

Plain language summary
The lysosomal degradation pathway of autophagy depends on a set of evolutionarily conserved autophagy‐related molecules (ATGs) bestowed with the power to direct membrane trafficking and biology. In this issue of EMBO Journal, Kakanj P et al reveal a surprising role for the autophagy machinery in cell fusion (Kakanj et al, 2022). Autophagy is physiologically required for cell syncytium formation through dismantling the lateral plasma membrane during wound healing, and unchecked autophagy can drive cell fusion in epithelial tissues without compromising epithelial integrity.
Subject Categories: Development, Membranes & Trafficking
Recent work shows a surprising role for the autophagy machinery in mediating cell–cell fusion and syncytium formation during Drosophila wound healing.
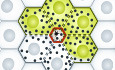
Drosophila represents a powerful animal model to investigate the molecular and cellular mechanisms of wound repair. The epidermis serves as a protective layer against the outside world that is uniquely experimentally accessible for injury manipulation and observation. Underneath the secreted cuticle, the epidermis consists of a monolayer of postmitotic epithelial cells on top of a basal lamina (Fig 1). In flies, wound healing occurs without cell division or apoptosis by two parallel cellular processes that execute wound closure (Tsai et al, 2018). Apical actomyosin assembly and contraction are coordinated with basal lamellipodia formation that together leads to effective cell elongation to close the wound gap. Earlier works have also shown that wounding additionally leads to plasma membrane fusion of cells surrounding the wound, resulting in a multinucleate syncytium in the wound vicinity (Fig 1A). Polyploidy, either in the form of multiple nuclei in a single syncytium or as a result of endoreplication within a single nucleus, is a widespread phenomenon across animals and tissues during tissue repair and regeneration (Bailey et al, 2021). A common belief is that polyploidization aids tissue repair. Although stress responses and molecular mechanisms of wound closure are emerging, how membrane events are initiated and controlled in injury‐induced cell fusion has remained enigmatic (Tsai et al, 2018).
Figure 1. Autophagy promotes syncytium formation during wound healing.
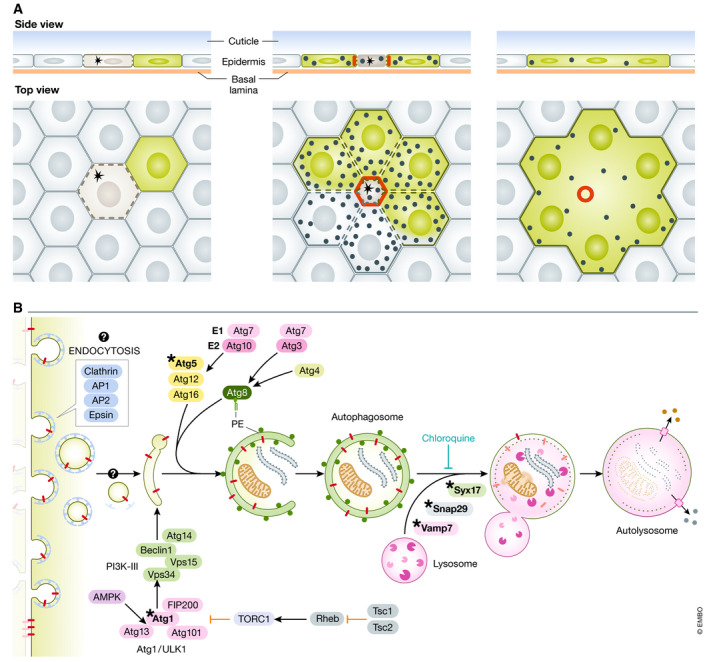
(A) Illustration with side view and top view of epidermal cells during single cell wounding (asterisk). Actomyosin ring contraction (red) contributes to wound closure. Parallel autophagy (black circles) engages in dismantling of lateral plasma membrane and junctional proteins (stippled lines). Cytoplasmic GFP in a cell in contact with wounded cells shares its cytoplasm visualized by GFP diffusion in the resulting syncytium. (B) Model showing how autophagy can utilize plasma membrane delivered through endocytosis for generation of phagophores. This may incorporate transmembrane junctional proteins (red). Proteins known to be required for autophagy pathway regulation and execution are shown. *Denotes components genetically tested in this study.
