

Abstract
Anti-Müllerian hormone (AMH) is a distinctive biomarker of the immature Sertoli cell. AMH expression, triggered by specific transcription factors upon fetal Sertoli cells differentiation independently of gonadotropins or sex steroids, drives Müllerian duct regression in the male, preventing the development of the uterus and Fallopian tubes. AMH continues to be highly expressed by Sertoli until the onset of puberty, when it is downregulated to low adult levels. FSH increases testicular AMH output by promoting immature Sertoli cell proliferation and individual cell expression. AMH secretion also showcases a differential regulation exerted by intratesticular levels of androgens and estrogens. In the fetus and the newborn, Sertoli cells do not express the androgen receptor, and the high androgen concentrations do not affect AMH expression. Conversely, estrogens can stimulate AMH production because estrogen receptors are present in Sertoli cells and aromatase is stimulated by FSH. During childhood, sex steroids levels are very low and do not play a physiological role on AMH production. However, hyperestrogenic states upregulate AMH expression. During puberty, testosterone inhibition of AMH expression overrides stimulation by estrogens and FSH. The direct effects of sex steroids on AMH transcription are mediated by androgen receptor and estrogen receptor α action on AMH promoter sequences. A modest estrogen action is also mediated by the membrane G-coupled estrogen receptor GPER. The understanding of these complex regulatory mechanisms helps in the interpretation of serum AMH levels found in physiological or pathological conditions, which underscores the importance of serum AMH as a biomarker of intratesticular steroid concentrations.
Keywords: androgen insensitivity, androgen response element, estradiol, estrogen response element, dihydrotestosterone, ketoconazole, testotoxicosis, triptorelin
1 Introduction
Anti-Müllerian hormone (AMH) is a glycoprotein hormone (1) that belongs to the transforming growth factor beta (TGFβ) superfamily (2). It is produced essentially by Sertoli cells of the testis and granulosa cells of the ovary: after castration, AMH is no longer detected in serum (3, 4). AMH is synthesized as a homodimeric precursor consisting of two identical polypeptide chains, with a large N-terminal pro-region of 110-kDa and a small C-terminal mature domain of 25-kDa. AMH is subjected to post-translational proteolytic processing (5); the resulting N-terminal and C-terminal dimers remain associated in a non-covalent complex that is biologically active (6, 7). The pro-region is displaced from the non-covalent complex upon binding to an AMH receptor homodimer (8). AMH transduces its signal through a specific type II receptor, AMHR2, that is expressed at the cell surface in target organs (9) and interacts with the non-specific type I receptors ACVR1 (initially called ALK2), BMPR1A (ALK3) or BMPR1B (ALK6) (reviewed in (10). The most widely recognized action of AMH takes place during male fetal development, where it provokes the regression of the Müllerian ducts –the anlagen of the Fallopian tubes, the uterus and the upper part of the vagina (reviewed in (11).
The human AMH gene is located on chromosome 19p13.3 (12) and consists of 5 exons spanning 2.8 kb (2). The 3’ end of the 5th exon encodes the biologically active C-terminal domain of the protein. In the testis, the major transcription initiation site is located 10 bp upstream of the ATG codon (13). Most of the best characterized binding sites for transactivating factors with a relevant role in fetal life lie within the proximal 500 bp ( Figure 1 ) (reviewed in (15), but other response elements for factors involved in AMH regulation in postnatal life are present in the distal promoter (16). Indeed, distal promoter sequences are necessary for the maintenance of AMH expression in the testis during postnatal life (17). While no canonical androgen response elements (AREs) have been described (18), a consensus sequence for estrogen receptor binding (ERE) is present at -1782 bp in the human AMH promoter (13, 19).
Figure 1.

Onset and upregulation of AMH expression. Transcription factors SOX9, SF1, GATA4 and WT1 bind to specific response elements in the proximal AMH promoter and trigger AMH expression in early fetal life, independently of gonadotropin or steroid action, resulting in Müllerian duct regression. From the second trimester of gestation, FSH increases testicular AMH production by 2 different mechanisms: it promotes cell proliferation and upregulates AMH transcription via the transduction pathway involving the heterotrimeric (α/β/γ) G protein, adenylyl cyclase (AC), cyclic AMP and protein kinase A (PKA) resulting in increased expression and nuclear translocation of transcription factors SOX9, SF1, AP2 and NFκB. Modified with permission from Grinspon and Rey (14), © 2010 S. Karger AG.
1.1 The Ontogeny of AMH Expression in the Testis
AMH is one of the earliest cell-specific proteins produced by Sertoli cells when the testis differentiates from the gonadal ridge in the XY embryo (20). AMH starts to be expressed at 12.5 days post-coitum in the mouse (21) and from the 8th week onwards in the human (22). Although its role in sex differentiation of the internal ducts takes place in the first phases of fetal development (see below), AMH continues to be produced by Sertoli cells at very high levels during the whole fetal life and, postnatally, until the onset of puberty ( Figure 2 ). During pubertal development, Sertoli cells progressively express less AMH (25), and the directional secretion switches from the basal to the adluminal compartment (26). Consequently, AMH concentration in serum is high during childhood but decreases during puberty, when it increases in seminal plasma [reviewed in ref (27)]. The decrease in AMH expression coincides with the establishment of the blood-testis barrier and the onset of germ cell meiosis ( Figure 3 ) (28–30), which are androgen-dependent processes. During adulthood, serum AMH is approximately ten- to twentyfold lower as compared to the prepubertal period in males (24), but still twofold higher than in females (31).
Figure 2.

Relationship between AMH production, androgen receptor (AR) expression and androgen levels in the human testis. During the postnatal period of activation of the hypothalamic-pituitary-testicular axis (0-6 months, first column, A–C), testosterone (T) levels are high (bottom graph, serum hormone levels); however, Sertoli cells do not show maturational changes because they do not express the AR (upper graph, second line, immunohistochemistry). Therefore, AMH expression is high (upper graph, third line, AMH immunohistochemistry) and germ cells do not enter meiosis (upper graph, first line, HE: hematoxylin-eosin staining). During the “quiescent” period of the gonadal axis (childhood, second column, D–F), androgen synthesis is negligible, which explains why Sertoli cells remain immature even though they have started to express the AR. In puberty and adulthood (third column, G–I), T increases, inducing Sertoli cell maturation, reflected in the decline of AMH expression and the onset of adult spermatogenesis. Bottom graph shows % AR+: percentage of Sertoli cells expressing the AR. AMH (pmol/l) and T (ng/dl): schematic representation of AMH and T serum levels from birth to adulthood in males.
Figure 3.

Classical (genomic) and non-classical (non-genomic) androgen signaling in Sertoli cells. In cells not exposed to androgens, the androgen receptor (AR) is bound to heat-shock proteins (HSP) in the cytoplasm. Testosterone and other androgens are steroids that easily cross the cell membrane and bind to the AR, that is released from HSP. The ligand-bound AR can either translocate to the nucleus and interact with androgen response elements (ARE) or with other transcription factors (TF), thus activating the classical/genomic pathway, or migrate to the inner side of the cell membrane and interact with Src, thus activating the non-classical/non-genomic pathway. CREB, cAMP response element binding protein; EGFR, Epidermal growth factor receptor; ERK, Extracellular signal-regulated kinase; MEK, Mitogen-activated protein kinase; Src, Steroid receptor coactivator. Reproduced with permission from Edelsztein and Rey (18) © 2019 The Authors.
1.2 Physiological Role of AMH in Male Development
In mammals, the earliest stages of intrauterine development are characterized by the existence of a sexual undifferentiated period where no differences can be observed, except at the chromosomal level, between XX and XY embryos. The gonadal ridges and the precursors of the external genitalia are identical in both sexes, and there are two sets of internal ducts: Wolffian ducts that give rise to the male internal genitalia, and the Müllerian ducts that differentiate into the female internal genitalia. The pioneering work by Alfred Jost (32) demonstrated the existence of a Mullerian inhibiting activity, specific to the fetal testis, explaining why the female internal organs do not develop in the male fetus. In the absence of AMH action, e.g. in individuals with mutations of the genes coding for AMH or its specific receptor AMHR2, the Fallopian tubes and the uterus are present [reviewed in ref (15)]. The absence of AMH action is also evidenced in 46,XY disorders of sex development (DSD) due to complete gonadal dysgenesis [reviewed in ref (33)]. Interestingly, the window of action of AMH on Müllerian ducts is limited to the earliest stages of intrauterine life (34); this explains why the development of internal genitalia is not disrupted by ovarian AMH production that starts when Müllerian ducts are already insensitive to its action (35). The fact that AMH continues to be secreted by the testis after the critical window of Müllerian duct regression and that it is produced by the ovaries at clearly detectable levels suggest that AMH may have other physiological roles. Nonetheless, individuals with AMH or AMHR2 null mutations resulting in complete loss of AMH expression or action do not show other evident phenotypes [reviewed in ref (11)].
