

Abstract
Protein phosphorylation is a central mechanism of cellular signal transduction in living organisms. Phosphoproteomic studies systematically catalogue and characterize alterations in phosphorylation states across multiple cellular conditions and are often incorporated into global proteomics experiments. Previously, we found that spin column-based Fe3+-NTA enrichment integrated well with our workflow but remained a bottleneck for methods that require higher throughput or a scale that is beyond the capacity of these columns. Here, we compare our well-established spin column-based enrichment strategy with one encompassing magnetic beads. Our data show little difference when using either method in terms of the number of identified phosphopeptides as well as their physicochemical properties. In all, we illustrate how the potentially scalable and automation-friendly magnetic Fe3+-NTA beads can seamlessly substitute spin column-based Fe3+-NTA agarose beads for global phosphoproteome profiling.
Keywords: automation, phosphoproteome, IMAC, phosphopeptide enrichment
Graphical Abstract
INTRODUCTION
Phosphorylation analysis is key to understanding protein signaling. Phosphoproteomics methodologies have enabled the global and unbiased characterization of protein phosphorylation states helping to broaden our understanding of associated signaling mechanisms [1, 2]. However, without enrichment, the abundance of phosphorylated peptides is orders of magnitude below that of unphosphorylated peptides. Enhancements in sample preparation, including efficient and potentially automated enrichment, may overcome the hurdles of low abundance associated with phosphopeptide identification and facilitate high throughput phosphoproteome profiling [3, 4]. Therefore, a need exists to establish improved methods aimed for the enrichment and subsequent analysis of phosphopeptides.
Phosphoproteomic studies are often hampered by the overall low stoichiometry of phosphorylated peptides, as phosphoproteins are typically at an exceptionally low abundance in any given cell. As such, phosphoproteomic analyses often require greater than 100 times more starting material than whole-proteome profiling as phosphopeptides comprise <2–3% of the peptides in a typical tryptic digest [5, 6]. This major obstacle has led to numerous innovations for the specific enrichment of phosphopeptides. Immobilized metal affinity chromatography (IMAC) [7] and metal oxide affinity chromatography (MOAC) [8, 9] methods have become the predominant means for enriching phosphorylated peptides. We have compared previously the MOAC affinity matrix, titanium dioxide (TiO2), with Fe3+-NTA-based spin columns and we have shown that these spin columns offer a compromise among recovery, enrichment, throughput, and efficiency for global phosphoproteomic analysis [10]. Moreover, these spin columns (which may process 50 μg to 5 mg of protein) have since been integrated into the StreamLined TMT (SL-TMT)-based “mini-phos” protocol [11].
The “mini-phos” step can be implemented following isobaric tag peptide labeling and pooling in a sample multiplexed quantitative proteomics experiment. As such, a single enrichment is performed on a pooled sample (consisting of up to 18 individual samples [12, 13]) which has been normalized at the protein level. Such “mini-phos” analyses typically yields 5,000 to over 10,000 phosphorylation sites, depending on the samples analyzed [11]. This enrichment strategy is simple to implement on labeled, multiplexed samples as it requires only a single enrichment on the pooled sample. Often, however, a deeper, more comprehensive snapshot of the phosphoproteome is sought [14], which would entail a greater starting amount of protein but preclude labeling prior to mixing due to the prohibitive cost and thus an enrichment step is needed for each sample. Moreover, as the level of sample multiplexing increases, so does the number of individual enrichments. For instance, a TMTpro18-plex experiment performed in triplicate would require 54 individual enrichments [12, 13]. As such, high-throughput and potentially automatable plate-based platforms would be preferable over many individual spin columns.
Here, we investigated an alternative phosphopeptide enrichment strategy which is not restricted by spin column capacity and is fully amenable to high-throughput processing without loss in phosphoproteome depth. We compared our standard Fe3+-NTA-based spin column enrichment method against one using NTA-based magnetic beads. We achieved similar depth of phosphopeptide identifications when using High-Select Fe3+-NTA magnetic beads compared to the spin columns. Together, these data show that High-Select Fe3+-NTA magnetic beads may facilitate the high-throughput enrichment of phosphopeptides and can be integrated seamlessly into the SL-TMT workflow.
MATERIAL AND METHODS
Materials.
Trypsin was purchased from Pierce Biotechnology (Rockford, IL) and LysC from Wako Chemicals (Richmond, VA). Water and organic solvents were from J.T. Baker (Center Valley, PA). Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) was from Life Technologies (Waltham, MA), penicillin-streptomycin antibiotics were from Gibco (Waltham, MA). High-Select Fe-NTA Phosphopeptide Enrichment Kit was purchased from Pierce Biotechnology (Rockford, IL). Magnetic Fe3+-NTA beads were a gift from Pierce Biotechnology (Rockford, IL), which is now commercially available (product number A52283 and A52284).
Sample Preparation.
