

Marquis and Sack show that the use-dependent inhibition of Kv2.1 by RY785 is consistent with gated access to a cavity within the channel that traps RY785. While voltage activation is required for access to the blocking site, channel opening itself is not required for access.
Abstract
Understanding the mechanism by which ion channel modulators act is critical for interpretation of their physiological effects and can provide insight into mechanisms of ion channel gating. The small molecule RY785 is a potent and selective inhibitor of Kv2 voltage-gated K+ channels that has a use-dependent onset of inhibition. Here, we investigate the mechanism of RY785 inhibition of rat Kv2.1 (Kcnb1) channels heterologously expressed in CHO-K1 cells. We find that 1 µM RY785 block eliminates Kv2.1 current at all physiologically relevant voltages, inhibiting ≥98% of the Kv2.1 conductance. Both onset of and recovery from RY785 inhibition require voltage sensor activation. Intracellular tetraethylammonium, a classic open-channel blocker, competes with RY785 inhibition. However, channel opening itself does not appear to alter RY785 access. Gating current measurements reveal that RY785 inhibits a component of voltage sensor activation and accelerates voltage sensor deactivation. We propose that voltage sensor activation opens a path into the central cavity of Kv2.1 where RY785 binds and promotes voltage sensor deactivation, trapping itself inside. This gated-access mechanism in conjunction with slow kinetics of unblock supports simple interpretation of RY785 effects: channel activation is required for block by RY785 to equilibrate, after which trapped RY785 will simply decrease the Kv2 conductance density.
Introduction
Ion channel inhibitors are used to investigate the physiological functions of their target proteins. Each ion channel inhibitor has a characteristic mechanism of action which determines whether the degree of inhibition will vary with local conditions such as membrane potential or permeant ion concentration (Sack and Eum, 2015; Dilly et al., 2011). To interpret the impact of an inhibitor in a physiological experiment, it is important to understand the mechanism of the inhibitor. Here, we investigate the mechanism of a small molecule that potently and selectively inhibits Kv2 voltage-gated K+ channels.
Kv2 channels are conserved from Cnidaria to Chordata (Li et al., 2015), suggesting they serve unique and fundamental purposes. Mammals have two Kv2 orthologs, Kv2.1 and Kv2.2, which are pore-forming protein subunits that can assemble as homotetramers or heterotetramers to form voltage-gated Kv2 channels (Frech et al., 1989; Kihira et al., 2010). Kv2 channels are expressed in nervous, muscular, and endocrine cell types (Bocksteins, 2016; Vacher et al., 2008). Kv2.1 is notably important in the brain, where it is highly and widely expressed in central neurons and forms the principal delayed rectifier current of many neuron types (Trimmer, 1991; Liu and Bean, 2014; Guan et al., 2007; Mandikian et al., 2014; Malin and Nerbonne, 2002; Du et al., 2000; Murakoshi and Trimmer, 1999; Pathak et al., 2016; Kimm et al., 2015). In mice and humans, mutations of Kv2.1 (KCNB1) result in severe neuropathologies, suggesting a fundamental importance in neuronal function (Bar et al., 2020; Speca et al., 2014; Hawkins et al., 2021; Thiffault et al., 2015; Torkamani et al., 2014). In neurons, Kv2 channels can mediate or suppress sustained, high-frequency action potential generation (Hönigsperger et al., 2017; Liu and Bean, 2014; Romer et al., 2019). In smooth muscle, Kv2.1 modulates myogenic tone (O’Dwyer et al., 2020; Amberg and Santana, 2006; Zhong et al., 2010). In pancreatic β cells, Kv2 channels suppress insulin secretion (Li et al., 2013; Jacobson et al., 2007). In photoreceptors, Kv2.1 contributes to the outward dark current (Fortenbach et al., 2021). Kv2.1 is subject to complex regulation by phosphorylation (Misonou et al., 2005; Murakoshi et al., 1997; Cerda and Trimmer, 2011; McCord and Aizenman, 2013; Misonou et al., 2004), SUMOylation (Dai et al., 2009; Plant et al., 2011), assembly with pore-forming KvS and auxiliary subunits (Bocksteins and Snyders, 2012; Bocksteins, 2016; Peltola et al., 2016, 2011), and membrane lipid composition (Delgado-Ramírez et al., 2018; Milescu et al., 2009; Ramu et al., 2006). These complex regulations make the voltage responses of Kv2 channels difficult to predict, highlighting the importance of inhibitors for determining the contributions of Kv2 channels to the wide range of physiological functions they participate in.
Recently, the contributions of Kv2 channels to electrical signaling have been identified using peptide toxins from spiders (Pathak et al., 2016; Kimm et al., 2015; Liu and Bean, 2014; Newkirk et al., 2021; Romer et al., 2019; Speca et al., 2014). Especially useful toxins include stromatoxin-1 and guangitoxin-1E (GxTX), with GxTX being more selective for Kv2 channels (Escoubas et al., 2002; Herrington et al., 2006). However, these peptides are not uniformly efficacious. Both stromatoxin-1 and GxTX are partial inverse agonists. GxTX acts by preferentially binding to resting voltage sensors on the channel, thereby stabilizing closed channels (Tilley et al., 2019). An aspect of this mechanism is that depolarization promotes toxin dissociation. GxTX inhibition can thus be overcome at more depolarized potentials or during high-frequency stimuli, and this negative use dependence can complicate interpretation (Tilley et al., 2014). Because of the difficulties of interpretation that stem from the allosteric mechanism of spider toxins, a Kv2-selective inhibitor without the complications of use dependence and gating modulation could enable more definitive physiology experiments.
A class of Kv2-selective inhibitors were developed in a small-molecule medicinal chemistry project at Merck (Herrington et al., 2011). Of these, an uncharged molecule, RY785, was the most potent Kv2.1 inhibitor, with a half-maximal inhibitory concentration (IC50) of 50 nM. Depolarization from a negative holding potential was required for the onset of inhibition by RY785, leading us to wonder whether this apparent use dependence would impact RY785’s performance as an inhibitor. Here, we determine how voltage activation of Kv2.1 interacts with inhibition by RY785.
Two mechanisms that can create use dependence are allosteric modulation and gated access to a binding site. Allosteric modulation can inhibit ion channels via stabilization of nonconducting conformations (Hille, 1977; Hondeghem and Katzung, 1977). This can result in modification of conductance kinetics or voltage dependence. A hallmark of allosteric inhibition is that channels in nonconducting conformations bind their ligands with higher affinity. A gated-access mechanism involves a conformational change that allows an inhibitor to access and exit its binding site. A gated-access mechanism controls intracellular quaternary ammonium ion block of voltage-gated K+ channels (Armstrong, 1971; Armstrong and Hille, 1972). Use dependence can also be produced by a combination of allosteric modulation and gated access, as with lidocaine block of voltage-gated Na+ channels (Nguyen et al., 2019; Hille, 1977; Vedantham and Cannon, 1999) or dofetilide block of hERG K+ channels (Wang et al., 2016; Wu et al., 2015; Ficker et al., 1998). Here, we investigate whether allosteric modulation or gated-access mechanisms undergird the use-dependent inhibition of Kv2.1 by RY785.
Materials and methods
Cell culture
A Chinese hamster ovary (CHO)-K1 cell line stably transfected with vectors enabling tetracycline-induced expression of the rat Kv2.1 channel (Trapani and Korn, 2003) was maintained in cell culture–treated polystyrene dishes (130180; Thermo BioLite) at 37°C in a 5% CO2 atmosphere in growth medium composed of Ham’s F-12 medium (11765-054; Gibco) containing 10% FBS (100-500; GemCell) and 1% penicillin–streptomycin solution (15140-122; Life Technologies). The CHO cell line was a gift from Stephen Korn, University of Connecticut, Storrs, CT. It was validated by tetracycline induction of Kv2.1-like delayed rectifier K+ currents and tested negative for mycoplasma (Lonza MycoAlert). Cells were cultured with 1 μg/ml blasticidin S HCl (A11139-03; Gibco) and 25 μg/ml zeocin (46-0509; Invitrogen) to retain transfected vectors. 1–2 h before experiments, 1 μg/ml minocycline HCl (ALX-380-109-M050; Enzo) was added to medium to induce channel expression. For voltage-clamp recording, cells were harvested by manual scraping in PBS with 0.48 mM EDTA (15040-066; Gibco) and pelleted by centrifugation at 1,000 g for 2 min. For K+ current experiments, cells were resuspended in the growth medium. For gating current experiments, cells were resuspended and pelleted three times in gating current external solution. Cells were slowly rotated in a polypropylene tube (05-408-134; Fisher) at room temperature until use. For K+ current experiments, aliquots of cell suspension were added to a recording chamber containing external solution, allowed to settle, and rinsed with external solution before recording. For gating current experiments, aliquots of cell suspension in gating current external solution were added to a dry recording chamber.
K+ current measurements
Patch-clamp experiments were performed at room temperature (22.0–23.5°C). Voltage clamp was achieved with an Axopatch 200B amplifier (Axon Instruments) run by Patchmaster v2x90.5 software (HEKA). The external bath solution contained (in mM) 155 NaCl, 10 HEPES, 1.5 CaCl2, 1 MgCl2, and 3.5 KCl, adjusted to pH 7.2 with NaOH. External solution was supplemented with 1:1,000 vol:vol DMSO as a vehicle control or, when indicated, 1 µM RY785 (19813; Cayman) as 1:1,000 1 mM RY785 in DMSO. In some experiments, 5 µM tetrodotoxin was added to suppress endogenous voltage-gated Na+ channels. We did not see any suggestion that tetrodotoxin or the rapidly inactivating endogenous Na+ channels impacted analyses of Kv2.1 currents, and results with and without tetrodotoxin were pooled. The internal pipette solution contained (in mM) 50 KF, 70 KCl, 35 KOH, 5 EGTA, and 50 HEPES, adjusted to pH 7.4 with KOH. Internal solution osmolarity was adjusted with sucrose in some experiments. Data with and without sucrose are pooled. Recording pipettes were pulled from thin-walled borosilicate glass (1.5-mm outer diameter, 1.1-mm inner diameter, with filament; Sutter Instrument) on a horizontal micropipette puller (Sutter P-87) using five or more heating cycles to achieve a taper to the tip over minimal length. Pipettes were typically coated with a silicone elastomer (Dow Corning Sylgard 184) and heat-cured. Data with and without Sylgard are pooled. Pipette-tip resistances with the above solutions were 0.96–2.41 MΩ with positive pressure applied to pipette.