1.3 AMH as a Biomarker in Physiological and Pathological States
Independently of its physiological role(s), AMH is a proven biomarker of the immature Sertoli cell population in normal and pathological conditions [reviewed in ref (36–39)]. Serum AMH reflects the mass of immature Sertoli cells, i.e. of functional testicular tissue, from fetal life to adulthood. In the fetus (22), newborn and child (40), detectable AMH in serum indicates the existence of testes, which is particularly useful for the differential diagnosis between bilateral cryptorchidism and anorchidism in boys with nonpalpable gonads. In patients with DSD, serum AMH levels are commensurate with the amount of testicular tissue, except for those with a Persistent Müllerian duct syndrome due to AMH mutations (41). Undetectable AMH is indicative of no testicular development in 46,XY girls with complete gonadal dysgenesis. Circulating levels are below the normal male range in patients with 46,XY DSD due to partial gonadal dysgenesis –where both Sertoli and Leydig cell development is impaired– but normal or high in 46,XY DSD due to isolated steroidogenic defects –where only Leydig cell function is affected– or to androgen insensitivity or 5α-reductase deficiency –where both Leydig and Sertoli cell development is preserved (33). In patients with 46,XX DSD, serum AMH above the female range indicates the existence of functional testicular tissue suggesting the diagnosis of ovotesticular or testicular DSD, in contrast with other forms of XX virilization, such as aromatase deficiency, congenital adrenal hyperplasia of androgen-secreting tumors, which present with serum AMH in the female range (42). In normally virilized boys, low serum AMH is seen in patients with primary (24, 43, 44) or central hypogonadism (45). Conversely, high AMH is suggestive of Sertoli cell tumors (46), excessive signaling downstream the FSH receptor pathway like in McCune-Albright syndrome (47), or hyperestrogenism (19). During pubertal development, the decline in serum AMH levels is an early sign of Sertoli cell maturation (48), while an excessive decrease to undetectable levels is typical of Klinefelter syndrome (49). Conversely, the persistence of elevated AMH levels despite an increase in testosterone production is indicative of androgen insensitivity (50).
2 Steroid-Independent Regulation of Testicular AMH Expression
2.1 AMH Expression During Early Fetal Development
The onset of AMH expression in the fetal testis is independent of gonadotrophin or steroid regulation ( Figure 1 ). SOX9, upregulated by SRY, triggers AMH expression by binding to a specific element on the proximal AMH promoter (51). Subsequently SF1 (51–53), GATA4 (54) and WT1 (55) cooperate to further upregulate AMH transcription whereas DAX1 represses the transcriptional cooperation between SF1 and GATA4 (56).
As mentioned, although its action takes place in the early stages of fetal sex differentiation, AMH continues to be produced by Sertoli cells throughout life ( Figure 2 ). The proximal AMH promoter plays a major role in the initiation of fetal expression, when AMH induces the regression of Müllerian ducts, but it proves insufficient to maintain AMH expression thereafter, when sequences farthest from the transcription start site are required (17).
2.2 AMH Regulation by FSH in Late Fetal and Postnatal Life
From the second half of gestation, FSH increases testicular AMH output through two different mechanisms ( Figure 1 ): it induces Sertoli cell proliferation and upregulates AMH expression at the individual cell level (16, 30). The continuous effect of high FSH levels on Sertoli cells for almost one year (the last 6 months of fetal life plus the 3-6 months of postnatal life induces a progressive increase in serum AMH (57), which peaks at 5-6 months of age (58). This explains why serum AMH levels are low, in coincidence with small testicular size, in boys with congenital central (hypogonadotrophic) hypogonadism (45, 59) and increase after FSH treatment (60, 61). The underlying molecular mechanisms include the classical cyclic AMP-dependent pathway triggered by the FSH receptor coupled to the Gαs protein (16), which activates SOX9 (62) and SF1 (62) acting on the proximal AMH promoter, but also NFκB and AP2, which bind to response elements lying approximately 2 kb upstream of the AMH start site (62). These molecular mechanisms explain the elevated AMH production observed in boys with McCune-Albright syndrome carrying a gain-of-function somatic mutation in the GNAS1 gene encoding the Gαs protein (47, 63).
2.3 Lack of Androgen-Mediated Regulation of Testicular AMH Expression in Fetal and Early Postnatal Life
The fetal period and the stage of postnatal activation of the hypothalamic-pituitary-testicular axis occurring during the first 3 to 6 months after birth, usually referred to as “mini-puberty”, are characterized by the coexistence of high testosterone levels and AMH expression in the testes ( Figure 2 ). The continuous exposure to high androgen levels for approximately one year neither affects AMH expression nor induces histologic maturational changes in Sertoli cells or spermatogenic onset. Immunohistochemical studies have clarified why: Sertoli cells do not express the AR and, therefore, they are physiologically insensitive to androgens until the end of the first year of life in humans ( Figure 2 ). Subsequently during infancy and childhood, AR expression increases progressively in Sertoli cells until full expression is observed by the age of approximately 8 years (64–66). An equivalent ontogeny can be observed in rodents (30, 67). Interestingly, boys with high intratesticular androgen concentration due to precocious puberty show normal serum AMH during the first year of life but declining levels thereafter, when the AR starts to be expressed in Sertoli cells (68).
3 Regulation of Testicular AMH Production By Androgens
3.1 Downregulation of Testicular AMH Expression at Puberty
Testosterone is a well-known inducer of Sertoli cell maturation during puberty (69). During childhood, the pituitary-gonadal axis is mostly quiescent: LH levels are very low or undetectable and typical Leydig cells are absent in the testis interstitial tissue. Therefore, although Sertoli cells already express the androgen receptor (AR), they are not exposed to androgens ( Figure 2 ). When the axis reactivates at the beginning of pubertal development, LH induces the differentiation of Leydig cells that start producing testosterone. Androgen concentration increases rapidly within the testis, even though this is not reflected in the circulating levels until more advanced pubertal stages. Intratesticular testosterone triggers a number of morphologic and functional changes in Sertoli cells [reviewed in ref (18)]. One distinctive change is the decrease in AMH production that occurs as one of the first clinical signs of pubertal development in boys (48), as well as in numerous other mammals (3, 25, 70, 71), in coincidence with the onset of germ cell meiosis (50). Results obtained in experimental mouse models support these observations (30). In the naturally occurring Tfm mice, carrying a functionally impaired AR (30), and in mice with an artificially mutated Ar gene (72), AMH expression does not wain at the expected age of puberty.
3.1.1 Molecular Mechanisms
Androgen signaling in target cells typically occurs through the AR engaging either the classical (or genomic) or the non-classical (or non-genomic) pathways ( Figure 3 ) (73). The classical pathway involves the intracellular AR acting as a transcription factor. In the inactive state, the AR is bound to cytoplasmic heat-shock proteins. When testosterone or the more potent and non-aromatizable androgen dihydrotestosterone (DHT) binds to the AR, a conformational change leads to the release of the AR from the heat-shock proteins, AR phosphorylation, homodimer formation, nuclear translocation and interaction with androgen response elements (ARE), specific DNA sequences in the regulatory regions of target genes (18). Typical ARE are palindromic: in humans, the consensus sequence is 5’-AGAACAnnnTGTTCT-3’ (74). The androgen-bound AR can also regulate target gene expression in the absence of ARE, by interacting with trans-activating factors that bind to their specific binding sequences (75). Androgen signaling through the genomic pathway requires 30-45 minutes to induce transcriptional regulation (76).