The authors previously observed upregulation of macroautophagy (autophagy hereafter) markers in cells immediately surrounding the wound (Kakanj et al, 2016). Autophagy (self‐eating) is an intracellular degradative pathway where an autophagic double isolation membrane, termed a phagophore, engulfs cytoplasmic material, which is degraded upon fusion with the lysosome (Fig 1B; Levine & Kroemer, 2019). Originally defined through genetic screening in yeast, a set of evolutionarily conserved proteins act to carry out isolation, nucleation, membrane elongation, autophagosome–lysosome fusion, and degradation (Fig 1B). Most of the genes encoding these components were for a long time thought to be exclusively acting in autophagy. Many components, however, doubles in a growing list of other cellular processes and membrane trafficking events, such as endo‐ and exocytosis, unconventional cytokine secretion, melanogenesis, LC3‐associated phagocytosis (LAP), and more (Galluzzi & Green, 2019).
Kakanj et al set out to investigate whether autophagy could be involved in wound healing using a refined and powerful video microscopy set up with anaesthetized larvae triggered by single‐cell laser‐induced wounding (Kakanj et al, 2022). This allows highly stereotypic manipulation, and detailed observational and genetic analyses of wound healing dynamics that is completed within 2–3 h. Using fluorescently tagged Atg8a that label the autophagic membranes, they verified that autophagy markers were increased in cells surrounding the wound and peaked at about 120 min after injury, when wound closure is nearly complete. Knocking down atg1 that is required for autophagy initiation, or atg5 of the Atg5/Atg12/Atg16 complex involved in phagophore elongation, strongly inhibited autophagosome formation. Consistent with the late appearance of Atg8a structures, inhibition of atg1 or atg5 RNAi did not significantly affect actin nucleation, dynamics, or rate of wound closure. But what about cell fusion that acts in parallel to wound closure? To visualize cell fusion in real time, the authors devised a clever strategy of expressing GFP in random epidermal cells and subsequently wounding the adjacent single neighboring cells. By observing the spread of GFP between cells, syncytium formation can be directly assessed in real time. GFP spread between wound‐adjacent epithelial cells showed that cytosol mixing and syncytium formation initiated from 1.5 h after wounding coinciding with maximal autophagy activity. Knockdown of atg1 or atg5 in GFP‐expressing cells completely inhibited syncytium formation, demonstrating that at least some autophagy genes are required cell autonomously to somehow open the plasma membrane.
To address whether autophagy activation is sufficient to cause epithelial cell fusion, the authors manipulated autophagy induction. Under homeostatic conditions, the autophagy‐initiating Atg1 complex (ULK1 in mammals) is kept inactive by TORC1 that integrates nutrient and growth regulatory signals. Acutely inducing autophagy by lifting TORC1 inhibition through overexpression of TSC1/TSC2, mimicking energy depletion by overexpression of activated AMPK, inactivating TORC1 components, or expressing Atg1 itself all strongly induced autophagy which was verified by electron microscopy analyses. Initial live imaging using markers for the plasma membrane‐associated marker, Src‐GFP, and nuclear DsRed2, indicated that all TORC1‐inhibiting situations resulted in plasma membrane disruption, altered epithelia morphology, and multinuclear syncytium formation, albeit with a slower kinetics than during wounding. To try and understand the nature of the apparent membrane dismantling, detailed follow‐up analyses were conducted using apical domain epithelial markers (GFP‐Bazooka/Par3), lateral membrane (fasciclin‐III, neuroglian, and GFP‐Dlg), adherence junctions (DE‐cadherin), and the basolateral membrane marker βPS‐integrin. Strikingly, TORC1 inhibition or Atg1 overexpression both led to the dismantling of plasma membrane junctional markers over time, whereas basal βPS‐integrin remained intact arguing that the lateral membranes disintegrated and cells fused into a syncytium. Indeed, ultrastructural electron microscopy analyses of the most penetrant manipulation upon Atg1 overexpression revealed adjacent nuclei not separated by plasma membranes. The ability of Atg1 to drive to plasma membrane dismantling was not limited to the epidermis but extended to airway epithelia (trachea), gut epithelium, and secretory epithelial cells of the salivary gland. Syncytical epithelia do, however, seem to be structurally and functionally intact as the basement membrane stayed in place, no leakage of GFP from epithelia was apparent, and the gut barrier function remained as assayed by a colorimetric gut leakage assay. Functionally, the epidermis could undergo wound healing after Atg1‐induced syncytium formation where cells were connected in clusters up to 10–14 cells prior to laser‐induced injury. Taken together, directed lateral plasma membrane disintegration can be forced by experimentally lowering TORC1 activity or Atg1 overexpression in multiple epithelia, recapitulating the physiological role in wound healing.
