

Abstract
Nucleoside diphosphate sugar (NDP-sugar) substrates provide the inspiration for nucleoside analog inhibitor scaffolds. By employing solid-phase synthesis, we provide a method to access a library of peptidouridine inhibitors with both minimal compound handling and purification steps. Specifically, this strategy is exemplified by generating uridine diphosphate sugar (UDP-sugar) mimics, which allows compound elaboration by altering the dipeptide composition, the N-terminal linkage, and a pendant aryl group. To exemplify the versatility 41 unique nucleoside analogs are presented.
Graphical Abstract
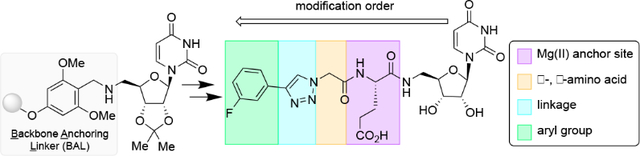
Nucleoside-containing natural products have provided a blueprint for inhibitor and antibiotic development. Many of these nucleoside-based inhibitors target glycan assembly pathways as these compounds structurally mimic the nucleoside diphosphate sugar (NDP-sugar) substrates. Specifically, uridine diphosphate (UDP)-sugar mimics (1) have shown inhibition towards glycosyl transferases, phosphoglycosyl transferases, and sugar-modifying enzymes (Figure 1).1 For example, muraymycin and analogs (2) inhibit MraY,2 a polytopic phosphoglycosyl transferase (PGT) responsible for transferring phospho-MurNAc-pentapeptide (Park’s nucleotide) to undecaprenyl phosphate in peptidoglycan biosynthesis.3 Additionally, analog libraries of sansanmycin (3) were found to target Mycobacterium tuberculosis (Mtb) MurX, an MraY-like enzyme in mycobacteria.4 However, the complexity of these uridine natural products, and the analogs that are based on them, make these targets synthetically cumbersome to access. Providing a streamlined strategy to access these scaffolds, while simultaneously providing opportunities for compound diversification, will expedite the production of high-value inhibitor libraries.
Figure 1.
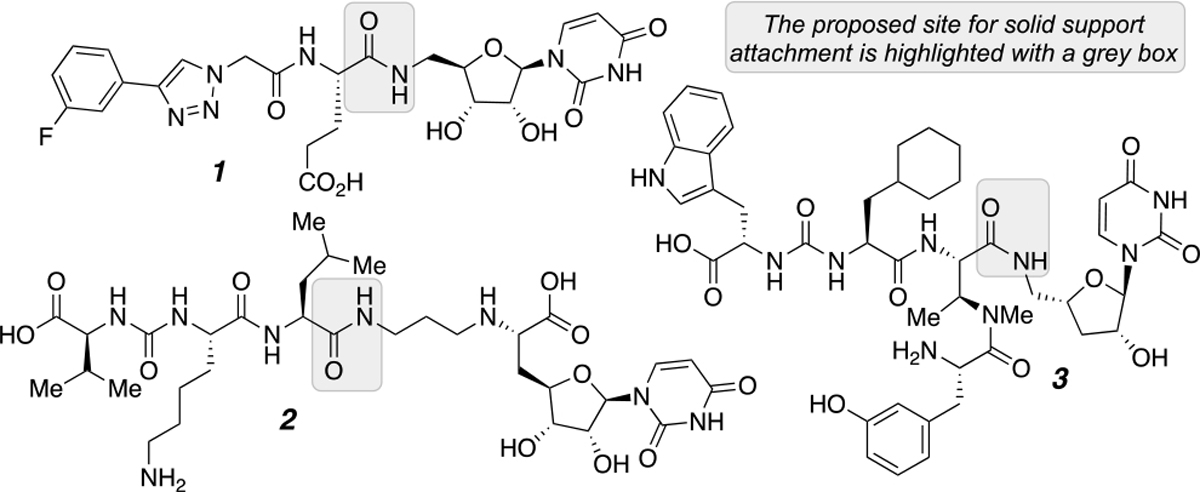
Select small molecule analogs derived from uridine natural products.