CHO cells with a round shape and smooth surface were selected for whole-cell voltage clamp. To minimize voltage errors at the cell membrane due to series resistance, several measures were taken. To control the magnitude of K+ currents, Kv2.1 expression was induced by incubation with minocycline for 128–155 min (experiments with tetraethylammonium [TEA]) and 42–110 min (other experiments). Series resistances were estimated by manual nulling of capacitance and were <11 MΩ. The series resistance compensation correction circuit was set to 60–90%. Lag was 10 μs. Data were excluded from analysis if the product of current amplitude and estimated series resistance remaining after compensation was >10 mV. The largest current analyzed in this dataset was 11.3 nA. Cell capacitances were 1.9–17.7 pF, resulting in cell membrane charging time constants of <54 μs before compensation, at least two orders of magnitude faster than time constants fitted to ionic currents. Currents for I–V relations were low-pass filtered at 10 kHz and digitized at 100 kHz. Currents for drug association/dissociation experiments were low-pass filtered at 5 kHz and digitized at 10 kHz. The holding potential was −100 mV. Remaining capacitance and Ohmic leak were subtracted offline using traces recorded during P/5 voltage protocols from holding potential. In sequences of voltage steps, at least 2 s elapsed between the start times of each recording. For vehicle and RY785 wash-in, solution was exchanged by flushing a volume of ≥200 μl through a recording chamber of <100 μl (Warner RC-24N). RY785 was applied after vehicle. Time courses of current inhibition (detailed below) were analyzed for artifacts resulting from solution exchange. We observed that, following wash-in of RY785 or vehicle, Kv2.1 current amplitudes would increase by 5.0 ± 2.9% (mean ± SEM, n = 32 cells) or 5.32 ± 0.70% (mean ± SEM, n = 4 cells), respectively. Current amplitudes in vehicle-washed cells would then slowly decay by as much as 23% (Fig. 3 B). This variability was deemed too minor to alter our interpretation or conclusions and therefore tolerated.
Figure 3.

RY785 inhibition rate is not proportional to K+ conductance. (A) Representative current traces from two cells subjected to the voltage protocol inset in B. Cells held at −100 mV were given a 20-ms step to +40 mV followed by a 30-ms step to −100 mV and then a 500-ms step to a voltage which varied between cells. 10 s elapsed during each cycle. Dashed lines indicate 0 current. Data from these exemplars are represented as stars in B and C. (B) Time courses of current inhibition from representative cells with postpulses to the indicated voltages. Mean currents from the final 1 ms of each test pulse (symbols) are normalized to the first test pulse in RY785. Stars correspond to exemplars shown in A. Arrows indicate time points shown in A. Function variables from fitted exponential function (Eq. 4, curves) ± SD; −80 mV, τ = 119.0 ± 01.2 s−1, I0 = 0.0211 ± 0.0016; −60 mV, τ = 116.0 ± 1.0 s−1, I0 = 0.0123 ± 0.0016; −40 mV, τ = 59.7 ± 02.0 s−1, I0 = 0.0034 ± 0.0085; −20 mV, τ = 14.13 ± 00.16 s−1, I0 = 0.0093 ± 0.0013; 0 mV, τ = 09.168 ± 00.063 s−1, I0 = 0.01323 ± 0.00083; +20 mV, τ = 08.71 ± 00.18 s−1, I0 = 0.0162 ± 0.0024; +40 mV, τ = 08.62 ± 00.59 s−1, I0 = 0.0017 ± 0.0080; vehicle control (gray), +20 mV, τ = 74 ± 21 s−1, I0 = 0.832 ± 0.023. (C) Apparent rates of inhibition (kapparent) plotted against postpulse voltage. kapparent ± SD from fits to individual cells. Stars and triangles correspond to exemplars shown in A and B. (D) Relation of RY785 inhibition rate during the postpulse (kpost) to postpulse voltage. Geometric mean rate ± SEM (circles) calculated from cells in C. Boltzmann function fitted to RY785 inhibition rates (curve). Function variables ± SD: x held at 1. Vhalf = −20.2 ± 4.9 mV, z = 1.97 ± 0.30 e0, A = 2.41 ± 0.24 s−1. (E) Comparison of RY785 inhibition rates to Kv2.1 G–V relation (yellow) and OFF gating charge–voltage (QOFF–V) relation (blue). Means ± SEM. G–V from cells in vehicle, n = 4 cells. QOFF–V replotted from a prior publication (Tilley et al., 2019). Amplitudes normalized to fitted Boltzmann functions (Eq. 2), shown as solid curves. Function variables ± SD; QOFF, x set to 1, Vhalf = −23.7 ± 1.6 mV, z = 1.81 ± 0.12 e0; G, x set to 4, Vhalf = −19.95 ± 0.73 mV, z = 0.968 ± 0.020 e0.
Gating current measurements
The following modifications from the whole-cell protocol were applied during gating-current recordings. Channel expression was induced by incubation with minocycline for 36–60 h. The external bath solution contained (in mM) 140 NMDG, 60 HEPES, 2 CaCl2, 2 MgCl2, 0.1 EDTA, 0.01 CsCl, and 84 methanesulfonic acid at pH 7.3. When indicated, the external solution was mixed 1,000:1 with DMSO or 1 mM RY785 in DMSO. The internal solution contained (in mM) 90 NMDG, 50 NMDG hydrofluoride, 1 NMDG hydrochloride, 60 HEPES, 5 EGTA, 5 sucrose, and 29 methanesulfonic acid at pH 7.3. Pipette-tip resistances with the above solutions were 2.7–7.7 MΩ with positive pressure applied to the pipette. All pipettes were coated with Sylgard and fire-polished. Series resistances were 5.5–13.8 MΩ, except for one 47-MΩ cell that responded well to compensation circuitry. Cell capacitances were 3.1–13.0 pF. To exclude artifacts of membrane charging, data were not analyzed until 200 μs after a voltage step. The series resistance prediction circuit was set to 70%, resulting in predicted membrane charging time constants of <45 µs. The series resistance compensation correction circuit was set to 70–75% with 10-µs lag. Currents were low-pass filtered at 10 kHz and digitized at 50 kHz. In sequences of voltage steps, 4 s elapsed between the start times of each recording. Vehicle or RY785 were manually added to a 200-μl recording chamber with a 5-min wait before recording. Vehicle and RY785 were applied in a blinded and randomized fashion with unblinding after completion of data analysis.
Monitoring current inhibition and recovery
The voltage protocols for determining the voltage dependence of Kv2.1 inhibition by and recovery from RY785 consisted of 10-s cycles containing two voltage steps from the holding potential of −100 mV. The first voltage step is a 20-ms test pulse to +40 mV to gauge the proportion of conductive channels. Afterward, the cell is returned to −100 mV holding potential for 30 ms, then given a postpulse to a voltage between −80 and +40 mV. The 30-ms interval between a test pulse and subsequent postpulse is 10 times the 3-ms deactivation time constant of Kv2.1 at −100 mV (Tilley et al., 2019), so deactivation is expected to be >99% complete by the end of the step. The postpulse was 500 ms for the inhibition protocols and 4 s for the recovery protocols. Testing of each cell went as follows: (1) a baseline was established in vehicle by recording 10 cycles of the inhibition protocol; (2) during a 2-min gap, the recording chamber solution was exchanged with 1 μM RY785 or vehicle control; (3) the inhibition protocol resumed until currents stabilized (15–80 cycles); (4) 10 cycles of the recovery protocol were recorded while the bath still contained 1 μM RY785; (5) during a 1-min gap, continuous flow was started of a solution without RY785; and (6) recording of the recovery protocol resumed with continuous solution flow until currents stabilized (71–155 cycles).
Analysis
Electrophysiology analysis, curve fitting, and plotting were performed with Igor Pro 8 (Wavemetrics), which uses a Levenberg–Marquardt algorithm for least-squares curve fitting. For presentation, gating current traces were Gaussian-filtered at 2 kHz.
Conductance (G) was calculated as ionic current divided by the K+ driving force:
| (1) |
where Vcell is membrane voltage; Vcommand is command voltage; fcompensated is fraction of series resistance compensated; Rseries is measured series resistance; I is measured current; VLJ is liquid-junction potential; and VLJ of 12 mV was calculated using Patchers Power Tools v2.15 (Mendez and Wurriehausen, 2009).
For the conductance–voltage (G–V) relation, conductance values were determined from current levels in the final 2 ms of 100-ms steps to the indicated voltage. Kv2.1-mediated currents were isolated by subtraction of currents remaining in 1 µM RY785. Kv2.1-mediated currents were divided by the driving force for K+ relative to the calculated Nernst potential of −97.4 mV. Conductance levels were plotted against command voltage and normalized to their mean from +80 to +100 mV. G–V relations were fitted with a Boltzmann function:
| (2) |
where A is maximum amplitude, z is valence in units of elementary charge (e0), F is the Faraday constant, R is the ideal gas constant, and T is absolute temperature. The variable x is the power, or order, of the Boltzmann function. When fitting the G–V, x was set to 4 (Tilley et al., 2019). Vhalf is the activation midpoint in units of millivolts. In Eq. 2, the voltage that produces half-maximal conductance, Vmid, when x = 4 is
| (3) |
Inhibition and recovery rate calculations
Rates of channel inhibition by RY785 for Fig. 3 were determined from the test pulses of the two-voltage-step protocol described above. The amplitudes of test pulse current were fitted with an exponential function (Eq. 4), starting with the first record after RY785 wash-in:
| (4) |
where I is current, I0 is current at time zero, A is equilibrium current amplitude, x is time, x0 is time 0, and τ is the time constant. The apparent inhibition rate (kapparent) is τ−1. To estimate an inhibition rate during the variable-potential postpulses (kpost), we assumed that no significant inhibition occurs when holding at −100 mV (Fig. 2 B) and applied a correction to account for inhibition during test pulses to +40 mV (ktest), weighted by pulse durations (dtest and dpost):
| (5) |
where dcycle is the length of one protocol cycle. ktest was determined from the mean kapparent with postpulses and test pulses to +40 mV such that ktest = kpost. For the estimates of inhibition rate during a single pulse (ksingle step) presented in Fig. 4, Eq. 4 was fitted to current decay during the first postpulse in RY785. As an attempt to correct for current decay in vehicle due to inactivation, current decay in vehicle was fitted with Eq. 4, and τ−1 in vehicle was subtracted from τ−1 in RY785.
