

Abstract
Ever since the discovery of the hypotensive and bradycardiac effects of adenosine, adenosine receptors continue to represent promising drug targets. First, this is due to the fact that the receptors are expressed in a large variety of tissues. In particular, the actions of adenosine (or methylxanthine antagonists) in the central nervous system, in the circulation, on immune cells, and on other tissues can be beneficial in certain disorders. Second, there exists a large number of ligands, which have been generated by introducing several modifications in the structure of the lead compounds (adenosine and methylxanthine), some of them highly specific. Four adenosine receptor subtypes (A1, A2A, A2B, and A3) have been cloned and pharmacologically characterized, all of which are G protein-coupled receptors. Adenosine receptors can be distinguished according to their preferred mechanism of signal transduction: A1 and A3 receptors interact with pertussis toxin-sensitive G proteins of the Gi and Go family; the canonical signaling mechanism of the A2A and of the A2B receptors is stimulation of adenylyl cyclase via Gs proteins. In addition to the coupling to adenylyl cyclase, all four subtypes may positively couple to phospholipase C via different G protein subunits. The development of new ligands, in particular, potent and selective antagonists, for all subtypes of adenosine receptors has so far been directed by traditional medicinal chemistry. The availability of genetic information promises to facilitate understanding of the drug–receptor interaction leading to the rational design of a potentially therapeutically important class of drugs. Moreover, molecular modeling may further rationalize observed interactions between the receptors and their ligands. In this review, we will summarize the most relevant progress in developing new therapeutic adenosine receptor antagonists.
Keywords: G protein-coupled receptor, adenosine receptor, antagonists
1. INTRODUCTION
The stimulation of cell surface adenosine receptors (ARs) is largely responsible for the broad variety of effects produced by adenosine throughout several organ systems. Based on the widespread and frequently beneficial effects, attributed to the accumulation of endogenously released adenosine, it has long been considered that regulation of ARs has substantial therapeutic potential. Incidentally, much recent focus has been on the cardioprotective1,2 and neuroprotective3,4 effects associated with AR activation during periods of cardiac and cerebral ischemia, respectively. On the other hand, it has been proposed recently that antagonists of distinct AR subtypes may be used in the treatment of asthma5,6 or certain neurological diseases such as Parkinson’s disease.6,7 Comprehensive reviews of the physiological roles of ARs and their potential as clinical targets in a variety of disease states have been published.6–11
ARs are members of the superfamily of G protein-coupled receptors (GPCRs), with four subtypes currently recognized, the A1AR, A2AAR, A2BAR, and A3AR.12 With the exception of the A3AR, the existence of AR subtypes in various tissues had been appreciated prior to their cloning as a result of pharmacological characterization.12
The cloning of the four AR subtypes has allowed for significant progresses to be made in the understanding of several facets of AR activity at a molecular level. A schematic representation of AR signaling pathways shown in Figure 1.11
Figure 1.
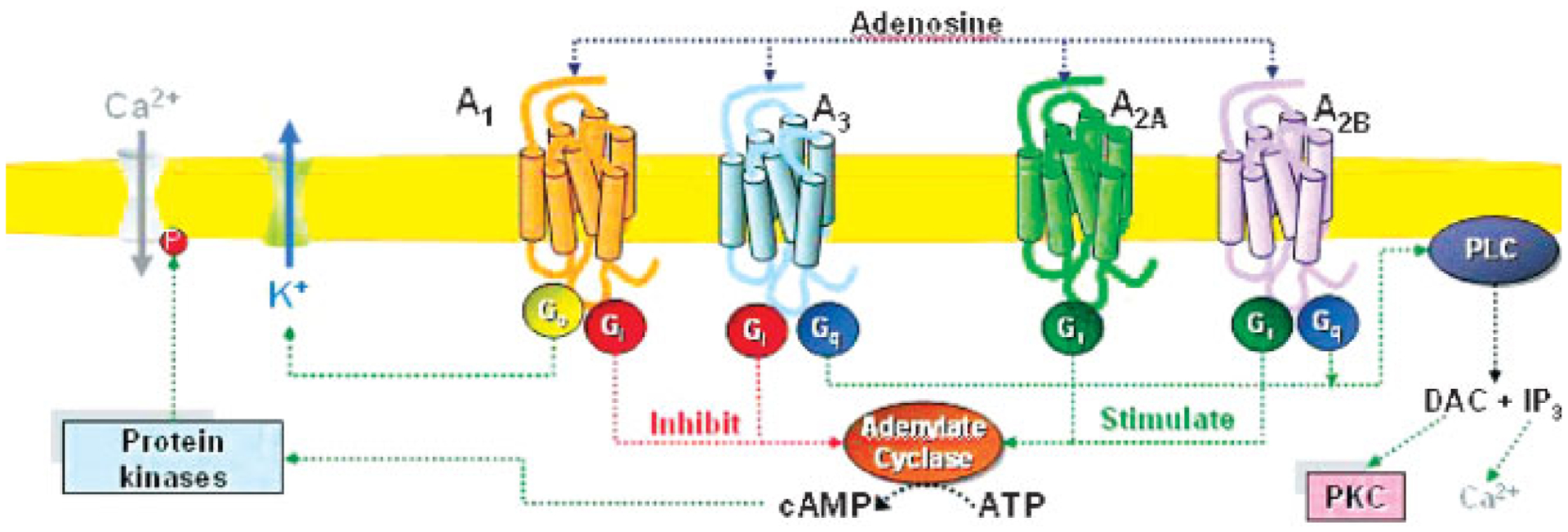
Signal transduction pathways associated with the activation of the human adenosine receptors. Abbreviations: α, α-subunit of G protein; βγ, βγ-subunits of G protein; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; Gi, Gi family of G proteins; Gs, Gs family of G proteins; Go, Go family of G proteins; Gq, Gq family of G proteins; IP3, inositol (1,4,5)-trisphosphate; P, phosphate moiety; PKC, proteinkinase C; PLC, phospholipase C.
Considering the overall protein structure, ARs display the topology typical of GPCRs. Many features of GPCR structure and function have been reviewed recently.12–15 Here we will highlight some fundamental features that may expand upon the classical view of GPCR structure and function. Sequence comparison between the different GPCRs revealed the existence of different receptor families sharing no sequence similarity even if specific fingerprints exist in all GPCR classes. However, all these receptors have in common a central core domain consisting of seven transmembrane helices (TM1-7), with each TM composed of 20–27 amino acids, connected by three intracellular (IL1, IL2, and IL3) and three extracellular (EL1, EL2, and EL3) loops. Two cysteine residues (one in TM3 and one in EL2), which are conserved in most GPCRs, form a disulfide link which is possibly crucial for the packing and for the stabilization of a restricted number of conformations of these seven TMs. Aside from sequence variations, GPCRs differ in the length and function of their N-terminal extracellular domain, their C-terminal intracellular domain, and their intracellular loops. Each of these domains provides very specific properties to these receptor proteins. Particularly, consensus sites for N-linked glycosylation exist on the extracellular regions of ARs, although the precise location of the sites for this post-translational modification varies amongst the AR subtypes.16–19 The carboxyl-terminal tails of the A1AR, A2BAR, and A3AR, but not A2AAR, possess a conserved cysteine residue that may putatively serve as a site for receptor palmitoylation and permit the formation of a fourth intracellular loop. However, site-directed mutagenesis of this residue has not been performed for any AR subtype, and no role for putative AR palmitoylation has been described.
The A1AR, A2BAR, and A3AR are very similar in regard to the number of amino acids composing their primary structure, and in general, these AR subtypes are among the smaller members of the GPCR family. For example, the human homologs of the A1AR, A2BAR, and A3AR consist of 326, 328, and 318 amino acid residues, respectively.20–22 Conversely, the human A2AAR is composed of 409 amino acids.23 All cloned species homologs of the A2AAR are of similar mass, and this relatively large size is manifested in the carboxyl-terminal tail of the receptor, which is much longer than that of the other AR subtypes. It should be noted that the size of ARs deduced from their primary amino acid structure frequently is not consistent with the mass estimated by polyacrylamide gel electrophoresis of the expressed proteins. The aforementioned post-translational glycosylation of ARs, which may vary in a cell type-dependent fashion, likely accounts for these discrepancies. The human A1AR and human A3AR display ca 49% overall sequence identity at the amino acid level, while the human A2AAR and human A2BAR are 45% identical. A general topology of all four receptor subtypes are shown in Figure 2.
Figure 2.
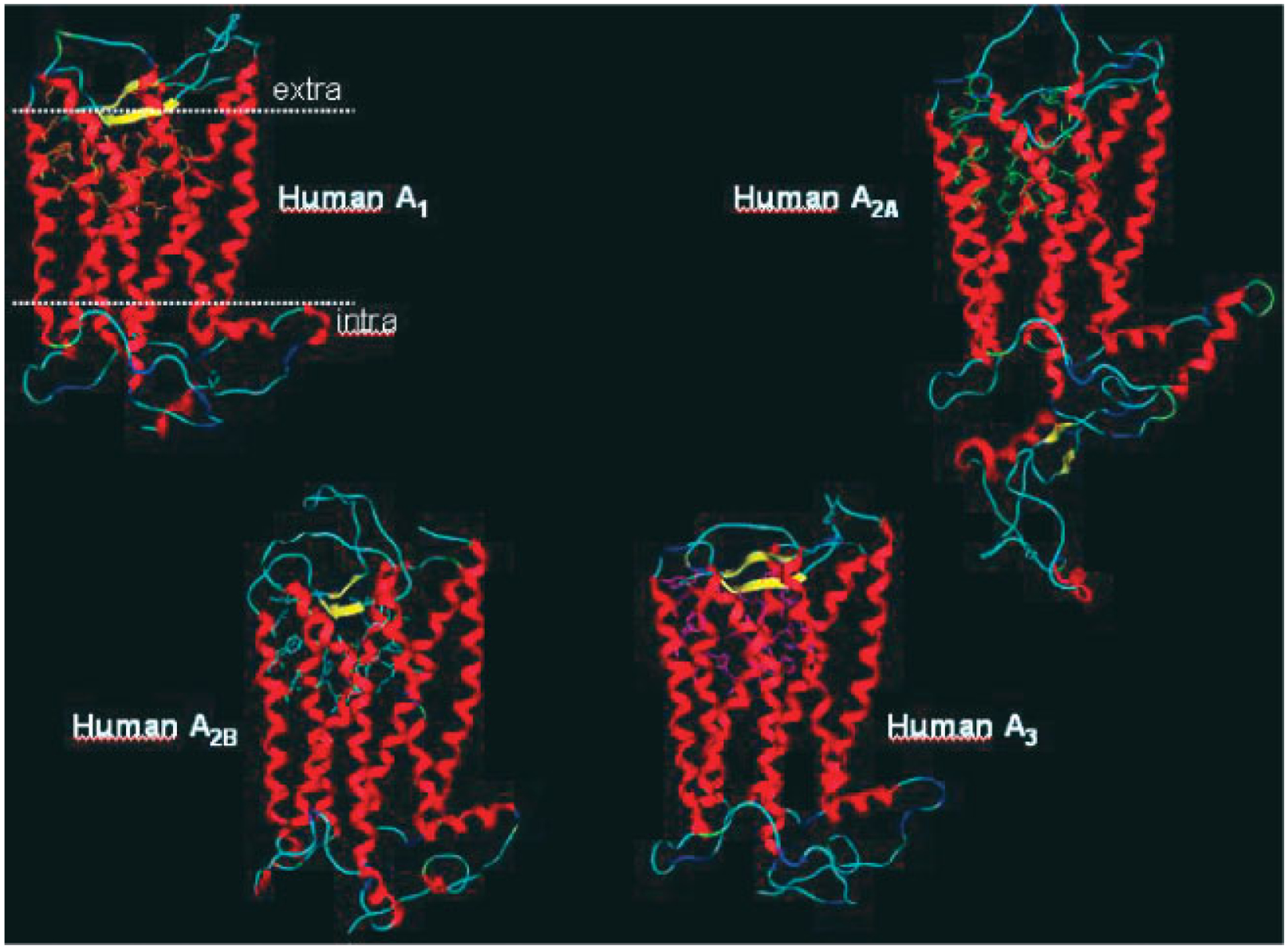
General topology of all adenosine receptors obtained using a rhodopsin-based homology modeling (modified from Moto et al.11).
Indeed, for all GPCRs, the identification of discrete receptor regions, or even single amino acids that are critical for ligand recognition and are responsible for discerning between agonist and antagonist ligands, has been an area of extensive investigation.11,24–27 In addition to a basic understanding of receptor activation, it has been hoped that a delineation of ligand–receptor interaction at a molecular level may provide the basis for rational drug design.11,24–27 As summarized below, both TMs and extracellular regions of ARs have been implicated as playing a role in the formation of the ligand-binding pocket.11,24–27 Key amino acids identified via mutagenesis studies as contributing to the ligand-binding properties of the ARs are briefly summarized in Table I.
Table I.
Amino Acids of ARs Implicated in Ligand Binding
| A1AR1 | ||
|---|---|---|
| H278L (bovine) | Nearly abolished both agonist and antagonist binding | Olah et al., 1992 [29] |
| H251L (bovine) | Decreased antagonist affinity 4-fold. No change in agonist affinity | Olah et al., 1992 [29] |
| T277A (human) | >400-fold decrease in affinity of NECA with slight changes in affinity for R-PIA and S-PIA. Substitution with serine significantly restored NECA affinity. Nature of residue in position 277 also involved in canine/bovine A1AR binding specificity Amino acid in position 270 contributes to canine/bovine A1AR binding specificity | Townsend-Nicholson and Schofield, 1994 [30] Tucker et al., 1994 [31] |
| I270M (bovine) M270I (canine) | Amino acid in position 270 contributes to canine/bovine A1AR binding specificity | Tucker et al., 1994 [31] |
| E16A (human) | Agonist affinity reduced 4- to 40-fold with little change in antagonist affinity | Barbhaiya et al., 1996 [32] |
| D55A (human) | Increase in agonist affinity with no change in antagonist affinity. Disrupted regulation of agonist binding by Na+ | Barbhaiya et al., 1996 [32] |
| S94A (human) | Loss of detectable agonist or antagonist binding. Restoration of binding by substitution with threonine | Barbhaiya et al., 1996 [32] |
| A2AAR1,2 | ||
| S281A | Loss of detectable agonist and antagonist radioligand binding; no agonist activity in functional assays; substitution with threonine enhanced affinity for most agonists. | Kim et al., 1995 [33] |
| H278A | Substantial loss of agonist and antagonist radioligand binding; functional assays indicated ~300-fold decrease in agonist potency | Kim et al., 1995 [33] |
| S277A | Substantial decrease in agonist affinity, but antagonist radioligand binding not altered; substitution with threonine restored agonist binding | Kim et al., 1995 [33] |
| I274A | Substantial decrease in agonist and antagonist radioligand binding; functional assays indicated ~30-fold decrease in agonist potency | Kim et al., 1995 [33] |
| N253A | Loss of detectable agonist and antagonist radioligand binding; radioligand binding not restored by replacement with glutamine | Kim et al., 1995 [33] |
| H250A | Loss of detectable agonist and antagonist radioligand binding; no agonist activity in functional assays; substitution with phenylalanine substantially restored binding | Kim et al., 1995 [33] |
| F182A | Loss of agonist and antagonist radioligand binding; replacement with tryptophan significantly restored binding of ligands | Kim et al., 1995 [33] |
| E151A | Loss of agonist and antagonist radioligand binding; functional assays indicated 1000-fold decrease in agonist potency; substitution with aspartic acid did not restore radioligand binding | Kim et al., 1996 [34] |
| T88A | Substantial decrease in agonist, but not antagonist, affinity; partially restored by substitution with threonine | Jiang et al., 1996 [35] |
| A3AR1,2 | ||
| H95A | Substantial decrease in agonist and antagonist affinity | Gao et al., 2002 [36] |
| K152A | Insignificantly affect the agonist binding but slightly decreased antagonist affinity | Gao et al., 2002 [36] |
| W243A W243F | Insignificantly affect the agonist binding but slightly decreased antagonist affinity. Trp243 is critical for receptor activation | Gao et al., 2002 [36] |
| N250A | Loss of agonist and antagonist radioligand binding | Gao et al., 2002 [36] |
Amino acids represented in single-letter code with position number shown. The first amino acid is that of the wild-type receptor, with the second residue that used for substitution. References are collected inside square brackets.
All studies referring A2AAR and A3AR examined the human cloned receptors.