Does the lateral membrane disintegration depend on the conventional autophagy–lysosomal degradation pathway or does it represent an autophagy‐independent role of autophagy‐regulating proteins, and is lowering TORC1 signaling physiologically relevant to plasma membrane dismantling or could other stimuli be involved?
To partially answer the first question, the authors conducted genetic epistasis analysis. Co‐depleting atg1 or atg5 upon strong TORC1 inhibition could completely prevent plasma membrane disruption, showing that Atg‐driven processes is the main, or only cellular process executing plasma membrane disruption upon TORC1 inhibition. Similarly, atg5 knockdown inhibited Atg1‐induced syncytium formation. In contrast, plasma membrane breakdown remained intact upon depletion of components required for autophagosome–lysosome fusion (Snap29, Vamp7, and Syx17) or pharmacological inhibition of lysosomal degradation using chloroquine administration in the same assays. From this, the authors conclude that later stages of autophagy are not required for membrane dismantling. This inference may not be that straight forward, however, as if the main role of autophagy in this context is to capture membrane components from a plasma membrane including junctions, it may be sufficient on a short time scale to keep the components secluded and not available at the plasma membrane even when they are not ultimately degraded.
The finding that TORC1 inhibition can cause lateral plasma membrane disruption through engaging the autophagy machinery is surprising. Is it a functional role of TORC1 signaling in syncytium formation during wounding? Perhaps contrary to the assumption, earlier studies showed that insulin signaling is elevated rather than reduced within minutes at the wound margin with consequent nuclear export of Foxo in wound‐adjacent cells (Kakanj et al, 2016). Insulin signaling positively regulates TORC1 that phosphorylates S6K and the translational repressor 4E‐BP. Both repression of Foxo (nuclear exclusion) and S6K activity was found to be required for the normal rate of wound closure. Coinciding with the end of wound closure (90–120 min), however, insulin signaling was again attenuated. Thus, it remains possible that elevated insulin‐TORC1 signaling is first required for efficient morphological wound closure, and thereafter TORC1 activity falls and facilitates syncytium formation through engaging the autophagy machinery. This two‐step scenario remains untested, likely due to technical challenges which makes the question experimentally hard to resolve in the tight timeframe that these processes dynamically evolve in. There are, however, other contenders for autophagy regulation during wounding. Activation of stress pathways such as JNK, JAK‐STAT, ROS, or calcium influx has been observed during wounding and all represent signaling pathways that are known to induce autophagy in other contexts (Tsai et al, 2018). Future work is needed to resolve what the inducing signal is during wounding.