Figure 2.

Voltage stimuli are required for RY785 to inhibit Kv2.1. (A) Current traces from a representative cell in vehicle (black) or 1 µM RY785 (red). Voltage protocol from a holding potential of −100 mV is a 20-ms step to +40 mV followed by a 30-ms step to −100 mV and then a 500-ms step to +40 mV. Arrows indicate time points labeled in B. (B) Mean current in the final 1 ms of the 20-ms test pulse (circles). Red bar indicates application of 1 µM RY785. Currents in RY785 are fitted with an exponential function (Eq. 4, red curve). Function variables ± SD, y0 = 0.0482 ± 0.0039 nA, A = 1.533 ± 0.012 nA, τ = 14.40 ± 0.24 s. Dotted blue line indicates steady state current before RY785 application.
Figure 4.
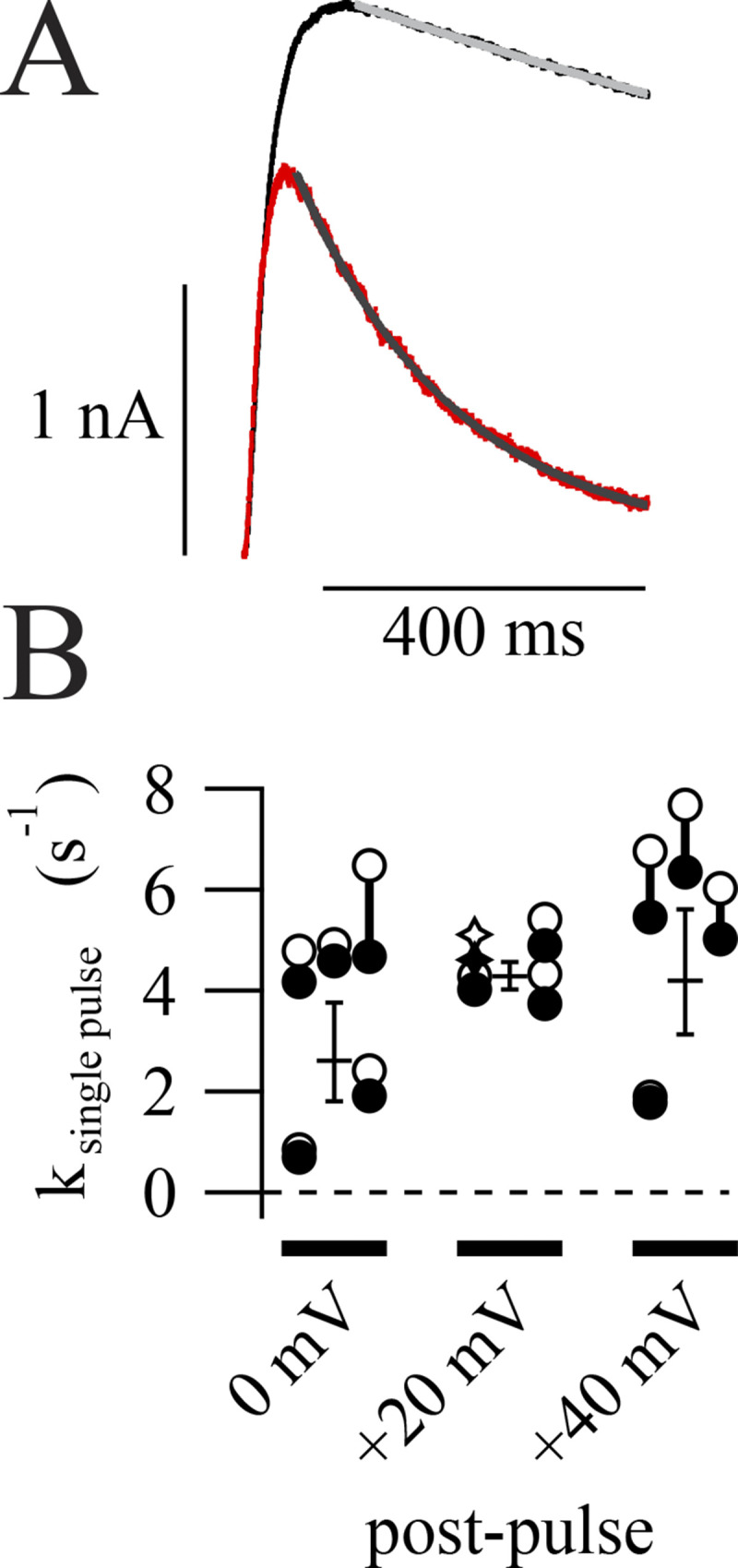
Rates of inhibition determined from single voltage steps. (A) Representative current decay during postpulses. First pulse in 1 µM RY785 (red) fitted with an exponential function (Eq. 4, gray curve). Function variables ± SD, I0 = 34.6 ± 1.4 pA, τ = 0.19591 ± 0.00051 s. Average of 10 traces in vehicle (black) and fit (light gray curve), τ ± SD = 7.4 ± 1.2 s, I0 held at 34.6 pA. (B) Category plot showing inhibition rates at three voltages. Hollow symbols represent rates of current decay in RY785 from individual cells. Filled symbols represent rates of current decay in RY785 corrected by subtracting rate of current decay in vehicle from the same cell. Stars correspond to the exemplar shown in A. Geometric means ± SEM of corrected rates are shown as horizontal lines. n = 4 or 5 per voltage.
Current recovery after RY785 wash-out with test pulses and postpulses to +40 mV was fitted with Eq. 4, yielding apparent recovery rates from which recovery rates (krecover) were determined using Eq. 5 as above, where ktest = kpost = krecover. For recovery experiments with postpulses to −80 mV, limited recovery occurred, and kapparent was estimated by linear regression. To calculate krecover using Eq. 5, ktest from the +40-mV recovery protocol was used. In one experiment, current did not recover following RY785 wash-out, and this cell was excluded from analysis.
Estimation of Kd
Rates of channel inhibition and recovery were used in Eq. 6 to estimate a dissociation constant (Kd) for RY785 binding to Kv2.1 at +40 mV:
| (6) |
where koff = krecover with postpulses to +40 mV and kon = kpost/[RY785] when kpost measured the rate of inhibition at +40 mV. The contribution of koff to the kinetics of inhibition at +40 mV was ignored as kpost ≫ koff. The parent compound from which RY785 was derived inhibits human Kv2.1 with a Hill coefficient of close to 1 (1.2; Herrington et al., 2011), suggesting that it is reasonable to ignore the possibility of cooperative binding in a Kd estimate.
Activation rate
Channel activation time constants were determined by fitting single-exponential functions (Eq. 4) to current amplitude time courses during the final 15 ms of the 20-ms test pulse to +40 mV. We did this for a randomly selected sample of 10 of the above inhibition rate experiments. Time constants were excluded if SD of fit was >15 ms. In Fig. S2 B, peak currents were means of the final 0.5 ms of the test pulse and were normalized to the first record after RY785 wash-in.
Figure S2.

RY785 does not affect Kv2.1 K+ current activation kinetics. (A) Representative K+ current traces during our two-pulse protocol in the presence of RY785. Overlaid traces from a representative cell during sequential steps to +40 mV from −100 mV (red traces), fitted with a single exponential function (Eq. 4, gray curves). Later traces are smaller in amplitude. Postpulse was to −60 mV in this cell. (B) Activation τ ± SD from fits as in A plotted against the corresponding trace’s peak current relative to that recorded during the first +40-mV step in RY785. Different symbols represent n = 10 cells. Stars correspond to the cell in A. Only measurements with >10% remaining current are presented, as fitted time constants became noisier at smaller current amplitudes.
Gating current analyses
Charge (Q) movement was quantified by integrating gating currents. ON gating currents were integrated from 0.2 to 6 ms after the start of the voltage step. OFF gating currents were integrated from 0.2 to 20 ms after the start of the voltage step. Currents were baseline-subtracted from 20 to 30 ms after the start of the voltage step. Charge movement data as functions of voltage were fitted with a Boltzmann function (Eq. 2) with f(V) = Q and the order, x, set to 1. Time constants were determined by fitting Eq. 4 to the decay phase of gating current traces. The voltage dependence of ON gating charge movement was determined by fitting gating current decay time constants as a function of voltage:
| (7) |
where τ is the time constant of ON gating current decay, τ0 mV is the time constant of ON gating current decay at 0 mV, V is voltage, z is valence in units of elementary charge (e0), F is the Faraday constant, R is the ideal gas constant, and T is absolute temperature. Traces with obvious leak artifacts were excluded from analysis.
Statistics
Statistical tests were performed in Igor Pro 8. Uncertainties reported with fit parameters are SDs. Arithmetic means and SEMs are reported for current amplitudes and charge movement. Geometric means and positive SEMs are reported for time constants and rates. Wilcoxon rank tests were performed assuming independent samples unless otherwise noted. Two-tailed P values are reported for the hypothesis that samples have identical medians.
Online supplemental material
Fig. S1 compares currents from untransfected CHO cells and Kv2.1-CHO cells in 1 µM RY785. Fig. S2 compares Kv2.1 activation kinetics during onset of inhibition by RY785. Fig. S3 is an alignment of expected cavity-lining residues of Kv2 and KvS channel subunits.
Figure S1.

