plate containing 100 μL media per well. The number of cells in the plate were assessed after 24 h. Wells with only one cell were marked. On day 5, wells that were marked at the initial assessment were re-evaluated. Only wells with one colony were considered monoclonal and were marked. After these wells were identified, colony numbers were verified every week until the well reached a high confluence. Single colonies/well were expanded by transferring to a 12-well tissue culture plate. Once the 12-well plate clones had expanded, they were passaged to a 6-well plate. Knockout efficiency was validated through immunoblotting using specific antibodies and functional analyses.
Mitochondria DNA subcellular fractionation, depletion, and membrane potential assay
Isolation and purification of cytoplasmic mitochondrial DNA was adopted and modified as previously described (West et al., 2015). In brief, C20 cells were lysed in digitonin lysis buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 25 μg/mL digitonin, supplemented with protease and phosphatase inhibitors cocktail, Roche) and incubated on a rotator at 4°C for 15 min. Samples were then centrifuged at speed of 4226 rpm for 10 min at 4°C. Supernatants were then transferred to fresh tubes and centrifuged three times at 13,363 rpm for 20 min at 4°C. Supernatants were transferred to fresh tubes between centrifugation steps to finally yield cytosolic fractions. The cytosolic fraction was split into two tubes (one for total DNA extraction and one for immunoblot analysis). The remaining pellet from the first spin was resuspended in ice-cold PBS to wash away the digitonin buffer. Samples were then centrifuged at 4226 rpm for five min at 4°C. The wash solution was aspirated, and samples were resuspended in NP-40 lysis buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 1% NP-40, protease, and phosphatase inhibitors) and incubated on ice for 30 min. Samples were centrifuged at 7,906 rpm for 10 min at 4°C to yield the crude mitochondria fraction for immunoblot analysis. The remaining pellet was resuspended in ice-cold PBS to remove away NP-40 buffer. Samples were divided into two tubes (one for total DNA extraction and one for cellular lysates). One tube was centrifuged at 4,226 rpm for 5 min at 4°C. The wash solution was aspirated and samples were resuspended in RIPA lysis buffer to yield nuclear fraction for immunoblot analysis. The DNA was subsequently extracted from the appropriate cytosolic and nuclear fractions using the QIAmp DNA Mini Kit (QIAGEN). The concentration of cytosolic DNA was measured via NanoDrop, and 10.0 ng were used for RT-qPCR analysis of mitochondrial DNA using gene-specific primers. The sequence of primers specifically targeting mitochondria DNA are provided in the Key resources table. Nuclear gene TERT was quantified from the respective nuclear fraction for normalization. To deplete mitochondria DNA, cells of interest were cultured with or without ethidium bromide or ddC for 4 days (Hashiguchi and Zhang-Akiyama, 2009). On day 4, cells were washed with sterile 1x PBS and trypsinized before counting and seeding for subsequent experiments in the absence of EtBr/ddC. Cytoplasmic mitochondria DNA was fractionated as described above. For quantification of mitochondrial membrane potential (MMP, ΔΨ), the MMP assay kit was used according to manual’s instructions.
ELISA, immunoblotting, and antibodies
For western blotting, cells were harvested in ice-cold Dulbecco’s phosphate-buffered saline (DPBS) using a sterile cell scraper. Cells were pelleted down by centrifugation and lysed in Modified Oncogene Science Lysis Buffer (MOSLB) (50 mM Na Pyrophosphate, 50 mM NaF, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 100 μM Na3VO4, 10 mM HEPES and 0.1% Triton X-100) containing protease inhibitor cocktail (Roche), followed by centrifugation at maximum speed for 15 min and ultracentrifugation at 100,000 rpm for 5 min. The protein concentration was measured using a Bradford assay (Bio-Rad). Whole cell extract was mixed with 1x sodium dodecyl sulfate (SDS) buffer, boiled at 100°C for 5 min before being immediately transferred on ice and incubated for 5 min. Sample volume corresponding to a protein amount of 50 μg was loaded into either 8%, 10% or 15% self-cast gel using standard SDS-polyacrylamide gel electrophoresis (PAGE) protocol before being transferred onto a nitrocellulose membrane (Bio-Rad). The membranes were blocked in 5% non-fat skim milk for 30 min at room temperature (RT), followed by incubation with primary antibody diluted in blocking buffer at 4°C overnight. Membranes were washed extensively with 1x TBST (mixture of Tris-buffered saline with 0.1% Tween20), followed by incubation with a conjugated secondary antibody for 1hat RT. After washing with 1x TBST, immunoreactive bands were visualized using alkaline phosphatase buffer containing 5′-bromo-4-chloro-3′-indolylphosphate (BCIP)-Nitro Blue Tetrazolium color development substrate. The list of primary antibodies used in this study are provided in the Key resources table. For the ELISA bioassay, all ELISA assays were performed using commercialized kit according to the manufacturer’s instruction. The absorbance was calculated based on a standard curve after subtracting the background.