Several human cell lines (SH-SY5Y, HEP-G2, Panc1 and HAP1) were propagated in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin until 70–90% confluent. The cells were washed twice with ice cold PBS, and harvested on-plate with 8 M urea, 200 mM EPPS pH 8.5 plus 1X Pierce protease and phosphatase inhibitor), and syringe lysed (10 times with 21-gauge needle). Lysates were pooled and stored at −80 °C until use.
Following a BCA assay to estimate protein concentration, all lysates were reduced (20 min with 5 mM TCEP at room temperature), alkylated (20 min with 10 mM iodoacetamide, at room temperature in the dark), and quenched (20 min with 10 mM DTT, at room temperature in the dark). Proteins were precipitated by single-pot, solid-phase-enhanced sample preparation (SP3), as described previously [15] [16]. Precipitated proteins were digested in 200 mM EPPS pH 8.5 (~1 mg/ml) with LysC for 3 hr 37°C shaking on a vortexer (speed =50%) followed by a 6 hr trypsin digestion at 37°C. Peptides were displaced from the beads and then desalted using a 1 g SepPak cartridge (Waters) prior to MS analysis. Peptides originating from Xenopus laevis eggs were prepared (and labeled with TMTpro) as described previously [17].
Bead-based phosphopeptide enrichment.
We resuspended the peptide pellets with 80% methanol, 0.1% TFA (trifluoroacetic acid) to 1 mg/mL. We also vortexed the beads until the solution was homogenously black in color from which we withdrew 2 μL beads in slurry per 100 μg of peptides. The samples were centrifuged briefly and set on a magnet to remove storage buffer. We washed the beads twice with 10X bead volume of 80% ACN (acetonitrile), 0.1% TFA. We resuspended the beads in 0.75X original bead volume of 80% ACN, 0.1% TFA. We then added the beads to the peptide solution. Beads were vortexed after solutions were added. We then incubated the samples for 30 min at room temperature with rotation. Following the incubation, the samples were placed on a magnetic stand and the flow-through was discarded. The beads were washed thrice with 10X bead volume of 80% methanol, 0.1% TFA. Next, we washed the beads once with 10X bead volume of water. Finally, we eluted twice with 10X bead volume of 5% ammonium hydroxide for 30–60 seconds each time. We then combined the elution along with 100 μL of 10% formic acid to neutralize the pH. The combined eluate was vacuum centrifuged to near dryness and desalted via StageTip [18] prior to mass spectrometry analysis.
Spin column-based phosphopeptide enrichment.
Phosphopeptides were also enriched using the High-Select Fe-NTA Phosphopeptide Enrichment Kit [10]. The only deviation from the manufacturer’s protocol was that we prepared an “elution collection tube” with 100 μL of 10% formic acid into which the eluates were eluted. The combined eluate was vacuum centrifuged to near dryness and desalted via StageTip [18] prior to mass spectrometry analysis.
Mass Spectrometric Data Acquisition and Data Analysis.
Peptides were resuspended in 3% ACN/5% formic acid at 1 mg/mL and loaded at 1 μg on an in-house pulled C18 column (30–35 cm, 2.6 um Accucore (Thermo Fisher), 100 μm ID), and eluted using a linear gradient from 3% to 30% Buffer B (95% ACN, 0.125% formic acid) which mixes with Buffer A (5% ACN, 0.125% formic acid).
For the human cell lysate experiment, peptides were analyzed by an Orbitrap Exploris 480 mass spectrometer over a 1.5 hr gradient. Data were collected in high-resolution MS2 (hrMS2) mode. The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 350–1200 Th, automatic gain control (AGC) target of 100%, maximum injection time 25 ms). The precursors selected for MS2 analysis were based on TopSpeed = 1 sec. MS2 analysis consisted of higher-energy collisional dissociation (HCD), MS2 AGC was set to standard, NCE (normalized collision energy) was 28%, resolution was 30,000, maximum injection time was 60 ms, and isolation window was 1.2 Th.
For the Xenopus egg lysate experiment, peptides were analyzed by a FAIMS-equipped Orbitrap Eclipse mass spectrometer over a 2 hr gradient. The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 400–1500 Th, automatic gain control (AGC) target of 100%, maximum injection time 50 ms). MS/MS data were collected in the ion trap (low-resolution MS2). The precursors selected for MS2 analysis were based on TopSpeed = 1 sec. MS2 analysis consisted of collision-induced dissociation (CID), MS2 AGC was set to standard, NCE (normalized collision energy) was 35%, scan rate to rapid, maximum injection time was 35 ms, and isolation window was 1.2 Th. MSA (multistage activation) was activated with neutral loss mass of 97.9763. FAIMS CVs were set to −35, −50, and −65V.
Mass spectrometry data analysis.