Residual currents in 1 µM RY785 resemble endogenous CHO currents. (A) Top: Voltage command. Bottom: Representative currents from Kv2.1-CHO cells or CHO cells without heterologous Kv2.1, in vehicle (black) and RY785 (red). Black arrow indicates time point used for current amplitude quantification in B. (B) Current amplitudes at +90 mV. Currents from 98 to 100 ms after stepping to +90 mV from a holding potential of −100 mV (symbols). Exemplars from A are shown as stars. Three cells had voltage clamp errors exceeding 10 mV at +90 mV (triangles). For these three cells, currents were analyzed from +20 to +30 mV, where voltage clamp errors were <10 mV, and extrapolated to +90 mV using the Boltzmann fit to the Kv2.1 G–V (Fig. 3 E). n = 8 Kv2.1-CHO cells and n = 4 CHO cells. Bars are arithmetic means ± SEM.
Figure S3.

Conservation of pore-lining residues of Kv2 orthologs and paralogs. The Rattus norvegicus Kv2.1 (RnKv2.1) sequence is used for residue numbering at top. Homology to Kv1.2 was used to infer the locations of pore-lining residues (purple highlights) in RnKv2.1. (A) Conservation of pore-lining residues of RnKv2.1 with Kv2-family orthologs. Alignments created in Jalview 2.11.1.4 (Waterhouse et al., 2009). Dots represent residues in protein subunits that are identical to corresponding residues in RnKv2.1. Residues forming the tetraalkylammonium binding site in RnKv1.2 (Lenaeus et al., 2014; Long et al., 2005) are shown in black. Species: Homo sapiens (Hs), Gallus gallus (Gg), Ophiophagus Hannah (Oh), Xenopus laevis (Xl), Danio rerio (Dr), Mytilus coruscus (Mc), Drosophila melanogaster (Dm), Caenorhabditis elegans (Ce), Nematostella vectensis (Nv). (B) Conservation of pore-lining residues of mammalian Kv2.1 with KvS silent subunits.
Results
At all physiological voltages, block by RY785 appears complete
Knowing how completely a drug can inhibit channels is important for interpretation of experiments and can reveal clues about the drug’s mechanism of action. We asked how completely RY785 inhibits Kv2 K+ currents. To study RY785 inhibition, we voltage-clamped homomeric channels composed of rat Kv2.1 subunits expressed in a stably transfected CHO-K1 cell line (Trapani and Korn, 2003). When 1 µM RY785 was applied and depolarizing steps were given until currents stabilized, RY785 eliminated voltage-activated outward currents (Fig. 1 A). Block by RY785 appeared complete, as currents with activation kinetics resembling Kv2.1 were no longer discernable. At +40 mV, 1 µM RY785 blocked 98.3 ± 0.4% (mean ± SEM) of outward current (Fig. 1 B). CHO-K1 cells have been found to lack endogenous voltage-gated K+ channel expression (Gamper et al., 2005), yet in untransfected CHO-K1 cells, endogenous outward currents were apparent above +40 mV. These endogenous CHO currents were not blocked by 1 µM RY785 and appeared similar to the residual currents of Kv2.1-CHO cells in 1 µM RY785 (Fig. S1 A). A comparison of current amplitudes at +90 mV, where endogenous CHO currents were detectable in all cells, revealed that the magnitude of residual currents of Kv2.1-CHO cells in 1 µM RY785 were within SEM of CHO cells without a Kv2.1 plasmid (Fig. S1 B). This suggests that most, if not all, residual outward current from Kv2.1-CHO cells in 1 µM RY785 (Fig. 1 C) is attributable to endogenous CHO currents, not Kv2.1. These findings indicate that 1 µM RY785 eliminates the Kv2.1 conductance in response to voltage steps up to at least +90 mV from a holding potential of −100 mV, a range spanning the physiologically relevant voltages set by typical Na+ and K+ reversal potentials.
Figure 1.

RY785 blocks Kv2.1. Displayed currents in 1 μM RY785 are following a train of depolarizations to allow RY785 to inhibit Kv2.1 currents. (A) Top: Voltage command. Bottom: Representative currents from a whole-cell voltage-clamped Kv2.1-CHO cell in vehicle (black) and RY785 (red). (B) Current remaining during inhibition by RY785 from eight cells (red symbols). Horizontal line is mean ± SEM. (C) Current as a function of command voltage. Currents are means 98–100 ms after the start of the activating voltage step. Symbols correspond to individual cells in vehicle (black) and RY785 (red). Points are excluded if the predicted voltage error of clamp is >10 mV. Inset: Structure of RY785.
RY785 inhibition requires voltage activation but not channel opening
After application of RY785 to human Kv2.1 channels, voltage activation is required for onset of inhibition, and K+ current progressively declines during repeated positive voltage steps (Herrington et al., 2011). We characterized how inhibition of rat Kv2.1 by RY785 responds to voltage activation. To measure the voltage dependence of the rate of inhibition by RY785, we designed a two-pulse protocol containing a brief test pulse to +40 mV followed by longer postpulse to a voltage which we later varied between experiments. In the presence of 1 µM RY785, Kv2.1 current amplitudes in response to a two-pulse protocol decay over time (Fig. 2 A). No inhibition occurs when cells are held at −100 mV (Fig. 2 B). The simple exponential kinetics of block in response to +40-mV voltage steps (Fig. 2 B, red curve) suggests that a single RY785 binding event is sufficient to completely inhibit Kv2.1. Additionally, when K+ conductance is only partially inhibited by RY785, the time constants associated with voltage activation do not vary (Fig. S2), consistent with all-or-nothing block of individual channels.
We next tested whether channel opening is required for RY785 inhibition. If so, then RY785 should inhibit Kv2.1 with a rate proportional to the channel’s open probability. We used the whole-cell Kv2.1 G–V relationship as a proxy for Kv2.1 open probability. Although the occurrence of subconducting conformations of rat Kv2.1 single channels (Chapman et al., 1997) makes the G–V relationship an imperfect proxy, the fully conducting conformation of a rat Kv2.1 variant has been used to effectively characterize its G–V relationship (Islas and Sigworth, 1999), and we have found subconducting conformations to occur <10% as often as full openings in this Kv2.1-CHO cell line (Tilley et al., 2019), suggesting that the G–V relationship is reasonably proportionate to open probability. We monitored the decay of Kv2.1 current amplitudes in the presence of RY785 and varied the voltage of the postpulse step between cells (Fig. 3, A and B). We then fitted an exponential function (Eq. 4) to the decay of Kv2.1 currents during repeated cycles of the two-pulse protocol (Fig. 3 B). In these fits, the apparent rate of inhibition seems to saturate below −60 mV and above 0 mV (Fig. 3 C). We estimated rates of inhibition during the postpulse steps (kpost; Eq. 5). A Boltzmann fit (Eq. 2) assigned half-maximal kpost at Vmid = −20.2 ± 4.9 mV (Fig. 3 D). However, G–V relations calculated from currents recorded in vehicle (Fig. 1) indicated that the voltage at which conductance was half maximal was Vmid = +23.8 ± 1.2 mV (Fig. 3 E), 44 mV more positive than the midpoint of the inhibition rate. It appears clear that the voltage dependence of block does not follow G. The coarse resolution and substantial cell-to-cell variability in the kpost–voltage relation precludes more detailed conclusions about the voltage dependence of the inhibition rate. Nevertheless, this finding is inconsistent with the hypothesis that RY785 binds only once channels open. To assess the validity of our estimates of kpost, we also determined RY785 inhibition rates by fitting exponential functions to current decay (Fig. 4 A). The inhibition rate was determined in this manner during 0-, +20-, and +40-mV pulses where Kv2.1 current decay could be reliably fitted. After applying a correction that attempts to account for current decay due to inactivation, the inhibition rates measured by fitting current decay during a single pulse (ksingle pulse, Fig. 4 B) were about two times faster than values measured from repeated two-pulse cycles (kpost, Fig. 3 E), which we consider to be roughly similar. RY785 inhibition rates measured by the method of Fig. 4 were not significantly different at 0, +20, or +40 mV (one-way ANOVA, P = 0.39). However, Kv2.1 conductance increases between 0 and +40 mV (one-way ANOVA, P = 1.7 × 10−10) by a factor of 3 (Fig. 3 E). Again, the onset of RY785 inhibition is responsive to voltage activation, but the rate is not correlated with the degree of channel opening. We noted that the Vmid of kpost was similar to the Vmid = −29.3 ± 2.8 mV of gating charge movement from Kv2.1 in the same CHO-K1 cell line (Fig. 3 E). This correlation suggests that the RY785 binding site is exposed by voltage-sensor activation, but not channel opening itself.
RY785 is trapped by the resting conformation of Kv2.1
We asked how membrane voltage affects relief of inhibition after wash-out of RY785 from the recording chamber. If RY785 is an allosteric modulator that simply prefers voltage-activated channel conformations, then channel deactivation at negative voltages would enhance drug dissociation. Alternatively, if voltage activation provides gated access to a binding site, channel deactivation could trap RY785 within the channel. To distinguish between these possibilities, we analyzed current recovery after drug wash-out as a proxy for RY785 dissociation and asked whether recovery from inhibition depends on voltage. Our recovery from inhibition protocol was performed following a subset of the 1 µM RY785 wash-in experiments of Fig. 3 (Fig. 5 A). During recovery experiments, we used a two-pulse protocol with 4-s postpulses and tested two postpulse voltages: −80 and +40 mV (Fig. 5 B). With a −80 mV postpulse, only 8.1 ± 1.9% (mean ± SEM) of current recovered after 600 s of pulsing (Fig. 5 C, left). However, with a +40 mV postpulse, 65.2 ± 8.3% of current recovered over the same duration (Fig. 5 C, right). Some current recovery time courses exhibited a delay before current amplitudes begin to rise. While this delay could be indicative of current recovery requiring multiple RY785 molecules to dissociate from each channel, the cell-to-cell variability in delay leads us to suspect that the delay results from a variable latency for RY785 to diffuse out from cells. From exponential fits (Eq. 4) of current recovery time courses, we estimate the rate of current recovery to be 0.0104 ± 0.0016 s−1 (mean ± SEM) during the pulses to +40 mV (Fig. 5 D). Current recovery was incomplete with postpulses to −80 mV. Using linear regression, we estimated the recovery rate to be 0.000134 ± 0.000031 s−1 (Fig. 5 D) during postpulses to −80 mV, 1% of the rate at +40 mV. These results suggest that dissociation of RY785 from Kv2.1 is highly dependent on voltage activation of the channel, occurring at a lower rate when the channel is deactivated. This result is inconsistent with a simple allosteric mechanism in which RY785 dissociates more rapidly from resting channels. Our observations instead suggest that RY785 has better access to and from its inhibitory site when channels are voltage activated.