Immunoprecipitation
For immunoprecipitation, soluble lysate was pre-cleared with protein G-sepharose beads for 1hat 4°C, and then incubated with control anti-mouse IgG or anti-S9.6 antibody plus protein G-sepharose beads at 4°C for 12 h. Beads, together with lysates were washed thrice with S9.6 immunoprecipitation buffer, thrice with S9.6 immunoprecipitation buffer containing 1 M NaCl, and thrice with S9.6 immunoprecipitation buffer, followed by elution in cytoplasmic lysis buffer containing RNase and DNase inhibitors.
Immunohistochemistry and histochemical staining
Paraffin-embedded sections (4.0 μm thick) were deparaffinized first through xylene and then with a gradually decreasing concentration of isopropanol (100%, 90% and 70%). Hematoxylin and eosin staining was performed to evaluate virus-induced brain damage. The sections were also subjected to immunohistochemical staining. Briefly, antigen retrieval was performed by heating the sections in citrate buffer pH 6.0 (Life Technologies Carlsbad, CA; 005000) for 25 minutes at 121°C. Slides then underwent peroxidase blocking using hydrogen peroxide (30 mg/mL), followed by washes with 1× TBS pH 7.6 (Tris-buffered saline, 0.05 M Tris, 0.155 M NaCl). After blocking, sections were incubated with primary antibodies overnight at 4°C. The MACH4 Universal HRP-Polymer Detection System (Biocare Medical, Pacheco CA; BRI4012H) was then used to detect the staining according to the manufacture’s protocol. At the end of the procedure, all slides were also counterstained with hematoxylin solution Gill II (Sigma-Aldrich, GHS232).
Mouse brain tissues were immunoassayed using the follow primary antibodies diluted in TBS-PBS buffer (1% BSA, 1.5 M NaCl, 0.5 M pH 7.6): GFAP (1:200 dilution); Iba1 (1:200 dilution); NK1.1 (1:300 dilution); CD4 (1:500 dilution); CD8 (1:500 dilution), and CD19 (1:500 dilution). Images were taken using the Aperio ScanScope AT (Digital slide scanner, Leica Biosystems Inc., Buffalo Grove, IL) or a Nikon Eclipse E600 microscope equipped with a SPOT Flex Model 15.2 64 Mp Shifting Pixel camera (Diagnostic Instrument Inc., Sterling Heights, MI).
Confocal microscopy imaging
For immunofluorescence staining, cells were washed in cold DPBS and fixed with 4% paraformaldehyde for 30 min at RT. After fixation, cells were permeabilized with 0.5% Triton X-100 in DPBS for 10 min, and blocked with Earle’s balanced salt solution containing 0.1% Triton X-100, 5% bovine serum albumin (BSA), 5% goat serum, and 0.1% saponin for 1hat RT. Primary antibodies were incubated at 4°C overnight and secondary antibodies were incubated for 1hat RT. Cells were washed three times with PBS containing 0.1% Triton X-100 and incubated for 15 min with four drops of Image-IT Fx signal enhancer. Coverslips were washed three times with PBST (mixture of 1x phosphate-buffered saline with 0.1% Tween®20), followed by counterstaining with DAPI (4,6-diamidino-2-phenylindole, Vector Laboratories; H-1200). Images were taken using the Zeiss LSM 980 with Airyan scan 2. While some antibodies are like those described in immunoblotting section, other antibodies specifically used for immunofluorescence staining included anti-p65, anti-IRF3, and anti-DNA-RNA Hybrid, clone S9.6.
Crystal violet, cell viability and luciferase reporter assay
For the crystal violet staining, cell culture medium from mock and infected cells was aspirated before washing with sterile DPBS, followed by incubation with 0.5% crystal violet solution at RT for 30 min (on a shaking platform). Cells was then rinsed with tap water several times. Plates were then inverted and with mild tapping the residual water was removed. Plates were air-dried at RT. Finally, methanol was added into each well and incubated on a shaking platform at RT for 20 min, the optical density at 540 nm wavelength (OD540nm) was measured. Lactate dehydrogenase (LDH) release assay were performed using the CyQUANTTM LDH cytotoxicity assay kit (Thermo Fisher) according to manufacturer’s protocol. The luciferase assay was performed with a dual luciferase reporter system (Promega) and pRL-TK (Promega) was used as the as the internal control.