Spectra were converted to mzXML via MSconvert [19]. Database searching included all entries from the human UniProt database (downloaded April 2021). The database was concatenated with one composed of all protein sequences for that database in the reversed order. Searches were performed using a 50-ppm precursor ion tolerance and a 0.03 Da product ion tolerance. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Comet searches and linear discriminant analysis [20, 21]. Carbamidomethylation of cysteine residues (+57.021 Da) was set as a static modification, while oxidation of methionine residues (+15.995 Da), deamidation (+0.984 Da) at glutamine and asparagine residues, and phosphorylation (+79.966 Da) were set as variable modifications. PSMs (Peptide-spectrum matches) were adjusted to a 1% FDR (false discovery rate) [22, 23]. PSM filtering was performed using a linear discriminant analysis, as described previously [21] and then assembled further to a final protein-level FDR of 1%. Data analysis and visualization was performed in Microsoft Excel or R Bioconductor [24] with the “peptides” package [25].
Data access.
RAW files will be made available upon request and the data have been deposited to the ProteomeXchange Consortium via the PRIDE [26] partner repository with the dataset identifier PXD031327. Username: reviewer_pxd031327@ebi.ac.uk; Password: hgHwo5YJ.
RESULTS and DISCUSSION
Magnetic bead-based enrichment resulted in data comparable to that of spin-column based enrichment for label-free samples.
We assessed the performance of Fe3+-NTA magnetic beads against Fe3+-NTA spin columns (with nonmagnetic IMAC-agarose beads) which we have employed extensively for “mini-phos” enrichments in SL-TMT-based workflows [11]. We first used a mixture of human cell lysates in efforts to maximize the total phosphopeptide population. Samples were lysed in 8 M urea and reduced and alkylated prior to digestion using the SP3 strategy [15, 27]. The samples were then divided into six 250 μg aliquots, with three enriched for phosphorylated peptides using Fe3+-NTA magnetic beads and the other three using the standard approach with Fe3+-NTA spin columns (Figure 1A). LC-MS/MS data were collected on an Exploris 480 mass spectrometer using higher energy collisional dissociated (HCD) and high-resolution MS2 analysis.
Figure 1: Comparison of magnetic bead and spin column workflows for phosphopeptide enrichment (label-free dataset).
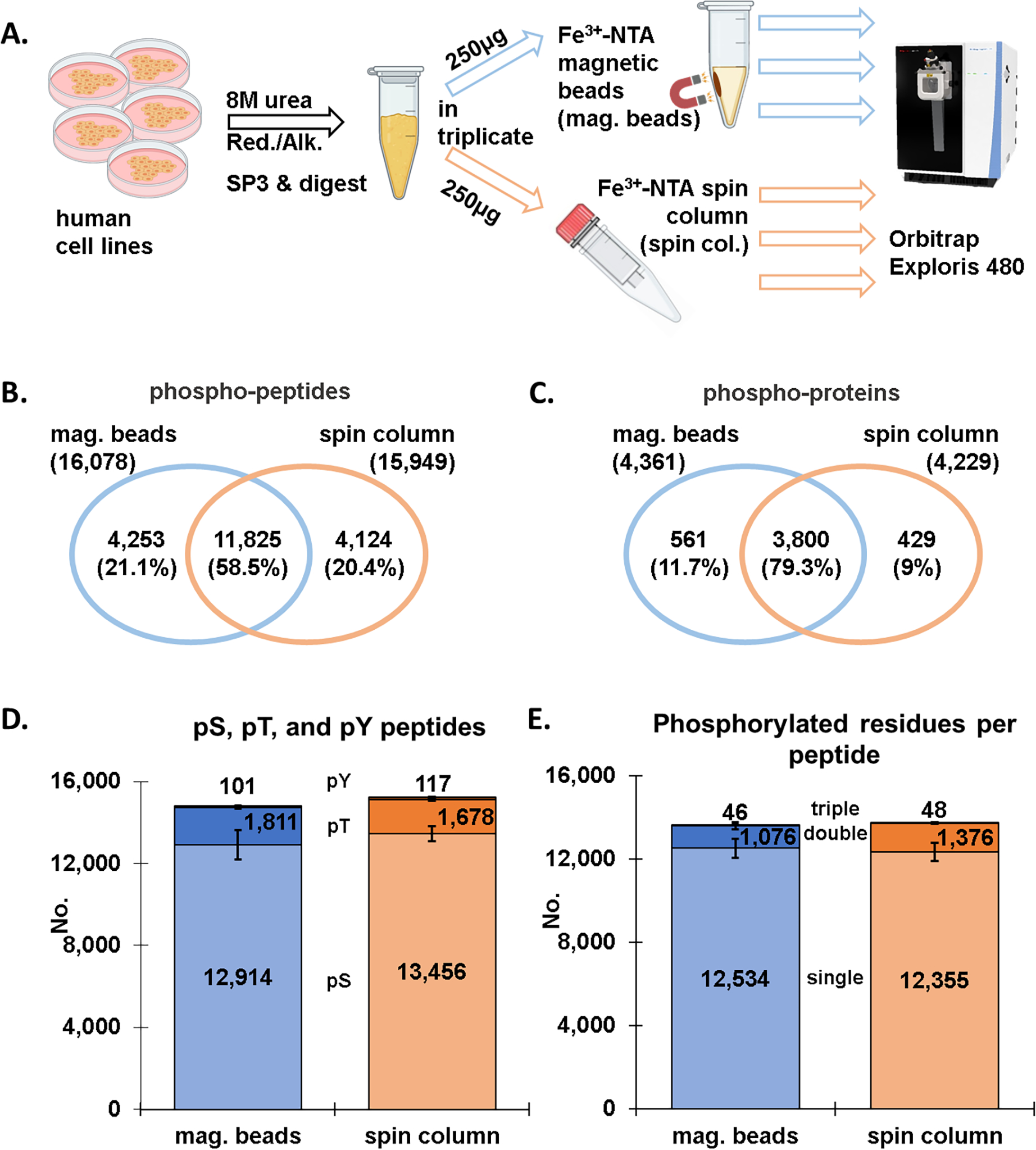
A) Overview of the phosphopeptide enrichment workflow in which aliquots of the identical sample were enriched by either Fe3+-NTA magnetic beads or Fe3+-NTA spin columns. Venn diagrams show the overlap in B) phosphopeptides and C) phosphoproteins between the Fe3+-NTA magnetic bead and the Fe3+-NTA spin column workflows. D) Bar graph illustrating the number of phosphorylated serine, threonine, and tyrosine (pSer, pThr, pTyr) for both enrichment methods. E) Bar graph depicting the number of singly, doubly, and triply phosphorylated peptides for the two enrichment methods. Error bars represent standard deviation.