Figure 5.

Negative membrane potential slows RY785 dissociation. (A) Cartoon timeline of experiments. The number of voltage protocol cycles used to inhibit and recover Kv2.1 currents varied between cells. (B) Representative current traces from two cells. Dashed lines indicate zero current. Data from these cells are represented as stars in C and D. (C) Time courses of current recovery with postpulses to −80 (left) or +40 (right) mV. Mean currents from the final 1 ms of each test pulse, normalized to before RY785 wash-in, are shown as symbols. Arrows indicate time points shown in B. Time courses at −80 mV (left, n = 4) are fitted with gray lines. Time courses at +40 mV (right, n = 6) are fitted with single exponential functions (Eq. 4, gray curves). (D) Category plot showing current recovery rates at two voltages. Rates were calculated from the fits in C. Geometric means ± SEM are shown as horizontal lines.
TEA competes with RY785
A gated-access mechanism was first used to explain open channel block by intracellular quaternary ammonium ions such as TEA (Armstrong and Hille, 1972). TEA binds within the central cavity of voltage-gated K+ channels, with access to the intracellular solution gated by the protein’s sixth transmembrane helix (S6; Armstrong and Hille, 1972; Choi et al., 1993; Zhou et al., 2001). If RY785 binds in the central cavity, then we would expect it to compete for its binding site with TEA. To test this, we asked whether the rate of inhibition by RY785 could be reduced by intracellular TEA. At +20 mV, the predicted IC50 of internally applied TEA against rat Kv2.1 is 0.25 mM (Taglialatela et al., 1991). We supplemented our pipette solution with 2 mM TEA, which is predicted to occupy the central cavity 89% of the time at +20 mV, then applied 1 µM RY785 and stimulated with a two-pulse protocol with postpulses to +20 mV. We found that pretreatment with TEA slowed RY785 inhibition by a factor of 4 (Fig. 6). Thus, although RY785 is not exclusively an open channel blocker, occupancy of the intracellular TEA binding site prevents RY785 from binding in the channel.
Figure 6.

Intracellular TEA competes with RY785. (A) Time courses of current inhibition by 1 µM RY785 from cells with postpulses to +20 mV. Mean currents from the final 1 ms of each test pulse (symbols), normalized to the first test pulse in RY785. Time courses from a representative control cell (open symbols) and a representative cell pretreated with 2 mM intracellular TEA (closed symbols). Time courses are fitted with exponential functions (Eq. 4). Data from these cells are represented as stars in B. (B) Apparent RY785 inhibition rates ± TEA. Rates ± SD from fits to individual cells (symbols). Geometric means ± SEM are shown as horizontal lines.
RY785 alters gating charge movement
To search for evidence of allosteric modulation by RY785, we measured Kv2.1 gating currents. After treatment with 1 µM RY785, both ON and OFF gating currents appeared to be altered (Fig. 7, A and B). RY785 diminished the amount of ON gating charge movement compared with vehicle controls (Fig. 7 C) and, to a greater extent, diminished OFF gating charge (Fig. 7 D), indicating that RY785 inhibits components of voltage sensor movement. We suspect that the discrepancy between the magnitude of diminishment of the ON and OFF gating charges is an artifact of our current integration procedures. While we were able to reliably integrate a fast component of ON gating charge movement, the final steps of Kv2.1 voltage activation are much slower, and these small gating currents were difficult to reliably differentiate from integration noise. Consequently, more charge is apparent in OFF gating charge movements after these slow steps have occurred, leading to an apparent QOFF/QON ratio >1 (Tilley et al., 2019). Here, the average apparent QOFF/QON ratio from 0 to +80 mV in vehicle controls was 1.6 ± 0.1 (SEM, n = 8). However, the QOFF/QON ratio drops to near unity in 1 µM RY785, 0.9 ± 0.2 (SEM, n = 4), indicating that that, in addition to the diminishment of fast ON charge movement, a slower component of ON charge movement is eliminated by RY785. To probe how gating charge dynamics are modulated by RY785, we analyzed gating current kinetics. The kinetics and voltage dependence of the decay of ON gating current remaining in RY785 are similar to vehicle controls (Fig. 7 E). However, RY785 accelerates OFF gating charge kinetics (Fig. 7 F), indicating that deactivation of channels inhibited by RY785 is accelerated. Altogether, the gating current modifications indicate that RY785 prevents movement of a component of gating charge and accelerates deactivation of the gating charge that remains mobile.
Figure 7.

RY785 modifies gating currents. Kv2.1 gating currents were elicited in K+-free solutions with 100-ms steps to voltages between −80 and +80 mV from a holding potential of −100 mV (ON gating currents) and during subsequent 50-ms steps to −140 mV (OFF gating currents). (A) Exemplar ON gating currents at +40 mV before (black) and after (red) application of RY785. Integration windows used for the analysis in C (highlighted) were 0.2–6 ms after the voltage step. (B) Exemplar OFF gating currents at −140 mV following the steps shown in A before (black) and after (red) application of RY785. Integration windows used for the analysis in D (highlighted) were 0.2–20 ms after the voltage step. (C) Mean ON gating charge transfer (QON) as a function of voltage from n = 8 vehicle-treated cells (blue) and n = 4 RY785-treated cells (red). Mean ± SEM. *, P < 0.05. Curve is fit of Eq. 2 with x held at 1. Function variables ± SD, vehicle: Vhalf = −20.1 ± 5.8 mV, z = 1.62 ± 0.57 e0, A = 0.79 ± 0.11. RY785 data was not well fitted by Eq. 2. (D) Mean OFF gating charge (QOFF) transfer as a function of voltage from vehicle-treated cells (blue) and RY785-treated cells (red). Mean ± SEM. *, P < 0.05. Vehicle: n = 7 overall, ≥6 at each voltage; RY785: n = 4 overall, ≥2 at each voltage. Curve is a fit of Eq. 2 with x held at 1. Function variables ± SD: Vhalf = −34.4 ± 9.0 mV, z = 1.7 ± 1.0 e0, A = 0.84 ± 0.21. RY785 was not well fitted by Eq. 2. (E) Mean time constants of activation (τON) determined by fitting Eq. 4 to the decay phase of ON gating current traces from n = 8 vehicle-treated cells (blue) and n = 4 RY785-treated cells (red). Mean ± SEM. Lines are fits of Eq. 7. Function variables ± SD; vehicle, τ = 3.2 ± 0.6 ms, z = 0.52 ± 0.09 e0; RY785, τ = 2.7 ± 0.5 ms, z = 0.41 ± 0.09 e0. (F) Mean time constants of deactivation (τOFF) determined by fitting Eq. 4 to the decay phase of OFF gating current traces from vehicle-treated cells (blue) and RY785-treated cells (red). Mean ± SEM. *, P < 0.05. Vehicle, n = 8 overall, ≥6 at each voltage; RY785, n = 4 overall, ≥2 at each voltage.
Discussion
The mechanism of inhibition
We conclude that intracellular TEA competes with RY785, that RY785 binds to and dissociates from an inhibitory site only accessible when voltage sensors are activated, that channel opening itself has little impact on access, and that RY785 binding prevents some gating charge movement and promotes voltage sensor deactivation. A physical model that might explain our observations is RY785 binding within a central cavity, with access available in activated-not-open and open states. Further, the accelerated OFF gating currents suggest that RY785 binding closes the cavity, trapping RY785 within the channel. Together, these results suggest that RY785 may act as a “closed-channel blocker” similar to 4-aminopyridine in Kv1 channels (Armstrong and Loboda, 2001). Trapping of blockers has been documented for Kv channels including Shaker Kv1 (Holmgren et al., 1997), and mammalian Kv3.1 (Kirsch and Drewe, 1993). When Kv channels close their intracellular gate, a constriction is formed that prevents access of molecules as small as Ag+ (del Camino and Yellen, 2001) and exit of blockers including quaternary ammonium ions and 4-aminopyridine (Kirsch and Drewe, 1993; Holmgren et al., 1997). We have found that RY785 bears the hallmarks of trapping: requirement for voltage activation for block or unblock. RY785 appears unique among these trapped blockers in that it eliminated the majority of OFF gating current, by 64 ± 15% in our experiments. Trapping of 4-aminopyridine in Shaker channels or TEA in Shaker I470C does not eliminate gating charge, but shifts charge movement to more positive voltages and accelerates OFF gating current (Loboda and Armstrong, 2001; Melishchuk and Armstrong, 2001).
Alternate interpretations
The observation that RY785 becomes trapped by negative membrane potentials is incompatible with a simple allosteric mechanism in which RY785 merely stabilizes voltage-activated channel conformations, and RY785 destabilizes activated conformations, as evidenced by diminished ON gating currents. While physical trapping of RY785 within the channel seems the most parsimonious explanation for our results, we cannot exclude more complicated models. Kv2.1 channels adopt inactivated and other nonconducting states (O’Connell et al., 2010; Fox et al., 2013; VanDongen et al., 1990). While it is possible that RY785 induces one of these states, this would offer no obvious explanation for RY785’s voltage dependence of inhibition or its effects on gating currents.
We interpreted RY785’s effects on gating current kinetics under the simplifying assumption that residual gating currents correspond to conformation changes that also occur in the unbound channel. However, multiple other possibilities exist if RY785 binding generates new voltage sensor conformations. Furthermore, differences in the solutions for K+ and gating current recordings could alter interactions with RY785. For example, intracellular NMDG+ is an open-channel blocker of Shaker K+ channels (Melishchuk and Armstrong, 2001), and if NMDG+ interacts similarly with Kv2.1, then acceleration of OFF gating current following RY785 application could be due to RY785 displacing NMDG+ from the channel cavity.