Solution-phase synthesis has been predominately employed to access uridine-based small molecules;2c, 5 however, the adaptation of a synthetic route to a solid-phase-based approach can improve yields by allowing the target to remain immobilized while excess reagents and solvents are washed away. To streamline inhibitor development, solid-phase approaches were developed by attaching uridine to the solid support via the C2’ and C3’ hydroxyls of the ribose,1a, 6 the uracil nitrogen,7 or by performing a semi-synthesis; synthesizing the peptide portion on resin, and attaching the uridine in solution.2d A disadvantage is that these strategies contain many manipulations to access key resin-bound intermediates to initiate inhibitor synthesis. More specifically, the previously reported solid-phase strategy that links uridine through the C2’ and C3’ hydroxyls contains multiple drawbacks.1a The amine source at the C5’ position is installed on resin through a Mitsunobu reaction with tetrachlorophthalimide. Once installed, the deprotection is executed with ethylenediamine; however, primary amines are known to prematurely cleave amino acids from Wang resin which can result in compound loss and lower yields.8 Moreover, the linker is attached to Ala-Wang resin. When treated with trifluoroacetic acid (TFA) to cleave the desired compound from the acetal linkage, simultaneous cleavage of the linker-Ala from the solid support could result in chromatography containing undesired side products. We envisioned designing a strategy that would circumvent these undesired side reactions. Furthermore, avoiding attachment at the ribose hydroxyls will have a greater impact on the nucleoside field as other nucleosides (e.g., thymidine) do not contain both C2’, C3’-hydroxyls required for attachment. At this time, we realized that many analogs based on UDP-sugars scaffolds (1),1a including muraymycin (2),2 and sansanmycin (3)4 contain an amide linkage proximal to the uridine core (Figure 1). This linkage site can be recapitulated in solid-phase synthesis by attaching a C5’-amino-nucleoside to the solid support.
Herein we present a solid-phase synthetic approach for uridine-linked small molecules using the backbone anchoring linker (BAL) resin (4, Scheme 1).9 This resin has been previously employed for backbone anchoring of peptides for C-terminal modifications and macrocyclization on resin.10 The backbone anchoring linker allows for minimal solution- and solid-phase manipulations to access the key intermediate for inhibitor elaboration. The 2’,3’-O-isopropylidene-C5’-aminouridine (5) was generated in solution from uridine in three steps. To attach the protected C5’-aminouridine to resin (4), compound 5 was dissolved in 1% AcOH/DMF and added to the solid support to form the corresponding imine. Subsequent addition of NaCNBH3 in MeOH overnight affords the resin-bound secondary amine 6. The conversion of the reductive amination reaction was monitored by the colorimetric nitrophenylhydrazine test.11 The secondary amine can be treated with standard solid-phase amide coupling conditions to elongate on resin. The final compound can be readily cleaved from the solid support with 95% TFA due to the labile nature of the linker.
Scheme 1.
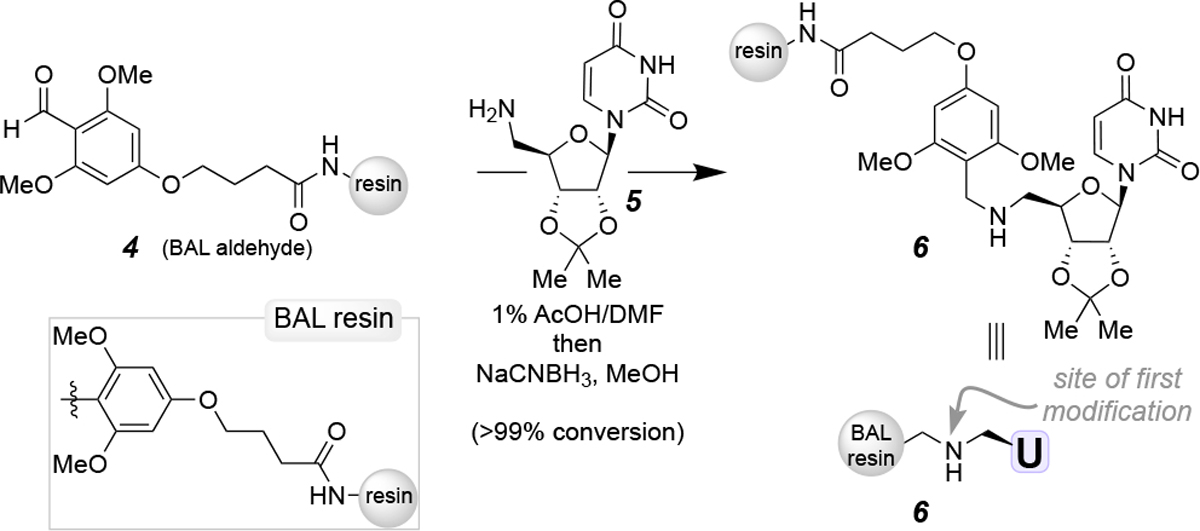
Attachment of C5’-aminouridine to the aldehyde-functionalized solid support.