Non-classical pathways induce cellular changes more rapidly, within seconds to minutes ( Figure 3 ). Androgen action leads the AR to localize near the plasma membrane, where it induces Src. This tyrosine kinase phosphorylates the epidermal growth factor receptor, which triggers the MAP kinase cascade ultimately resulting in the phosphorylation of specific transcription factors (73). Alternatively, the Zn2+ transporter ZIP9 has been proposed as a membrane AR that induces the phosphorylation of Erk1/2 and the transcription factors CREB and ATF1 (77).
Traditionally, maturation changes induced by androgens were believed to result from upregulation of target genes. However, high-throughput techniques have revealed similar amounts of up-regulated and down-regulated genes in response to androgen action in the maturing testis (78). However, the pathways underlying androgen-mediated downregulation have been barely explored. Using the mouse prepubertal Sertoli cell line SMAT1 (79), we have recently clarified the molecular mechanisms involved in AMH downregulation during puberty (80). The decrease in AMH production is a direct effect of androgens on Sertoli cells, not involving negative feedback on pituitary gonadotrophins or down-regulation of activating transcription factors. Indeed, FSH and testosterone have opposing effects on AMH expression at puberty, with the inhibitory effect of androgens largely exceeding the stimulatory effect of FSH. In SMAT1 cells, both testosterone and the nonaromatizable androgen DHT decrease the activity of a 3-kb human AMH promoter in the presence, but not in the absence, of the AR ( Figures 4A, B ), and this inhibition is prevented by the antiandrogen bicalutamide. Using human AMH promoters of different lengths in luciferase assays, we demonstrated that androgen-mediated downregulation involved the promoter sequences lying within the proximal 430 bp ( Figure 4A ). No canonical ARE can be found in the AMH promoter, and intact SF1 response elements are required for the negative regulation by androgens to occur ( Figure 4B ). This inhibitory effect on AMH expression could be mediated by a direct interaction between the ligand-bound AR and the SF1 elements, i.e. blockage by competition, or alternatively due to a protein-protein interaction between the ligand-bound AR and promoter-bound SF1, i.e. blockage by interaction. In any case, the AR prevents SF1 from upregulating AMH promoter activity ( Figures 4C, D ).
Figure 4.

Molecular mechanisms involved in androgen-induced downregulation of AMH expression in Sertoli cells. A and B: luciferase assays in SMAT1 Sertoli cells transfected with a luciferase plasmid under control of the human AMH promoter (pGL2B-hAMH) of different lengths and co-transfected with an expression plasmid of the androgen receptor (pSG5-AR) or pSG5 devoid of the AR. Cells were exposed to testosterone or DHT 10-7 M, or vehicle, and results were expressed as relative luciferase units (RLU) comparing cells exposed to androgens and those exposed to vehicle (* p<0.05, ** p<0.01, *** p<0.001). A 100% level indicates the basal AMH promoter activity. Testosterone and DHT induce an inhibition of AMH promoter activity when the AR is present but not in its absence, when 433 to 3076 bp of the promoter are present but not when the proximal 1916-bp sequences are lacking (A). The inhibition induced by androgen persists even if the binding sites for AP1 or GATA4 are mutated, but not when SF1 sites are mutated (B). (C, D) schematic of androgen regulation of AMH expression. In fetal and postnatal periods before the onset of puberty, the lack of androgen action allows the AMH promoter activity induced by SOX9, SF1 and GATA4, resulting in high AMH production. During puberty and adulthood, the steroid-bound AR represses AMH promoter activity by competition or by interaction with SF1. Modified with permission from Edelsztein et al. (80) © 2018 The Authors and Edelsztein and Rey (18) © 2019 The Authors.
3.1.2 Clinical Implications
Clinical studies have shown that AMH decline is not merely associated with chronological age but rather with the maturation status of the testes. Indeed, a decrease in serum AMH is an early sign of an increase in intratesticular testosterone concentration reflecting the activation of LH pulses occurring with pubertal onset. The decrease in serum AMH occurs already in Tanner 2 stage of puberty in boys, that is earlier than the increase in serum testosterone, taking place in Tanner 3 stage (24, 48, 81, 82). Furthermore, AMH is low for age in boys aged 2 to 8 years with precocious puberty, independently of gonadotrophin levels, and recovers prepubertal levels when testosterone production is effectively curtailed by treatment ( Figures 5A, B ) (48, 68). Serum AMH can be used as a marker of effective treatment. In fact, for Sertoli cells to resume their prepubertal status and increase AMH production, intratesticular androgen levels should remain continuously low for at least 6 months. If adherence to treatment is erratic and intratesticular testosterone concentration shows intermittent variations, serum AMH never increases to prepubertal levels ( Figure 5C ) (48).
Figure 5.
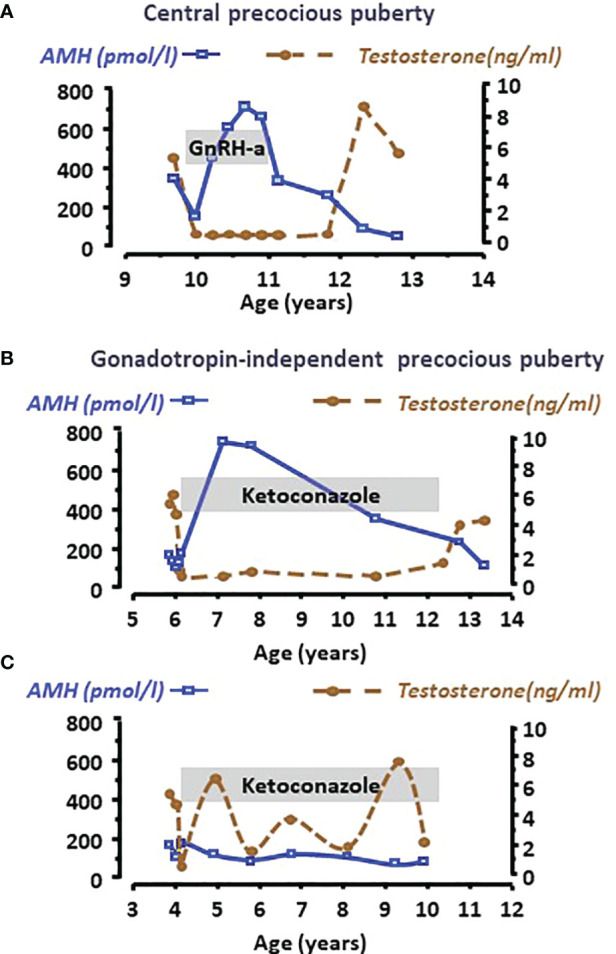
Serum AMH as a biomarker of intratesticular androgen concentration in boys with central precocious puberty (A) or with gonadotropin-independent precocious puberty (B, C). Serum AMH is low at diagnosis, indicating the inhibitory effect of high androgen concentration reflected in high serum testosterone levels in all cases. When testosterone production is effectively curtailed by the adequate treatment, such as a GnRH analogue in the patient with central precocious puberty (A) or ketoconazole in the patient with gonadotropin-independent precocious puberty (B), Sertoli cells recover their immature status and increase AMH production until treatment is discontinued. Conversely, when adherence to treatment is erratic (C), intratesticular and serum testosterone concentration does not remain at prepubertal levels and Sertoli cells do not revert their pubertal status, which is reflected in low serum AMH. Modified with permission from Rey et al. (48) © 1993 The Endocrine Society.
On the other hand, testicular AMH production remains high in adolescents and adults with androgen insensitivity due to mutations in the AR gene (50, 83–85). The increase in AMH expression has been ascribed to SOX9 upregulation in Sertoli cells (86).
4 Regulation of Testicular AMH Production By Estrogens
Like for androgens, the testis is both a source and a target organ of estrogens, synthesized from androgens by the cytochrome P450 enzyme aromatase encoded CYP19A1. Aromatase expression is present in the postnatal testis (64) and is stimulated by FSH action on Sertoli cells (87). Estrogen signaling involves 3 different receptors. The canonical nuclear estrogen receptors α (ERα) and β (ERβ) act through their binding capacity on DNA sequences known as estrogen response elements (ERE). ERα and ERβ show a 97% identity in their DNA-binding domains, while their ligand-binding domains have an identity of 60%. Both ERs bind to estrogens with a similar affinity (88). The membrane ER, named GPR30, GPER1 or simply GPER, is a seven-transmembrane domain, G protein-coupled receptor, predominantly present in the endoplasmic reticulum, that mediates rapid cellular responses involving second messengers, ion channels and kinase activities (89).