Site-directed mutagenesis studies in parallel with different molecular modeling approaches have been recently used as powerful strategy to design potent and selective GPCR ligand.11,36–39 Of course, the evolution of the field of computer-aided design of ligands (both agonists and antagonists) for GPCRs, including adenosine receptors, has depended on the availability of suitable molecular receptor templates. In fact, due to technical difficulties, which complicate experimental X-ray diffraction and NMR structure determination of GPCRs, the 3D structure of most GPCRs is still unknown. The only known GPCR structure, a 2.8 Å resolution structure of rhodopsin, was published only recently by Palczewski and collaborators.40 However, a structure-based approach to GPCR drug discovery in the absence, but probably also in the presence, of the real structures requires a multidisciplinary approach, where molecular models represent a structural context to efficiently integrate experimental data and inferences derived from molecular biological, biophysical, bioinformatic, pharmacological and organic chemical methods. Although not always achievable, the success of a synergistic effect among these disciplines is highly dependent on the experimental design. Synergy is best achieved when mutations are structurally interpretable, structural hypotheses are experimentally testable, ligands are well characterized pharmacologically, and the necessary chemical modifications of the ligands are feasible.11
In recent decades, numerous medicinal chemistry groups have made intense efforts in searching for ideal ligands for these receptor subtypes.9–11,41–45 In particular, the search for selective antagonists held greater appeal than selective agonists, not only for their potential therapeutic applications but also considering the fact that antagonists are preferred molecular probes for pharmacological characterization of receptors. Considering all of these aspects, the search for potent and selective adenosine receptor antagonists has been one of the most highly investigated areas in medicinal chemistry in recent years. It should be emphasized that for all the receptor subtypes the alkylxanthines (e.g., theophylline, caffeine), which are natural antagonists for the adenosine receptors, have represented the starting point for the discovery of potent and selective antagonists.41–45 Following multiple modifications of the xanthine nucleus, various potent and selective antagonists have been found. Nevertheless, xanthine derivatives have several physico-chemical limitations, including low water solubility. For this reason, several research groups have focused on compounds having a non-xanthine structure for improving the water solubility and consequently bioavailability.41–45
The purpose of this review is to summarize the most recent developments made in the field of adenosine receptor antagonists, which for all classes could be subdivided into two large families: (i) xanthine derivatives; (ii) polyheterocyclic derivatives.
2. A1 ADENOSINE RECEPTOR ANTAGONISTS
The A1AR could be considered the best-characterized member of the adenosine receptor family. Several antagonists are currently under clinical investigations, and are recently reviewed.10,41–43
A. Xanthine Derivatives
A large number of modifications on the xanthine core at the 1, 3, and 8 positions led to the discovery of 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 1) which was highly potent and selective at the adenosine A1AR in a rat model,46 while at the human A1AR it was 10-fold less potent with a consequent reduction of selectivity versus the other receptor subtypes. Also, DPCPX displays considerable affinity at the human A2BAR.47 For these reasons the search for a truly selective A1AR adenosine receptor antagonist in the human model represented a new appealing goal to be achieved (Fig. 3).
Figure 3.
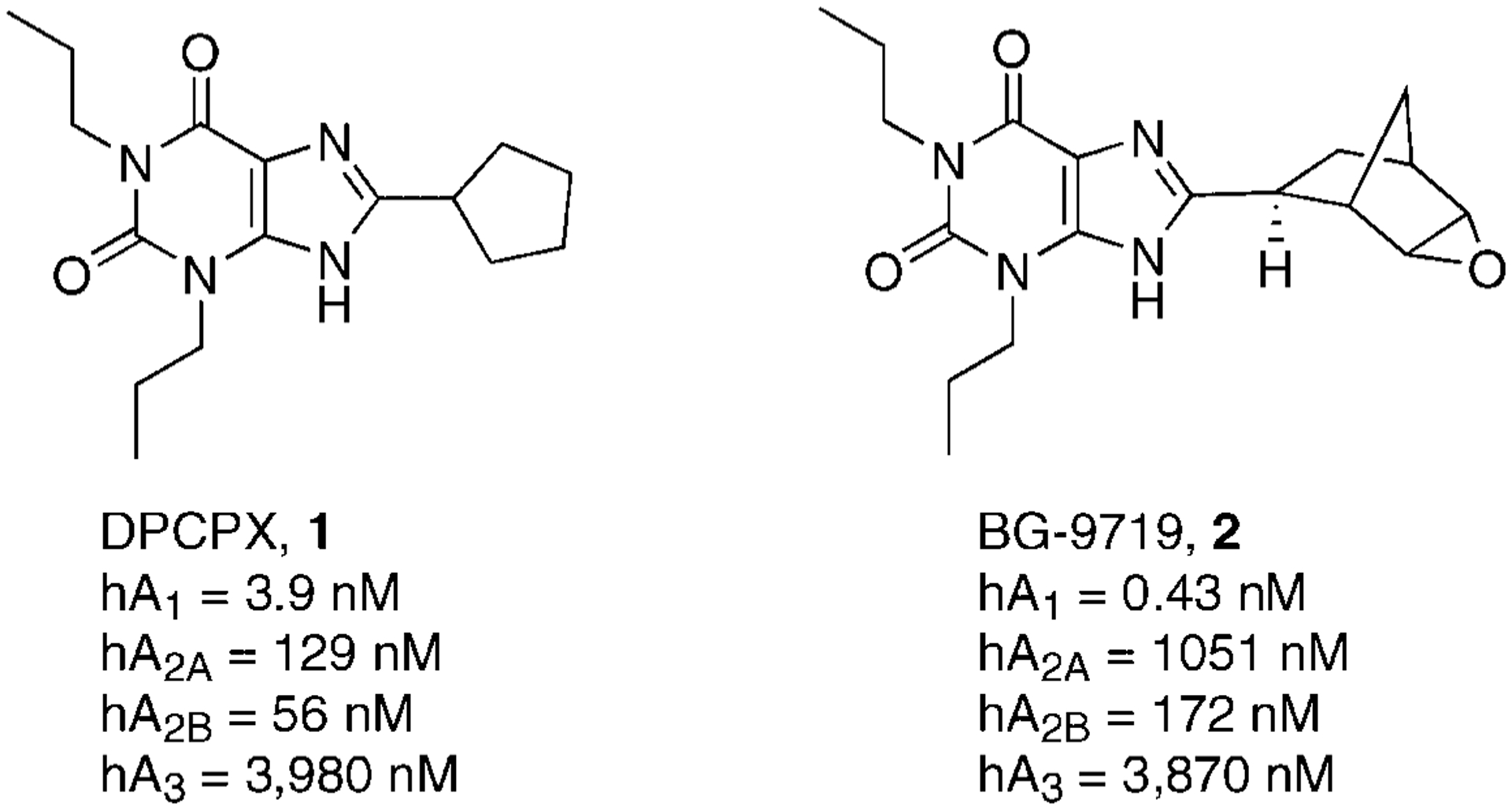
Structure and binding affinities of xanthines as A1AR antagonists.
In addition to DPCPX, other substituted xanthines have been proposed as A1AR antagonists, in particular, by introducing chiral substituents to demonstrate the importance of the stereochemistry, or by insertion of polar moieties. The introduction at the 8-position on the 1,3-dipropylxanthine nucleus of a [2-(5,6-epoxy)norbornyl] moiety led to the discovery of BG-9719 (2) which was highly potent and selective at the A1AR in a human model.48 A small stereochemical effect on the affinity was present with this compound, such that the R-isomer was twofold less potent (Ki hA1 = 0.80 nM) than the S-isomer (Fig. 3).
Recently, a fluorescent derivative of the xanthine amine functionalized congener (XAC)49 was shown to be useful for visualizing the A1AR in small areas of cell membranes using fluorescence correlation spectroscopy.50
B. Polyheterocyclic Derivatives
Numerous classes of heterocyclic derivatives were shown to bind to the A1AR. Among the first such derivatives were the 1H-imidazo[4,5-c]quinolin-4-amines, which were synthesized based on a prediction from early ligand modeling.51 Most of these derivatives could be considered an extension of the xanthine structure. More recently, some synthetic triazolo-purinones (3,4), clearly derived as tricyclic extensions of the xanthine nucleus, showed very promising affinity at the A1AR subtype with significant degree of selectivity versus the A2AAR subtype (Fig. 4).52
Figure 4.
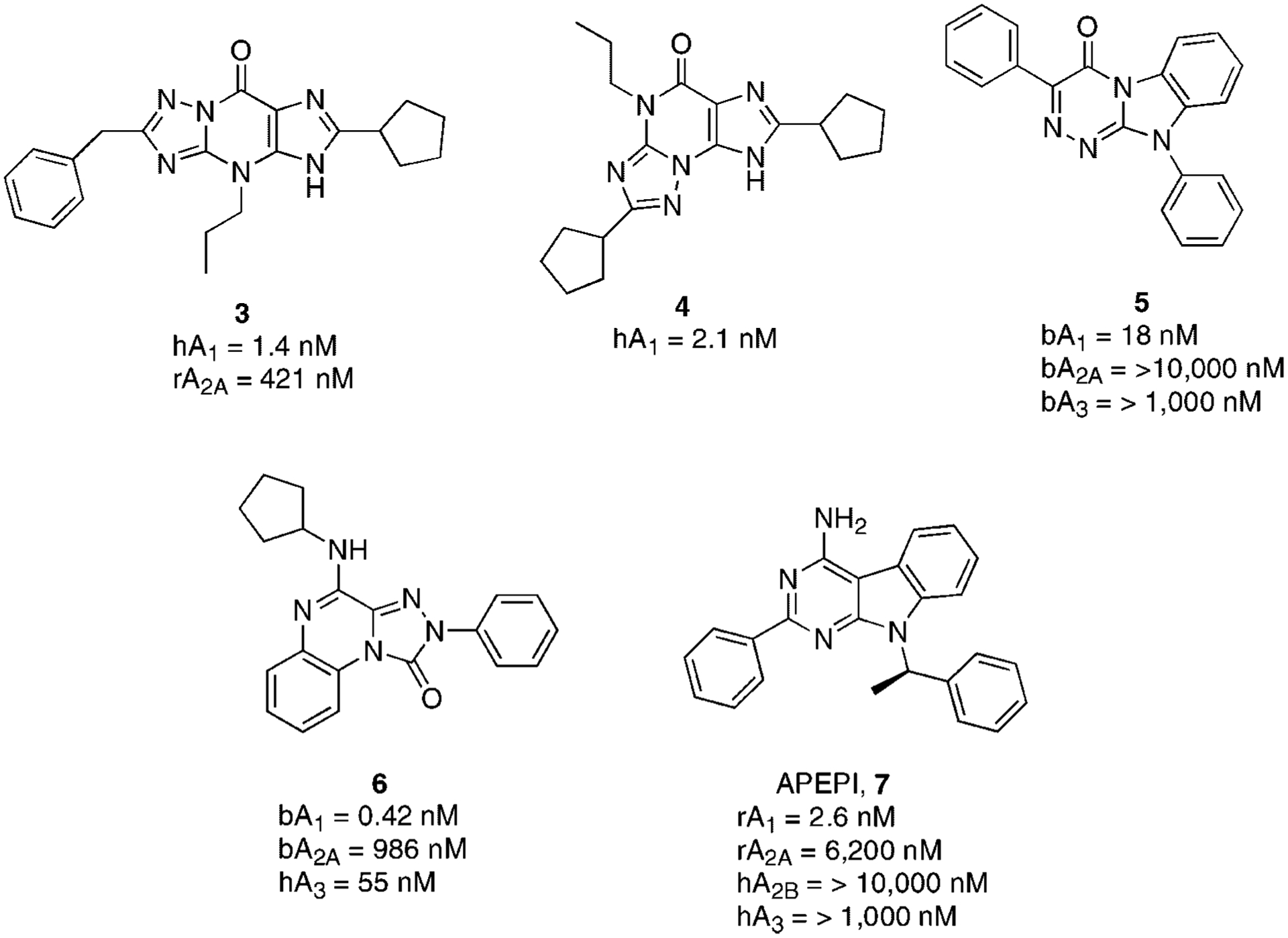
Structures and binding affinities of non-xanthine adenosine A1AR antagonists.
Another class of A1AR antagonists is represented by 3-aryl-[1,2,4]triazino[4,3-a]-benzimidazol(10H)-4-one derivatives. In particular, compound 5 displayed high affinity at the bovine A1AR and significant selectivity in comparison to the A2AAR and A3AR subtypes (Fig. 4).53 A related A1AR antagonist could be considered the triazolo-quinoxaline 6 which displays high potency and good selectivity (Fig. 4).54 Isosteres of 6, such as pyrazolo-quinolines or imidazo-quinoxalines have been also reported as A1AR antagonists. Although the affinity at A1AR was in the nanomolar range, none of the reported compounds were found to be highly selective.55,56
A series of non-xanthine heterocycles displaying high potency at A1AR and selectivity versus all the other subtypes is represented by 7-deaza-adenines. One particular derivative 7 (APEPI) proved to be highly potent and selective (Fig. 4). The A1AR affinity is prevalently due to the R-enantiomer. Many other modifications have been made on this nucleus (e.g., replacement of phenyl ring at the 2 position or structure simplification to an indole nucleus), but none of these variations improved both affinity and selectivity.56,57
Very simplified heterocyclic derivatives as adenosine receptor antagonists are represented by thiazole and thiadiazole derivatives (Fig. 5). In particular, thiadiazole 8 (LUF5437) has been considered the starting point for this new class of compounds.58 In fact, complete hydrogenation of phenyl ring led to derivative 9 (LUF5472), which was less potent at the A1AR but more selective. Replacement of the thiadiazole nucleus with a thiazole moiety seemed to be well tolerated at the A1 receptor.58,59
Figure 5.
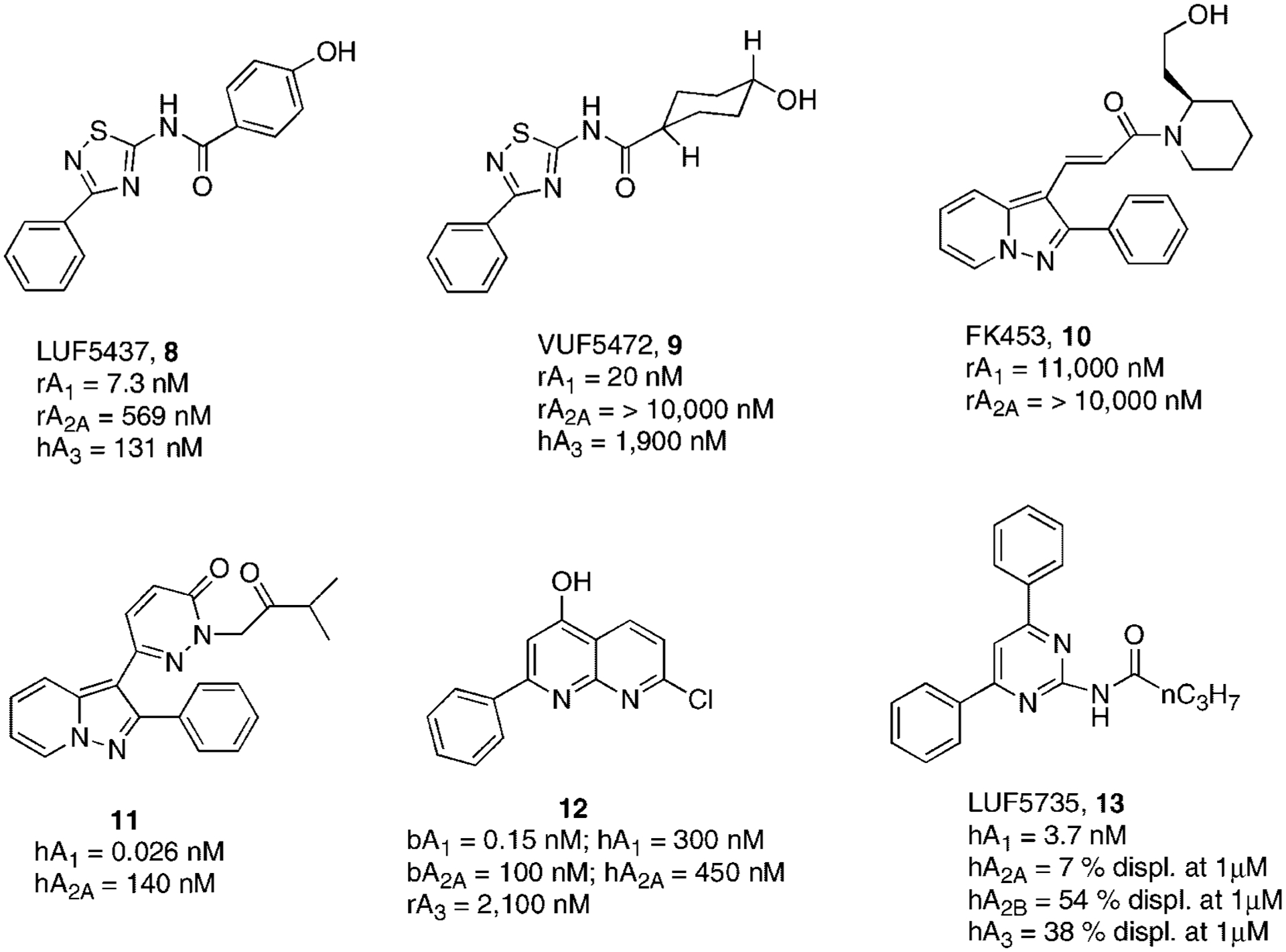
Structures and binding affinities of simplified A1AR antagonists.