RNA isolation and quantitative RT-PCR
RNA was harvested from cells using the RNeasy Mini extraction kit (Qiagen) according to the manufacturer’s instructions. A total of 100 ng of purified RNA was used as a template for gene expression analysis using POWER SYBR Green RNA-to-CTTM One-Step kit (Thermo Fisher). All gene expression analyses were then processed using the 2−ΔΔCt relative quantitative method and compared to control samples. Primers were all customized and purchased from Integrated DNA Technologies. The primer sequences for all RT-qPCR are provided in the Key resources table.
NanoString analysis
Total RNA was isolated from the brain and spinal cord of WT and G85R-mice, and 100 ng was used to determine the absolute levels of gene expression through an nCounter mouse neuroinflammation panel (∼770 genes). Hybridization and nCounter were performed by the Molecular Genomics Core of Heart Lung Innovation research institute, UBC, according to the manufacturer’s protocol (NanoString Technologies). In brief, reactions were hybridized for 20hat 65°C, after which the products were run on the nCounter preparation station for the removal of excess probes. Data were collected with the nCounter digital analyzer by counting individual barcodes. Data generated from the nCounter digital analyzer were examined using nSolver Analysis software 3.0 (NanoString Technologies). The data were normalized to the geometric means of spiked-in positive controls for assay efficiency and spiked-in negative controls to normalize background. The data were further normalized to the housekeeping genes Gapdh, Hprt, and β-actin, and reported as normalized RNA counts (means ± standard error of the mean [SEM]). For further analysis, differential expression of genes was performed using the Advanced Analysis module. For each gene, a single linear regression was fit using all selected covariates to predict expression. The output is shown with a non-adjusted p-value as well as Benjamini–Hochberg false discovery rate. Expression values within all heat-maps were converted either to Log-2 or Log-10 scale and presented as original normalized expression values.
Flow cytometry analysis
Trypsinized cells were maintained and stained in PBS containing 2 mM EDTA, 5% FBS. Flow cytometry was performed on FACSAria (BD Biosciences) and LSRFortessa (BD Biosciences) and was analyzed using FlowJo software version 10 and FACS Calibur CellQuest software (BD Biosciences). Values were plotted using GraphPad Prism version 8.0.
Quantification and statistical analysis
Mice were randomized to groups and the analysis of mice and tissue samples was performed blinded to the treatment or the genetic background of the animals. Sample size (N) representing biological replicates can be found on the figure legends. No statistical methods were used to predetermine sample sizes or to test for a normal distribution. The numbers of biological replicates were chosen based on the nature of the experiments and published papers describing similar experiments. Quantification of immunohistochemistry images was performed using ImageJ (version 1.0) with the combination of Color Deconvolution Plugin (version 1.5) to generate optical density values based on the intensity of the staining as described (Xue et al., 2018). Statistical tests for each assay are indicated in the figure legends. Data are presented as means and error bars represent SEMs. No statistical method was used to determine whether the data met assumptions of the statistical approach. Prism version 8.0 (GraphPad) was used for all statistical analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 was considered statistically significant.
Acknowledgments
We thank Tomomi Furihata (Tokyo University of Pharmacy and Life Sciences, Japan), Neil Cashman (University of British Columbia, Canada), Takashi Fujita (Kyoto University, Japan), David Alvarez-Carbonell (Case Western Reserve University, USA), Christine Vande Velde (Université deMontréal, Canada), Leigh Anne Swayne (University of Victoria, Canada), and Nelson O. Gekara (Stockholm University, Sweden) for kindly providing valuable materials. H.Y.T. (XMUMRF/2020-C5/ITCM/0003) and K.Y.Y. (XMUMRF/2018-C2/ILAB/0001) is supported by a research grant from Xiamen University Malaysia. T.F. is supported by funding from the Japan Society for the Promotion of Science Kakenhi (#19K07214). Y.C.X is the recipient of Canadian Institutes of Health Research Doctoral Fellowship. Y.C.X. are recipients of Four-Year Doctoral Fellowships from University of British Columbia. H.L. is the recipient of fellowship from VIROGIN Biotech through Mitacs-Accelerate program.
Author contributions
Conceptualization and developed research plan: H.Y.T., Y.K.Y., and C.S.N.; Methodology and Investigation: H.Y.T, Y.K.Y., Y.C.X., H.L., T.F., and C.S.N.; Intellectual input: E.M.S.; Manuscript preparation, writing, review, and editing: H.Y.T., Y.K.Y., E.M.S., and C.S.N.