The 3 ERs are expressed in the testis, but there are differences in the timing and cellular types according to species (90). Sertoli cells of the prepubertal and pubertal human testis express ERα (19) and ERβ (19, 64). In rodents, ERα expression predominates in the prepubertal testis while ERβ is more abundant in the adult (91). GPER has been identified in Sertoli cells from the onset of puberty (92).
4.1 Upregulation of Testicular AMH Expression in Postnatal Life
At the onset of puberty, testicular AMH expression increases concomitantly with FSH and estradiol (E2) levels in normal boys (93). This simultaneous elevation in the 3 hormones is also observed in patients with androgen insensitivity, in whom the inhibitory effect of androgens on AMH is disrupted. Patients with androgen insensitivity show increased aromatization of androgens to estrogens, resulting in the development of breasts. As already discussed, AMH upregulation can be explained by a direct action of FSH on Sertoli cell proliferation and on AMH transcription. However, AMH is also elevated in boys with Peutz-Jeghers syndrome (46), in whom Sertoli cell proliferations produce high estrogen levels leading to suppressed FSH (46). Interestingly, the human AMH promoter contains a half ERE ( Figure 1 ) (13), and E2 regulates AMH expression in the ovary (94).
We have recently tested the hypothesis that E2 upregulates AMH expression in the prepubertal and pubertal testis, using experimental mouse models. Treatment of 4-day-old mice with ICI 182780, an antagonist of ERα and ERβ, resulted in significant decrease in serum AMH ( Figure 6A ), indicating that abolishing ER signaling results in a decreased testicular AMH production. Nuclear ERs (91) and GPER (95) are involved in Sertoli cell proliferation, which could in part explain the increase in AMH output by the testes exposed to high estrogen levels.
Figure 6.

Molecular mechanisms involved in estrogen-induced upregulation of AMH expression in Sertoli cells. (A–D) Luciferase assays in SMAT1 Sertoli cells transfected with a luciferase plasmid under control of the human AMH promoter (pGL2B-hAMH) of different lengths and co-transfected with an expression plasmid of the estrogen receptor alfa (ERα) or the G protein-coupled estrogen receptor (GPER) or the control plasmids devoid of the estrogen receptor sequences. Cells were exposed to estradiol (E2), the ERα/β antagonist ICI 182780, the ERα-specific agonist PPT or antagonist MPP, or the GPER-specific agonist G1 or antagonist G15. Results were expressed as relative luciferase units (RLU) comparing cells exposed to estrogens, agonists and/or antagonists (* p<0.05, *** p<0.001). A 100% level indicates the basal AMH promoter activity. E2 induce an upregulation of AMH promoter activity when the ERα or the GPER is present but not in their absence (A–C), and when the estrogen response element (ERE) at -1782 of the promoter is conserved (D). (E) Proposed model for E2 regulation of AMH expression in Sertoli cells. E2 upregulates AMH transcription via ERα binding to the ERE. GPER also upregulates AMH expression more modestly. The increased AMH expression results in a higher testicular AMH production. Another potential mechanism is the increase in Sertoli cell proliferation induced by membrane-bound ERα signaling through the PI3K/Akt pathway, and/or GPER via MAPK signaling. Reproduced with permission from Valeri et al. (19)© 2020 The Authors.
4.1.1 Molecular Mechanisms
The existence of an ERE on the AMH promoter prompted us to test whether estrogens could have a direct effect on AMH expression at the individual Sertoli cell level. Luciferase assays clearly indicated that the activity of a 3-kb human AMH promoter was increased after exposure to E2 of SMAT1 Sertoli cells transfected with any of the 3 ERs. However, to elicit a significant response ERβ-transfected SMAT1 needed to be incubated with E2 10-7 M (19), a concentration 100-fold higher than that observed in the testes of adolescents (96), while ERα- and GPER-transfected SMAT1 cells showed a maximal response to E2 at the physiological concentration of 10-9 M (19). These observations suggest that ERα and GPER, but not ERβ, are involved in the upregulation of AMH expression by E2. Further support was provided by experiments showing that PPT, a potent and selective ERα agonist, increased AMH promoter activity while MPP, a specific ERα antagonist, inhibited the AMH promoter activity ( Figure 6B ). The effect induced by E2 via GPER was more modest. Its specificity was confirmed when SMAT1 cells were exposed to the potent and selective GPER agonist G1 or to the selective GPER antagonist G-15 ( Figure 6C ).
The upregulation induced by E2 in SMAT1 cells on the 3-kb AMH promoter was also observed when a 1926-bp promoter with an intact ERE present at position -1782 was transfected, but no estrogenic activity was detected when using AMH promoter constructs consisting of the proximal 433 bp or of sequences lying between -1926 and -2590 or between -1926 and -3078, which are devoid of the half-ERE ( Figure 6D ) (19). Furthermore, the response to E2 was abolished by a mutation introduced in the half-ERE at -1782 by site-directed mutagenesis. A direct interaction between ERα and the half-ERE was confirmed in electro mobility shift assays. Complex formation with ERα was observed when ERE wild-type probes, but not with mutant ERE probes, were used. Altogether, these results indicate that E2 at physiological concentrations increases the activity of the AMH promoter through ERα binding to the half-ERE site present at position -1782 ( Figure 6E ). The intracellular pathway involved in the more modest AMH promoter upregulation induced through the GPER still needs to be elucidated.
4.1.2 Clinical Implications
The cellular and molecular mechanisms involving ERα and, more modestly, GPER in E2 induction of the activity of the AMH promoter (19) and of Sertoli cell proliferation (91) provide biological plausibility to explain the increase in serum AMH levels observed in patients with androgen insensitivity (50, 84) or with Peutz-Jeghers syndrome (46). It could also explain why serum AMH is low despite elevated circulating E2 in estrogen insensitivity caused by defective ERα function (97).
5 Concluding Remarks
AMH production by Sertoli cells reflects the differential regulation exerted by intratesticular levels of androgens and estrogens in the prepubertal testis, with specific variations according to the developmental stage. In the fetus and the newborn, the high intratesticular androgen concentrations do not regulate AMH expression because the AR is not yet expressed in Sertoli cells, whereas intratesticular estradiol can moderately increase AMH production since ERs are present in Sertoli cells and aromatase activity resulting in androgen conversion to estrogens is stimulated by FSH. During childhood in humans, or the quiescent period of the hypothalamic-pituitary-gonadal axis described in most mammals, the extremely low steroid levels do not exert any physiological regulation on AMH production ( Figure 7 ). However, precocious androgen synthesis results in AMH downregulation since the AR is expressed in Sertoli cells from the second year of life. On the contrary, testis-borne hyperestrogenic states provoke an increase in AMH expression ( Figure 7 ). Finally, during puberty the inhibitory effect of intratesticular testosterone levels overcome the stimulatory effects of estrogens and FSH ( Figure 7 ). Apart from the indirect regulation of testicular function that they exert through their feedback on gonadotropins, both androgens and estrogens have a direct effect on AMH gene transcription. These direct effects are mediated by the classical mechanism involving nuclear AR and ER receptor activity on target gene promoters and, more modestly, by estrogen action through the membrane GPER ( Figure 7 ). The comprehension of these complex regulatory mechanisms helps in the interpretation of serum AMH levels found in physiological or pathological conditions, highlighting the capacity of serum AMH as a biomarker of intratesticular steroid concentrations, which are not always reflected in serum levels.
Figure 7.