We identified a comparable number of phosphorylated peptides with either method, specifically, a total of 16,078 peptides were identified with the magnetic bead workflow, while 15,949 with the spin column workflow (Figure 1B). Of these 11,825 phosphorylated peptides overlapped, amounting to 58.5%, which was in line with the overlap between two of the three replicates in either set (Figure S1A and B). This finding was also represented by an UpSet plot in which the replicates were displayed separately (Figure S1C). We note that the category with the most phosphopeptides was one represented by phosphorylated peptides that were identified in all six samples (grey bar). Findings at the phosphoprotein level were similar to those at phosphorylated peptide level. Specifically, a comparable number of phosphoproteins (n=4,361 for the magnetic bead and n=4,229 for the spin column strategy) were mapped back from the phosphorylated peptides, with an overlap of ~80% (Figure 1C). The data once again showed very little difference between the two phosphopeptide enrichment methods.
In addition, we tallied the phosphorylated peptide population according to the type of phosphorylated residue (i.e., serine, threonine, or tyrosine) (Figure 1D). We found a similar proportion of pSer, pThr and pTyr residues were identified with respect to the phosphopeptide enrichment method. Over 80% of the phosphorylated residues were serine and much fewer for the other two residues for both enrichment strategies. We noted redundancy in this tally as peptides may contain more than one type of phosphorylated residue. Similarly, we also tallied singly, doubly, and triply phosphorylated peptides (Figure 1E). We again observed little difference in the number of phosphorylated residues, with the majority of peptides being singly phosphorylated, about 10% doubly phosphorylated, and only a few dozen triply phosphorylated. We also separated the data according to the number of phosphorylated residues per peptide for an alternative visualization of the same result (Figure S1C).
We acknowledge that only a small population of the total phosphoproteome will be sampled when performing single-shot analyses [28]. We attempted to address bias of the different strategies with respect to phosphopeptide properties. As such, we examined the dataset further by isolating the phosphopeptides that were unique to each enrichment method (n=4,253 for magnetic beads and n=4,124 for spin column strategies (Figure 1B)). Specifically, we examined several characteristics and physicochemical properties of these peptides, including, peptide length (Figure 2A), m/z (Figure 2B), cross correlation (XCorr) score (Figure 2C), the grand average hydropathy (GRAVY) index (Figure 2D), the instability index (Figure 2E), and the aliphatic index (Figure 2F). Similar XCorr values reflect comparable data quality between enrichment strategies in terms of how well peptides were matched. We noted trends indicating that the spin column strategy identified peptides that were longer, coincidentally had higher m/z, lower GRAVY index, and lower aliphatic index. Although not statistically significant, these data do reveal that certain peptides may favor one enrichment strategy over another, but globally, little difference was observed between the two phosphopeptide populations that were enriched with different methods.
Figure 2: Peptide characteristics of workflow-specific label-free phosphopeptides.