Declaration of interests
The authors declared that the study was conducted in the absence of any commercial relationships that could be construed as a potential conflict of interest. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Published: June 17, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104404.
Supplemental information
Data and code availability
-
•
NanoString gene expression profiling data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albalawi F., Lu W., Beckel J.M., Lim J.C., McCaughey S.A., Mitchell C.H. The P2X7 receptor primes IL-1β and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front. Cell Neurosci. 2017;11:227. doi: 10.3389/fncel.2017.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almine J.F., O’Hare C.A.J., Dunphy G., Haga I.R., Naik R.J., Atrih A., Connolly D.J., Taylor J., Kelsall I.R., Bowie A.G., et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017;8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber G.N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers D.R., Henkel J.S., Xiao Q., Zhao W., Wang J., Yen A.A., Siklos L., McKercher S.R., Appel S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., Cleveland D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Brenner D., Sieverding K., Bruno C., Lüningschrör P., Buck E., Mungwa S., Fischer L., Brockmann S.J., Ulmer J., Bliederhäuser C., et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J. Exp. Med. 2019;216:267–278. doi: 10.1084/jem.20180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker S.W., Gauthier A.E., Mills E.W., Ingolia N.T., Kagan J.C. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156:800–811. doi: 10.1016/j.cell.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L., Cleveland D.W. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Bruijn L.I., Houseweart M.K., Kato S., Anderson K.L., Anderson S.D., Ohama E., Reaume A.G., Scott R.W., Cleveland D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Burdette D.L., Monroe K.M., Sotelo-Troha K., Iwig J.S., Eckert B., Hyodo M., Hayakawa Y., Vance R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas C., Herrando-Grabulosa M., Manzano R., Mancuso R., Osta R., Navarro X. Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 2013;3:145–158. doi: 10.1002/brb3.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman N.R., Durham H.D., Blusztajn J.K., Oda K., Tabira T., Shaw I.T., Dahrouge S., Antel J.P. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Chen S., Sayana P., Zhang X., Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener. 2013;8:28. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q., Yang Y., Gao J. Infectivity of human coronavirus in the brain. EBioMedicine. 2020;56:102799. doi: 10.1016/j.ebiom.2020.102799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A., Logroscino G., Traynor B.J., Collins J., Simeone J.C., Goldstein L.A., White L.A. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A., Mazzini L., D’Alfonso S., Corrado L., Canosa A., Moglia C., Manera U., Bersano E., Brunetti M., Barberis M., et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. 2018;91:e635–e642. doi: 10.1212/WNL.0000000000005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui R., Dorovini-Zis K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J. Neuroinflammation. 2010;7:1. doi: 10.1186/1742-2094-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli E.T., Lasseigne B.N., Petrovski S., Sapp P.C., Dion P.A., Leblond C.S., Couthouis J., Lu Y.-F., Wang Q., Krueger B.J., et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowl J.T., Gray E.E., Pestal K., Volkman H.E., Stetson D.B. Intracellular nucleic acid detection in autoimmunity. Annu. Rev. Immunol. 2017;35:313–336. doi: 10.1146/annurev-immunol-051116-052331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R., Tuo M., Li P., Zhou C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol. Sci. 2018;39:811–820. doi: 10.1007/s10072-018-3246-0. [DOI] [PubMed] [Google Scholar]
- Du M., Chen Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–709. doi: 10.1126/science.aat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas J.C., Mandemakers W., Rogers M., Ibrahim A., Daneman R., Barres B.A. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J. Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo F., Komine O., Yamanaka K. Neuroinflammation in motor neuron disease. Clin. Exp. Neuroimmunol. 2016;7:126–138. doi: 10.1111/cen3.12309. [DOI] [Google Scholar]
- Foo L.C. Purification of rat and mouse astrocytes by immunopanning. Cold Spring Harb. Protoc. 2013;2013 doi: 10.1101/pdb.prot074211. pdb.prot074211–pdb.prot074432. [DOI] [PubMed] [Google Scholar]