Another class of simplified analogs structurally related to the xanthine core consists of derivatives of the pyrazolo[1,5-a]pyridine nucleus. Compound 10 (FK453) represents the lead compound of this series showing favorable affinity and selectivity for the A1AR compared to the A2AAR (Fig. 5). Various modifications have been performed on this nucleus but when acryloyl amide was constrained into a pyridazinone nucleus and nitrogen was substituted with an isobutyryl moiety (compound 11) a significant increase of potency and selectivity was obtained (Fig. 5).60–62
Also, a naphthyridine nucleus has been investigated for A1AR antagonists. In a naphthyridine series, compound 12 proved to be a promising antagonist displaying affinity in a sub-nanomolar range and high levels of selectivity in a bovine model, while unfortunately in human the compound dramatically lost potency and consequently selectivity.63,64
Very recently, IJzerman and coworkers synthesized a large number of pyrimidine derivatives designed with the help of molecular modeling. This study permitted to identify the compound 13 that was potent and selective as a human A1AR antagonist (Fig. 5).65
As clearly described, chemically diverse classes of compounds have been identified as A1AR antagonists. Nevertheless, considering that many have not yet been examined at all four adenosine receptor subtypes and the species differences are evident, most of these synthetic compounds should be reexamined in a human model for better understanding and for consideration as clinical candidates.
C. Biological Actions of A1 Adenosine Receptor Antagonists
Peripheral applications envisioned for A1 receptor antagonists include kidney protection and cardiac anti-arrhythmic agents.42,43 Since caffeine is best known for its stimulant activity in the central nervous system, adenosine antagonists of the A1 receptor and other subtypes have been of interest in cognitive disorders. A novel, potent, and selective adenosine A1 receptor antagonist FR194921 exerts both cognitive-enhancing and anxiolytic activity, suggesting the therapeutic potential of such compounds for dementia and anxiety disorders.66
3. A2A ADENOSINE RECEPTOR ANTAGONISTS
Both xanthines and non-xanthines have been developed as selective A2AAR antagonists. A2AAR antagonists proved to be attractive for the treatment of several diseases of the central nervous system, such as motor dysfunctions, due to the clearly demonstrated interaction between A2AAR and D2 dopamine receptors (both at the protein and second messenger levels) in the basal ganglia.67 For this reason, A2AAR antagonists could be considered potential drugs for the treatment of neurodegenerative disorders such as Parkinson’s disease.
A. Xanthine Derivatives
The first xanthine analog, which displayed good potency at the A2AAR subtype (100 nM) and significant selectivity in comparison to the A1AR (45-fold) was the 8-unsubstituted 1-propargylxanthine (14) (Fig. 6).68
Figure 6.
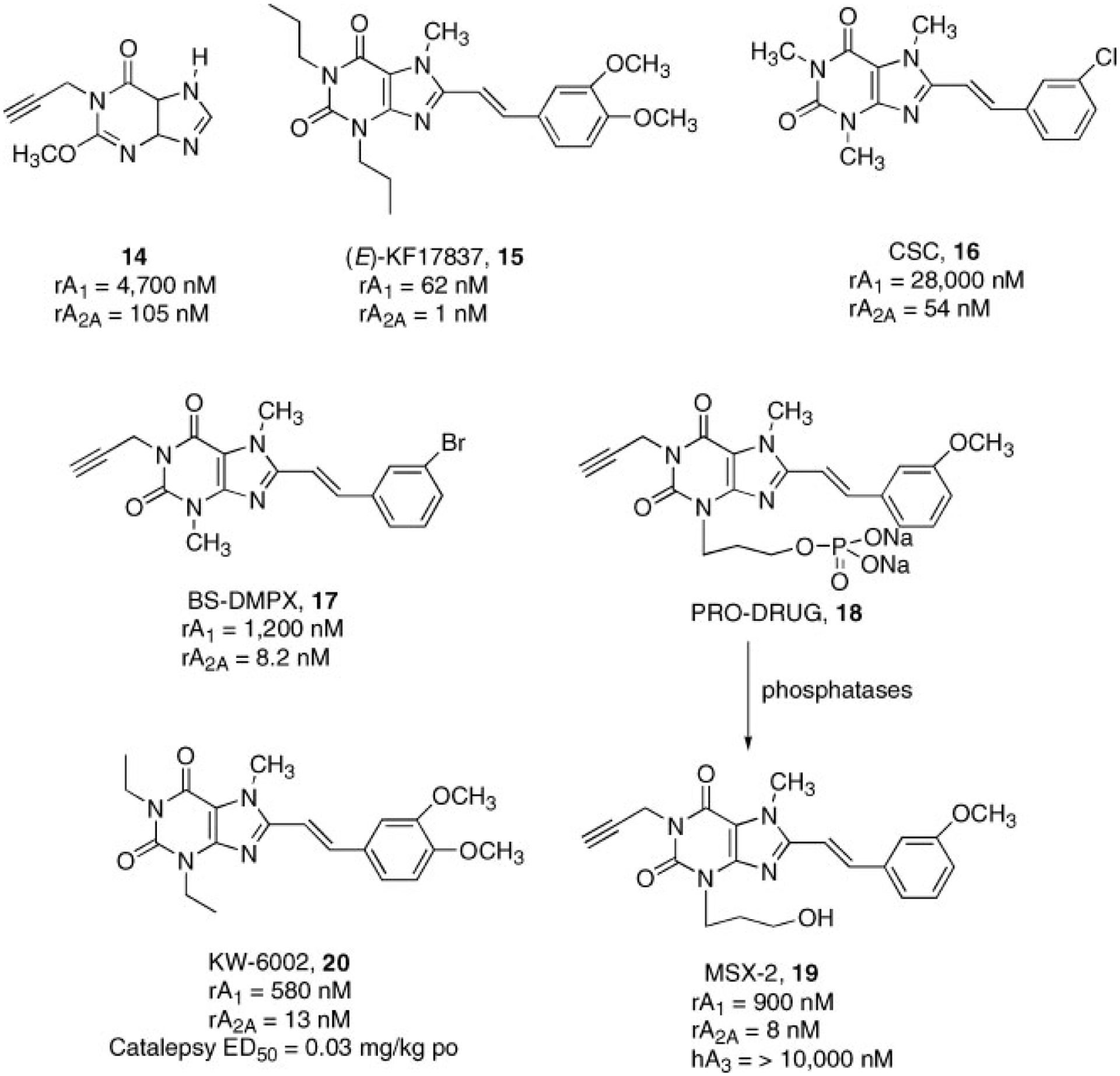
Structures and binding affinities of xanthines as A2AAR antagonists.
Starting from this observation, a program to screen various 1-, 3-, 8-substituted xanthines led to the discovery of the first very potent and selective A2AAR antagonist, 1,3-dipropyl-7-methyl-8-(3, 4-dimethoxystyryl)xanthine (KF17837, 15), which proved to be potent in the nanomolar range at the A2AAR subtype (1 nM) and significantly selective in comparison to A1AR (62-fold) (Fig. 3).69,70 In a detailed SAR study on this class of compounds, the 3-chlorostyrylcaffeine (CSC, 16) was identified as being less potent than 15 at the A2AAR (54 nM) but with an increased selectivity in comparison to the A1AR subtype (560-fold).71
Two major problems have limited the use of these xanthine derivatives as pharmacological tools for studying the A2AAR subtype: (a) the low water solubility;72 (b) the rapid photoisomerization which they undergo when exposed to daylight in dilute solution.73 It should be noted that this isomerization process is not relevant when styrylxanthines are administered orally as solid substances. In an attempt to avoid this problem, the styryl moiety has been replaced with different functional groups (e.g., triple bond, cyclopropyl, or diazo group) or constrained structure. However, none of this isosteric substitutions has led to an improvement in the pharmacological profile, but rather in many cases to a complete loss of affinity.68,74
Instead, the introduction of a propargyl at the 1-position in combination with the 8-styryl group by Müller and coworkers seemed to increase affinity at the A2AAR subtypes with the retention of selectivity. These studies led to the discovery of the BS-DMPX (3,7-dimethyl-1-propargyl-8-(3-bromostyryl)xanthine 17, which could be considered a lead compound of a new series.75 However, at the 3 and 7 positions, methyl substitution seemed to be desirable for achieving both affinity and selectivity at the A2AAR subtype (Fig. 6).76–78 2A Regarding the substitutions at the 8-position, it has been clearly demonstrated that an aromatic ring attached to an ethenyl group is a fundamental requirement for both affinity and selectivity at the A2AAR.77,79
For the improvement of water solubility of styryl xanthines, two different approaches have been utilized: (a) introduction of polar groups on the phenyl ring; (b) preparation of phosphate pro-drugs. The introduction of a sulfonate group on the phenyl ring of styryl moiety produces a significant reduction of affinity (20- to 30-fold) at the A2AAR but with retention of selectivity.80
More interesting results have been obtained using phosphate ester pro-drugs. In fact, the pro-drug 18, which was stable in aqueous solution but readily cleaved by phosphatases to liberate MSX-2 (3-(3-hydroxypropyl)-8-(3-methoxystyryl)-1-propargylxanthine, showed a very high affinity and selectivity for the A2AAR (19, Fig. 6).81
All these studies, performed by several laboratories, have strongly suggested reconsidering the xanthine family as A2AAR antagonists. In fact, such an antagonist, KW-6002 (1,3-diethyl-8-(3-methoxystyryl)-7-methilxanthine, 20, is already in phase II clinical trials for the treatment of basal ganglia disorders such as Parkinson’s disease.82
Unfortunately, very recently, more detailed studies performed on MSX-2 (19), in contrast with previous studies, clearly demonstrated that styryl xanthines at the solid state upon light irradiation led to dimmer derivatives which are almost inactive at the A2AAR. This should be considered a further limit of clinical use of styryl xanthine derivatives.83
B. Polyheterocyclic Derivatives
The first promising A2AAR antagonist with a non-xanthine structure was CGS 15943 (21, 9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine),84,85 which showed affinity but not selectivity versus A1AR, A2BAR, and A3AR (Fig. 7).86 Nevertheless, it has represented the starting point for developing new non-xanthine structures as A2AAR adenosine antagonists. A few years later, bioisosteric replacement of the phenyl ring of CGS15943 with an N7-substituted pyrazole led to the family of N8-substituted pyrazolo-triazolo-pyrimidines. Two selected compounds of this family named SCH 58261 (22, 5-amino-7-(β-phenylethyl)-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine) and SCH 63390 (23, 5-amino-7-(3-phenylpropyl)-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine) proved to be potent and selective A2AAR antagonists both in rat and human models (Fig. 7).87,88
Figure 7.
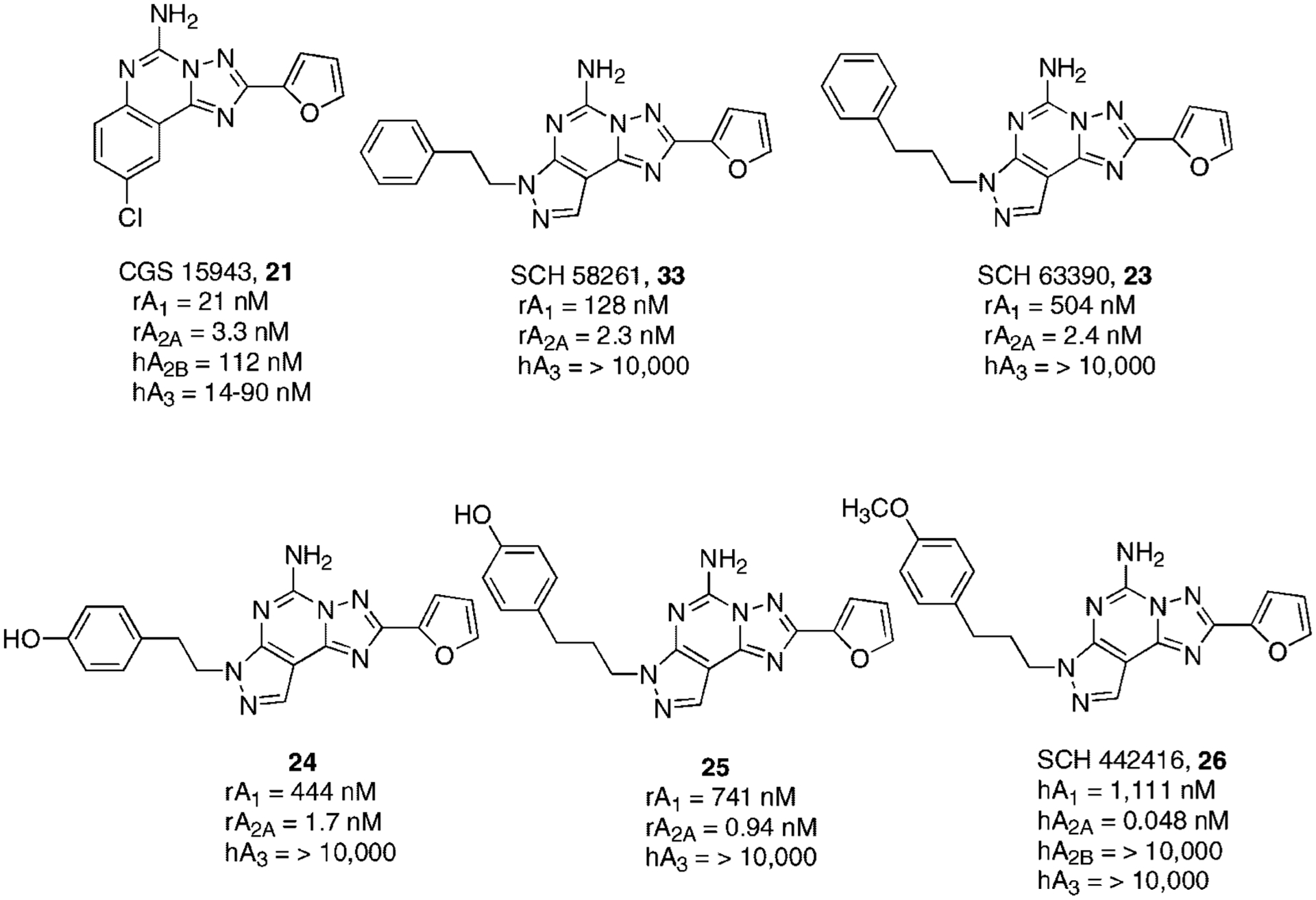
Structures and binding affinities of triazolo-quinazoline and pyrazolo-triazolo-pyrimidines as A2AAR antagonists.
However, a major problem of this class of compounds is related to their low water solubility and consequently poor bioavailability. The introduction of a hydroxyl group at the para position on the phenyl ring of compounds 22 and 23 led to derivatives 24 (5-amino-7-[β-(4-hydroxyphenyl)ethyl]-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine) and 25 (5-amino-7-[3-(4-hydroxyphenyl)-propyl]-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine), which not only displayed greater hydrophilic character, but also a significant increase of both affinity and selectivity at the A2AAR subtype, most probably due to hydrogen bond formation (Fig. 7). Therefore, to understand the nature of the hydrogen bond, compound SCH 442416 (26, 5-amino-7-[3-(4-methoxyphenyl)-propyl]-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine) was synthesized. This derivative showed a surprising increase of affinity for the A2A adenosine receptor to qualify it as a candidate tool for PET studies in its 11C labeled form. The high affinity was consistent with the compound acting as a hydrogen bond acceptor (Fig. 7).89
However, the introduction of oxygenated groups on the phenyl ring of the side chain was not sufficient to confer the necessary water solubility, and the introduction of additional functionality resulted in a compelling need to address this problem. Toward this purpose, carboxylic and sulfonic moieties were introduced, which contributed greatly to the water solubility especially in the case of the sulfonic moiety, but a great loss of affinity was observed.90
A partial resolution to this problem was obtained by the former Zeneca group in proposing a compound named ZM 241385 (27, 4-[2-[[7-amino-2-(2-furyl) [1,2,4]-triazolo[2,3-a] [1,3,5]triazin-5-yl]amino]ethyl]phenol), which proved to be one of the most potent A2AAR antagonists ever reported and having favorable water solubility (Fig. 8).91
Figure 8.