Implications regarding channel gating
Taken together, competition with TEA and the requirement of voltage sensor activation for block imply that the channel creates a pathway for RY785 to enter the central cavity of activated-not-open channels. What might this activated-not-open conformation be? When Kv2.1 activates in response to positive voltage steps, pore opening is far slower than voltage-sensor activation, and the G–V is shifted to voltages more positive than the fourth power of the Q–V (Tilley et al., 2019; Islas and Sigworth, 1999; Scholle et al., 2004). This behavior resembles that of the ILT mutant of the Shaker voltage-gated K+ channel (Smith-Maxwell et al., 1998; Ledwell and Aldrich, 1999). However, the central cavity of the activated-not-open Shaker ILT channel is not open to the cytosol (del Camino et al., 2005). If the S6 gate in activated-not-open Kv2.1 is also closed, and RY785 indeed binds within the central cavity, then RY785 would need to gain access through a voltage-dependent gate which is distinct from the intracellular S6 movements that gate access to K+ and TEA. Such a gate exists in voltage-gated Na+ channels that have membrane-buried fenestrations in their pores through which lipophilic drug molecules may enter (Hille, 1977; Nguyen et al., 2019). However, no fenestrations are apparent in the open state of Kv1.2, the closest relative of Kv2.1 with a high-resolution structure (Long et al., 2005). As such, RY785 access to the central cavity with a closed S6 intracellular gate seems unlikely.
It seems more plausible that Kv2.1 adopts an activated-not-open conformation with the central cavity open to the cytosol, providing RY785 and intracellular TEA a route of access and subsequent egress. It is not clear whether widening of the intracellular S6 gate is the final opening step in the Kv2.1 activation path. BK and CNG channels, which are structurally related to Kv channels, gate access of blocking molecules to the intracellular cavity, yet gate ionic permeation with the selectivity filter (Wilkens and Aldrich, 2006; Contreras et al., 2008; Contreras and Holmgren, 2006; Tang et al., 2009; Thompson and Begenisich, 2012; Yan et al., 2016). If selectivity filter opening is the final step in the Kv2.1 activation pathway, then intracellular S6 movements could gate access of RY785. This mechanism would also explain voltage-gated access of 4-aminopyridine to closed states of Kv2.1 (Kirsch and Drewe, 1993). Selectivity filter gating in Kv2.1 channels has been studied in the context of inactivation and operates differently than in Kv1 channels (Klemic et al., 1998; Cheng et al., 2011; Coonen et al., 2020; Immke et al., 1999; Andalib et al., 2004). We note that a selectivity filter mutation that abolishes selectivity filter gating also abolishes the slow step in Kv2.1 activation gating and shifts the G–V to more negative voltages (Coonen et al., 2020). These behaviors are consistent with gating at the selectivity filter underlying the separation of gating-charge movement from conductance in both time and voltage. Considering these features of Kv2 gating, we propose an explanation for gated access of RY785: upon voltage sensor activation, Kv2.1 adopts an activated-not-open conformation in which the intracellular S6 gate has opened to the cytosol, yet the selectivity filter is in a nonconducting state (Scheme 1). The gating current modifications could potentially be accounted for by RY785 accelerating deactivation from the activated-not-open conformation (red arrows) or other modifications to gating kinetics. As deactivation traps RY785 and RY785 accelerates voltage sensor deactivation, the slow unblock kinetics may result from “self-trapping” upon RY785 binding.
Scheme 1.

Proposed mechanism of inhibition. State diagram depicting a Kv2.1 channel gating in the presence of RY785 (red rectangle). RY785 has access only to voltage-activated conformations of the channel. RY785 promotes deactivation and is trapped by deactivation of voltage sensors to a resting state.
Pharmacological implications
We find that 1 µM RY785 almost completely inhibits the K+ conductance mediated by rat Kv2.1 channels heterologously expressed in CHO-K1 cells. Extrapolating from the block and recovery kinetics with 1 µM RY785 and postpulses to +40 mV, we estimate a Kd of 6.2 ± 1.2 nM (SEM) for the rat Kv2.1 channels expressed in CHO-K1 cells. This appears more potent than the reported IC50 of 50 nM for RY785 against human Kv2.1 expressed in CHO-K1 cells (Herrington et al., 2011). However, we do not know whether there is a difference in affinities between human and rat Kv2.1 or whether the difference is due to distinct measurement methods or other differences in experiments, e.g., temperature, voltage clamp configuration, or voltage protocols. We did not measure an IC50 from concentration-effect experiments and note that slow kinetics of block at nanomolar concentrations, slow kinetics of unblock, time-dependent changes in Kv2.1 gating after whole-cell break, voltage dependence of RY785 access, and allosteric effects all pose challenges to accurate measurement of a meaningful IC50.
The process of RY785 block appears to simply decrease K+ conductance density, and a simple decrease in Kv2 conductance density is the expected effect of RY785 in other experimental conditions. As RY785 appears to alter gating charge equilibria, the principle of coupled equilibria requires that RY785 affinity for Kv2.1 be affected by gating. As such, the extensive regulation of Kv2 channel gating in vivo could alter the affinity and dynamics of RY785 inhibition. Because RY785 stabilizes a deactivated conformation, more positive membrane potentials could weaken RY785 affinity. However, any changes in membrane potential within the physiological voltage range would be expected to only slowly impact RY785 blockade. The estimated dissociation rates at −80 mV and +40 mV, of 0.0001 s−1 and 0.01 s−1, respectively, suggest that any unblock induced by more positive voltages would equilibrate far too slowly to impact K+ conductance kinetics on the millisecond timescale of a typical action potential.
As TEA competes with RY785, it is possible that residues of the intracellular cavity, in which TEA binds, also form the RY785 binding site. If so, sequence similarities suggest that RY785 will be effective at blocking the Kv2 channels of many animals. Residues lining the intracellular cavity are identical between Kv2.1 and Kv2.2 and among several deuterostome species with divergence occurring among protostomes (Fig. S3 A). Consistent with this sequence conservation, human Kv2.1 and Kv2.2 have similar affinities for the parent compound from which RY785 was derived (Herrington et al., 2011). Kv1.2 is inhibited by this parent compound with 61-times lower affinity and no apparent use dependence (Herrington et al., 2011). The apparent lack of use dependence could result from a separate mechanism of inhibition, or use dependence could be obscured by rapid equilibration of RY785 with its Kv1.2 binding site. Kv2 subunits can heteromultimerize with Kv5, Kv6, Kv8, and Kv9 subunits, which are collectively known as KvS or silent subunits. The pore regions of KvS subunits are poorly conserved with Kv2 (Fig. S3 B). Some, but not all, Kv2/KvS heteromers have altered TEA pharmacology (Moreno-Domínguez et al., 2009; Zhu et al., 1999), leaving open the question of how Kv2/KvS heteromers will be affected by RY785.
While Kv2 channel heterogeneity may complicate how RY785 impacts endogenous Kv2 currents, we expect the fundamentals of the RY785 gated-access trapping mechanism to apply generally. We suggest that understanding the trapping mechanism presented here could aid interpretation of experiments using RY785 to block endogenous Kv2 channels: channel activation is required for block by RY785 to equilibrate, after which, trapped RY785 will simply decrease the Kv2 conductance density.
Acknowledgments
Christopher J. Lingle served as editor.
We thank Rebecka Sepela, Tsung-Yu Chen, Jie Zheng, Bruce Bean, and Sooyeon Jo for critical feedback.
This research was supported by US National Institutes of Health grants R01NS096317 and R01HL128537.
The authors declare no competing financial interests.
Author contributions: M.J. Marquis: Conceptualization, Formal analysis, Investigation, Methodology, Visualization, Writing - original draft, Writing - reviewing & editing. J.T. Sack: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Visualization, Writing - original draft, Writing - reviewing & editing.