- Freischmidt A., Wieland T., Richter B., Ruf W., Schaeffer V., Müller K., Marroquin N., Nordin F., Hübers A., Weydt P., et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Furihata T., Ito R., Kamiichi A., Saito K., Chiba K. Establishment and characterization of a new conditionally immortalized human astrocyte cell line. J. Neurochem. 2016;136:92–105. doi: 10.1111/jnc.13358. [DOI] [PubMed] [Google Scholar]
- Gal J., Kuang L., Barnett K.R., Zhu B.Z., Shissler S.C., Korotkov K.V., Hayward L.J., Kasarskis E.J., Zhu H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016;132:563–576. doi: 10.1007/s00401-016-1601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P., Ascano M., Wu Y., Barchet W., Gaffney B.L., Zillinger T., Serganov A.A., Liu Y., Jones R.A., Hartmann G., et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mesa Y., Jay T.R., Checkley M.A., Luttge B., Dobrowolski C., Valadkhan S., Landreth G.E., Karn J., Alvarez-Carbonell D. Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017;23:47–66. doi: 10.1007/s13365-016-0499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag S.M., Gulen M.F., Reymond L., Gibelin A., Abrami L., Decout A., Heymann M., van der Goot F.G., Turcatti G., Behrendt R., Ablasser A. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269–273. doi: 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- Hammitt L.L., Kazungu S., Welch S., Bett A., Onyango C.O., Gunson R.N., Scott J.A.G., Nokes D.J. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J. Clin. Microbiol. 2011;49:2318–2320. doi: 10.1128/JCM.02605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U.-K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hardiman O., Al-Chalabi A., Chio A., Corr E.M., Logroscino G., Robberecht W., Shaw P.J., Simmons Z., van den Berg L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- Harraz M.M., Marden J.J., Zhou W., Zhang Y., Williams A., Sharov V.S., Nelson K., Luo M., Paulson H., Schöneich C., Engelhardt J.F. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A., Erttmann S.F., Raffi F.A., Schmalz A.M., Resch U., Anugula S., Lienenklaus S., Nilsson L.M., Kröger A., Nilsson J.A., et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Hashiguchi K., Zhang-Akiyama Q.-M. Establishment of human cell lines lacking mitochondrial DNA. Methods Mol. Biol. 2009;554:383–391. doi: 10.1007/978-1-59745-521-3_23. [DOI] [PubMed] [Google Scholar]
- Hong S., Hwang I., Gim E., Yang J., Park S., Yoon S.-H., Lee W.-W., Yu J.-W. Brefeldin A-sensitive ER-Golgi vesicle trafficking contributes to NLRP3-dependent caspase-1 activation. FASEB J. 2019;33:4547–4558. doi: 10.1096/fj.201801585R. [DOI] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., Skaug B., Jiang Q.-X., Chen Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D., Haasdijk E.D., Grashorn J.A., Hawkins R., van Duijn W., Verspaget H.W., London J., Holstege J.C. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol. Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Jaarsma D., Rognoni F., van Duijn W., Verspaget H.W., Haasdijk E.D., Holstege J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jacomy H., Fragoso G., Almazan G., Mushynski W.E., Talbot P.J. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology. 2006;349:335–346. doi: 10.1016/j.virol.2006.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Xue X., Panda S., Kawale A., Hooy R.M., Liang F., Sohn J., Sung P., Gekara N.O. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 2019;38:e102718. doi: 10.15252/embj.2019102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai Y., Takamatsu C., Tokuda K., Okamoto M., Irita K., Takahashi S. Rapid and random turnover of mitochondrial DNA in rat hepatocytes of primary culture. Mitochondrion. 2006;6:299–304. doi: 10.1016/j.mito.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Kao L.-P., Ovchinnikov D., Wolvetang E. The effect of ethidium bromide and chloramphenicol on mitochondrial biogenesis in primary human fibroblasts. Toxicol. Appl. Pharmacol. 2012;261:42–49. doi: 10.1016/j.taap.2012.03.009. [DOI] [PubMed] [Google Scholar]
- Kasereka M.C., Hawkes M.T. Neuroinvasive potential of human coronavirus OC43: case report of fatal encephalitis in an immunocompromised host. J. Neurovirol. 2021;27:340–344. doi: 10.1007/s13365-020-00926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T., Barry B., Hickey W.F. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain Abscesses1. J. Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kitamura K., Ito R., Umehara K., Morio H., Saito K., Suzuki S., Hashimoto M., Saito Y., Anzai N., Akita H., et al. Differentiated HASTR/ci35 cells: a promising in vitro human astrocyte model for facilitating CNS drug development studies. J. Pharmacol. Sci. 2018;137:350–358. doi: 10.1016/j.jphs.2018.06.013. [DOI] [PubMed] [Google Scholar]
- Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O’Loughlin E., Xu Y., Fanek Z., et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langereis M.A., Rabouw H.H., Holwerda M., Visser L.J., van Kuppeveld F.J.M. Knockout of cGAS and STING rescues virus infection of plasmid DNA-transfected cells. J. Virol. 2015;89:11169–11173. doi: 10.1128/JVI.01781-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-Y., Jeon G.S., Sung J.-J. ALS-Linked Mutant SOD1 associates with TIA-1 and alters stress granule dynamics. Neurochem. Res. 2020;45:2884–2893. doi: 10.1007/s11064-020-03137-5. [DOI] [PubMed] [Google Scholar]
- Lee K.-G., Kim S.Y., Kui L., Voon D.C.-C., Mauduit M., Bist P., Bi X., Pereira N.A., Liu C., Sukumaran B., et al. Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep. 2015;10:1055–1065. doi: 10.1016/j.celrep.2015.01.039. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch M.A. The type I interferonopathies. Annu. Rev. Med. 2017;68:297–315. doi: 10.1146/annurev-med-050715-104506. [DOI] [PubMed] [Google Scholar]
- Lehmann M.H., Torres-Domínguez L.E., Price P.J.R., Brandmüller C., Kirschning C.J., Sutter G. CCL2 expression is mediated by type I IFN receptor and recruits NK and T cells to the lung during MVA infection. J. Leukoc. Biol. 2016;99:1057–1064. doi: 10.1189/jlb.4MA0815-376RR. [DOI] [PubMed] [Google Scholar]
- Liao B., Zhao W., Beers D.R., Henkel J.S., Appel S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman A.W., Leskov I.L., Butcher J.T., Johnstone S.R., Stokes T.A., Begandt D., DeLalio L.J., Best A.K., Penuela S., Leitinger N., et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat. Commun. 2015;6:7965. doi: 10.1038/ncomms8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenkova H.P., Shah S.B., Shestopalov V.I. The two faces of pannexins: new roles in inflammation and repair. J. Inflamm. Res. 2018;11:273–288. doi: 10.2147/JIR.S128401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankan A.K., Schmidt T., Chauhan D., Goldeck M., Höning K., Gaidt M., Kubarenko A.V., Andreeva L., Hopfner K.-P., Hornung V. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 2014;33:2937–2946. doi: 10.15252/embj.201488726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei L.M., Cotmore S.F., Li L., Tattersall P., Iwasaki A. Toll-like receptor 9 in plasmacytoid dendritic cells fails to detect parvoviruses. J. Virol. 2013;87:3605–3608. doi: 10.1128/JVI.03155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley M.E., O’Rourke J.G., Yáñez A., Markman J.L., Ho R., Wang X., Chen S., Lall D., Jin M., Muhammad A.K.M.G., et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585:96–101. doi: 10.1038/s41586-020-2625-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J., Trudel G.C., Shaw I.T., Antel J.P., Cashman N.R. A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J. Neurobiol. 1995;26:283–293. doi: 10.1002/neu.480260212. [DOI] [PubMed] [Google Scholar]
- Mejzini R., Flynn L.L., Pitout I.L., Fletcher S., Wilton S.D., Akkari P.A. ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 2019;13:1310. doi: 10.3389/fnins.2019.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfopoulou S., Brown J.R., Davies E.G., Anderson G., Virasami A., Qasim W., Chong W.K., Hubank M., Plagnol V., Desforges M., et al. Human coronavirus OC43 associated with fatal encephalitis. N. Engl. J. Med. 2016;375:497–498. doi: 10.1056/NEJMc1509458. [DOI] [PubMed] [Google Scholar]
- Mori M., Yoneyama M., Ito T., Takahashi K., Inagaki F., Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- Narita R., Takahasi K., Murakami E., Hirano E., Yamamoto S.P., Yoneyama M., Kato H., Fujita T. A novel function of human Pumilio proteins in cytoplasmic sensing of viral infection. PLoS Pathog. 2014;10:e1004417. doi: 10.1371/journal.ppat.1004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson I., Hanna M.G., Wood N.W., Harding A.E. Depletion of mitochondrial DNA by ddC in untransformed human cell lines. Somat. Cell Mol. Genet. 1997;23:287–290. doi: 10.1007/BF02674419. [DOI] [PubMed] [Google Scholar]
- Ng C.S., Jogi M., Yoo J.-S., Onomoto K., Koike S., Iwasaki T., Yoneyama M., Kato H., Fujita T. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 2013;87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C.S., Kasumba D.M., Fujita T., Luo H. Spatio-temporal characterization of the antiviral activity of the XRN1-DCP1/2 aggregation against cytoplasmic RNA viruses to prevent cell death. Cell Death Differ. 2020;27:2363–2382. doi: 10.1038/s41418-020-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson A., Edner N., Albert J., Ternhag A. Fatal encephalitis associated with coronavirus OC43 in an immunocompromised child. Infect. Dis. (Lond) 2020;52:419–422. doi: 10.1080/23744235.2020.1729403. [DOI] [PubMed] [Google Scholar]