Interaction between androgens, estrogens and FSH on the regulation of AMH expression in Sertoli cells. During childhood, the hypothalamic-pituitary-gonadal axis is quiescent, and the extremely low steroid levels do not exert any physiological regulation on AMH production that is mostly hormone-independent. At puberty, the reactivation of the GnRH neuron and the gonadotropes result in higher LH and FSH levels. LH induces testosterone secretion by testicular Leydig cells. FSH acts on its receptor in the Sertoli cell membrane, resulting in a direct upregulation of AMH expression, through the cyclic AMP-PKA pathway involving transcription factors SOX9, SF1, AP2 and NFκB, and in an indirect upregulation of AMH by inducing aromatase expression. Aromatase converts androgens into estrogens, which can upregulate AMH directly by binding to the nuclear ERα or indirectly acting through the GPER expressed in the membrane of the endoplasmic reticulum. Nonetheless, the inhibitory effect of androgens overrides the stimulatory effect of FSH and estrogens on AMH expression, resulting in a decreased AMH secretion. In hyperestrogenic states with abrogated androgen action, such as the androgen insensitivity syndrome, the inhibitory effect of androgens does not exist, FSH and LH increase resulting in high testosterone that is converted to estradiol. Consequently, AMH production is substantially boosted.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
Until 2020, RR received royalties for the development of an AMH ELISA kit and honoraria for technology services using the AMH ELISA.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Picard JY, Tran D, Josso N. Biosynthesis of Labelled Anti-Müllerian Hormone by Fetal Testes: Evidence for the Glycoprotein Nature of the Hormone and for Its Disulfide-Bonded Structure. Mol Cell Endocrinology (1978) 12:17–30. doi: 10.1016/0303-7207(78)90098-9 [DOI] [PubMed] [Google Scholar]
- 2. Cate RL, Mattaliano RJ, Hession C, Tizard R, Farber NM, Cheung A, et al. Isolation of the Bovine and Human Genes for Müllerian Inhibiting Substance and Expression of the Human Gene in Animal Cells. Cell (1986) 45:685–98. doi: 10.1016/0092-8674(86)90783-x [DOI] [PubMed] [Google Scholar]
- 3. Vigier B, Tran D, du Mesnil du Buisson F, Heyman Y, Josso N. Use of Monoclonal Antibody Techniques to Study the Ontogeny of Bovine Anti-Müllerian Hormone. J Reprod Fertil (1983) 69:207–14. doi: 10.1530/jrf.0.0690207 [DOI] [PubMed] [Google Scholar]
- 4. Rey RA, Lhommé C, Marcillac I, Lahlou N, Duvillard P, Josso N, et al. Antimüllerian Hormone as a Serum Marker of Granulosa Cell Tumors of the Ovary: Comparative Study With Serum Alpha-Inhibin and Estradiol. Am J Obstet Gynecol (1996) 174:958–65. doi: 10.1016/s0002-9378(96)70333-2 [DOI] [PubMed] [Google Scholar]
- 5. Pepinsky RB, Sinclair LK, Chow EP, Mattaliano RJ, Manganaro TF, Donahoe PK, et al. Proteolytic Processing of Mullerian Inhibiting Substance Produces a Transforming Growth Factor-Beta-Like Fragment. J Biol Chem (1988) 263:18961–4. doi: 10.1016/S0021-9258(18)37375-7 [DOI] [PubMed] [Google Scholar]
- 6. Wilson CA, di Clemente N, Ehrenfels C, Pepinsky RB, Josso N, Vigier B, et al. Müllerian Inhibiting Substance Requires its N-Terminal Domain for Maintenance of Biological Activity, a Novel Finding Within the Transforming Growth Factor-Beta Superfamily. Mol Endocrinol (1993) 7:247–57. doi: 10.1210/mend.7.2.8469238 [DOI] [PubMed] [Google Scholar]
- 7. Nachtigal MW, Ingraham HA. Bioactivation of Mullerian Inhibiting Substance During Gonadal Development by a Kex2/Subtilisin-Like Endoprotease. Proc Natl Acad Sci United States America (1996) 93:7711–6. doi: 10.1073/pnas.93.15.7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cate RL, di Clemente N, Racine C, Groome NP, Pepinsky RB, Whitty A. The Anti-Mullerian Hormone Prodomain is Displaced From the Hormone/Prodomain Complex Upon Bivalent Binding to the Hormone Receptor. J Biol Chem (2022) 298:101429. doi: 10.1016/j.jbc.2021.101429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. di Clemente N, Wilson C, Faure E, Boussin L, Carmillo P, Tizard R, et al. Cloning, Expression, and Alternative Splicing of the Receptor for Anti-Müllerian Hormone. Mol Endocrinol (1994) 8:1006–20. doi: 10.1210/mend.8.8.7997230 [DOI] [PubMed] [Google Scholar]
- 10. di Clemente N, Racine C, Pierre A, Taieb J. Anti-Müllerian Hormone in Female Reproduction. Endocr Rev (2021) 42:753–82. doi: 10.1210/endrev/bnab012 [DOI] [PubMed] [Google Scholar]
- 11. Josso N. Women In Reproductive Science: Anti-Mullerian Hormone: A Look Back and Ahead. Reproduction (2019) 158:F81–9. doi: 10.1530/REP-18-0602 [DOI] [PubMed] [Google Scholar]
- 12. Cohen-Haguenauer O, Picard JY, Mattei MG, Serero S, Nguyen VC, de Tand MF, et al. Mapping of the Gene for Anti-Mullerian Hormone to the Short Arm of Human Chromosome 19. Cytogenet Cell Genet (1987) 44:2–6. doi: 10.1159/000132332 [DOI] [PubMed] [Google Scholar]
- 13. Guerrier D, Boussin L, Mader S, Josso N, Kahn A, Picard JY. Expression of the Gene for Anti-Müllerian Hormone. JReprodFertil (1990) 88:695–706. doi: 10.1530/jrf.0.0880695 [DOI] [PubMed] [Google Scholar]
- 14. Grinspon RP, Rey RA. Anti-Mullerian Hormone and Sertoli Cell Function in Paediatric Male Hypogonadism. Horm Res Paediatr (2010) 73:81–92. doi: 10.1159/000277140 [DOI] [PubMed] [Google Scholar]
- 15. Josso N, Picard JY. Genetics of Anti-Mullerian Hormone and its Signaling Pathway. Best Pract Res Clin Endocrinol Metab (2022) 36:101634. doi: 10.1016/j.beem.2022.101634 [DOI] [PubMed] [Google Scholar]
- 16. Lukas-Croisier C, Lasala C, Nicaud J, Bedecarrás P, Kumar TR, Dutertre M, et al. Follicle-Stimulating Hormone Increases Testicular Anti-Müllerian Hormone (AMH) Production Through Sertoli Cell Proliferation and a Nonclassical Cyclic Adenosine 5'-Monophosphate-Mediated Activation of the AMH Gene. Mol Endocrinol (2003) 17:550–61. doi: 10.1210/me.2002-0186 [DOI] [PubMed] [Google Scholar]
- 17. Beau C, Vivian N, Münsterberg A, Dresser DW, Lovell-Badge R, Guerrier D. In Vivo Analysis of the Regulation of the Anti-Müllerian Hormone, as a Marker of Sertoli Cell Differentiation During Testicular Development, Reveals a Multi-Step Process. Mol Reprod Dev (2001) 59:256–64. doi: 10.1002/mrd.1030 [DOI] [PubMed] [Google Scholar]
- 18. Edelsztein NY, Rey RA. Importance of the Androgen Receptor Signaling in Gene Transactivation and Transrepression for Pubertal Maturation of the Testis. Cells (2019) 8:1–17. doi: 10.3390/cells8080861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valeri C, Lovaisa MM, Racine C, Edelsztein NY, Riggio M, Giulianelli S, et al. Molecular Mechanisms Underlying AMH Elevation in Hyperoestrogenic States in Males. Sci Rep (2020) 10:15062. doi: 10.1038/s41598-020-71675-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tran D, Meusy-Dessolle N, Josso N. Anti-Müllerian Hormone is a Functional Marker of Foetal Sertoli Cells. Nature (1977) 269:411–2. doi: 10.1038/269411a0 [DOI] [PubMed] [Google Scholar]