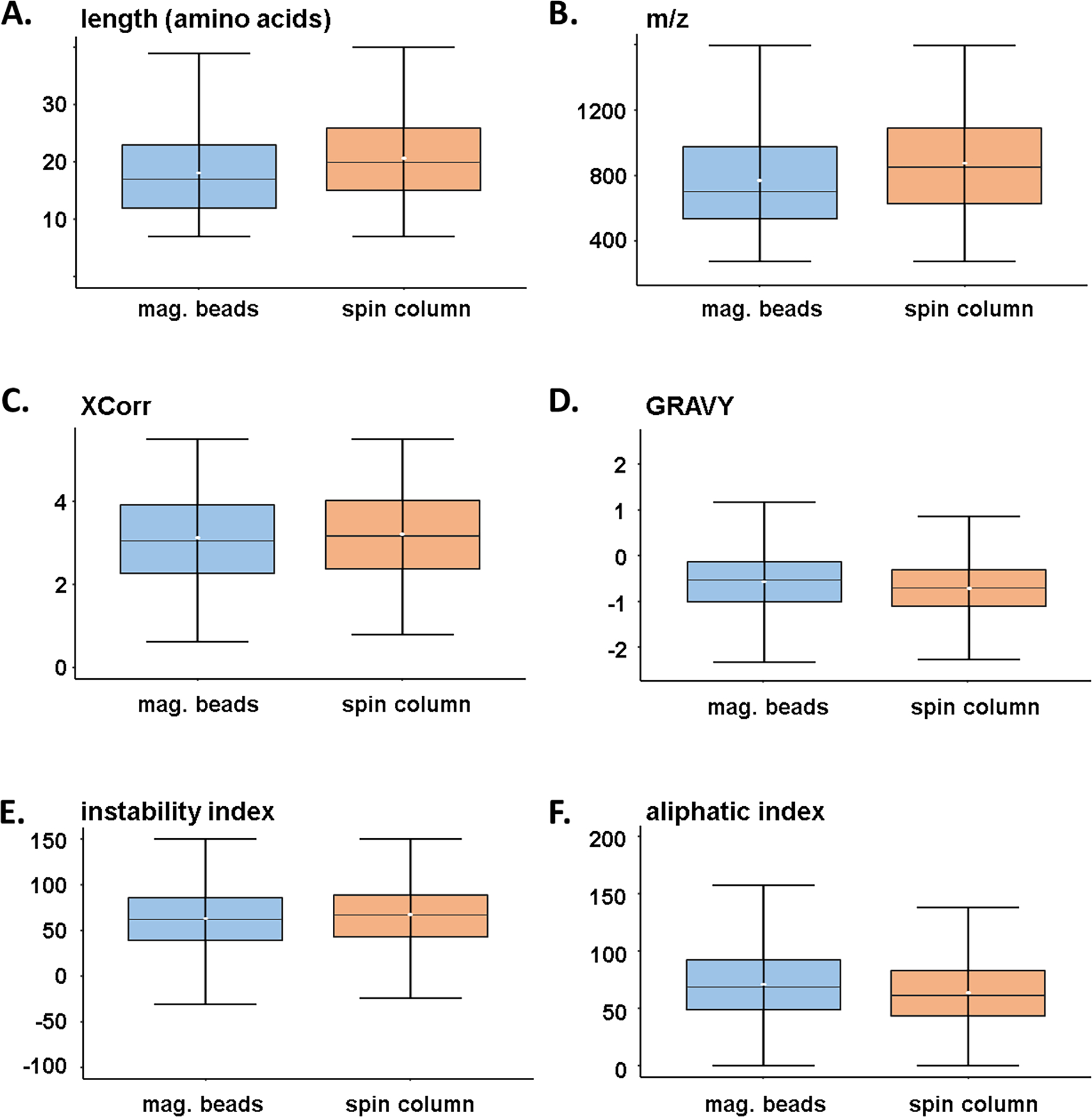
Box-and-whisker plots illustrating the distribution of A) peptide length, B) mass-to-charge ratio, C) XCorr, D) GRAVY (Grand Average of Hydropathy) index, E) instability index, and F) aliphatic index. Only phosphopeptides that were exclusive to a given workflow were compared (n=4,254 for the magnetic bead strategy and n=4,124 for the spin column strategy). The center line specifies the median; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles and the white dot in the box indicates the mean value.
Similar to the label-free data, magnetic bead-based enrichment was comparable to that of the spin column strategy for TMTpro-labeled samples.