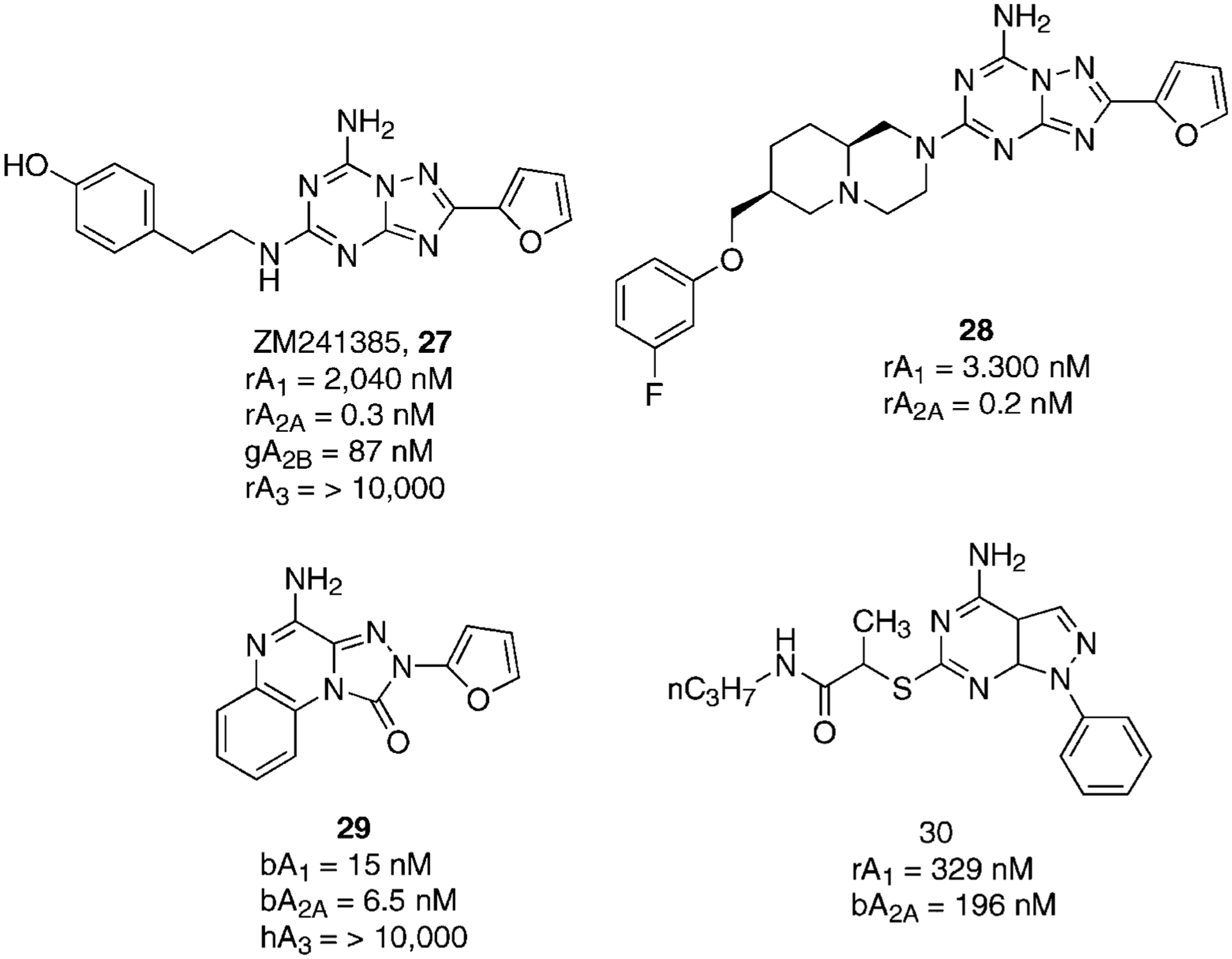
Structures and binding affinities of triazolo-triazine and new tools as A2AAR antagonists.
However, ZM 241385, which could be considered a simplified analog of the pyrazolo-triazolo-pyrimidine series, was found to bind also with good affinity at the human A2BAR. In fact, its tritiated form is actually used in radioligand binding studies of this receptor subtype as well as at the A2AAR.92
Recently, a large series of derivatives bearing various substituents at the 5-position on the triazolo-triazine nucleus and the related triazolo-pyrimidine nucleus have been synthesized. In particular, derivative 28 showed great potency and selectivity for the A2AAR as compared with the A1AR (Fig. 8). Nevertheless, the lack of binding data at the A2B and A3 prevents a comparison of the derivatives with other fully characterized derivatives. Some of these derivatives, although not displaying exceptional high potency in binding studies, showed good oral efficacy in a rodent catalepsy model of Parkinson’s disease.93–97
Over the last few years, other classes of compounds have been investigated with the aim of obtaining new antagonist tools for studying A2AAR. Unfortunately, none of the reported compounds showed a better profile than the above-mentioned derivatives. Only two classes of compounds, the triazolo-quinoxaline98 and some pyrazolo-pyrimidines,99 seem to possess promising requirements as A2A adenosine receptor antagonists (Fig. 8).
In the triazolo-quinoxaline series, only one compound 29 showed interesting A2AAR affinity. Unfortunately, this nucleus seemed to be very sensitive to any kind of modification. In fact, alkylation of the amino group, or its replacement with a carbonyl group, or substitution of the phenyl ring was detrimental in terms of affinity at the A2AAR. In some cases, the affinity at the human A3AR was predominant. Instead, in the pyrazolo-pyrimidine series, only one 30 showed a promising binding profile, but was nevertheless of low potency and low selectivity for the A2AAR.
C. Biological Actions of A2A Adenosine Receptor Antagonists
The A2AAR antagonists that is furthest advanced in clinical trials is KW6002, as described above, and other antagonists of this subtype are under development.82,97,100 The interest in CNS action of A2AAR antagonists also extends to impeding the neurodegenerative process4,7,8,10 and possibly the treatment of stroke.101 The peripheral actions of A2AAR antagonists might be complicated by a proinflammatory effect,102 but might be therapeutically useful for cancer treatment.103
4. A2B ADENOSINE RECEPTOR ANTAGONISTS
Most of the high affinity receptor antagonists thus far reported have been xanthine derivatives. Consideration of the potential therapeutic applications of A2BAR antagonists, particularly their possible use an anti-asthmatic agents,104,105 has stimulated many research groups to search for potent and selective antagonists for this subtype. Recognition of the possibility that the mechanism of action of the anti-asthmatic drugs theophylline (1,3-dimethylxanthine) and enprofylline (3-propylxanthine) might involve the A2B adenosine receptor has spurred this research.105
A. Xanthine Derivatives
A large number of substitutions at the 1, 3, and 8 positions of the xanthine core have been performed with the aim of describing an SAR profile for the A2BAR subtype. In particular, it has been observed that 1,3-unsubstituted xanthine derivatives bearing a phenyl ring at the 8-position possesses good selectivity but poor potency at the A2BAR subtype.106 An optimization of this structure led to the discovery of 1-propyl-8-(4-sulphonyl)phenyl xanthine PSB 1115 (31) and some related pro-drugs, such as a 4-nitrophenylester, which were found to be potent and selective A2BAR antagonists (Fig. 9).107,108
Figure 9.
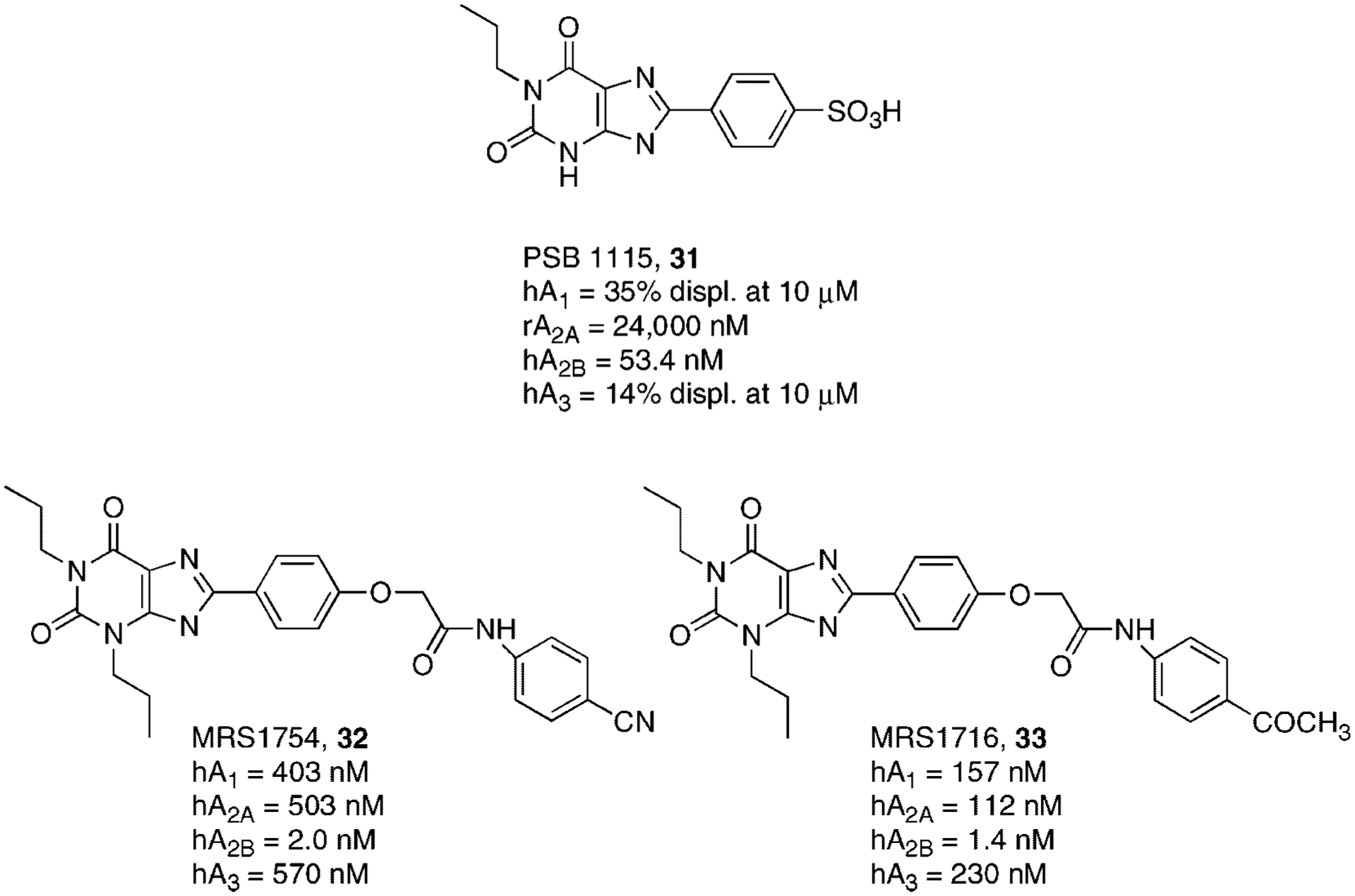
Structures and binding affinities of xanthine derivatives as A2BAR antagonists.
In the series of 8-phenyl xanthine derivatives, a large number of amides derived from the 8-{4-[(carboxymethyl)oxy]phenyl}-1,3-dipropylxanthine have been prepared and tested as A2BAR antagonists.109,110 This study led to the discovery of the ([N-(4-cyanophenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy] acetamide] (32, MRS1754) and ([N-(4-acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy] acetamide] (33, MRS 1716), which proved to be the most potent and selective human A2BAR antagonists.111 In fact, the tritium labeled form of derivative 32 has been prepared and utilized in radioligand binding studies112 (Fig. 9).
Phenyl replacement with a pyrazole moiety led to compounds which showed a quite similar A2BAR affinity with respect to the phenyl series.113,114 Recently, Zablocki and coworkers have reported an extended series of 8-aryl xanthines as selective A2BAR antagonists.115
In the xanthine family, a new class of deaza-analogs has recently been reported, which displayed affinity at A2BAR in the micromolar range, but poor selectivity versus the A2AAR subtype.116
B. Polyheterocyclic Derivatives
Within this category of adenosine antagonists, quite varied structures have been introduced and modified in the search for new A2BAR antagonists.
Starting from the experimental observation that the non-selective A2AAR antagonist CGS15943 (21) (Fig. 7) also proved to be an effective A2BAR antagonist both in functional and binding studies, a large number of acyl moieties have been placed at the N5 position (Fig. 10).117
Figure 10.
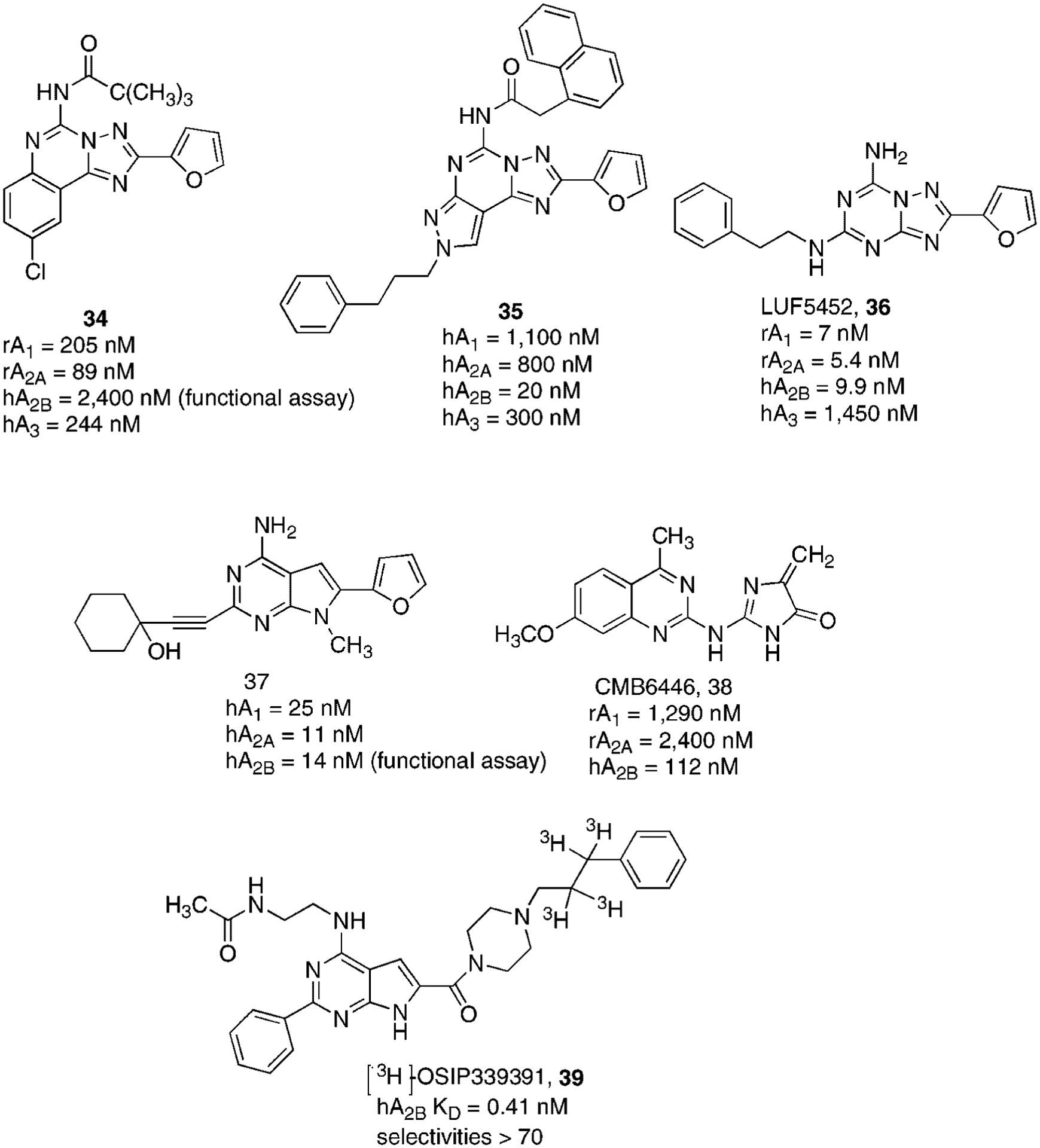
Structures and binding affinities of polyheterocyclic derivatives as A2BAR antagonists.
The introduction of apolar chains such as the N5-pivaloyl group resulted in compound 34, which displayed significant selectivity but not high potency at the A2BAR (Fig. 10).117
A similar approach has been utilized with the pyrazolo-triazolo-pyrimidine nucleus, whose potency at the A2BAR was obtained while a complete loss of selectivity was observed.118 Only when bulky substituents at both N5 and N8 positions were present (compound 35), was a significantly potent and selective A2BAR antagonist obtained (Fig. 10).119
Considering that the potent and selective A2AAR antagonist ZM241385, 27 (Fig. 8) proved to be also quite potent at the A2BAR, its tritiated form is usually utilized in radioligand binding studies,120 several modifications at the 5-position of the triazolo-triazine nucleus have been performed. It has been observed that the hydroxyl group replacement (36) enhanced the A2BAR affinity although the selectivity was poor (Fig. 10).121
Very promising results at this receptor subtype were obtained upon modification of adenine nucleus.122 A detailed investigation on this class of compounds permitted the partial optimization of the substitution of adenine nucleus to enhance both potency and selectivity for the A2BAR subtype. In particular, the presence of alkynyl moiety at the 2-position and the presence of a furyl ring at the 7-position led to a very promising potent and selective A2BAR antagonist (37).123 These data suggest that further optimization of the pattern of substitutions at this third position could lead to the discovery of a highly potent and selective A2BAR antagonist (Fig. 10).