- Onoguchi K., Onomoto K., Takamatsu S., Jogi M., Takemura A., Morimoto S., Julkunen I., Namiki H., Yoneyama M., Fujita T. Virus-infection or 5’ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010;6:e1001012. doi: 10.1371/journal.ppat.1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onomoto K., Jogi M., Yoo J.-S., Narita R., Morimoto S., Takemura A., Sambhara S., Kawaguchi A., Osari S., Nagata K., et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One. 2012;7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablo J., Banack S.A., Cox P.A., Johnson T.E., Papapetropoulos S., Bradley W.G., Buck A., Mash D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand. 2009;120:216–225. doi: 10.1111/j.1600-0404.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- Palomino-Segura M., Perez L., Farsakoglu Y., Virgilio T., Latino I., D’Antuono R., Chatziandreou N., Pizzagalli D.U., Wang G., García-Sastre A., et al. Protection against influenza infection requires early recognition by inflammatory dendritic cells through C-type lectin receptor SIGN-R1. Nat. Microbiol. 2019;4:1930–1940. doi: 10.1038/s41564-019-0506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadeas S.T., Kraig S.E., O’Banion C., Lepore A.C., Maragakis N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. U S A. 2011;108:17803–17808. doi: 10.1073/pnas.1103141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzych K., Zetterqvist A.V., Wright W.R., Kirkby N.S., Mitchell J.A., Paul-Clark M.J. Differential role of pannexin-1/ATP/P2X7 axis in IL-1β release by human monocytes. FASEB J. 2017;31:2439–2445. doi: 10.1096/fj.201600256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrini S., Sau D., Guareschi S., Bogush M., Brown R.H., Naniche N., Kia A., Trotti D., Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 2010;19:2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrishevsky E., Hong R.H., Mackenzie I.R., Cashman N.R. Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS One. 2017;12:e0184384. doi: 10.1371/journal.pone.0184384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi N., Shi Y., Zhang R., Zhu W., Yuan B., Li X., Wang C., Zhang X., Hou F. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat. Commun. 2017;8:15676. doi: 10.1038/ncomms15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer C., Weihe E., Schütz B. SOD1G93A mutant mice develop a neuroinflammation-independent dendropathy in excitatory neuronal subsets of the olfactory bulb and retina. J. Neuropathol. Exp. Neurol. 2017;76:769–778. doi: 10.1093/jnen/nlx057. [DOI] [PubMed] [Google Scholar]
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O’Regan J.P., Deng H.X., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Roy E.R., Wang B., Wan Y.w., Chiu G., Cole A., Yin Z., Propson N.E., Xu Y., Jankowsky J.L., Liu Z., et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 2020;130:1912–1930. doi: 10.1172/jci133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi S., Stauffer J.E., Schulte D.J., Ravits J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 2015;33:855–876. doi: 10.1016/j.ncl.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Malovic E., Plante B., Zenitsky G., Jin H., Anantharam V., Kanthasamy A., Kanthasamy A.G. Rapid and refined CD11b magnetic isolation of primary microglia with enhanced purity and versatility. J. Vis. Exp. 2017;122:55364. doi: 10.3791/55364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E., Suadicani S.O., Dahl G., Spray D.C. Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol. 2007;3:199–208. doi: 10.1017/S1740925X08000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler S., Gagné M., Garg P., Pickles S.R., Baudouin C., Hamon-Keromen E., Destroismaisons L., Khalfallah Y., Chaineau M., Caron E., et al. TNF receptor-associated factor 6 interacts with ALS-linked misfolded superoxide dismutase 1 and promotes aggregation. J. Biol. Chem. 2020;295:3808–3825. doi: 10.1074/jbc.RA119.011215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple B.D., Kossmann T., Morganti-Kossmann M.C. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010;30:459–473. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu R.C.F., Smirnova E., Brown C.A., Zoidl C., Spray D.C., Donaldson L.W., Zoidl G. Structural and functional consequences of connexin 36 (Cx36) interaction with calmodulin. Front. Mol. Neurosci. 2016;9:120. doi: 10.3389/fnmol.2016.