We analyzed a second sample type to illustrate the utility of the Fe3+-NTA magnetic bead workflow for isobarically-labeled peptides. In contrast to the label-free phosphopeptide enrichment from human cell lysates highlighted above, we enriched from TMTpro-labeled peptides originating from Xenopus eggs [17]. Evaluating these magnetic beads with TMT-labeled samples was necessary as phosphopeptide enrichment of TMT-labeled peptides was integral to the “mini-phos” step in the SL-TMT protocol [11]. Following this protocol, the Xenopus samples were chloroform-methanol precipitated prior to digestion, labeled with TMTpro, and desalted prior to phosphopeptide enrichment. We collected LC-MS/MS data on an Orbitrap Eclipse mass spectrometer using collision-induced dissociation (CID) and low-resolution MS2 analysis (Figure S2A). For quantification, TMT-labeled samples typically require HCD energy and high-resolution MS2 or MS3 analysis. Our goal here, however, was not to evaluate quantification of the Xenopus proteome across multiple conditions, and instead we focused on protein identification. SPS-MS3 analysis typically uses lrMS2 data acquisition. Here, we did not collect MS3 scans as we limited our focus on phosphopeptide identification rather than quantification. The data acquisition strategy used here leveraged the relatively higher speed of ion trap low-resolution MS2 (lrMS2) analysis to maximize our phosphopeptide coverage. Although we presumed that quantification of isobaric-tagged peptides will not be altered by a different enrichment method, further investigation will be needed to clearly demonstrate this assumption.
A comparable number of phosphorylated peptides were identified (i.e.,11,253 with the magnetic bead, and 11,170 with the spin column strategies) with an overlap of ~57% (Figure S2B). This overlap was similar to that of the cell lysate-based analysis (Figure 1B). We again showed that the inferred proteins reflected the phosphopeptide data as 3,316 proteins were identified using the magnetic bead protocol, while a comparable number (n=3,280) were identified when using the spin column protocol with an overlap of ~75% (Figure S2C). As done previously, we tallied the phosphorylated serine, threonine, or tyrosine residues and showed that a similar proportion of these residues were identified by each enrichment strategy (Figure S2D). In addition, we again tallied the number of phosphorylated residues per peptide and showed a similar proportion among singly, doubly, and triply phosphorylated peptides between the two enrichment strategies (Figure S2E).
We again examined several characteristics and physicochemical properties for peptides identified more readily by one method than the other by interrogating peptide length (Figure S3A), m/z (Figure S3B), cross correlation (XCorr) (Figure S3C), the grand average hydropathy (GRAVY) index (Figure S3D), the instability index (Figure S3E), and the aliphatic index (Figure S3F). Consistent with the label-free data, we observed trends indicating that the spin column strategy identified peptides that were slightly longer, had higher m/z, lower GRAVY index, and lower aliphatic index, although none were statistically significant. These data supported, as noted with human cell lysate-based data, that certain peptide may favor one enrichment strategy over another, but globally, little difference was observed between the two peptide populations. Overall similar conclusions could be drawn from these two experiments which differed in sample type and labeling status. These data support the idea that the magnetic Fe3+-NTA beads can seamlessly substitute spin column-based Fe3+-NTA for global phosphopeptide enrichment analysis.
Conclusion.
Here we have evaluated magnetic Fe3+-NTA beads as a substitute for Fe3+-NTA spin columns. We showed no loss in identified proteins or obvious bias towards certain classes of peptides (e.g., multiply phosphorylated, residue-specificity, hydrophobicity) at the global phosphopeptide level. The magnetic Fe3+-NTA beads are preferable over the Fe3+-NTA spin column under some circumstances. First, magnetic Fe3+-NTA beads had better scalability in that larger or smaller amounts of starting material can be used with no limitation due to column capacity. As such, the need to divide and then recombine samples using multiple spin columns for large amount of stating material or conversely removing some resin into separate column (or a fritted pipette tip) for smaller-scale enrichment, is unnecessary. Second, the magnetic bead-based enrichment platform lends itself to being amenable to high-throughput sample processing and automation. To date, many proteomics workflows have taken advantage of magnetic bead-based sample processing workflows, including the epMotion 5073m liquid handling platform [29], the Agilent Bravo [30], the Kingfisher Flex [31], Proteograph [32] and the opentrons OT-2 liquid handler [33–35]. Lastly, the beads offer a more environmentally sustainable option, with less plastic waste associated with the recommended single use of the spin columns. We note the availability of other magnetic-IMAC beads - with metals such as gallium [36, 37], titanium [38, 39], or zirconium [40, 41] - which may be evaluated against the magnetic Fe3+-NTA beads as presented here. Moreover, antibody-based phosphopeptide enrichment strategies (e.g., PTMscan [42]) will similarly have their own benefits and caveats and merit future evaluation against the magnetic Fe3+-NTA bead workflow [43]. In all, these data illustrated how the potentially scalable and automation-friendly magnetic Fe3+-NTA bead-based workflow is a flexible alternative enrichment strategy for phosphoproteome profiling experiments.
Supplementary Material
Highlights.
Fe3+-NTA magnetic beads offer potentially scalable and automation-friendly phosphopeptide enrichment
Spin column-based and magnetic Fe3+-NTA beads enrich similar numbers of phosphopeptides
Both methods enrich peptides with similar physicochemical properties
Results for both methods are similar for label-free and TMTpro-labeled samples
SIGNIFICANCE.