Another unresolved question is how the autophagy machinery contributes to the dismantling of the lateral plasma membrane, and moreover, whether this represents classical autophagy or an autophagy‐related process. Live imaging during wounding revealed that lateral membranes (FasIII‐GFP) or a neutral plasma membrane transmembrane marker, mCD8‐GFP, dissolves into small vesicle‐like structures that rapidly coincide with mCherry‐Atg8a. Based on existing literature from mammalian studies, two autophagy gene‐dependent membrane trafficking pathways may explain the removal of plasma membrane that could include cell junctions: (1) LC3‐associated phagocytosis (LAP) and (2) autophagy utilizing plasma membrane as a source for the isolation membrane. As the name implies, LAP engulfs extracellular material such as bacteria, whole cells (entosis), or cell debris, thus potentially being a process to remove injured cells or cell fragments during wounding. Single membrane phagocytic vacuoles become decorated by the Atg8 family protein, LC3, and are directed to the lysosome for degradation. This is distinct from autophagy by that the cargo is extracellular rather than intracellular, the enclosed cargo is within a single vs. a double membrane in autophagy. Functionally, LAP relies on a subset of autophagy genes including Beclin1/Atg6, Vps34, UVRAG, and Vps15 of the PI3K‐III complex required for endocytic and phagocytic trafficking as well as ATG7, ATG3, ATG5, ATG12, and ATG16L. It does not, however, require ATG1/ULK1. The autophagic isolation membranes can arise from several sources including ER, TGN, mitochondria, and the plasma membrane. Although not considered to constitute the most common site of autophagosome formation, plasma membrane‐derived authophagy has been shown to arise from small endocytic vesicles that are recruited to Atg16L structures which proceed to form a flattened membrane structure that comprises the phagophore (Ravikumar et al, 2010). Mechanistically, this process depends on clathrin‐mediated endocytosis (clathrin, AP1, AP2, and Eps1), and Atg16L which binds to clathrin at plasma membrane sites (Cuervo, 2010; Ravikumar et al, 2010). There is indeed precedent for turnover of transmembrane junctional proteins through plasma membrane‐derived autophagy. The gap junction protein, Connexin 43, is targeted for autophagy through Nedd4‐mediated ubiquitinylation and subsequent Eps15 recruitment resulting in autophagy‐mediated internalization and degradation through autophagy (Bejarano et al, 2012). Based on these findings, it has been suggested that autophagosomes forming at the plasma membrane could mainly be destined for the turnover of the plasma membrane itself, perhaps resulting from primary damage of the plasma membrane that trigger maximal activation from this site (Cuervo, 2010). How autophagy would be targeted specifically to intercellular lateral plasma membranes upon wounding and more importantly, how it would lead to opening up the intercellular membranes remain open questions. Electron microscopy analyses do not provide conclusive evidence. Upon Atg1 overexpression, 500‐nm‐sized vesicular structures are abundant at the plasma membrane abutting electron dense areas that are consistent with cell junctions. Whether these vesicles represent classical autophagosomes is unresolved as they do contain ribosomes consistent with isolation of ER‐containing cytosol like that of classical autophagosomes, but only a single membrane as opposed to two is clearly evident in many vesicles. Correlative light and electron microscopy analysis would be needed to verify that these structures are Cherry‐Atg8a‐positive vesicles. Caution must also be exercised when extrapolating the findings between Atg1 overexpression and wound‐induced cell fusion, as the two stimuli may differ in important ways of membrane sources and topology.
The elegant studies by Kakanj P et al firmly establish a physiological instance where autophagy executes degradation of the lateral plasma membrane and syncytial formation in the physiological response of wound healing. This is likely executed by autophagy‐utilizing plasma membrane as a source for phagophore formation and through this mode degrades plasma membrane that include cell junctions. This opens up for mechanistic studies in a powerful genetic model that may inform other cell fusion events in muscle, pancreas, and other organs. The power of autophagy has expanded yet again; this time to fuse those cells!
Acknowledgements
RK and TER are supported by grants #262652 and #276070 from the Norwegian Research Council (to TER).
The EMBO Journal (2022) 41: e111424
See also: P Kakanj et al (June 2022)
References
- Bailey EC, Kobielski S, Park J, Losick VP (2021) Polyploidy in tissue repair and regeneration. Cold Spring Harb Perspect Biol 13: a040881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM (2012) Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin‐dependent manner. Mol Biol Cell 23: 2156–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM (2010) The plasma membrane brings autophagosomes to life. Nat Cell Biol 12: 735–737 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Green DR (2019) Autophagy‐independent functions of the autophagy machinery. Cell 177: 1682–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakanj P, Bhide S, Moussain B, Leptin M (2022) Autophagy‐mediated plasma membrane removal promotes the formation of epithelial syncytia. EMBO J 41: e109992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakanj P, Moussian B, Grönke S, Bustos V, Eming SA, Partridge L, Leptin M (2016) Insulin and TOR signal in parallel through FOXO and S6K to promote epithelial wound healing. Nat Commun 7: 12972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre‐autophagosomal structures. Nat Cell Biol 12: 747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CR, Wang Y, Galko MJ (2018) Crawling wounded: molecular genetic insights into wound healing from Drosophila larvae. Int J Dev Biol 62: 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]