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter D.A., Martinez J., Hao L., Chen X., Sun N., Fischer T.D., Burman J.L., Li Y., Zhang Z., Narendra D.P., et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrou S., Aguilera A.N., Blouch K., Ross S.R. DDX41 recognizes RNA/DNA retroviral reverse transcripts and is critical for in vivo control of murine leukemia virus infection. MBio. 2018;9:e00923–e01018. doi: 10.1128/mBio.00923-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Ko J.S., Heidmann T., Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Sun B., Sundström K.B., Chew J.J., Bist P., Gan E.S., Tan H.C., Goh K.C., Chawla T., Tang C.K., Ooi E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017;7:3594. doi: 10.1038/s41598-017-03932-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Taylor J.P., Brown R.H., Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A., Xu Z., Hayward L.J. Aberrantly increased hydrophobicity shared by mutants of Cu, Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2005;280:29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- Turner M.R., Cagnin A., Turkheimer F.E., Miller C.C.J., Shaw C.E., Brooks D.J., Leigh P.N., Banati R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Turner M.R., Swash M. The expanding syndrome of amyotrophic lateral sclerosis: a clinical and molecular odyssey. J. Neurol. Neurosurg. Psychiat. 2015;86:667–673. doi: 10.1136/jnnp-2014-308946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S., et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bosch L. Genetic rodent models of amyotrophic lateral sclerosis. J. Biomed. Biotechnol. 2011;2011:1–11. doi: 10.1155/2011/348765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J., Adura C., Gao P., Luz A., Lama L., Asano Y., Okamoto R., Imaeda T., Aida J., Rothamel K., et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017;8:750. doi: 10.1038/s41467-017-00833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S., Westeneng H.-J., Al-Chalabi A., Van Den Berg L.H., Talman P., Kiernan M.C. Amyotrophic lateral sclerosis as a multi-step process: an Australia population study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019;20:532–537. doi: 10.1080/21678421.2018.1556697. [DOI] [PubMed] [Google Scholar]
- Wang H., Xing Y., Mao L., Luo Y., Kang L., Meng G. Pannexin-1 influences peritoneal cavity cell population but is not involved in NLRP3 inflammasome activation. Protein Cell. 2013;4:259–265. doi: 10.1007/s13238-013-2114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Yang B., Zhang D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia. 2011;59:946–958. doi: 10.1002/glia.21167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., Bestwick M., Duguay B.A., Raimundo N., MacDuff D.A., et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesekera L.C., Nigel Leigh P. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.C., Ruller C.M., Fung G., Mohamud Y., Deng H., Liu H., Zhang J., Feuer R., Luo H. Enteroviral infection leads to transactive response DNA-binding protein 43 pathology in vivo. Am. J. Pathol. 2018;188:2853–2862. doi: 10.1016/j.ajpath.2018.08.013. [DOI] [PubMed] [Google Scholar]
- Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H., Takahashi R., Misawa H., Cleveland D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S., Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015;4:13. doi: 10.1186/s40035-015-0036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Delalio L.J., Best A.K., Macal E., Milstein J., Donnelly I., Miller A.M., McBride M., Shu X., Koval M., et al. Endothelial pannexin 1 channels control inflammation by regulating intracellular calcium. J. Immunol. 2020;204:2995–3007. doi: 10.4049/jimmunol.1901089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T., Ishigaki S., Yamamoto M., Liang Y., Niwa i., Takeuchi H., Doyu M., Sobue G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 2002;80:158–167. doi: 10.1046/j.0022-3042.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- Yu C.-H., Davidson S., Harapas C.R., Hilton J.B., Mlodzianoski M.J., Laohamonthonkul P., Louis C., Low R.R.J., Moecking J., De Nardo D., et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636–649.e18. doi: 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Shi Y., Wei X., Yang Y., Zhou Y., Hao X., Zhang N., Niu R. Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol. Lett. 2007;170:83–93. doi: 10.1016/j.toxlet.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Yuzefovych L.V., Musiyenko S.I., Wilson G.L., Rachek L.I. Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS One. 2013;8:e54059. doi: 10.1371/journal.pone.0054059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K.Q., Green C.R., Bennet L., Gunn A.J., Davidson J.O. The role of connexin and pannexin channels in perinatal brain injury and inflammation. Front. Physiol. 2019;10:141. doi: 10.3389/fphys.2019.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z., Sun J., Kang Z., Wang Y., Zhao H., Zhu K., Wang J. Tyrosine kinase receptors axl and MerTK mediate the beneficial effect of electroacupuncture in a cuprizone-induced demyelinating model. Evid. base Compl. Alternative Med. 2020;2020:1–13. doi: 10.1155/2020/3205176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
NanoString gene expression profiling data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.