Protein phosphorylation plays a key role in regulating a multitude of biological processes and can lead to insights into disease pathogenesis. Methodologies which can efficiently enrich phosphopeptides in a scalable and high-throughput manner is essential for profiling dynamic phosphoproteomes. Here we compare two phosphopeptide enrichment workflows, a well-established spin column-based strategy with agarose Fe3+-NTA beads and a strategy using magnetic Fe3+-NTA beads. Our data suggest that the scalable and automation-friendly magnetic bead-based workflow is an equivalent, but more flexible, enrichment strategy for phosphoproteome profiling experiments.
ACKNOWLEDGEMENTS
We would like to thank the members of the Gygi Lab at Harvard Medical School. This work was funded in part by the Cell Biology Education and Fellowship Fund (V.R.) and NIH/NIGMS grants R01 GM132129 (J.A.P.) and GM67945 (S.P.G.).
Footnotes
Supplementary data
Supplementary material
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Liu X, Fields R, Schweppe DK, Paulo JA, Strategies for mass spectrometry-based phosphoproteomics using isobaric tagging, Expert Rev Proteomics 18(9) (2021) 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Paulo JA, Schweppe DK, Advances in quantitative high-throughput phosphoproteomics with sample multiplexing, Proteomics 21(9) (2021) e2000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Qiu W, Evans CA, Landels A, Pham TK, Wright PC, Phosphopeptide enrichment for phosphoproteomic analysis - A tutorial and review of novel materials, Anal Chim Acta 1129 (2020) 158–180. [DOI] [PubMed] [Google Scholar]
- [4].Urban J, A review on recent trends in the phosphoproteomics workflow. From sample preparation to data analysis, Anal Chim Acta 1199 (2022) 338857. [DOI] [PubMed] [Google Scholar]
- [5].Manning G, Plowman GD, Hunter T, Sudarsanam S, Evolution of protein kinase signaling from yeast to man, Trends Biochem Sci 27(10) (2002) 514–20. [DOI] [PubMed] [Google Scholar]
- [6].Moorhead GB, De Wever V, Templeton G, Kerk D, Evolution of protein phosphatases in plants and animals, Biochem J 417(2) (2009) 401–9. [DOI] [PubMed] [Google Scholar]
- [7].Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ, Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns, Anal Chem 76(14) (2004) 3935–43. [DOI] [PubMed] [Google Scholar]
- [8].Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck AJ, Zou H, Mohammed S, Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography, Nat Protoc 8(3) (2013) 461–80. [DOI] [PubMed] [Google Scholar]
- [9].Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM, Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae, Nat Biotechnol 20(3) (2002) 301–5. [DOI] [PubMed] [Google Scholar]
- [10].Paulo JA, Navarrete-Perea J, Erickson AR, Knott J, Gygi SP, An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies, J Am Soc Mass Spectrom 29(7) (2018) 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Navarrete-Perea J, Yu Q, Gygi SP, Paulo JA, Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3, J Proteome Res 17(6) (2018) 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li J, Cai Z, Bomgarden RD, Pike I, Kuhn K, Rogers JC, Roberts TM, Gygi SP, Paulo JA, TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing, J Proteome Res 20(5) (2021) 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Van Vranken JG, Pontano Vaites L, Schweppe DK, Huttlin EL, Etienne C, Nandhikonda P, Viner R, Robitaille AM, Thompson AH, Kuhn K, Pike I, Bomgarden RD, Rogers JC, Gygi SP, Paulo JA, TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples, Nat Methods 17(4) (2020) 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Popow O, Liu X, Haigis KM, Gygi SP, Paulo JA, A Compendium of Murine (Phospho)Peptides Encompassing Different Isobaric Labeling and Data Acquisition Strategies, J Proteome Res (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, Krijgsveld J, Single-pot, solid-phase-enhanced sample preparation for proteomics experiments, Nature Protocols 14(1) (2019) 68–85. [DOI] [PubMed] [Google Scholar]
- [16].Paulo JA, Gygi SP, A comprehensive proteomic and phosphoproteomic analysis of yeast deletion mutants of 14–3-3 orthologs and associated effects of rapamycin, Proteomics 15(2–3) (2015) 474–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rossio V, Paulo JA, Chick J, Brasher B, Gygi SP, King RW, Proteomics of broad deubiquitylase inhibition unmasks redundant enzyme function to reveal substrates and assess enzyme specificity, Cell Chem Biol 28(4) (2021) 487–502 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rappsilber J, Ishihama Y, Mann M, Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics, Anal Chem 75(3) (2003) 663–70. [DOI] [PubMed] [Google Scholar]
- [19].Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P, A cross-platform toolkit for mass spectrometry and proteomics, Nat Biotechnol 30(10) (2012) 918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP, A probability-based approach for high-throughput protein phosphorylation analysis and site localization, Nature biotechnology 24(10) (2006) 1285–92. [DOI] [PubMed] [Google Scholar]