- 21. Münsterberg A, Lovell-Badge R. Expression of the Mouse Anti-Müllerian Hormone Gene Suggests a Role in Both Male and Female Sexual Differentiation. Development (1991) 113:613–24. doi: 10.1242/dev.113.2.613 [DOI] [PubMed] [Google Scholar]
- 22. Josso N, Lamarre I, Picard JY, Berta P, Davies N, Morichon N, et al. Anti-Müllerian Hormone in Early Human Development. Early Hum Dev (1993) 33:91–9. doi: 10.1016/0378-3782(93)90204-8 [DOI] [PubMed] [Google Scholar]
- 23. Rey RA, Musse M, Venara M, Chemes HE. Ontogeny of the Androgen Receptor Expression in the Fetal and Postnatal Testis: Its Relevance on Sertoli Cell Maturation and the Onset of Adult Spermatogenesis. Microscopy Res Technique (2009) 72:787–95. doi: 10.1002/jemt.20754 [DOI] [PubMed] [Google Scholar]
- 24. Grinspon RP, Bedecarrás P, Ballerini MG, Iñíguez G, Rocha A, Mantovani Rodrigues Resende EA, et al. Early Onset of Primary Hypogonadism Revealed by Serum Anti-Müllerian Hormone Determination During Infancy and Childhood in Trisomy 21. Int J Androl (2011) 34:e487–98. doi: 10.1111/j.1365-2605.2011.01210.x [DOI] [PubMed] [Google Scholar]
- 25. Tran D, Meusy-Dessolle N, Josso N. Waning of Anti-Müllerian Activity: An Early Sign of Sertoli Cell Maturation in the Developing Pig. Biol Reprod (1981) 24:923–31. doi: 10.1095/biolreprod24.4.923 [DOI] [PubMed] [Google Scholar]
- 26. Josso N, Picard JY, Dacheux JL, Courot M. Detection of Anti-Müllerian Activity in Boar Rete Testis Fluid. J Reprod Fertil (1979) 57:397–400. doi: 10.1530/jrf.0.0570397 [DOI] [PubMed] [Google Scholar]
- 27. Rey R, Lukas-Croisier C, Lasala C, Bedecarrás P. AMH/MIS: What We Know Already About the Gene, the Protein and Its Regulation. Mol Cell Endocrinology (2003) 211:21–31. doi: 10.1016/j.mce.2003.09.007 [DOI] [PubMed] [Google Scholar]
- 28. Hirobe S, He WW, Lee MM, Donahoe PK. Mullerian Inhibiting Substance Messenger Ribonucleic Acid Expression in Granulosa and Sertoli Cells Coincides With Their Mitotic Activity. Endocrinology (1992) 131:854–62. doi: 10.1210/endo.131.2.1639028 [DOI] [PubMed] [Google Scholar]
- 29. Rey R, Al-Attar L, Louis F, Jaubert F, Barbet P, Nihoul-Fékété C, et al. Testicular Dysgenesis Does Not Affect Expression of Anti-Mullerian Hormone by Sertoli Cells in Premeiotic Seminiferous Tubules. Am J Pathology (1996) 148:1689–98. [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Attar L, Noël K, Dutertre M, Belville C, Forest MG, Burgoyne PS, et al. Hormonal and Cellular Regulation of Sertoli Cell Anti-Müllerian Hormone Production in the Postnatal Mouse. J Clin Invest (1997) 100:1335–43. doi: 10.1172/JCI119653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hagen CP, Aksglæde L, Sorensen K, Main KM, Boas M, Cleemann L, et al. Serum Levels of Anti-Mullerian Hormone as a Marker of Ovarian Function in 926 Healthy Females From Birth to Adulthood and in 172 Turner Syndrome Patients. J Clin Endocrinol Metab (2010) 95:5003–10. doi: 10.1210/jc.2010-0930 [DOI] [PubMed] [Google Scholar]
- 32. Jost A. Problems of Fetal Endocrinology: The Gonadal and Hypophyseal Hormones. Recent Prog Hormone Res (1953) 8:379–418. [Google Scholar]
- 33. Grinspon RP, Bergadá I, Rey RA. Male Hypogonadism and Disorders of Sex Development. Front Endocrinol (Lausanne) (2020) 11:211. doi: 10.3389/fendo.2020.00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taguchi O, Cunha GR, Lawrence WD, Robboy SJ. Timing and Irreversibility of Müllerian Duct Inhibition in the Embryonic Reproductive Tract of the Human Male. DevBiol (1984) 106:394–8. doi: 10.1016/0012-1606(84)90238-0 [DOI] [PubMed] [Google Scholar]
- 35. Kuiri-Hänninen T, Kallio S, Seuri R, Tyrvainen E, Liakka A, Tapanainen J, et al. Postnatal Developmental Changes in the Pituitary-Ovarian Axis in Preterm and Term Infant Girls. J Clin Endocrinol Metab (2011) 96:3432–9. doi: 10.1210/jc.2011-1502 [DOI] [PubMed] [Google Scholar]
- 36. Josso N, Rey RA, Picard JY. Anti-Müllerian Hormone: A Valuable Addition to the Toolbox of the Pediatric Endocrinologist. Int J Endocrinol (2013) 2013:674105. doi: 10.1155/2013/674105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Condorelli RA, Cannarella R, Calogero AE, La Vignera S. Evaluation of Testicular Function in Prepubertal Children. Endocrine (2018) 62:274–80. doi: 10.1007/s12020-018-1670-9 [DOI] [PubMed] [Google Scholar]
- 38. Rey RA. Biomarkers of Male Hypogonadism in Childhood and Adolescence. Adv Lab Med (2020) 2020:24. doi: 10.1515/almed-2020-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanakatti Shankar R, Dowlut-McElroy T, Dauber A, Gomez-Lobo V. Clinical Utility of Anti-Mullerian Hormone in Pediatrics. J Clin Endocrinol Metab (2022) 107:309–23. doi: 10.1210/clinem/dgab687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee MM, Donahoe PK, Silverman BL, Hasegawa T, Hasegawa Y, Gustafson ML, et al. Measurements of Serum Müllerian Inhibiting Substance in the Evaluation of Children With Nonpalpable Gonads. New Engl J Med (1997) 336:1480–6. doi: 10.1056/NEJM199705223362102 [DOI] [PubMed] [Google Scholar]
- 41. Josso N, Rey RA. What Does AMH Tell Us in Pediatric Disorders of Sex Development? Front Endocrinol (Lausanne) (2020) 11:619. doi: 10.3389/fendo.2020.00619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grinspon RP, Rey RA. Molecular Characterization of XX Maleness. Int J Mol Sci (2019) 20:6089. doi: 10.3390/ijms20236089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Misra M, MacLaughlin DT, Donahoe PK, Lee MM. Measurement of Mullerian Inhibiting Substance Facilitates Management of Boys With Microphallus and Cryptorchidism. J Clin Endocrinol Metab (2002) 87:3598–602. doi: 10.1210/jcem.87.8.8742 [DOI] [PubMed] [Google Scholar]
- 44. Grinspon RP, Gottlieb S, Bedecarras P, Rey RA. Anti-Müllerian Hormone and Testicular Function in Prepubertal Boys With Cryptorchidism. Front Endocrinol (Lausanne) (2018) 9:182:182. doi: 10.3389/fendo.2018.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grinspon RP, Castro S, Brunello FG, Sansó G, Ropelato MG, Rey RA. Diagnosis of Male Central Hypogonadism During Childhood. J Endocrine Society (2021) 5:1–8. doi: 10.1210/jendso/bvab145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Venara M, Rey R, Bergadá I, Mendilaharzu H, Campo SM, Chemes H. Sertoli Cell Proliferations of the Infantile Testis: An Intratubular Form of Sertoli Cell Tumor? Am J Surg Pathol (2001) 25:1237–44. doi: 10.1097/00000478-200110000-00003 [DOI] [PubMed] [Google Scholar]
- 47. Rey RA, Venara M, Coutant R, Trabut JB, Rouleau S, Lahlou N, et al. Unexpected Mosaicism of R201H-GNAS1 Mutant-Bearing Cells in the Testes Underlie Macro-Orchidism Without Sexual Precocity in McCune-Albright Syndrome. Hum Mol Genet (2006) 15:3538–43. doi: 10.1093/hmg/ddl430 [DOI] [PubMed] [Google Scholar]