We have exemplified this solid-phase synthetic strategy by developing inhibitors that may target bacterial PGTs. PGTs are responsible for catalyzing the initial membrane-committed step in the biosynthesis of N-linked glycoproteins associated with bacterial virulence.12 Previous work has identified key features for analog development, including (1) uridine, (2) a carboxylate to substitute or coordinate a metal ion at the active site, (3) a side chain to occupy the carbohydrate-binding site, and (4) an alkyl chain to mimic a prenyl moiety.13 These features ultimately identified the m-fluoro-phenyl-substituted analog (1) which exhibited the most encouraging inhibition of a monotopic PGT from Campylobacter concisus (IC50 of 72 ± 7 μM), a potential emerging pathogen of the human intestinal tract. 14 Herein, this compound will be referred to as the benchmark inhibitor (1). These results set an excellent starting point for strategic modifications of uridine analogs, thus we utilized this solid-phase strategy to provide an opportunity for structure-activity relationship (SAR) exploration. Keeping the essential uridine, we focused on exploring the Mg(II) anchor site by altering the spatial orientation (D or L) of the amino-acid side chain or the distance (Asp or Glu) of the carboxylate from the peptide backbone. Moreover, the composition of the dipeptide in the peptidouridine can be altered to explore enhanced substrate mimetic properties. To introduce pharmacologically-relevant aryl moieties, we focused on the ease of the Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC),15 noting that an amine, amide, and urea linkage could also be installed through other elongation strategies. Lastly, we wanted to diversify the terminal aryl group as a surrogate for the carbohydrate portion of the UDP-sugar; carbohydrate binding sites are frequently lined with aromatic amino acid side chains.16
As the glutamic acid was previously reported to be important for binding,1a we first modified the length and spatial orientation of the amino-acid side chain to strategically diversify the Mg(II) anchor site. Starting from the C5’ of uridine the carboxyl-modified amino acid is the first site for variation of the benchmark inhibitor 1 (Scheme 2). The secondary amine 6 was coupled to various Fmoc-protected amino acids using standard coupling conditions, followed by Fmoc deprotection with 20% piperidine/DMF (7). The free N-terminus was coupled to azido acetic acid to poise 8 for a CuAAC.15 After the cycloaddition (9), the resin was treated with TFA to furnish the desired purified small molecules in 12–17% yield (10a-e). In addition to carboxylate amino acids, a carboxamide (Gln, 10d, 13% yield) and methyl ester (Glu(OMe), 10e, 7% yield) were prepared.