Recently, in a screening program focused on the searching of new tools as adenosine receptor antagonists, a quinazoline derivative, named CMB 6446 ((4-methyl-7-methoxyquinazolyl-2-(2′-amino-4′-imidazolinone)) (38), proved to be quite potent and selective at the A2BAR subtype with a binding Ki value of 112 nM.124 (Fig. 10). However, further efforts to enhance the A2BAR affinity of 38 failed even extensive synthetic modification was made on this class of compounds.
Very recently, a tritiated form of a pyrrolo-pyrimidine derivative, named [3H]OSIP-339391 (39) has been proposed as a very promising radioligand for studying A2BAR. In fact, it showed a KD value of 0.41 nM and selectivities versus the other receptor subtypes higher than 70.125
C. Biological Actions of A2B Adenosine Receptor Antagonists
The therapeutic potential-based peripheral actions of A2B adenosine receptor antagonists might include treatment of asthma.104,105 The anti-asthma drugs, theophylline and enprofylline, are used therapeutically to treat asthma at concentrations to block A2BAR.104,105 A2BAR antagonists may also serve as novel drugs for type-II diabetes,126 Alzheimer’s disease127, and cystic fibrosis.128
5. A3 ADENOSINE RECEPTOR ANTAGONISTS
In the last years, many efforts have been made to search for potent and selective human A3AR antagonists.11 The interest in blocking this class of receptors arose after the discovery of their involvement in cellular growth.129
A. Xanthine Derivatives
Natural xanthines, such as caffeine and theophylline that are considered the natural antagonists for adenosine receptors, show in general very low affinity for the A3AR subtype (in the high micromolar range).130 Nevertheless, very recent SAR studies on these compounds indicated that a cyclization between the 7 and 8 positions led to pyridopurine-2,4-dione derivative (40) as potent A3 adenosine receptor antagonists131 (Fig. 11).
Figure 11.
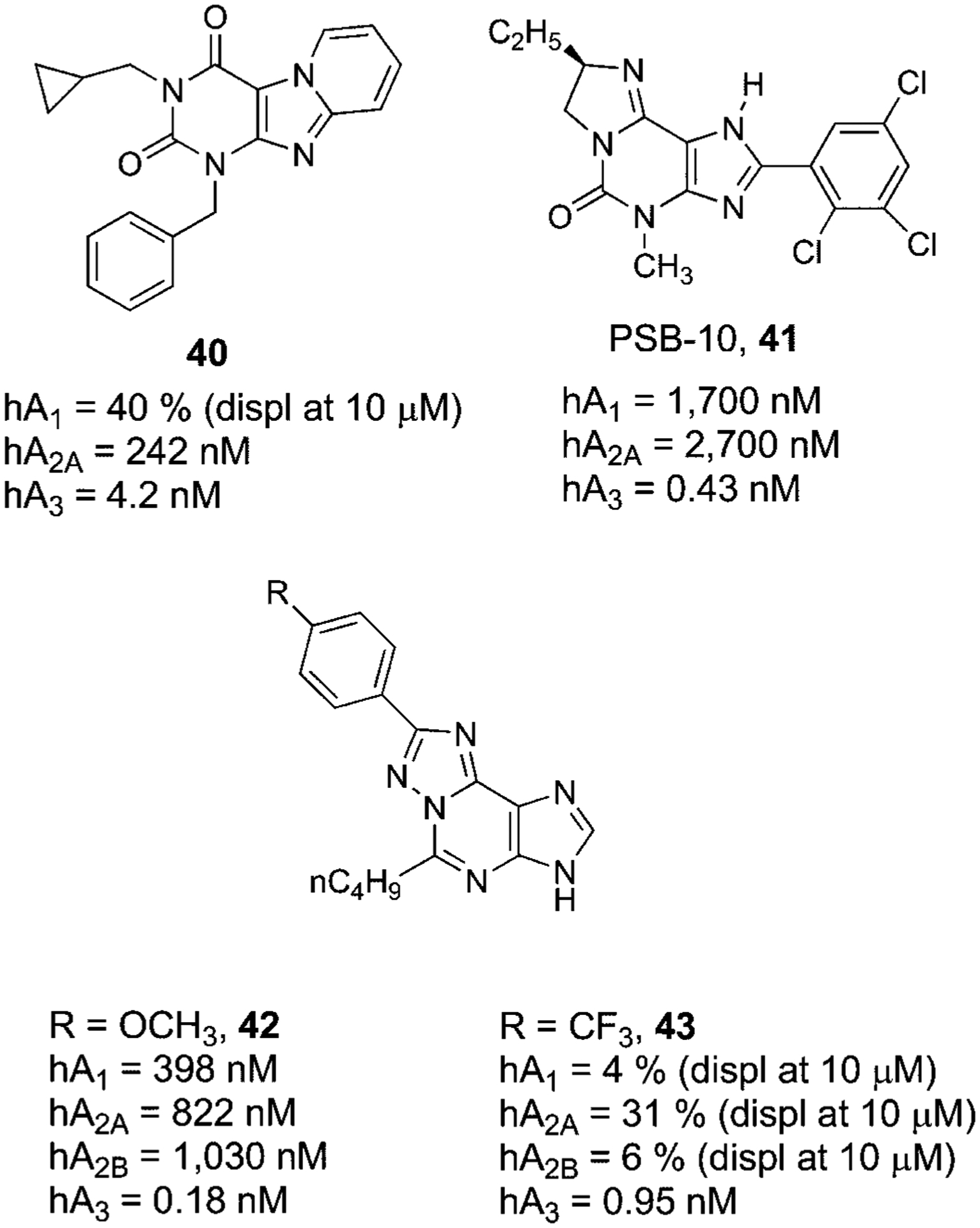
Structures and binding affinities of xanthine derivatives as human A3AR antagonists.
Other positions of the xanthine core have been modified with the aim of improving A3AR affinity. The discovery of 2-phenylimidazopurin-5-ones as water soluble derivative of xanthines led to PSB-10 (41) a highly potent and selective human A3AR antagonist (Fig. 11).132 The tritiated form of a related compound named PSB-11 has been used as a high affinity radioligand at this subtype with favorably low non-specific membrane binding.133
Following these observations, other structural classes in which the xanthine structure was extended have been reported as A3AR antagonists. One such class was the triazolo-purines (42,43), which proved to be quite potent and selective human A3AR antagonists (Fig. 11).134,135
B. Polyheterocyclic Derivatives
In this class of compounds, different heterocyclic moieties have been identified as potential A3AR, and extensively reviewed, which can be classified in six families of derivatives: (i) flavonoids; (ii) 1,4-dihydropyridines and pyridines; (iii) triazolo-quinazolines; (iv) isoquinoline and quinazolines; (v) pyrazolo-triazolo-pyrimidines; (vi) various.11,136 In Figures 12–14, representative members of these family of compounds are presented.
Figure 12.
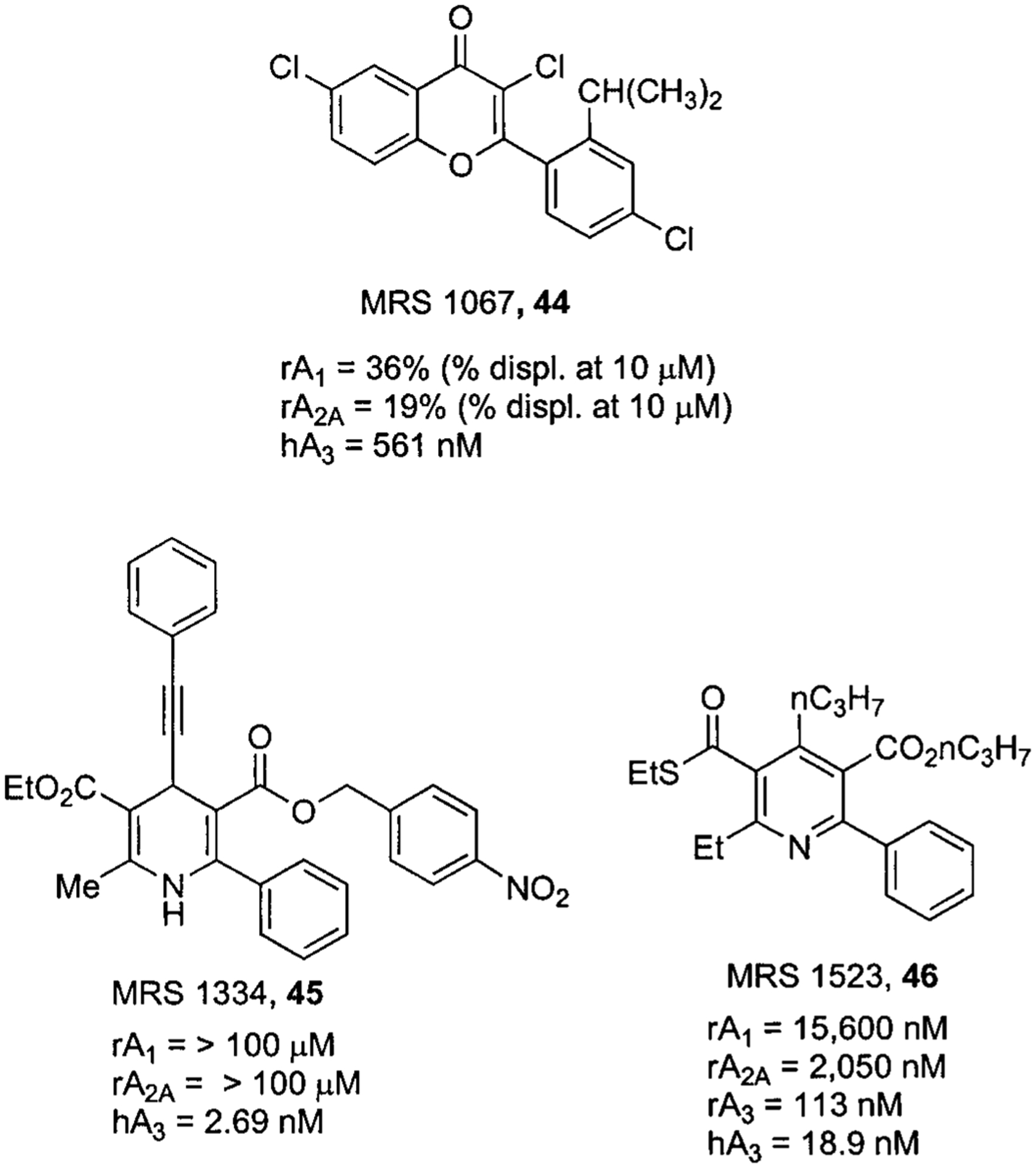
Structures and binding affinities of flavonoid, dihydropyridine, and pyridine derivatives as human A3AR antagonists.
Figure 14.
Structures and binding affinities of other heterocyclic derivatives as human A3AR antagonists.
The discovery of flavonoids as human A3AR antagonists was initiated by a broad screening of phytochemicals, in which it was shown that some flavonoid derivatives possess micromolar affinity at human A3AR. Optimization of reference compounds, through a classical structure-activity relationship study and with the help of molecular modeling approach, led to MRS 1067 (44), which proved to be the most potent (Ki, 561 nM) and selective compound of this series at the human A3AR subtype (Fig. 12).137MRS 1067 was the first reported antagonist suitable also for use with the rat A3 adenosine receptor,138 although it has since been superseded by more potent compounds.
A very similar approach was utilized for studying the SAR at the human A3AR of 1,4-dihydropyridines, which are typically antagonists of the L-type calcium channel. Initially, it was necessary to eliminate binding to these ion channels, which was accomplished through the introduction of extended arylalkynyl groups at the 4-position of the dihydropyridine nucleus in combination with phenyl substituents at the 6-position. These changes not only prevented the recognition at the calcium channel but also significantly improved the affinity at the human A3AR. In particular, a nitro derivative MRS1334 (45) proved to be the most potent analog of this series (Fig. 12).139
Simultaneously, the same authors studied the affinity of the pyridines, derived from the oxidation of the corresponding 1,4 dihydropyridines. In this class of compounds, to retain affinity and selectivity at human A3 adenosine receptor, small groups at the 4-position were found to be essential. This effect could be attributed to the change of the C4-hybridization from sp3 to sp2, with a consequent variation of the C5-C4-R4 angle from 68.1° to 0.2°. This study strongly supported by theoretical studies led to the discovery of MRS1523 (46), which showed favorable affinity at the human A3AR (18 nM) and was also the first derivative to possess submicromolar affinity at the rat A3AR subtype (Fig. 12).140 For various structural classes, most antagonists described as having high potency at the human A3AR subtype were consistently found to be weak or ineffective at the rat A3AR. This pronounced species difference could be correlated with the relatively modest sequence similarity (only 74%) that exists between rat and human A3AR sequences.141
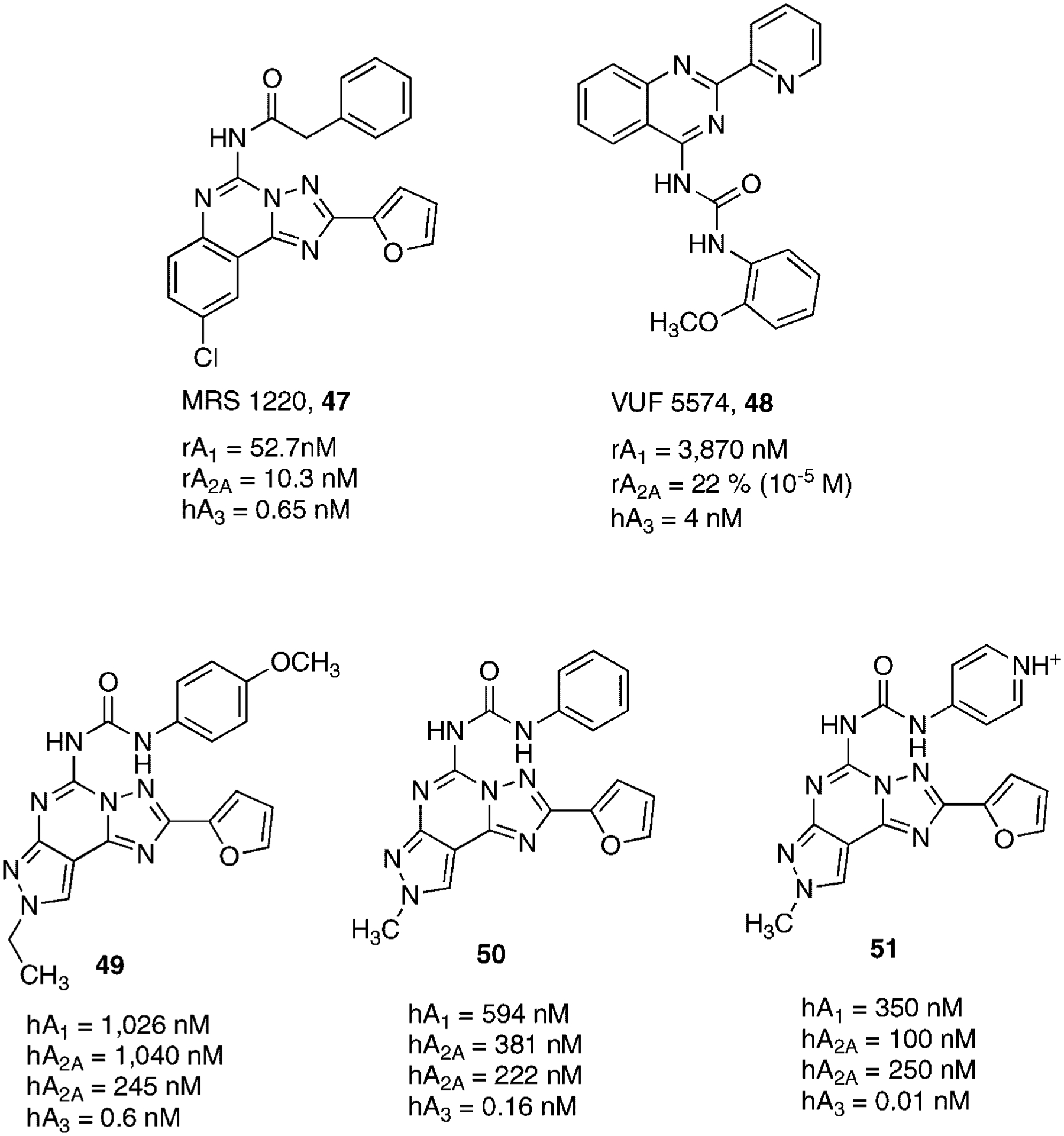
The triazolo-quinazoline derivative CGS 15943 (21, Fig. 7), a classic non-selective adenosine receptor antagonist, has been a starting point for searching new potent and selective human A3AR antagonists. Its acylation led to the discovery of MRS1220 (47) a highly potent (0.65 nM) and quite selective human A3AR antagonist (Fig. 13).142
Figure 13.