- 48. Rey R, Lordereau-Richard I, Carel JC, Barbet P, Cate RL, Roger M, et al. Anti-Müllerian Hormone and Testosterone Serum Levels Are Inversely Related During Normal and Precocious Pubertal Development. J Clin Endocrinol Metab (1993) 77:1220–6. doi: 10.1210/jc.2005-2564 [DOI] [PubMed] [Google Scholar]
- 49. Bastida MG, Rey RA, Bergadá I, Bedecarrás P, Andreone L, del Rey G, et al. Establishment of Testicular Endocrine Function Impairment During Childhood and Puberty in Boys With Klinefelter Syndrome. Clin Endocrinol (Oxf) (2007) 67:863–70. doi: 10.1111/j.1365-2265.2007.02977.x [DOI] [PubMed] [Google Scholar]
- 50. Rey R, Mebarki F, Forest MG, Mowszowicz I, Cate RL, Morel Y, et al. Anti-Müllerian Hormone in Children With Androgen Insensitivity. J Clin Endocrinol Metab (1994) 79:960–4. doi: 10.1210/jcem.79.4.7962305 [DOI] [PubMed] [Google Scholar]
- 51. Arango NA, Lovell-Badge R, Behringer RR. Targeted Mutagenesis of the Endogenous Mouse Mis Gene Promoter: In Vivo Definition of Genetic Pathways of Vertebrate Sexual Development. Cell (1999) 99:409–19. doi: 10.1016/s0092-8674(00)81527-5 [DOI] [PubMed] [Google Scholar]
- 52. Shen WH, Moore CC, Ikeda Y, Parker KL, Ingraham HA. Nuclear Receptor Steroidogenic Factor 1 Regulates the Müllerian Inhibiting Substance Gene: A Link to the Sex Determination Cascade. Cell (1994) 77:651–61. doi: 10.1016/0092-8674(94)90050-7 [DOI] [PubMed] [Google Scholar]
- 53. Schteingart HF, Picard JY, Valeri C, Marshall I, Treton D, di Clemente N, et al. A Mutation Inactivating the Distal SF1 Binding Site on the Human Anti-Mullerian Hormone Promoter Causes Persistent Mullerian Duct Syndrome. Hum Mol Genet (2019) 28:3211–8. doi: 10.1093/hmg/ddz147 [DOI] [PubMed] [Google Scholar]
- 54. Viger RS, Mertineit C, Trasler JM, Nemer M. Transcription Factor GATA-4 is Expressed in a Sexually Dimorphic Pattern During Mouse Gonadal Development and Is a Potent Activator of the Müllerian Inhibiting Substance Promoter. Development (1998) 125:2665–75. doi: 10.1242/dev.125.14.2665 [DOI] [PubMed] [Google Scholar]
- 55. Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan JN, Hammer GD, Ingraham HA. Wilms' Tumor 1 and Dax-1 Modulate the Orphan Nuclear Receptor SF-1 in Sex-Specific Gene Expression. Cell (1998) 93:445–54. doi: 10.1016/S0092-8674(00)81172-1 [DOI] [PubMed] [Google Scholar]
- 56. Tremblay JJ, Viger RS. Nuclear Receptor Dax-1 Represses the Transcriptional Cooperation Between GATA-4 and SF-1 in Sertoli Cells. Biol Reprod (2001) 64:1191–9. doi: 10.1095/biolreprod64.4.1191 [DOI] [PubMed] [Google Scholar]
- 57. Bergadá I, Milani C, Bedecarrás P, Andreone L, Ropelato MG, Gottlieb S, et al. Time Course of the Serum Gonadotropin Surge, Inhibins, and Anti-Mullerian Hormone in Normal Newborn Males During the First Month of Life. J Clin Endocrinol Metab (2006) 91:4092–8. doi: 10.1210/jc.2006-1079 [DOI] [PubMed] [Google Scholar]
- 58. Busch AS, Ljubicic ML, Upners EN, Fischer MB, Raket LL, Frederiksen H, et al. Dynamic Changes of Reproductive Hormones in Male Minipuberty: Temporal Dissociation of Leydig- and Sertoli-Cell Activity. J Clin Endocrinol Metab (2022). doi: 10.1210/clinem/dgac115 [DOI] [PubMed] [Google Scholar]
- 59. Braslavsky D, Grinspon RP, Ballerini MG, Bedecarrás P, Loreti N, Bastida G, et al. Hypogonadotropic Hypogonadism in Infants With Congenital Hypopituitarism: A Challenge to Diagnose at an Early Stage. Horm Res Paediatr (2015) 84:289–97. doi: 10.1159/000439051 [DOI] [PubMed] [Google Scholar]
- 60. Young J, Chanson P, Salenave S, Noel M, Brailly S, O'Flaherty M, et al. Testicular Anti-Mullerian Hormone Secretion Is Stimulated by Recombinant Human FSH in Patients With Congenital Hypogonadotropic Hypogonadism. J Clin Endocrinol Metab (2005) 90:724–8. doi: 10.1210/jc.2004-0542 [DOI] [PubMed] [Google Scholar]
- 61. Bougnères P, François M, Pantalone L, Rodrigue D, Bouvattier C, Demesteere E, et al. Effects of an Early Postnatal Treatment of Hypogonadotropic Hypogonadism With a Continuous Subcutaneous Infusion of Recombinant Follicle-Stimulating Hormone and Luteinizing Hormone. J Clin Endocrinol Metab (2008) 93:2202–5. doi: 10.1210/jc.2008-0121 [DOI] [PubMed] [Google Scholar]
- 62. Lasala C, Schteingart HF, Arouche N, Bedecarrás P, Grinspon RP, Picard JY, et al. SOX9 and SF1 are Involved in Cyclic AMP-Mediated Upregulation of Anti-Mullerian Gene Expression in the Testicular Prepubertal Sertoli Cell Line SMAT1. Am J Physiol Endocrinol Metab (2011) 301:E539-547. doi: 10.1152/ajpendo.00187.2011 [DOI] [PubMed] [Google Scholar]
- 63. Mamkin I, Philibert P, Anhalt H, Ten S, Sultan C. Unusual Phenotypical Variations in a Boy With McCune-Albright Syndrome. Horm Res Paediatr (2010) 73:215–22. doi: 10.1159/000284365 [DOI] [PubMed] [Google Scholar]
- 64. Berensztein EB, Baquedano MS, González CR, Saraco NI, Rodriguez J, Ponzio R, et al. Expression of Aromatase, Estrogen Receptor Alpha and Beta, Androgen Receptor, and Cytochrome P-450scc in the Human Early Prepubertal Testis. Pediatr Res (2006) 60:740–4. doi: 10.1203/01.pdr.0000246072.04663.bb [DOI] [PubMed] [Google Scholar]
- 65. Chemes HE, Rey RA, Nistal M, Regadera J, Musse M, González-Peramato P, et al. Physiological Androgen Insensitivity of the Fetal, Neonatal, and Early Infantile Testis Is Explained by the Ontogeny of the Androgen Receptor Expression in Sertoli Cells. J Clin Endocrinol Metab (2008) 93:4408–12. doi: 10.1210/jc.2008-0915 [DOI] [PubMed] [Google Scholar]
- 66. Boukari K, Meduri G, Brailly-Tabard S, Guibourdenche J, Ciampi ML, Massin N, et al. Lack of Androgen Receptor Expression in Sertoli Cells Accounts for the Absence of Anti-Mullerian Hormone Repression During Early Human Testis Development. J Clin Endocrinol Metab (2009) 94:1818–25. doi: 10.1210/jc.2008-1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Majdic G, Millar MR, Saunders PT. Immunolocalisation of Androgen Receptor to Interstitial Cells in Fetal Rat Testes and to Mesenchymal and Epithelial Cells of Associated Ducts. J Endocrinol (1995) 147:285–93. doi: 10.1677/joe.0.1470285 [DOI] [PubMed] [Google Scholar]
- 68. Grinspon RP, Andreone L, Bedecarrás P, Ropelato MG, Rey RA, Campo SM, et al. Male Central Precocious Puberty: Serum Profile of Anti-Mullerian Hormone and Inhibin B Before, During, and After Treatment With GnRH Analogue. Int J Endocrinol (2013) 2013:823064. doi: 10.1155/2013/823064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rey RA. The Role of Androgen Signaling in Male Sexual Development at Puberty. Endocrinology (2021) 162:bqaa215. doi: 10.1210/endocr/bqaa215 [DOI] [PubMed] [Google Scholar]
- 70. Racine C, Pask AJ, Wijayanti GE, di Clemente N, Picard JY, Shaw G, et al. Early Expression of the Androgen Receptor in the Sertoli Cells of a Marsupial Coincides With Downregulation of Anti-Mullerian Hormone at the Time of Urogenital Virilization. Sex Dev (2009) 3:317–25. doi: 10.1159/000273263 [DOI] [PubMed] [Google Scholar]