Scheme 2.
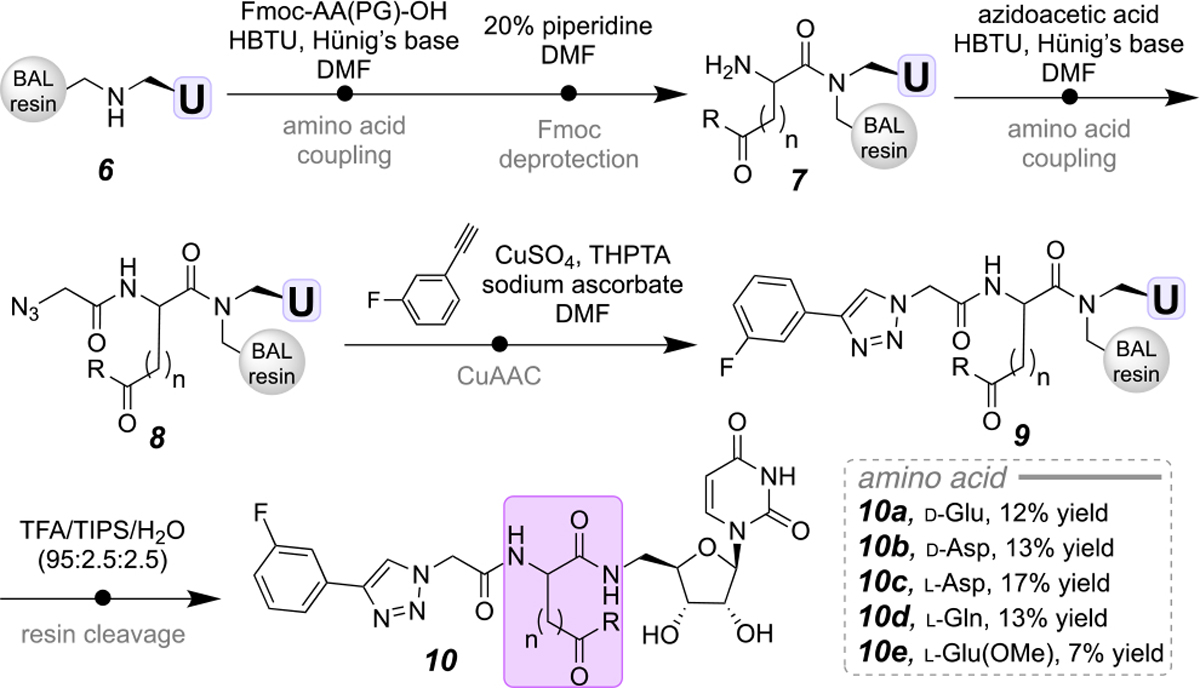
Modification of the metal-binding site amino acid in the PGT benchmark inhibitor.
The scaffold of benchmark inhibitor 1 was modified by substituting the second amino acid from the C-terminus (Scheme 3). To avoid synthesizing each amino acid with the N-terminal azide prior to coupling, the azide was installed on resin. This transformation was achieved using a diazotransfer reagent, imidazole sulfonyl azide, to conveniently convert the primary amine of the amino acid (11) to an azide in 2 h in the presence of Hünig’s base and DMSO.17 After the azide is installed, the CuAAC reaction was performed to produce the penultimate molecule (12) in Series 1. Subsequent resin cleavage with 95% TFA furnished the deprotected, purified compounds (13a-l, Series 1) in 6–27% yield over 8 steps. Figure 2 represents the excellent compound purity after reverse-phase HPLC (RP-HPLC) purification.
Scheme 3.
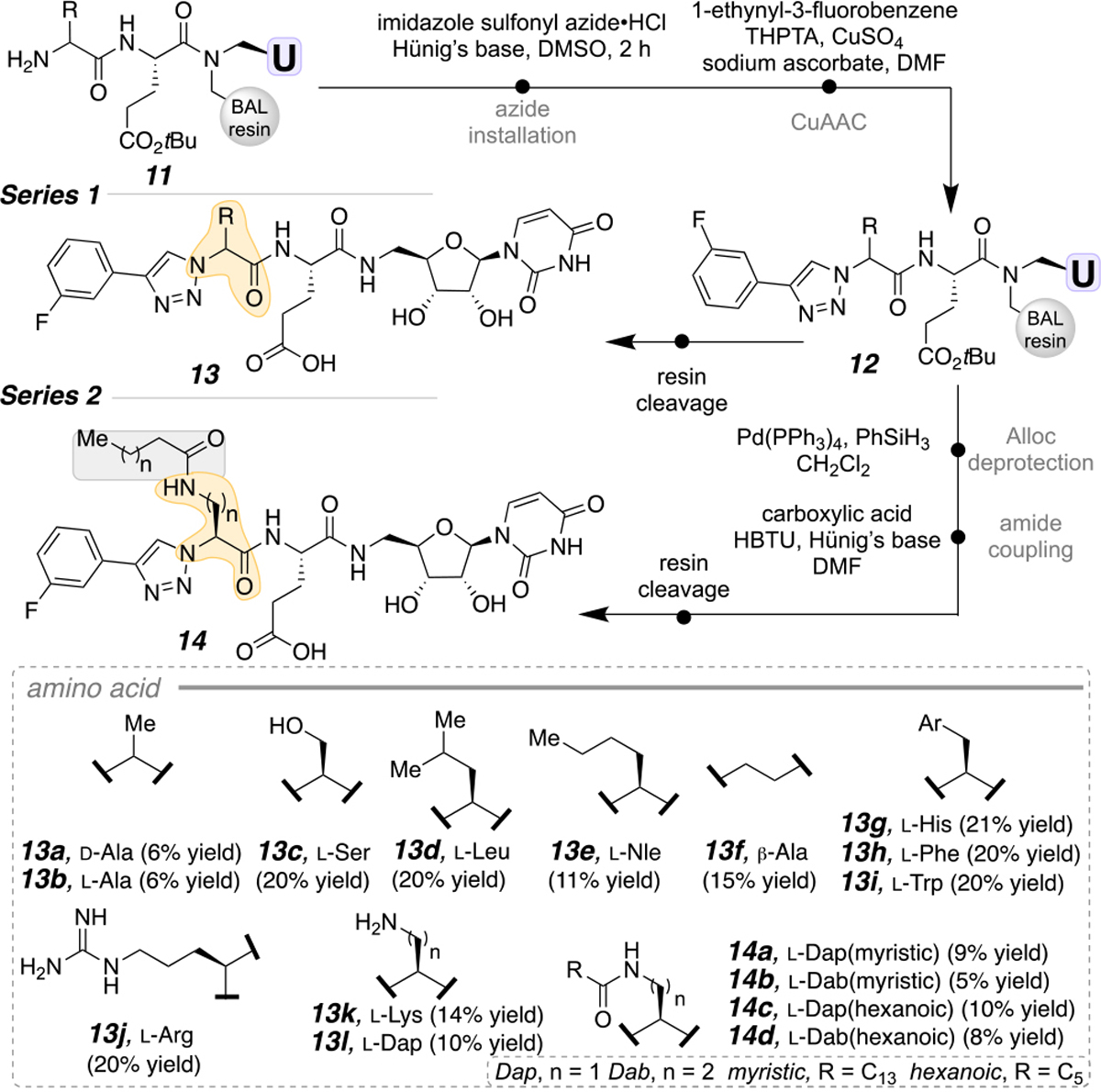
Glutamic acid-containing inhibitors with various modifications to the dipeptide scaffold.
Figure 2.
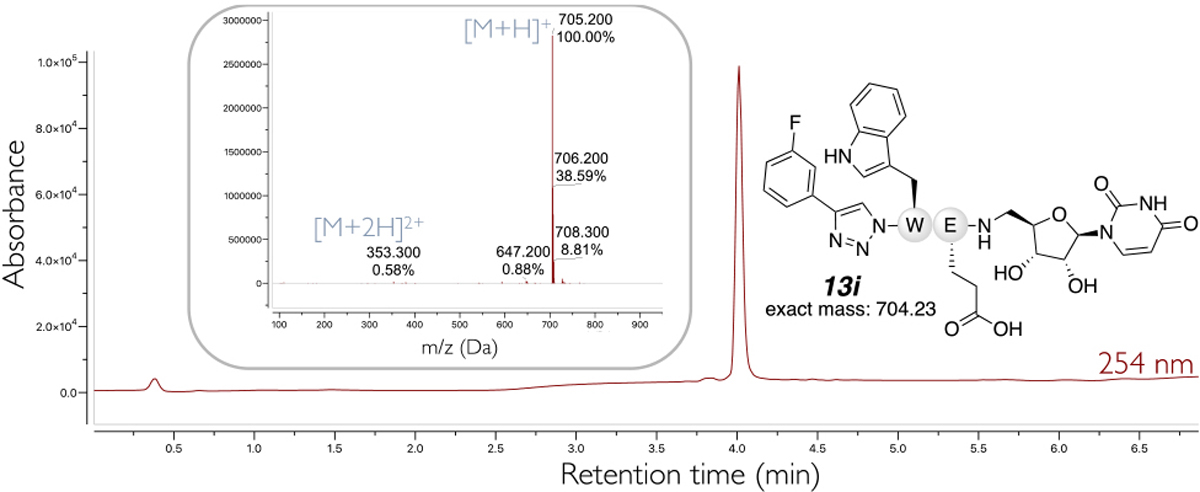
LCMS trace at 254 nm and ESI-MS spectrum of purified compound 13i from Scheme 3.