Structures and binding affinities of polyheterocyclic systems as human A3AR antagonists.
In a program of screening compound libraries by IJzerman and coworkers, it has been found that a series of 3-(2-pyridinyl)-isoquinoline derivatives possessed adenosine A3AR affinity.143 The synthesis of related quinazoline derivatives, with a classical bioisosteric substitution of carbon with nitrogen and the substitution of amide spacer with an urea moiety led to a compound, VUF5574 (48), which had improved affinity at human A3AR while being entirely inactive at A1AR and A2AAR receptor subtypes144 (Fig. 13).
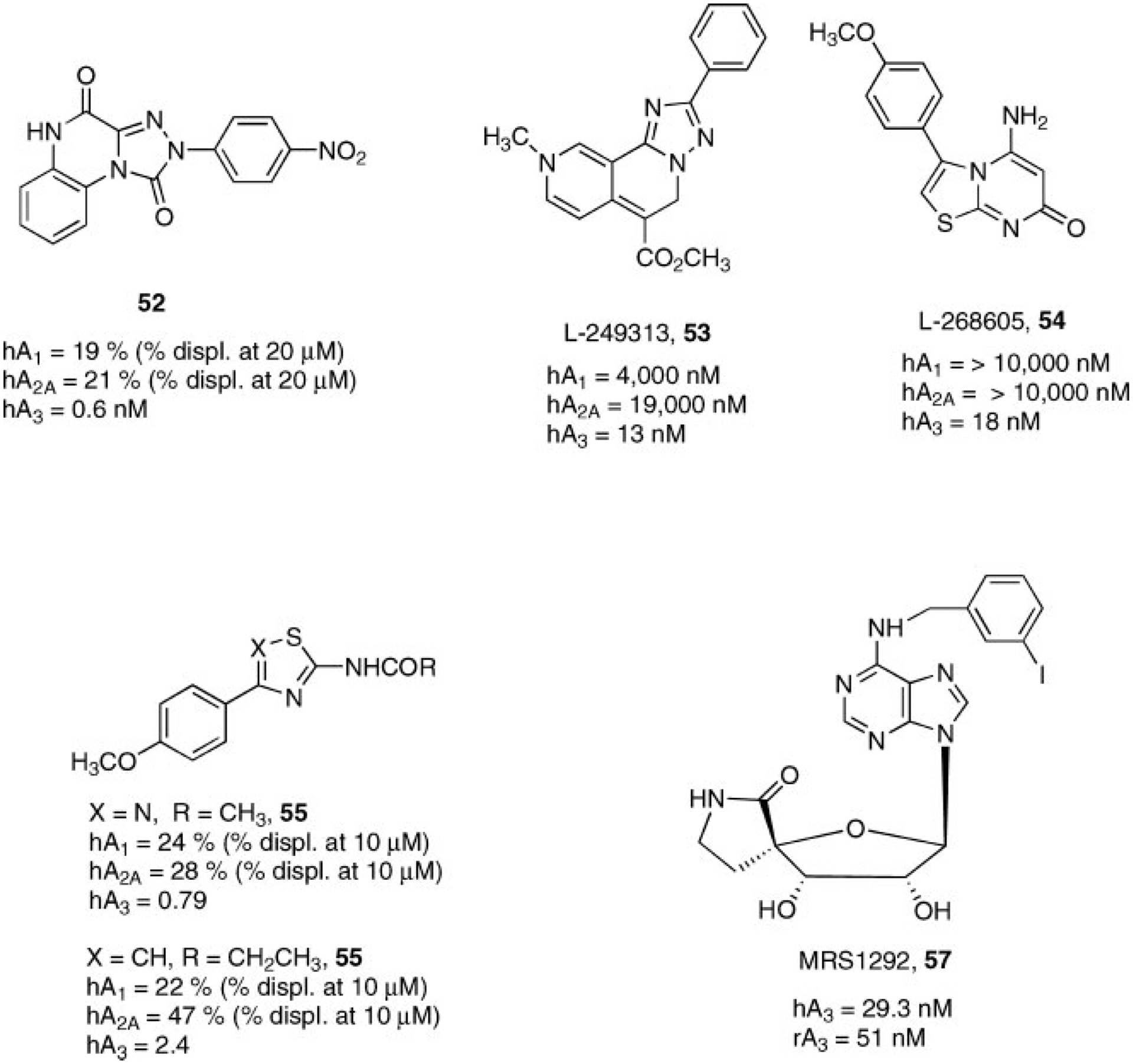
The discovery of pyrazolo-triazolo-pyrimidines as human A3AR antagonists, was based on the creation of a hybrid molecule between antagonists and agonists of this subtype. Specifically, the triazolo-pyrazolo-pyrimidine core, typical of classic adenosine receptor antagonists, was substituted at the N5 position a 4-methoxy phenyl carbamoyl moiety, which resulted to be optimal for having A3 affinity when introduced at the N8 position of NECA.130 This combination led to compound 49 which was one of the most potent and selective human A3AR antagonist ever reported. A classic SAR study combined with molecular modeling simulation permitted the identification of the structural requirements indispensable for receptor recognition. In particular, small substituents (e.g., methyl group) at the N8 position and at the 5-position unsubstituted phenyl ring seemed well tolerated. This resulted in compound 50, which displayed an increased affinity at the human A3AR.145 These results permitted the synthesis of a completely water soluble (15 mM) derivative 51, in which the phenyl ring was replaced by a pyridinium salt. The introduction of a nitrogen not only improved water solubility but also a significant increased affinity at the human A3AR. This observation suggested that electrostatic interactions were strongly involved in receptor recognition in this region (Fig. 13).146 Other derivatives structurally related to this family have been reported, including the triazolo-quinoxalines. Within this structural class, several compounds have been synthesized as antagonists for different adenosine receptor subtypes. These SAR studies permitted the identification of compound 52 as one of the most potent and selective human A3AR (Fig. 14).147,148
Other A3AR antagonists were the result of library screening in which novel heterocyclic derivatives with high affinity were identified, such as L-249313 (53) and L-268605 (54) (Fig. 14), however, no detailed SAR has been provided.149
Structurally simplified A3AR antagonists have been reported. The thiadiazole (55) and the bioisostere thiazole derivative (56) seem to be the promising agents, considering their very straightforward synthetic pathway and their low hydrophobic character (Fig. 14).150
Since nearly all of the reported A3AR antagonists showed significant potency and selectivity at the A3 adenosine receptor only in the human model, pre-clinical studies in vitro and in vivo in other species were severely limited. This aspect has been partially avoided by working on the adenosine core, that is, converting a selective A3AR receptor agonist into an antagonist. This was particularly effective in designing species-independent A3AR antagonists, since the nucleosides tend to bind well at this subtype across species. In general, adenosine receptor agonism correlates with the presence of a 9-ribose moiety on the adenine structure, while other adenine derivatives (such as 9-methyl or ethyl) are usually adenosine receptor antagonists. A3AR homology modeling combined with mutagenesis and SAR studies indicated that the ribose moiety also had a requirement of flexibility, particularly in the 5′-region, to fully activate the receptor. Consistent with this finding, a spirolactam analog, that is in which the ribose ring was sterically constrained (MRS1292, 57)151 proved to be a potent A3AR antagonist both in human and rat models (Fig. 14). MRS1292 contained the 5′-amide group, typical of potent agonists such as NECA, however the bicyclic constraint precluded receptor activation. Other modifications of nucleosides, such as the introduction of extended substituents at the 8-position and various substitutions of the N6 and 2 positions23,24 tended to convert agonists into antagonists.152
C. Biological Actions of A3 Adenosine Receptor Antagonists
A3AR antagonists appear to be of use in reducing intraocular pressure, which would be useful in the treatment of glaucoma.153 The A3AR promotes flow into the aqueous humor by coupling in a positive fashion to chloride inflow in non-pigmented ciliary epithelial cells.153 A3AR antagonists have also been of interest in possibly treating allergic conditions and inflammation.6,9–11
6. CONCLUSIONS
Potent and selective antagonists have been developed for the four subtypes of adenosine receptors. These advances have been based on both empirical methods and semi-rational design approaches, such as QSAR and receptor homology modeling. Both xanthines and non-xanthines have filled this need. The first non-xanthine heterocycles to attain nanomolar affinity were designed for the A2A receptor, by a series of studies in various laboratories in which the xanthine nucleus was elaborated and ring modified. The screening of chemically diverse libraries has resulted in novel chemical classes of A3 receptor antagonists, and also an intensive SAR studies on xanthines led to potent and selective A3AR antagonists. Now at all four subtypes, non-xanthine classes have been introduced as antagonists and optimized through substitution of functional groups and pendant moieties. The low aqueous solubility seen with many of these optimized compounds has been partially overcome with the introduction of polar or charged groups. Thus, the introduction of selective adenosine antagonists for the therapeutic treatment of a variety of diseases remains hopeful.
ACKNOWLEDGMENTS
The authors are very grateful for the financial support of the University of Padova and the Italian Ministry for University and Research (MIUR), Rome, Italy.
Contract grant sponsor: University of Padova;Contract grant sponsor: Italian Ministry for University and Research, Rome, Italy
Biographies
Stefano Moro is an assistant professor of medicinal chemistry at Padova University, Italy. He received his B.Sc. degree in Medicinal Chemistry and his Ph.D. degree in Physical Organic Chemistry from the University of Padova, Italy, and was a postdoctoral fellow at the National Institutes of Health in Bethesda, MD. He was a visiting professor at ETH of Zurich, Switzerland. He is the chief of the Molecular Modeling Section at Padova University. His main interest concerning the application of computational approach to medicinal chemistry. He is the recipient of the National Award in Medicinal Chemistry for Excellent Research (2002).
Zhan-Guo Gao is a senior scientist at the Molecular Recognition Section, National Institute of Diabetes, Digestive, and Kidney Diseases, at the National Institutes of Health in Bethesda, MD. He is an expert in adenosine receptor molecular pharmacology and site-directed mutagenesis experiments.
Kenneth A. Jacobson is chief of the Molecular Recognition Section and Director of the Chemical Biology Laboratory, National Institute of Diabetes, Digestive, and Kidney Diseases, at the National Institutes of Health in Bethesda, MD. He is a medicinal chemist with interests in the structure and pharmacology of G protein-coupled receptors, particularly receptors for the purine nucleosides and nucleotides. His interdisciplinary approach involves both synthesis of small molecule ligands and characterization of their protein targets (receptors).
Giampiero Spalluto received his degree in Chemistry and Pharmaceutical Technology in 1987 from the University of Ferrara. He obtained his Ph.D. degree in Organic Chemistry from the University of Parma in 1992. Between 1995 and 1998, he was an assistant professor of Medicinal Chemistry at the University of Ferrara. Since November1998 to the present, he has held the position of associate professor of Medicinal Chemistry at the University of Trieste. His scientific interests have focused on the medicinal chemistry of ligands for adenosine receptor subtypes.
REFERENCES
- 1.Downey JM, Cohen MV, Ytrehus K, Liu Y. Cellular mechanisms in ischemic preconditioning: The role of adenosine and protein kinase Ca. Ann NY Acad Sci 1994;723:82–98. [PubMed] [Google Scholar]
- 2.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: Cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol 1997;52:846–860. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson KA, von Lubitz DKJE, Daly JW, Fredholm BB. Adenosine receptor ligands: Differences with acute versus chronic treatment. Trends Pharmacol Sci 1996;17:108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fredholm BB. Adenosine and neuroprotection. Int Rev Neurobiol 1997;40:259–280. [PubMed] [Google Scholar]
- 5.Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Adenosine A2B receptors: A novel therapeutic target in asthma? Trends Pharmacol Sci 1998;19:148–153. [DOI] [PubMed] [Google Scholar]
- 6.Fozard JR, McCarthy C. Adenosine receptor ligands as potential therapeutics in asthma. Curr Opin Investig Drugs 2002;3:69–77. [PubMed] [Google Scholar]
- 7.Richardson PJ, Kase H, Jenner PG. Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol Sci 1997;18:338–344. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro JA, Sebastiao AM, de Mendonca A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog Neurobiol 2002;68:377–392. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, Burbiel JC, Maass A, Muller CE. Adenosine receptor agonists: From basic medicinal chemistry to clinical development. Expert Opin Emerg Drugs 2003;8:537–576. [DOI] [PubMed] [Google Scholar]
- 10.Fredholm BB. Adenosine receptors as targets for drug development. Drug News Perspect 2003;16:283–289. [DOI] [PubMed] [Google Scholar]
- 11.Moro S, Spalluto G, Jacobson KA. Techniques: Recent developments in computer-aided engineering of GPCR ligands using the human A3 adenosine receptor as an example. Trends Pharmacol Sci 2005;26:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 2000;362:364–374. [DOI] [PubMed] [Google Scholar]
- 13.Fishman P, Bar-Yehuda S. Pharmacology and therapeutic applications of A3 receptor subtype. Curr Top Med Chem 2003;3:463–469. [DOI] [PubMed] [Google Scholar]
- 14.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 2003;63:1256–1272. [DOI] [PubMed] [Google Scholar]
- 15.Lefkowitz RJ. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci 2004;25:413–422. [DOI] [PubMed] [Google Scholar]
- 16.Olah ME, Jacobson KA, Stiles GL. Purification and characterization of bovine cerebral cortex A1 adenosine receptor. Arch Biochem Biophys 1990;283:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrington WW, Jacobson KA, Stiles GL. Glycoprotein nature of the A2 adenosine receptor binding subunit. Mol Pharmacol 1990;38:177–183. [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer TM, Jacobson KA, Stiles GL. Immunological identification of A2 adenosine receptors by two antipeptide antibody preparations. Mol Pharmacol 1992;42:391–397. [PMC free article] [PubMed] [Google Scholar]
- 19.Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle JH, Riv-kees SA, Fink JS, Reppert SM. Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol Pharmacol 1993;44:524–532. [PubMed] [Google Scholar]
- 20.Libert F, Van Sande J, Lefort A, Czernilofsky A, Dumont JE, Vassart G, Ensinger HA, Mendla KD. Cloning and functional characterization of a human A1 adenosine receptor. Biochem Biophys Res Commun 1992;187:919–926. [DOI] [PubMed] [Google Scholar]
- 21.Pierce KD, Fulong TJ, Selbie LA, Shine J. Molecular cloning and expression of an adenosine A2b receptor from human brain. Biochem Biophys Res Commun 1992;187:86–93. [DOI] [PubMed] [Google Scholar]
- 22.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3adenosine receptor. Proc Natl Acad Sci USA 1993;90:10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furlong TJ, Pierce KD, Selbie LA, Shine J. Molecular characterization of a human brain adenosine A2 receptor. Mol Brain Res 1992;15:62–66. [DOI] [PubMed] [Google Scholar]
- 24.Fong TM, Strader CD. Functional mapping of the ligand binding sites of G-protein coupled receptors. Med Res Rev 1994;14:387–399. [DOI] [PubMed] [Google Scholar]
- 25.Moro S, Jacobson KA. Molecular modeling as a tool to investigate molecular recognition in P2Y receptors. Curr Pharm Des 2002;8:2401–2413. [DOI] [PubMed] [Google Scholar]
- 26.Pauwels PJ, Wurch T. Review: Amino acid domains involved in constitutive activation of G protein-coupled receptors. Mol Neurobiol 1998;17:109–135. [DOI] [PubMed] [Google Scholar]
- 27.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J 1999;7:1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: Similarities to rhodopsin. Biochemistry 2003;42:2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olah ME, Ren H, Ostrowski J, Jacobson KA, Stiles GL. Cloning, expression, and characterization of the unique bovine A1 adenosine receptor: Studies on the ligand binding site by site-directed mutagenesis. J Biol Chem 1992;267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- 30.Townsend-Nicholson A, Schofield PR. A threonine residue in the seventh transmembrane domain of the human A1 adenosine receptor mediates specific agonist binding. J Biol Chem 1994;269:2373–2376. [PubMed] [Google Scholar]
- 31.Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J. A1 adenosine receptors: Two amino acids are responsible for species differences in ligand recognition. J Biol Chem 1994;269: 27900–27906. [PubMed] [Google Scholar]
- 32.Barbhaiya H, McClain R, Ijzerman A, Rivkees SA. Site-directed mutagenesis of the human A1 adenosine receptor: Influences of acidic and hydroxy residues in the first four transmembrane domains on ligand binding. Mol Pharmacol 1996;50:1635–1642. [PubMed] [Google Scholar]
- 33.Kim J, Wess J, van Rhee AM, Schöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J Biol Chem 1995;270:13987–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson KA. Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol Pharmacol 1996;49:683–691. [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Q, van Rhee AM, Kim J, Yehle S, Wess J, Jacobson KA. Hydrophilic side chains in the third and seventh transmembrane helical domains of human A2A adenosine receptors are required for ligand recognition. Mol Pharmacol 1996;50:512–521. [PMC free article] [PubMed] [Google Scholar]