- 71. Almeida J, Conley AJ, Mathewson L, Ball BA. Expression of Anti-Mullerian Hormone, Cyclin-Dependent Kinase Inhibitor (CDKN1B), Androgen Receptor, and Connexin 43 in Equine Testes During Puberty. Theriogenology (2012) 77:847–57. doi: 10.1016/j.theriogenology.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 72. Chang C, Chen YT, Yeh SD, Xu Q, Wang RS, Guillou F, et al. Infertility With Defective Spermatogenesis and Hypotestosteronemia in Male Mice Lacking the Androgen Receptor in Sertoli Cells. Proc Natl Acad Sci USA (2004) 101:6876–81. doi: 10.1073/pnas.0307306101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith LB, Walker WH. The Regulation of Spermatogenesis by Androgens. Semin Cell Dev Biol (2014) 30:2–13. doi: 10.1016/j.semcdb.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Denayer S, Helsen C, Thorrez L, Haelens A, Claessens F. The Rules of DNA Recognition by the Androgen Receptor. Mol Endocrinol (2010) 24:898–913. doi: 10.1210/me.2009-0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Heckert LL, Wilson EM, Nilson JH. Transcriptional Repression of the {Alpha}-Subunit Gene by Androgen Receptor Occurs Independently of DNA Binding But Requires the DNA-Binding and Ligand-Binding Domains of the Receptor. Mol Endocrinol (1997) 11:1497–506. doi: 10.1210/mend.11.10.9996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shang Y, Myers M, Brown M. Formation of the Androgen Receptor Transcription Complex. Mol Cell (2002) 9:601–10. doi: 10.1016/S1097-2765(02)00471-9 [DOI] [PubMed] [Google Scholar]
- 77. Bulldan A, Dietze R, Shihan M, Scheiner-Bobis G. Non-Classical Testosterone Signaling Mediated Through ZIP9 Stimulates Claudin Expression and Tight Junction Formation in Sertoli Cells. Cell Signal (2016) 28:1075–85. doi: 10.1016/j.cellsig.2016.04.015 [DOI] [PubMed] [Google Scholar]
- 78. Gautam M, Bhattacharya I, Rai U, Majumdar SS. Hormone Induced Differential Transcriptome Analysis of Sertoli Cells During Postnatal Maturation of Rat Testes. PloS One (2018) 13:e0191201. doi: 10.1371/journal.pone.0191201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dutertre M, Rey R, Porteu A, Josso N, Picard JY. A Mouse Sertoli Cell Line Expressing Anti-Müllerian Hormone and Its Type II Receptor. Mol Cell Endocrinology (1997) 136:57–65. doi: 10.1016/s0303-7207(97)00214-1 [DOI] [PubMed] [Google Scholar]
- 80. Edelsztein NY, Racine C, di Clemente N, Schteingart HF, Rey RA. Androgens Downregulate Anti-Mullerian Hormone Promoter Activity in the Sertoli Cell Through the Androgen Receptor and Intact SF1 Sites. Biol Reprod (2018) 99:1303–12. doi: 10.1093/biolre/ioy152 [DOI] [PubMed] [Google Scholar]
- 81. Hero M, Tommiska J, Vaaralahti K, Laitinen EM, Sipila I, Puhakka L, et al. Circulating Antimullerian Hormone Levels in Boys Decline During Early Puberty and Correlate With Inhibin B. Fertil Steril (2012) 97:1242–7. doi: 10.1016/j.fertnstert.2012.02.020 [DOI] [PubMed] [Google Scholar]
- 82. Grinspon R, Chemes H, Rey RA. Decline in Serum Antimullerian Hormone Due to Androgen Action in Early Puberty in Males. Fertil Steril (2012) 98:e23. doi: 10.1016/j.fertnstert.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 83. Rajpert-De Meyts E, Jørgensen N, Græm N, Müller J, Cate RL, Skakkebæk NE. Expression of Anti-Müllerian Hormone During Normal and Pathological Gonadal Development: Association With Differentiation of Sertoli and Granulosa Cells. J Clin Endocrinol Metab (1999) 84:3836–44. doi: 10.1210/jcem.84.10.6047 [DOI] [PubMed] [Google Scholar]
- 84. Rey RA, Belville C, Nihoul-Fékété C, Michel-Calemard L, Forest MG, Lahlou N, et al. Evaluation of Gonadal Function in 107 Intersex Patients by Means of Serum Antimüllerian Hormone Measurement. J Clin Endocrinol Metab (1999) 84:627–31. doi: 10.1210/jcem.84.2.5507 [DOI] [PubMed] [Google Scholar]
- 85. Liu Q, Yin X, Li P. Clinical, Hormonal and Genetic Characteristics of Androgen Insensitivity Syndrome in 39 Chinese Patients. Reprod Biol Endocrinol (2020) 18:34. doi: 10.1186/s12958-020-00593-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lan KC, Chen YT, Chang C, Chang YC, Lin HJ, Huang KE, et al. Up-Regulation of SOX9 in Sertoli Cells From Testiculopathic Patients Accounts for Increasing Anti-Mullerian Hormone Expression via Impaired Androgen Receptor Signaling. PloS One (2013) 8:e76303. doi: 10.1371/journal.pone.0076303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dorrington JH, Armstrong DT. Follicle-Stimulating Hormone Stimulates Estradiol-17beta Synthesis in Cultured Sertoli Cells. Proc Natl Acad Sci USA (1975) 72:2677–81. doi: 10.1073/pnas.72.7.2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of Estrogen Action. Physiol Rev (2001) 81:1535–65. doi: 10.1152/physrev.2001.81.4.1535 [DOI] [PubMed] [Google Scholar]
- 89. Prossnitz ER, Barton M. Estrogen Biology: New Insights Into GPER Function and Clinical Opportunities. Mol Cell Endocrinol (2014) 389:71–83. doi: 10.1016/j.mce.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev (2017) 97:995–1043. doi: 10.1152/physrev.00018.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lucas TFG, Lazari MFM, Porto CS. Differential Role of the Estrogen Receptors ESR1 and ESR2 on the Regulation of Proteins Involved With Proliferation and Differentiation of Sertoli Cells From 15-Day-Old Rats. Mol Cell Endocrinol (2014) 382:84–96. doi: 10.1016/j.mce.2013.09.015 [DOI] [PubMed] [Google Scholar]
- 92. Lucas TF, Royer C, Siu ER, Lazari MF, Porto CS. Expression and Signaling of G Protein-Coupled Estrogen Receptor 1 (GPER) in Rat Sertoli Cells. Biol Reprod (2010) 83:307–17. doi: 10.1095/biolreprod.110.084160 [DOI] [PubMed] [Google Scholar]
- 93. Grinspon RP, Urrutia M. The Importance of Follicle-Stimulating Hormone in the Prepubertal and Pubertal Testis. Curr Opin Endocrine Metab Res (2020) 14:137–44. doi: 10.1016/j.coemr.2020.07.007 [DOI] [Google Scholar]
- 94. Grynberg M, Pierre A, Rey R, Leclerc A, Arouche N, Hesters L, et al. Differential Regulation of Ovarian Anti-Mullerian Hormone (AMH) by Estradiol Through Alpha- and Beta-Estrogen Receptors. J Clin Endocrinol Metab (2012) 97:E1649-1657. doi: 10.1210/jc.2011-3133 [DOI] [PubMed] [Google Scholar]
- 95. Yang WR, Zhu FW, Zhang JJ, Wang Y, Zhang JH, Lu C, et al. PI3K/Akt Activated by GPR30 and Src Regulates 17beta-Estradiol-Induced Cultured Immature Boar Sertoli Cells Proliferation. Reprod Sci (2017) 24:57–66. doi: 10.1177/1933719116649696 [DOI] [PubMed] [Google Scholar]
- 96. Matthiesson KL, Stanton PG, O'Donnell L, Meachem SJ, Amory JK, Berger R, et al. Effects of Testosterone and Levonorgestrel Combined With a 5alpha-Reductase Inhibitor or Gonadotropin-Releasing Hormone Antagonist on Spermatogenesis and Intratesticular Steroid Levels in Normal Men. J Clin Endocrinol Metab (2005) 90:5647–55. doi: 10.1210/jc.2005-0639 [DOI] [PubMed] [Google Scholar]
- 97. Bernard V, Kherra S, Francou B, Fagart J, Viengchareun S, Guechot J, et al. Familial Multiplicity of Estrogen Insensitivity Associated With a Loss-Of-Function ESR1 Mutation. J Clin Endocrinol Metab (2017) 102:93–9. doi: 10.1210/jc.2016-2749 [DOI] [PMC free article] [PubMed] [Google Scholar]