The successful incorporation of amino acids that contain nucleophilic side chains (Lys, Dap) inspired us to further functionalize those sites (14, Scheme 3, Series 2). To perform a bi-directional synthesis on the terminal amino acid, a non-acid/base labile protecting group was required to avoid premature resin cleavage. Thus, for Series 2 compounds, an allyl-based (allyloxycarbonyl, Alloc) protecting group was used. This protecting group was removed using a Pd-based catalyst (Pd(PPh3)4) in the presence of a reducing agent, PhSiH3, in CH2Cl2.18 The free amine of the amino acid side chain was coupled to both short- and long-chain carboxylic acids. Treating the resin with TFA and RP-HPLC purification furnished the acyl-modified molecules (14a-d) in 5–10% purified yield over 10 steps. Traditionally, these lipidic modifications, in particular the long-chain analogs, are challenging to incorporate in solution due to poor solubility. By immobilizing on the solid support, these solubility issues are circumvented by avoiding work-up and chromatography after each iteration.
In addition to coupling via CuAAC to furnish a triazole, we wanted to showcase the versatility of the solid-phase strategy with three additional elongation reactions (Scheme 4). These linkages are commonly found in natural products antibiotics.19 In this case, the aryl installation is achieved by coupling a carboxylic acid, performing a reductive amination, or reacting the terminal amine with a phenyl isocyanate, to ultimately produce amide-, amine-, or urea-linked small molecules, respectively. Specifically, the immobilized peptidouridine (11) was treated with a 3-F-Ph-R group with the appropriate elongation conditions.8 For Series 3, the resin was treated with TFA to afford compounds 15a-c in 3–18% purified yield. Series 4 included the installation of the lipidic myristoyl tail (16a-e), performed in a similar fashion to the triazole series from Scheme 3. First the Alloc-protecting group was removed using Pd, followed by subsequent coupling with myristic acid. The compounds were then cleaved from the resin providing the amide (15a, 16a, 16d), urea (15b, 16b, 16e), and amine (15c, 16c) in 3–18% yield after RP-HPLC purification.
Scheme 4.
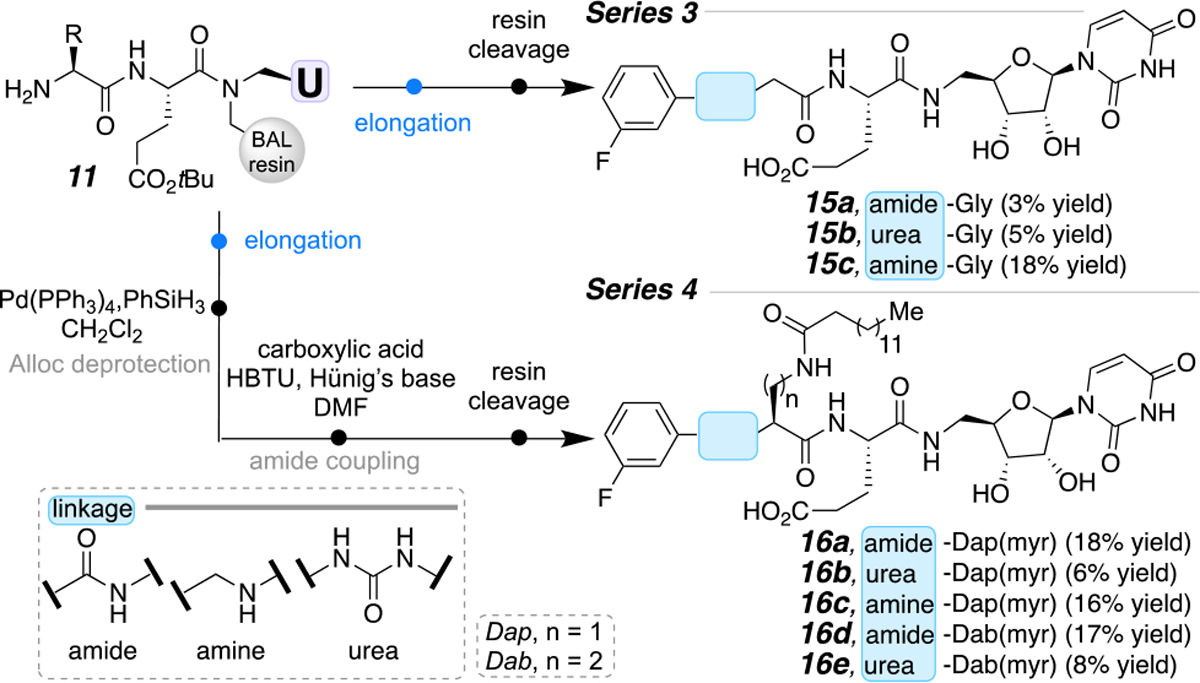
Modifying the aryl-connective moiety for both Gly and lipidic-containing dipeptides.