- 36.Gao ZG, Chen A, Barak D, Kim SK, Müller CE, Jacobson KA. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J Biol Chem 2002;277:19056–19063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moro S, Deflorian F, Spalluto G, Pastorin G, Cacciari B, Kim SK, Jacobson KA. Demystifying the three dimensional structure of G protein-coupled receptors (GPCRs) with the aid of molecular modeling. Chem Commun (Camb) 2003;21:2949–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem 2002;3:928–944. [DOI] [PubMed] [Google Scholar]
- 39.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. G protein-coupled receptors: In silico drug discovery in 3D. Proc Natl Acad Sci USA 2004;101:11304–11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000;289:739–745. [DOI] [PubMed] [Google Scholar]
- 41.Klotz KN. Adenosine receptors and their ligands. Naunyn Schmiedebergs Arch Pharmacol 2000;362: 382–391. [DOI] [PubMed] [Google Scholar]
- 42.Dhalla AK, Shryock JC, Shreeniwas R, Belardinelli L. Pharmacology and therapeutic applications of A1 adenosine receptor ligands. Curr Top Med Chem 2003;3:369–385. [DOI] [PubMed] [Google Scholar]
- 43.Soudijn W, van Wijngaarden I, IJzerman AP. Medicinal chemistry of adenosine A1 receptor ligands. Curr Top Med Chem 2003;3:355–367. [DOI] [PubMed] [Google Scholar]
- 44.Cristalli G, Lambertucci C, Taffi S, Vittori S, Volpini R. Medicinal chemistry of adenosine A2A receptor agonists. Curr Top Med Chem 2003;3:387–401. [DOI] [PubMed] [Google Scholar]
- 45.Muller CE. Medicinal chemistry of adenosine A3 receptor ligands. Curr Top Med Chem 2003;3:445–462. [DOI] [PubMed] [Google Scholar]
- 46.Lohse MJ, Klotz KN, Lindenborn-Fotinos J, Reddington M, Schwabe U, Olsson RA. 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX): A selective high affinity antagonist radioligand for A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 1987;336:204–210. [DOI] [PubMed] [Google Scholar]
- 47.Robeva AS, Woodard R, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Molecular characterization of recombinant human adenosine receptors. Drug Dev Res 1996;36:243–252. [Google Scholar]
- 48.Pfister JR, Belardinelli L, Lee G, Lum MT, Milner P, Stanley WC, Linden J, Baker SP, Schreiner G. Synthesis and biological evaluation of the enantiomers of the potent and selective A1 adenosine antagonist 1,3-dipropyl-8-[2-(5,6-epoxy)norbornyl]xanthine. J Med Chem 1997;40:1773–1778. [DOI] [PubMed] [Google Scholar]
- 49.Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW. Molecular probes for extracellular adenosine receptors. Biochem Pharmacol 1987;36:1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briddon SJ, Middleton RJ, Cordeaux Y, Flavin FM, Weinstein JA, George MW, Kellam B, Hill SJ. Quantitative analysis of the formation and diffusion of A1-adenosine receptor-antagonist complexes in single living cells. Proc Nat Acad Sci USA 2004;101:4673–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Galen PJ, Nissen P, van Wijngaarden I, IJzerman AP, Soudijn W. 1H-imidazo[4,5-c]quinolin-4-amines: Novel non-xanthine adenosine antagonists. J Med Chem 1991;34:1202–1206. [DOI] [PubMed] [Google Scholar]
- 52.Boehringer Ingelheim Pharm. K.G. Triazolopurines, process for their preparation and their use as medicaments. EP 0978517, 2000; Imidazotriazolopyrimidines. WO oo12511, 2000. [Google Scholar]
- 53.Da Settimo F, Primofiore G, Taliani S, Marini AM, La Motta C, Novellino E, Greco G, Lavecchia A, Trincavelli L, Martini C. 3-Aryl[1,2,4]triazino[4,3-a]benzimidazol-4(10H)-ones: A new class of selective A1 adenosinereceptor antagonists. J Med Chem 2001;44:316–327. [DOI] [PubMed] [Google Scholar]
- 54.Colotta V, Catarzi D, Varano F, Cecchi L, Filacchioni G, Martini C, Trincavelli L, Lucacchini A. 1,2,4-triazolol[4,3-a]quinoxalin-1-one: A versatile tool for the synthesis of potent and selective adenosine receptor antagonists. J Med Chem 2000;43:1158–1164. [DOI] [PubMed] [Google Scholar]
- 55.Colotta V, Catarzi D, Varano F, Cecchi L, Filacchioni G, Martini C, Trincavelli L, Lucacchini A. Synthesis and structure-activity relationships of a new set of 2-arylpyrazolo [3,4-c] quinoline derivatives as adenosine receptor antagonists. J Med Chem 2000;43:3118–3124. [DOI] [PubMed] [Google Scholar]
- 56.Ceccarelli S, D’Alessandro A, Prinzivalli M, Zanarella S. Imidazo[1,2-a]quinoxalin-4-amines: A novel class of non-xanthine A1 adenosine receptor antagonists. Eur J Med Chem 1998;33:943–955. [Google Scholar]
- 57.Hess A, Müller CE, Frobenius W, Reith U, Klotz KN, Eger K. 7-Deazaadenines bearing polar substituents: Structure activity relathioships of new A1 and A3 adenosine receptor antagonists. J Med Chem 2000;43: 4636–4646. [DOI] [PubMed] [Google Scholar]
- 58.Van Muijlwijk-Koezen JE, Timmerman E, Vollinga RC, Von Frijtag Drabbe Künzel JK, De Groote M, Visser S, IJzerman AP. Thiazole and thiadiazole analogues as a novel class of adenosine receptor antagonists. J Med Chem 2001;44:749–762. [DOI] [PubMed] [Google Scholar]
- 59.Van Tilburg EW, Van der Klein PAM, De Groote M, Beukers MW, IJzerman AP. Substituted 4-phenyl-2-(phenylcarboxamido)-1,3-thiazole derivatives as antagonists for the A1 adenosine receptors. Bioorg Med Chem Lett 2001;11:2017–2019. [DOI] [PubMed] [Google Scholar]
- 60.Kuroda S, Akahane A, Itani H, Nishimura S, Durkin K, Kinoshita T, Tenda Y, Sakane K. Discovery of FR166124, a novel water soluble pyrazolo[1,5-a]pyridine as adenosine A1 receptor antagonist. Bioorg Med Chem Lett 1999;9:1979–1984. [DOI] [PubMed] [Google Scholar]
- 61.Kuroda S, Akahane A, Itani H, Nishimura S, Durkin K, Kinoshita T, Tenda Y, Sakane K. Novel adenosine A1 receptor antagonists, synthesis and structure activity relationships of a novel series of 3-(2-Cyclohexenyl-3-oxo-2,3-dihydropyridazin-6-yl)-2-phenylpyrazolo[1,5-a]pyridines. Bioorg Med Chem 2000;8:55–64. [DOI] [PubMed] [Google Scholar]
- 62.Kuroda S, Takamura F, Tenda Y, Itani H, Tomisha Y, Akahane A, Sakane K. Design, synthesis and biological evaluation of a novel series of potent, orally active adenosine A1 receptor antagonists with high blood–brain barrier permeability. Chem Pharm Bull 2001;49:988–998. [DOI] [PubMed] [Google Scholar]
- 63.Ferrarini PL, Mori C, Manera C, Martinelli A, Mori F, Saccomanni G, Barili PL, Botti L, Giannaccini G, Trincavelli L, Lucacchini A. A novel class of highly potent and selective A1 adenosine antagonists: Structure-affinity profile of a series of 1,8-naphthyridine derivatives. J Med Chem 2000;43:2814–2823. [DOI] [PubMed] [Google Scholar]
- 64.Ferrarini PL, Betti L, Cavallini T, Gianaccini G, Lucacchini A, Manera C, Martinelli A, Ortore G, Saccomanni G, Tuccinardi T. Study on affinity profile toward native human and bovine adenosine receptors of a series of 1,8-naphthyridine derivatives. J Med Chem 2004;47:3019–3031. [DOI] [PubMed] [Google Scholar]
- 65.Chang LCW, Spanjersberg RF, Von Frijtag Drabbe Künzel JK, Mulder-Krieger T, van den Hout G, Beukers MW, Brussee J, IJzerman AP. 2,4,6-Trisubstituted pyrimidines as a new class of selective adenosine A1 receptor antagonists. J Med Chem 2004;47:6529–6540. [DOI] [PubMed] [Google Scholar]
- 66.Maemoto T, Tada M, Mihara T, Ueyama N, Matsuoka H, Harada K, Yamaji T, Shirakawa K, Kuroda S, Akahane A, Iwashita A, Matsuoka N, Mutoh S. Pharmacological characterization of FR194921, a new potent, selective, and orally active antagonist for central adenosine A1 receptors. J Pharmacol Sci 2004;96:42–52. [DOI] [PubMed] [Google Scholar]
- 67.Dassesse D, Massie A, Ferrari R, Ledent C, Parmentier M, Arckens L, Zoli M, Schiffmann SN. Functional striatal hypodopaminergic activity in mice lacking adenosine A(2A) receptors. J Neurochem 2001;78: 183–198. [DOI] [PubMed] [Google Scholar]
- 68.Müller CE, Stein B. Adenosine receptor antagonists: Structures and potential therapeutic applications. Curr Pharm Des 1996;2:501–530. [Google Scholar]
- 69.Shimada J, Suzuki F, Nonaka H, Ishii A, Ichikawa S. (E)-1,3-dialkyl-7-methyl-8-(3,4,5-trimethoxy-styryl)xanthines: Potent and selective adenosine A2A antagonists. J Med Chem 1992;35:2342–2345. [DOI] [PubMed] [Google Scholar]
- 70.Nonaka H, Ichimura M, Takeda M, Nonaka Y, Shimada J, Suzuki F, Yamaguchi K, Kase H. KF17837 ((E)-8-(3,4-dimethoxystyryl)-1,3-dipropyl-7-methylxanthine), a potent and selective adenosine A2A receptor antagonist. Eur J Pharmacol Mol Pharm Sect 1994;267:335–341. [DOI] [PubMed] [Google Scholar]
- 71.Jacobson KA, Gallo-Rodriguez C, Melman N, Fischer B, Maillard M, van Bergen A, van Galen PJM, Karton YJ. Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J Med Chem 1993;36:1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jackson EK, Herzer WA, Suzuki F. KF17837 is an A2 adenosine receptor antagonist in vivo. J Pharmacol Exp Ther 1993;267:1304–1310. [PubMed] [Google Scholar]
- 73.Nonaka Y, Shimada J, Nonaka H, Koike N, Aoki N, Kobayashi H, Kase H, Kase H, Yamaguchi K, Suzuki F. Photoisomerization of a potent and selective adenosine A2 antagonist, (E)-1,3-Dipropyl-8-(3, 4-dimethoxystyryl)-7-methylxanthine. J Med Chem 1993;34:3731–3733. [DOI] [PubMed] [Google Scholar]
- 74.Müller CE, Schobert U, Hipp J, Geis U, Frobenius W, Pawlowski M. Configurationally stable analogs of styrylxanthines as A2A adenosine receptor antagonists. Eur J Med Chem 1997;32:709–719. [Google Scholar]
- 75.Müller CE, Geis U, Hipp J, Schobert U, Frobenius W, Pawlowski M, Suzuki F, Sandoval-Ramirez J. Synthesis and structure-activity relationship of 3,7-Dimethyl-1-propargylxanthine derivatives, A2A-selective adenosine receptor antagonists. J Med Chem 1997;40:4396–4405. [DOI] [PubMed] [Google Scholar]
- 76.Shamin MT, Ukena D, Padgett WL, Daly JW. Effects of 8-phenyl and 8-cycloalkyl substituents on the activity of mono-, di-, and trisubstituted alkylxanthines with substitution at the 1-, 3-, and 7-positions. J Med Chem 1989;32:1231–1237. [DOI] [PubMed] [Google Scholar]
- 77.Del Giudice MR, Borioni A, Mustazza C, Gatta F, Dionisotti S, Zocchi C, Ongini E. (E)-1-(heterocyclyl or cyclohexyl)-2-[1,3,7-trisubstituted(xanthin-8-yl)] ethenes as A2A adenosine receptor antagonists. Eur J Med Chem 1996;31:59–63. [Google Scholar]
- 78.Erickson RH, Hiner RN, Feeney SW, Blake PR, Rzeszotarski WJ, Hicks P, Costello DG, Abren ME. 1,3,8-trisubstituted xanthines. Effects of substitution pattern upon adenosine receptor A1/A2 affinity. J Med Chem 1991;34:1431–1435. [DOI] [PubMed] [Google Scholar]
- 79.Jacobson KA, Shi D, Gallo-Rodriguez C, Manning M, Jr., Müller C, Daly JW, Neumeyer JL, Kiriasis L, Pfleiderer W. Effect of trifluoromethyl and other substituents on activity of xanthines at adenosine receptors. J Med Chem 1993;36:2639–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Müller CE, Sandoval-Ramirez J, Schobert U, Geis U, Frobenius W, Klotz KN. 8-(Sulfostyryl)xanthines: Water soluble A2A-selective adenosine receptor antagonists. Bioorg Med Chem 1998;6:707–719. [DOI] [PubMed] [Google Scholar]
- 81.Sauer R, Maurinsh J, Reith U, Fulle F, Klotz KN, Müller CE. Water-soluble phosphate pro-drugs of 1-propargyl-8-styrylxanthine derivatives, A2A-selective adenosine receptor antagonists. J Med Chem 2000; 43:440–448. [DOI] [PubMed] [Google Scholar]
- 82.Knutsen LJ, Weiss SM. KW-6002 (Kyowa Hakko Kogyo). Curr Opin Invest Drugs 2001;2:668–673. [PubMed] [Google Scholar]
- 83.Hockenmeyer J, Burbiel JC, Muller CE. Multigram scale syntheses, stability and photoreaction of A2A adenosine receptor antagonists with 8-styrylxanthine structure: Potential drugs for Parkinson’s disease. J Org Chem 2004;69:3308–3318. [DOI] [PubMed] [Google Scholar]
- 84.Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P, Friedmann RC, Atkins C, Warren V, Furness P, Hyun JL, Stone GA, Desai M, Williams M. Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J Med Chem 1988;31:1014–1020. [DOI] [PubMed] [Google Scholar]
- 85.Williams M, Francis J, Ghai G, Braunwalder A, Psychoyos S, Stone GA, Cash WD. Biochemical characterization of the triazoloquinazoline, CGS 15943, a novel, non-xanthine adenosine antagonist. J Pharmacol Exp Ther 1987;241:415–420. [PubMed] [Google Scholar]
- 86.Kim YC, Ji X, Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS 15943) are selective for the human A3 receptor subtype. J Med Chem 1996;39:4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baraldi PG, Cacciari B, Spalluto G, Pineda MJ, Zocchi C, Dionisotti S, Ongini E. Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives: Potent and selective A2A adenosine antagonists. J Med Chem 1996;39:1164–1171. [DOI] [PubMed] [Google Scholar]
- 88.Baraldi PG, Cacciari B, Spalluto G, Bergonzoni M, Dionisotti S, Ongini E, Varani K, Borea PA. Design, synthesis, and biological evaluation of a second generation of pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines as potent and selective A2A adenosine receptor antagonists. J Med Chem 1998;41:2126–2133. [DOI] [PubMed] [Google Scholar]
- 89.Todde S, Moresco RM, Simonelli P, Baraldi PG, Cacciari B, Spalluto G, Varani K, Monopoli A, Matarrese M, Carpimnelli A, Magni F, Galli Kienle M, Fazio F. Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A2A receptor system using positron emission tomography. J Med Chem 2000;43:4359–4362. [DOI] [PubMed] [Google Scholar]
- 90.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Monopoli A, Ongini E, Varani K, Borea PA. 7-Substituted 5-amino-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as A2A adenosine receptor antagonists: A study on the importance of modifications at the side chain on the activity and solubility. J Med Chem 2002;45:115–126. [DOI] [PubMed] [Google Scholar]
- 91.Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, Collis MG. The in vitro pharmacology of ZM 241385, a potent, non-xanthine, A2A selective adenosine receptor antagonist. Br J Pharmacol 1995;115:1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ji XD, Jacobson KA. Use of triazolotriazine [3H]-ZM241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Des Discov 1999;16:217–226. [PMC free article] [PubMed] [Google Scholar]
- 93.Vu CB, Pan D, Peng B, Kumaravel G, Phadke D, Engber T, Huamg C, Reilly J, Tam S, Petter RC. Studies on adenosine A2A receptor antagonists: Comparison of three core heterocycles. Bioorg Med Chem Lett 2004;14:4831–4834. [DOI] [PubMed] [Google Scholar]
- 94.Vu CB, shields P, Peng B, Kumaravel G, Jin X, Phadke D, wang J, Engber T, Ayyub E, Petter RC. Triamino derivatives of triazolo triazine and triazolopyrimidines as adenosine A2A receptor antagonists. Bioorg Med Chem Lett 2004;14:4835–4838. [DOI] [PubMed] [Google Scholar]