References
- Amberg, G.C., and Santana L.F.. 2006. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am. J. Physiol. Cell Physiol. 291:C348–C356. 10.1152/ajpcell.00086.2006 [DOI] [PubMed] [Google Scholar]
- Andalib, P., Consiglio J.F., Trapani J.G., and Korn S.J.. 2004. The external TEA binding site and C-type inactivation in voltage-gated potassium channels. Biophys. J. 87:3148–3161. 10.1529/biophysj.104.046664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. 10.1085/jgp.58.4.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and Hille B.. 1972. The inner quaternary ammonium ion receptor in potassium channels of the node of ranvier. J. Gen. Physiol. 59:388–400. 10.1085/jgp.59.4.388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and Loboda A.. 2001. A model for 4-aminopyridine action on K channels: Similarities to tetraethylammonium ion action. Biophys. J. 81:895–904. 10.1016/S0006-3495(01)75749-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar, C., Barcia G., Jennesson M., Le Guyader G., Schneider A., Mignot C., Lesca G., Breuillard D., Montomoli M., Keren B., et al. 2020. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies: 27 new patients and overview of the literature. Hum. Mutat. 41:69–80. 10.1002/humu.23915 [DOI] [PubMed] [Google Scholar]
- Bocksteins, E. 2016. Kv5, Kv6, Kv8, and Kv9 subunits: No simple silent bystanders. J. Gen. Physiol. 147:105–125. 10.1085/jgp.201511507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocksteins, E., and Snyders D.J.. 2012. Electrically silent Kv subunits: Their molecular and functional characteristics. Physiology 27:73–84. 10.1152/physiol.00023.2011 [DOI] [PubMed] [Google Scholar]
- Cerda, O., and Trimmer J.S.. 2011. Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J. Biol. Chem. 286:28738–28748. 10.1074/jbc.M111.251942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M.L., VanDongen H.M., and VanDongen A.M.. 1997. Activation-dependent subconductance levels in the drk1 K channel suggest a subunit basis for ion permeation and gating. Biophys. J. 72:708–719. 10.1016/s0006-3495(97)78707-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y.M., Azer J., Niven C.M., Mafi P., Allard C.R., Qi J., Thouta S., and Claydon T.W.. 2011. Molecular determinants of U-type inactivation in Kv2.1 channels. Biophys. J. 101:651–661. 10.1016/j.bpj.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.L., Mossman C., Aubé J., and Yellen G.. 1993. The internal quaternary ammonium receptor site of shaker potassium channels. Neuron. 10:533–541. 10.1016/0896-6273(93)90340-W [DOI] [PubMed] [Google Scholar]
- Contreras, J.E., and Holmgren M.. 2006. Access of quaternary ammonium blockers to the internal pore of cyclic nucleotide-gated channels: Implications for the location of the gate. J. Gen. Physiol. 127:481–494. 10.1085/jgp.200509440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras, J.E., Srikumar D., and Holmgren M.. 2008. Gating at the selectivity filter in cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 105:3310 LP–3314. 10.1073/pnas.0709809105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonen, L., Mayeur E., De Neuter N., Snyders D.J., Cuello L.G., and Labro A.J.. 2020. The selectivity filter is involved in the U-type inactivation process of Kv2.1 and Kv3.1 channels. Biophys. J. 118:2612–2620. 10.1016/j.bpj.2020.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X.Q., Kolic J., Marchi P., Sipione S., and MacDonald P.E.. 2009. SUMOylation regulates Kv2.1 and modulates pancreatic β-cell excitability. J. Cell Sci. 122:775–779. 10.1242/jcs.036632 [DOI] [PubMed] [Google Scholar]
- del Camino, D., and Yellen G.. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 32:649–656. 10.1016/S0896-6273(01)00487-1 [DOI] [PubMed] [Google Scholar]
- del Camino, D., Kanevsky M., and Yellen G.. 2005. Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J. Gen. Physiol. 126:419–428. 10.1085/jgp.200509385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Ramírez, M., De Jesús-Pérez J.J., Aréchiga-Figueroa I.A., Arreola J., Adney S.K., Villalba-Galea C.A., Logothetis D.E., and Rodríguez-Menchaca A.A.. 2018. Regulation of Kv2.1 channel inactivation by phosphatidylinositol 4,5-bisphosphate. Sci. Rep. 8:1769. 10.1038/s41598-018-20280-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilly, S., Lamy C., V Marrion N., Liégeois J.F., Liégeois J., and Seutin V.. 2011. Ion-channel modulators: More diversity than previously thought. ChemBioChem. 12:1808–1812. 10.1002/cbic.201100236 [DOI] [PubMed] [Google Scholar]
- Du, J., Haak L.L., Phillips-Tansey E., Russell J.T., and McBain C.J.. 2000. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J. Physiol. 522 Pt 1:19–31. 10.1111/j.1469-7793.2000.t01-2-00019.xm [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas, P., Diochot S., Célérier M.L., Nakajima T., and Lazdunski M.. 2002. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol. Pharmacol. 62:48–57. 10.1124/mol.62.1.48 [DOI] [PubMed] [Google Scholar]
- Ficker, E., Jarolimek W., Kiehn J., Baumann A., and Brown A.M.. 1998. Molecular determinants of dofetilide block of HERG K+ channels. Circ. Res. 82:386–395. 10.1161/01.RES.82.3.386 [DOI] [PubMed] [Google Scholar]
- Fortenbach, C., Allina G.P., Shores C.M., Karlen S.J., Miller E.B., Bishop H., Trimmer J.S., Burns M.E., and Pugh E.N.. 2021. Loss of the K+ channel Kv2.1 greatly reduces outward dark current and causes ionic dysregulation and degeneration in rod Photoreceptors. J. Gen. Physiol. 153:e202012687. 10.1085/JGP.202012687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, P.D., Loftus R.J., and Tamkun M.M.. 2013. Regulation of Kv2.1 K+ conductance by cell surface channel density. J. Neurosci. 33:1259 LP–1270. 10.1523/JNEUROSCI.3008-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech, G.C., VanDongen A.M., Schuster G., Brown A.M., and Joho R.H.. 1989. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 340:642–645. 10.1038/340642a0 [DOI] [PubMed] [Google Scholar]
- Gamper, N., Stockand J.D., and Shapiro M.S.. 2005. The use of Chinese hamster ovary (CHO) cells in the study of ion channels. J. Pharmacol. Toxicol. Methods. 51:177–185. 10.1016/j.vascn.2004.08.008 [DOI] [PubMed] [Google Scholar]
- Guan, D., Tkatch T., Surmeier D.J., Armstrong W.E., and Foehring R.C.. 2007. Kv2 subunits underlie slowly inactivating potassium current in rat neocortical pyramidal neurons. J. Physiol. 581:941–960. 10.1113/jphysiol.2007.128454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, N.A., Misra S.N., Jurado M., Kang S.K., Vierra N.C., Nguyen K., Wren L., George A.L., Trimmer J.S., and Kearney J.A.. 2021. Epilepsy and neurobehavioral abnormalities in mice with a dominant-negative KCNB1 pathogenic variant. Neurobiol. Dis. 147:105141. 10.1016/j.nbd.2020.105141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington, J., Zhou Y.P., Bugianesi R.M., Dulski P.M., Feng Y., Warren V.A., Smith M.M., Kohler M.G., Garsky V.M., Sanchez M., et al. 2006. Blockers of the delayed-rectifier potassium current in pancreatic β-cells enhance glucose-dependent insulin secretion. Diabetes. 55:1034 LP–1042. 10.2337/diabetes.55.04.06.db05-0788 [DOI] [PubMed] [Google Scholar]
- Herrington, J., Solly K., Ratliff K.S., Li N., Zhou Y.P., Howard A., Kiss L., Garcia M.L., McManus O.B., Deng Q., et al. 2011. Identification of novel and selective Kv2 channel inhibitors. Mol. Pharmacol. 80:959–964. 10.1124/mol.111.074831 [DOI] [PubMed] [Google Scholar]
- Hille, B. 1977. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. 10.1085/jgp.69.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., Smith P.L., and Yellen G.. 1997. Trapping of organic blockers by closing of voltage-dependent K+ channels: Evidence for a trap door mechanism of activation gating. J. Gen. Physiol. 109:527–535. 10.1085/jgp.109.5.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem, L.M., and Katzung B.G.. 1977. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 472:373–398. 10.1016/0304-4157(77)90003-x [DOI] [PubMed] [Google Scholar]
- Hönigsperger, C., Nigro M.J., and Storm J.F.. 2017. Physiological roles of Kv2 channels in entorhinal cortex layer II stellate cells revealed by guangxitoxin-1E. J. Physiol. 595:739–757. 10.1113/JP273024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke, D., Wood M., Kiss L., and Korn S.J.. 1999. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J. Gen. Physiol. 113:819–836. 10.1085/jgp.113.6.819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas, L.D., and Sigworth F.J.. 1999. Voltage sensitivity and gating charge in shaker and shab family potassium channels. J. Gen. Physiol. 114:723–742. 10.1085/jgp.114.5.723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, D.A., Kuznetsov A., Lopez J.P., Kash S., Ämmälä C.E., and Philipson L.H.. 2007. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 6:229–235. 10.1016/j.cmet.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihira, Y., Hermanstyne T.O., and Misonou H.. 2010. Formation of heteromeric Kv2 channels in mammalian brain neurons. J. Biol. Chem. 285:15048–15055. 10.1074/JBC.M109.074260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm, T., Khaliq Z.M., and Bean B.P.. 2015. Differential regulation of action potential shape and burst-frequency firing by BK and Kv2 channels in substantia nigra dopaminergic neurons. J. Neurosci. 35:16404–16417. 10.1523/JNEUROSCI.5291-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, G.E., and Drewe J.A.. 1993. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J. Gen. Physiol. 102:797–816. 10.1085/jgp.102.5.797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic, K.G., Shieh C.C., Kirsch G.E., and Jones S.W.. 1998. Inactivation of Kv2.1 potassium channels. Biophys. J. 74:1779–1789. 10.1016/S0006-3495(98)77888-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledwell, J.L., and Aldrich R.W.. 1999. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113:389–414. 10.1085/jgp.113.3.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus, M.J., Burdette D., Wagner T., Focia P.J., and Gross A.. 2014. Structures of KcsA in complex with symmetrical quaternary ammonium compounds reveal a hydrophobic binding site. Biochemistry. 53:5365–5373. 10.1021/bi500525s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X.N., Herrington J., Petrov A., Ge L., Eiermann G., Xiong Y., Jensen M.V., Hohmeier H.E., Newgard C.B., Garcia M.L., et al. 2013. The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J. Pharmacol. Exp. Therapeut. 344:407 LP–416. 10.1124/jpet.112.199083 [DOI] [PubMed] [Google Scholar]
- Li, X., Liu H., Luo J.C., Rhodes S.A., Trigg L.M., Van Rossum D.B., Anishkin A., Diatta F.H., Sassic J.K., Simmons D.K., et al. 2015. Major diversification of voltage-gated K+ channels occurred in ancestral parahoxozoans. Proc. Natl. Acad. Sci. USA. 112:E1010–E1019. 10.1073/pnas.1422941112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P.W., and Bean B.P.. 2014. Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci. 34:4991–5002. 10.1523/JNEUROSCI.1925-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loboda, A., and Armstrong C.M.. 2001. Resolving the gating charge movement associated with late transitions in K channel activation. Biophys. J. 81:905–916. 10.1016/S0006-3495(01)75750-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, S.B., Campbell E.B., and MacKinnon R.. 2005. Crystal structure of a mammalian voltage-dependent shaker family K+ channel. Science. 309:897–903. 10.1126/science.1116269 [DOI] [PubMed] [Google Scholar]