- [21].Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP, A tissue-specific atlas of mouse protein phosphorylation and expression, Cell 143(7) (2010) 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Elias JE, Gygi SP, Target-decoy search strategy for mass spectrometry-based proteomics, Methods Mol Biol 604 (2010) 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Elias JE, Gygi SP, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry, Nat Methods 4(3) (2007) 207–14. [DOI] [PubMed] [Google Scholar]
- [24].Reimers M, Carey VJ, Bioconductor: an open source framework for bioinformatics and computational biology, Methods Enzymol 411 (2006) 119–34. [DOI] [PubMed] [Google Scholar]
- [25].Osorio D, Rondón-Villarreal P, Torres Sáez R, Peptides: A Package for Data Mining of Antimicrobial Peptides, The R Journal 7 (2015) 4–14. [Google Scholar]
- [26].Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Perez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaino JA, The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res 47(D1) (2019) D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Paulo JA, Navarrete-Perea J, Gygi SP, Multiplexed proteome profiling of carbon source perturbations in two yeast species with SL-SP3-TMT, J Proteomics 210 (2020) 103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lawrence RT, Searle BC, Llovet A, Villén J, Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry, Nat Methods 13(5) (2016) 431–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Waas M, Pereckas M, Jones Lipinski RA, Ashwood C, Gundry RL, SP2: Rapid and Automatable Contaminant Removal from Peptide Samples for Proteomic Analyses, J Proteome Res 18(4) (2019) 1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Muller T, Kalxdorf M, Longuespee R, Kazdal DN, Stenzinger A, Krijgsveld J, Automated sample preparation with SP3 for low-input clinical proteomics, Mol Syst Biol 16(1) (2020) e9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leutert M, Rodriguez-Mias RA, Fukuda NK, Villen J, R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies, Mol Syst Biol 15(12) (2019) e9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Blume JE, Manning WC, Troiano G, Hornburg D, Figa M, Hesterberg L, Platt TL, Zhao X, Cuaresma RA, Everley PA, Ko M, Liou H, Mahoney M, Ferdosi S, Elgierari EM, Stolarczyk C, Tangeysh B, Xia H, Benz R, Siddiqui A, Carr SA, Ma P, Langer R, Farias V, Farokhzad OC, Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona, Nature Communications 11(1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Han Y, Thomas CT, Wennersten SA, Lau E, Lam MPY, Shotgun Proteomics Sample Processing Automated by an Open-Source Lab Robot, J Vis Exp (176) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liang Y, Acor H, McCown MA, Nwosu AJ, Boekweg H, Axtell NB, Truong T, Cong Y, Payne SH, Kelly RT, Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling, Anal Chem 93(3) (2021) 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu X, Gygi SP, Paulo JA, A Semiautomated Paramagnetic Bead-Based Platform for Isobaric Tag Sample Preparation, J Am Soc Mass Spectrom (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aryal UK, Olson DJ, Ross AR, Optimization of immobilized gallium (III) ion affinity chromatography for selective binding and recovery of phosphopeptides from protein digests, J Biomol Tech 19(5) (2008) 296–310. [PMC free article] [PubMed] [Google Scholar]
- [37].Sun Z, Hamilton KL, Reardon KF, Evaluation of quantitative performance of sequential immobilized metal affinity chromatographic enrichment for phosphopeptides, Anal Biochem 445 (2014) 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang X, Yu J, Yang H, Shen J, Liu H, Zhou J, A new Ti-based IMAC nanohybrid with high hydrophilicity and enhanced absorption capacity for the selective enrichment of phosphopeptides, J Chromatogr B Analyt Technol Biomed Life Sci 1179 (2021) 122851. [DOI] [PubMed] [Google Scholar]
- [39].Zhang K, Hao Y, Hu D, Deng S, Jin Y, Wang X, Liu H, Liu Y, Xie M, Development of dual-ligand titanium (IV) hydrophilic network sorbent for highly selective enrichment of phosphopeptides, J Chromatogr A 1659 (2021) 462648. [DOI] [PubMed] [Google Scholar]
- [40].Arribas Diez I, Govender I, Naicker P, Stoychev S, Jordaan J, Jensen ON, Zirconium(IV)-IMAC Revisited: Improved Performance and Phosphoproteome Coverage by Magnetic Microparticles for Phosphopeptide Affinity Enrichment, J Proteome Res 20(1) (2021) 453–462. [DOI] [PubMed] [Google Scholar]
- [41].Dai J, Wang M, Liu H, Highly selective enrichment of phosphopeptides using Zr(4+)-immobilized Titania nanoparticles, Talanta 164 (2017) 222–227. [DOI] [PubMed] [Google Scholar]
- [42].Stokes MP, Farnsworth CL, Moritz A, Silva JC, Jia X, Lee KA, Guo A, Polakiewicz RD, Comb MJ, PTMScan direct: identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS, Mol Cell Proteomics 11(5) (2012) 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Possemato AP, Paulo JA, Mulhern D, Guo A, Gygi SP, Beausoleil SA, Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events, J Proteome Res 16(4) (2017) 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.