- 95.Vu CB, Peng B, Kumaravel G, Smits G, Jin X, Phadke D, Enber T, Huang C, Reilly J, Tam S, Grant D, Hetu G, Chen L, Zhang J, Petter RC. Piperazine derivatives of [1,2,4]triazolo[1,5-a] [1,3,5]triazine as potent and selective adenosine A2A receptor antagonists. J Med Chem 2004;47:4291–4299. [DOI] [PubMed] [Google Scholar]
- 96.Peng B, Kumaravel G, Yao G, Sha L, Van Vlijmen H, Bohnert T, Huang C, Vu CB, Ensinger CL, Chang H, Engber TM, Whalley E, Petter RC. Novel bictclic piperazine derivatives of triazolotriazine and triazolopyrimidins as highly potent and selective Adenosine A2A receptor antagonists. J Med Chem 2004;47:6218–6229. [DOI] [PubMed] [Google Scholar]
- 97.Vu CB, Pan D, Kumaravel G, Smits G, Jin X, Phadke D, Engber T, Huang C, Reilly J, Tam S, Grant D, Hetu G, Petter RC. Novel diamino derivatives of [1,2,4]triazolo[1,5-a] [1,3,5]triazine as potent and selective adenosine A2A receptor antagonists. J Med Chem 2005;48:2009–2018. [DOI] [PubMed] [Google Scholar]
- 98.Colotta V, Catarzi D, Varano F, Cecchi L, Filacchioni G, Martini C, Trincavelli L, Lucacchini A. 1,2,4-Triazolo[4,3-a]quinoxalin-1-one: A versatile tool for the synthesis of potent and selective adenosine receptor antagonists. J Med Chem 2000;43:1158–1164. [DOI] [PubMed] [Google Scholar]
- 99.Chebib M, McKeveney D, Quinn RJ. 1-Phenylpyrazolo[3,4-d]pyrimidines; structure-activity relationships for C6 substituents at A1 and A2A adenosine receptors. Bioorg Med Chem 2000;8:2581–2590. [DOI] [PubMed] [Google Scholar]
- 100.Matasi JJ, Caldwell JP, Hao JS, Neustadt B, Arik L, Foster CJ, Lachowicz J, Tulshian DB. The discovery and synthesis of novel adenosine receptor A2A antagonists. Bioorg Med Chem Lett 2005;15:1333–1336. [DOI] [PubMed] [Google Scholar]
- 101.von Lubitz DK. Adenosine in the treatment of stroke: Yes, maybe, or absolutely not? Expert Opin. Investig Drugs 2001;10:619–632. [DOI] [PubMed] [Google Scholar]
- 102.Scholz-Pedretti K, Pfeilschifter J, Kaszkin M. Potentiation of cytokine induction of group IIA phospholipase A(2) in rat mesangial cells by ATP and adenosine via the A2A adenosine receptor. Br J Pharmacol 2001;132:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jacobson KA, Hoffmann C, Cattabeni F, Abbracchio MP. Adenosine-induced cell death: Evidence for receptor-mediated signalling. Apoptosis 1999;4:197–211. [DOI] [PubMed] [Google Scholar]
- 104.Landells LJ, Jensen MW, Orr LM, Spina D, O’Connor BJ, Page CP. The role of adenosine receptors in the action of theophylline on human peripheral blood mononuclear cells from healthy and asthmatic subjects. Br J Pharmacol 2000;129:1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest 1995;96:1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim SA, Marshall MA, Melman N, Kim SH, Müller CE, Linden J, Jacobson KA. Structure-activity relationships at human and rat adenosine receptors of xanthine derivatives substituted at the 1-, 3-, 7-, and 8-positions. J Med Chem 2002;45:2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hayallah AM, Sandoval-Ramirez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE. 1, 8-Disubstituted xanthine derivatives: Synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem 2002;45:1500–1510. [DOI] [PubMed] [Google Scholar]
- 108.Yan L, Müller CE. Preparation, properties and adenosine receptor affinities of sulfophenylxanthine nitrophenyl esters: Toward the development of sulfonic acid prodrugs with peroral bioavailability. J Med Chem 2004;47:1031–1043. [DOI] [PubMed] [Google Scholar]
- 109.Jacobson KA, IJzerman AP, Linden J. 1,3-Dialkylxanthine derivatives having high potency as antagonists at human A2B adenosine receptors. Drug Dev Res 1999;47:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim YC, Karton Y, Ji XD, Melman N, Linden J, Jacobson KA. Acyl-hydrazide derivatives of a xanthine carboxylic congener (XCC) as selective antagonists at human A2B adenosine receptors. Drug Dev Res 1999;47:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim YC, Ji XD, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J Med Chem 2000;43:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ji XD, Kim YC, Ahern DG, Linden J, Jacobson KA. [3H]MRS 1754, a selective antagonist radioligand for A2B adenosine receptors. Biochem Pharmacol 2001;61:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baraldi PG, Tabrizi MA, Preti D, Bovero A, Romagnoli R, Fruttarolo F, Zaid NA, Moorman AR, Varani K, Gessi S, Merighi S, Borea PA. Design, synthesis and biological evaluation of new 8-heterocyclic xanthine derivatives as highly potent and selective human A2B adenosine receptor antagonists. J Med Chem 2004;47:1434–1447. [DOI] [PubMed] [Google Scholar]
- 114.Baraldi PG, Tabrizi MA, Preti D, Bovero A, Romagnoli R, Fruttarolo F, Moorman AR, Gessi S, Merighi S, Varani K, Borea PA. [3H]-MRE 2029-F20, a selective antagonist radioligand for the human A2B adenosine receptors. Bioorg Med Chem Lett 2004;14:3607–3610. [DOI] [PubMed] [Google Scholar]
- 115.Zablocki J, Kalla R, Perry T, Palle V, Varkhedkar V, Xiao D, Piscopio A, Maa T, Gimbel A, Hao J, Chu N, Leung K, Zeng D. The discovery of a selective, high affinity A2B adenosine receptor antagonist for the potential treatment of asthma. Bioorg Med Chem Lett 2005;15:609–612. [DOI] [PubMed] [Google Scholar]
- 116.Carotti A, Stefanachi A, Ravina E, Sotelo E, Loza MI, Cadavid MI, Centeno NB, Nicolotti O. 8-Substituted-9-deazaxanthines as adenosine receptor ligands: Design, synthesis and structure-affinity relationships at A2B. Eur J Med Chem 2004;39:879–887. [DOI] [PubMed] [Google Scholar]
- 117.Kim YC, de Zwart M, Chang L, Moro S, Jacobien K, Frijtag DK, Melman N, IJzerman AP, Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) having high potency at the human A2B and A3 receptor subtypes. J Med Chem 1998;41:2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baraldi PG, Cacciari B, Romagnoli R, Klotz KN, Spalluto G, Varani K, Gessi S, Merighi S, Borea PA. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as adenosine receptor ligands: A starting point for searching A2B adenosine receptor antagonists. Drug Dev Res 2001;53:225–235. [Google Scholar]
- 119.Pastorin G, Da Ros T, Spalluto G, Deflorian F, Moro S, Cacciari B, Baraldi PG, Varani K, Gessi S, Borea PA. Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as adenosine receptor antagonists. Influence of the N5 substituent on the affinity at the human A3 and A2B adenosine receptor subtypes: A molecular modeling investigation. J Med Chem 2003;46:4287–4296. [DOI] [PubMed] [Google Scholar]
- 120.Ji XD, Jacobson KA. [3H]-ZM241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Des Discov 1999;16:217–226. [PMC free article] [PubMed] [Google Scholar]
- 121.deZwart M, Vollinga RC, Beukers MW, Sleegers DF, von Frijtag Drabbe Künzel JK, de Groote M, IJzerman AP. Potent antagonist for the human adenosine A2B receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM241385 with high affinity. Drug Dev Res 1999;48:95–103. [Google Scholar]
- 122.Camaioni E, Costanzi S, Vittori S, Volpini R, Klotz KN, Cristalli G. New substituted 9-alkylpurines as adenosine receptor ligands. Bioorg Med Chem 1998;6:523–533. [DOI] [PubMed] [Google Scholar]
- 123.Lambertucci C, Camaioni E, Costanzi S, Kachler S, Klotz KN, Volpini R, Cristalli G, Vittori S. A bromine atom in C8 position of 9-substituted adenines enhances A2A affinity. Drug Dev Res 2000;50:67. [Google Scholar]
- 124.Webb TR, Lvovskiy D, Kim S-A, Ji X-D, Melman N, Linden J, Jacobson KA. Quinazolines as adenosine receptor antagonists: SAR and selectivity for A2B receptors. Bioorg Med Chem 2003;11:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stewart M, Steining AG, Ma C, Song JP, McKibben B, Castelhano AL, MacLennan SJ. [3H]OSIP339391, a selective, novel, and high affinity antagonist radioligand for adenosine A2B receptors. Biochem Pharmacol 2004;68:315–322. [DOI] [PubMed] [Google Scholar]
- 126.Volpini R, Costanzi S, Vittori S, Cristalli G, Klotz KN. Medicinal chemistry and pharmacology of A2B adenosine receptors. Curr Top Med Chem 2003;3:427–443. [DOI] [PubMed] [Google Scholar]
- 127.Rosi S, McGann K, Hauss-Wegrzyniak B, Wenk GL. The influence of brain inflammation upon neuronal adenosine A2B receptors. J Neurochem 2003;86:220–227. [DOI] [PubMed] [Google Scholar]
- 128.Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci USA 2001;98:14120–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceschi C, Kim Y, Jacobson KA, Klotz KN, Lohse MJ, Abbracchio MP. Activation of the A3 adenosine receptor effects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch Pharmacol 2000;361:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baraldi PG, Cacciari B, Romagnoli R, Merighi S, Varani K, Borea PA, Spalluto G. A3 Adenosine receptor ligands; history and perspectives. Med Res Rev 2000;20:103–128. [DOI] [PubMed] [Google Scholar]
- 131.Priego EM, von Frijtag Drabbe Kuenzel J, Ijzerman AP, Camarasa MJ, Perez Perez MJ. Pyrido[2,1-f]purine-2,4-dione derivatives as a novel class of highly potent human A3 adenosine receptor antagonists. J Med Chem 2002;45:3337–3344. [DOI] [PubMed] [Google Scholar]
- 132.Ozola V, Thorand M, Diekmann M, Qurishi R, Schumacher B, Jacobson KA, Müller CE. 2-Phenylimidazo [2,1-i]purin-5-ones: Structure–activity relationships and characterization of potent and selective inverse agonists at human A3 adenosine receptors. Bioorg Med Chem 2003;11:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Müller CE, Diekmann M, Thorand M, Ozola V. [(3)H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([(3)H]PSB-11), a novel high-affinity antagonist radioligand for human A(3) adenosine receptors. Bioorg Med Chem Lett 2002;12:501–503. [DOI] [PubMed] [Google Scholar]
- 134.Okamura T, Kurogi Y, Nishikawa H, Hashimoto K, Fujiwara H, Nagao Y. 1,2,4-Triazolo[5,1-i]purine derivatives as highly potent and selective human adenosine A3 receptor ligands. J Med Chem 2002;45: 3703–3708. [DOI] [PubMed] [Google Scholar]
- 135.Okamura T, Kurogi Y, Hashimoto K, Nishikawa H, Nagao Y. Facile synthesis of fused 1,2,4-triazolo [1,5-c] pyrimidine derivatives as human adenosine A3 receptor ligands. Bioorg Med Chem Lett 2004;14: 2443–2446. [DOI] [PubMed] [Google Scholar]
- 136.Jacobson KA, Tchilibon S, Joshi BV, Gao ZG. A3 adenosine receptors. In: Doherty AM, editor. Annual report in medicinal chemistry, 1st edition.: Elsevier; 2003;38:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moro S, van Rhee AM, Sanders LH, Jacobson KA. Flavonoid derivatives as adenosine receptor antagonists: A comparison of the hypothetical receptor binding site based on a comparative molecular field analysis model. J Med Chem 1998;41:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 1997;36:1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiang J-L, van Rhee AM, Chang L, Patchornik A, Ji X-D, Evans P, Melman N, Jacobson KA. Structure-activity relationships of 4-(phenylethynyl)-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J Med Chem 1997;40:2596–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li A-N, Moro S, Forsyth N, Melman N, Ji X-D, Jacobson KA. Synthesis, CoMFA analysis, and receptor docking of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem 1999;42:706–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ji XD, Von Lubitz D, Olah ME, Stiles GL, Jacobson KA. Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev Res 1994;33:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim YC, Ji XD, Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS 15943) are selective for the human A3 receptor subtype. J Med Chem 1996;39:4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Muijlwijk-koezen JE, Timmerman H, Link R, van der Goot H, IJzerman AP. A novel class of adenosine A3 receptor ligands. 2. Stucture affinity profile of a series of isoquinoline and quinazoline compounds. J Med Chem 1998;41:3994–4000. [DOI] [PubMed] [Google Scholar]
- 144.van Muijlwijk-Koezen JE, Timmerman H, van der Goot H, Menge WMPB, von Frijtag von Drabbe Künzel JK, de Groote M, Ijzerman AP. Isoquinoline and quinazoline urea analogues as antagonists for the human adenosine A3 receptor. J Med Chem 2000;43:2227–2238. [DOI] [PubMed] [Google Scholar]
- 145.Baraldi PG, Cacciari B, Moro S, Spalluto G, Pastorin G, Da Ros T, Klotz K-N, Varani K, Gessi S, Borea PA. Synthesis, biological activity, and molecular modeling investigation of new pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as human A3 adenosine receptor antagonists. J Med Chem 2002;45: 770–780. [DOI] [PubMed] [Google Scholar]
- 146.Maconi A, Pastorin G, Da Ros T, Spalluto G, Gao ZG, Jacobson KA, Baraldi PG, Cacciari B, Varani K, Moro S, Borea PA. Synthesis, biological properties and molecular modeling investigation of the first potent, selective and water soluble human A3 adenosine receptor antagonist. J Med Chem 2002;45:3579–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Colotta V, Catarzi D, Varano F, Romana Calabri F, Lenzi O, Filacchioni G, Martini C, Trincavelli L, Deflorian F, Moro S. 1,2,4-triazolo[4,3-a]quinoxalin-1-one moiety as an attractive scaffold to develop new potent and selective human A3 adenosine receptor antagonists: Synthesis, pharmacological, and ligand-receptor modeling studies. J Med Chem 2004;47:3580–3590. [DOI] [PubMed] [Google Scholar]
- 148.Catarzi D, Colotta V, Varano F, Calabri FR, Lenzi O, Filacchioni G, Trincavelli L, Martini C, Tralli A, Montopoli C, Moro S. 2-aryl-8-chloro-1,2,4-triazolo[1,5-a]quinoxalin-4-amines as highly potent A1 and A3 adenosine receptor antagonists. Bioorg Med Chem 2005;13:705–715. [DOI] [PubMed] [Google Scholar]
- 149.Jacobson MA, Chakravarty PK, Johnson RG, Norton R. Novel selective non-xanthine A3 adenosine receptor antagonists. Drug Dev Res 1996;37:131. [Google Scholar]
- 150.Jung KY, Kim SK, Gao ZG, Gross AS, Melman N, Jacobson KA, Kim YC. Structure-activity relationships of thiazole and thiadiazole derivatives as potent and selective human adenosine A3 receptor antagonists. Bioorg Med Chem 2004;12:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem 2002;45:4471–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Costanzi S, Lambertucci C, Vittori S, Volpini R, Cristalli G. 2-and 8-alkynyladenosines: Conformational studies and docking to human adenosine A(3) receptor can explain their different biological behavior. J Mol Graph Mod 2003;21:253–262. [DOI] [PubMed] [Google Scholar]
- 153.Okamura T, Kurogi Y, Hashimoto K, Sato S, Nishikawa H, Kiryu K, Nagao Y. Structure-activity relationships of adenosine A3 receptor ligands: New potential therapy for the treatment of glaucoma. Bioorg Med Chem Lett 2004;14:3775–3779. [DOI] [PubMed] [Google Scholar]