- Malin, S.A., and Nerbonne J.M.. 2002. Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J. Neurosci. 22:10094–10105. 10.1523/JNEUROSCI.22-23-10094.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandikian, D., Bocksteins E., Parajuli L.K., Bishop H.I., Cerda O., Shigemoto R., and Trimmer J.S.. 2014. Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol. 522:3555–3574. 10.1002/cne.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord, M.C., and Aizenman E.. 2013. Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2.1 K+ currents. Proc. Natl. Acad. Sci. USA. 110:13988–13993. 10.1073/pnas.1306238110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melishchuk, A., and Armstrong C.M.. 2001. Mechanism underlying slow kinetics of the OFF gating current in Shaker potassium channel. Biophys. J. 80:2167–2175. 10.1016/S0006-3495(01)76189-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez, F., and Wurriehausen F.. 2009. Patcher’s Power Tools. Max-Planck-Institut für biophysikalische Chemie, Göttingen, Germany. http://www3.mpibpc.mpg.de/groups/neher/index.php?page=aboutppt [Google Scholar]
- Milescu, M., Bosmans F., Lee S., Alabi A.A., Kim J.I., and Swartz K.J.. 2009. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16:1080–1085. 10.1038/nsmb.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou, H., Mohapatra D.P., Park E.W., Leung V., Zhen D., Misonou K., Anderson A.E., and Trimmer J.S.. 2004. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7:711–718. 10.1038/nn1260 [DOI] [PubMed] [Google Scholar]
- Misonou, H., Mohapatra D.P., and Trimmer J.S.. 2005. Kv2.1: A voltage-gated K+ channel critical to dynamic control of neuronal excitability. NeuroToxicology. 26:743–752. 10.1016/j.neuro.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Moreno-Domínguez, A., Cidad P., Miguel-Velado E., López-López J.R., and Pérez-García M.T.. 2009. De novo expression of Kv6.3 contributes to changes in vascular smooth muscle cell excitability in a hypertensive mice strain. J. Physiol. 587:625–640. 10.1113/jphysiol.2008.165217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi, H., and Trimmer J.S.. 1999. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J. Neurosci. 19:1728–1735. 10.1523/JNEUROSCI.19-05-01728.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi, H., Shi G., Scannevin R.H., and Trimmer J.S.. 1997. Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol. Pharmacol. 52:821 LP–828. 10.1124/mol.52.5.821 [DOI] [PubMed] [Google Scholar]
- Newkirk, G.S., Guan D., Dembrow N., Armstrong W.E., Foehring R.C., and Spain W.J.. 2022. Kv2.1 potassium channels regulate repetitive burst firing in extratelencephalic neocortical pyramidal neurons. Cereb. Cortex. 32:1055–1076. 10.1093/cercor/bhab266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, P.T., DeMarco K.R., Vorobyov I., Clancy C.E., and Yarov-Yarovoy V.. 2019. Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl. Acad. Sci. USA. 116:2945–2954. 10.1073/pnas.1817446116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell, K.M.S., Loftus R., and Tamkun M.M.. 2010. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl. Acad. Sci. USA. 107:12351–12356. 10.1073/pnas.1003028107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dwyer, S.C., Palacio S., Matsumoto C., Guarina L., Klug N.R., Tajada S., Rosati B., McKinnon D., Trimmer J.S., and Santana L.F.. 2020. Kv2.1 channels play opposing roles in regulating membrane potential, Ca2+ channel function, and myogenic tone in arterial smooth muscle. Proc. Natl. Acad. Sci. USA. 117:3858–3866. 10.1073/pnas.1917879117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak, D., Guan D., and Foehring R.C.. 2016. Roles of specific Kv channel types in repolarization of the action potential in genetically identified subclasses of pyramidal neurons in mouse neocortex. J. Neurophysiol. 115:2317–2329. 10.1152/jn.01028.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola, M.A., Kuja-Panula J., Lauri S.E., Taira T., and Rauvala H.. 2011. AMIGO is an auxiliary subunit of the Kv2.1 potassium channel. EMBO Rep. 12:1293–1299. 10.1038/embor.2011.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola, M.A., Kuja-Panula J., Liuhanen J., Võikar V., Piepponen P., Hiekkalinna T., Taira T., Lauri S.E., Suvisaari J., Kulesskaya N., et al. 2016. AMIGO-Kv2.1 potassium channel complex is associated with schizophrenia-related phenotypes. Schizophrenia Bull. 42:191–201. 10.1093/schbul/sbv105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant, L.D., Dowdell E.J., Dementieva I.S., Marks J.D., and Goldstein S.A.N.. 2011. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol. 137:441–454. 10.1085/jgp.201110604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu, Y., Xu Y., and Lu Z.. 2006. Enzymatic activation of voltage-gated potassium channels. Nature. 442:696–699. 10.1038/nature04880 [DOI] [PubMed] [Google Scholar]
- Romer, S.H., Deardorff A.S., and Fyffe R.E.W.. 2019. A molecular rheostat: Kv2.1 currents maintain or suppress repetitive firing in motoneurons. J. Physiol. 597:3769–3786. 10.1113/JP277833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, J.T., and Eum K.S.. 2015. Ion channel inhibitors. In Handbook of Ion Channels. Zheng J., and Trudeau M.C., editors. 1st ed. Florida: Taylor & Francis Group, Boca Raton. 10.1201/b18027 [DOI] [Google Scholar]
- Scholle, A., Dugarmaa S., Zimmer T., Leonhardt M., Koopmann R., Engeland B., Pongs O., and Benndorf K.. 2004. Rate-limiting reactions determining different activation kinetics of Kv1.2 and Kv2.1 channels. J. Membr. Biol. 198:103–112. 10.1007/s00232-004-0664-0 [DOI] [PubMed] [Google Scholar]
- Smith-Maxwell, C.J., Ledwell J.L., and Aldrich R.W.. 1998. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 111:421–439. 10.1085/jgp.111.3.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speca, D.J., Ogata G., Mandikian D., Bishop H.I., Wiler S.W., Eum K., Wenzel H.J., Doisy E.T., Matt L., Campi K.L., et al. 2014. Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Gene Brain Behav. 13:394–408. 10.1111/gbb.12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela, M., Vandongen A.M., Drewe J.A., Joho R.H., Brown A.M., and Kirsch G.E.. 1991. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol. Pharmacol. 40:299–307 [PubMed] [Google Scholar]
- Tang, Q.Y., Zeng X.H., and Lingle C.J.. 2009. Closed-channel block of BK potassium channels by BbTBA requires partial activation. J. Gen. Physiol. 134:409–436. 10.1085/jgp.200910251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiffault, I., Speca D.J., Austin D.C., Cobb M.M., Eum K.S., Safina N.P., Grote L., Farrow E.G., Miller N., Soden S., et al. 2015. A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J. Gen. Physiol. 146:399–410. 10.1085/jgp.201511444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J., and Begenisich T.. 2012. Selectivity filter gating in large-conductance Ca2+-activated K+ channels. J. Gen. Physiol. 139:235–244. 10.1085/jgp.201110748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley, D.C., Eum K.S., Fletcher-Taylor S., C Austin D., Dupré C., Patrón L.A., Garcia R.L., Lam K., Yarov-Yarovoy V., Cohen B.E., and Sack J.T.. 2014. Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc. Natl. Acad. Sci. USA. 111:E478–E4796. 10.1073/pnas.1406876111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley, D.C., Angueyra J.M., Eum K.S., Kim H., Chao L.H., Peng A.W., and Sack J.T.. 2019. The tarantula toxin GxTx detains K+ channel gating charges in their resting conformation. J. Gen. Physiol. 151:292 LP–315. 10.1085/jgp.201812213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani, A., Bersell K., Jorge B.S., Bjork R.L. Jr., Friedman J.R., Bloss C.S., Cohen J., Gupta S., Naidu S., Vanoye C.G., et al. 2014. De novo KCNB1 mutations in epileptic encephalopathy. Ann. Neurol. 76:529–540. 10.1002/ana.24263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani, J.G., and Korn S.J.. 2003. Control of ion channel expression for patch clamp recordings using an inducible expression system in mammalian cell lines. BMC Neurosci. 4:15. 10.1186/1471-2202-4-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer, J.S. 1991. Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl. Acad. Sci. USA. 88:10764–10768. 10.1073/pnas.88.23.10764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher, H., Mohapatra D.P., and Trimmer J.S.. 2008. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88:1407–1447. 10.1152/physrev.00002.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDongen, A.M.J., Frech G.C., Drewe J.A., Joho R.H., and Brown A.M.. 1990. Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron. 5:433–443. 10.1016/0896-6273(90)90082-Q [DOI] [PubMed] [Google Scholar]
- Vedantham, V., and Cannon S.C.. 1999. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J. Gen. Physiol. 113:7–16. 10.1085/jgp.113.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Guo J., Perissinotti L.L., Lees-Miller J., Teng G., Durdagi S., Duff H.J., and Noskov S.Y.. 2016. Role of the PH in state-dependent blockade of HERG currents. Sci. Rep. 6:32536. 10.1038/srep32536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, A.M., Procter J.B., Martin D.M.A., Clamp M., and Barton G.J.. 2009. Jalview version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics. 25:1189–1191. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkens, C.M., and Aldrich R.W.. 2006. State-independent block of BK channels by an intracellular quaternary ammonium. J. Gen. Physiol. 128:347–364. 10.1085/jgp.200609579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W., Gardner A., and Sanguinetti M.C.. 2015. The link between inactivation and high-affinity block of HERG1 channels. Mol. Pharmacol. 87:1042–1050. 10.1124/mol.115.098111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, J., Li Q., and Aldrich R.W.. 2016. Closed state-coupled C-type inactivation in BK channels. Proc. Natl. Acad. Sci. USA. 113:6991–6996. 10.1073/pnas.1607584113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X.Z., Abd-Elrahman K.S., Liao C.H., El-Yazbi A.F., Walsh E.J., Walsh M.P., and Cole W.C.. 2010. Stromatoxin-sensitive, heteromultimeric Kv2.1/Kv9.3 channels contribute to myogenic control of cerebral arterial diameter. J. Physiol. 588:4519–4537. 10.1113/jphysiol.2010.196618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M., Morais-Cabral J.H., Mann S., and MacKinnon R.. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. 10.1038/35079500 [DOI] [PubMed] [Google Scholar]
- Zhu, X.R., Netzer R., Böhlke K., Liu Q., and Pongs O.. 1999. Structural and functional characterization of Kv6.2, a new γ-subunit of voltage-gated potassium channel. Recept. Channel. 6:337–350 [PubMed] [Google Scholar]