Lastly, the terminal aryl diversity was expanded to mimic the carbohydrate-portion of the UDP-sugar substrate by implementing the triazole linkage (Scheme 5). A significant proportion of biologically-active and, importantly, FDA-approved small molecules contain heterocycles.20 The incorporation of these heterocyclic components aids in compound lipophilicity, polarity, solubility, and hydrogen bonding capacity.20 For these reasons, both N- (17a, 17d-l) and S-containing (17b) heteroaromatic groups were incorporated (4–37% yield). Additionally, heteroatom-appended aryl groups were successfully installed (17a, 17c). These derivatives expanded on the uridinyl library data set previously obtained in our laboratory.1a Through these efforts we have established a routine to prepare ample amounts of purified materials to make 10 mM stock solutions in DMSO for plate-based inhibitor screening and distribution to other laboratories.
Scheme 5.
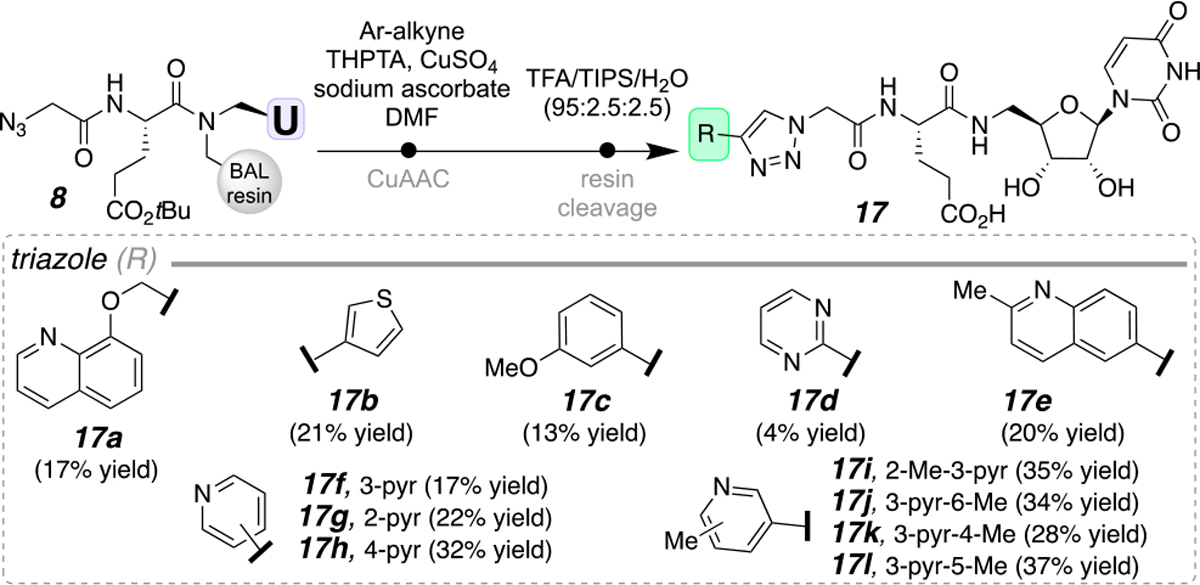
Terminal aryl modifications using the triazole connective moiety.
Although most compounds in this study were predicted to have optimal physicochemical properties,21 compound aggregation can be a problem in small molecule libraries.22 However, we hypothesize that the peptidyl-nucleoside structural core will prevent undesired colloid formation. As these undesired molecular features tend to be observed with flat, aromatic-rich molecules.23 Even so, this inhibitor promiscuity will be investigated with our library and reported in due course.
In addition to uridine, this strategy can be applied with other natural or pseudo nucleosides,1a, 24 including analogs with highly modified ribose moieties. Due to the structural similarities to uridine, thymidine and pseudouridine will be easy to employ. We have previously demonstrated the compatibility of pseudouridine with solid phase synthesis without the need of nucleobase protecting groups.1a Solution- phase synthesis to access C5’-substituted cytidine and analogs has been established,25 suggesting this solid-phase approach is feasible. Although also conceivable, guanosine and adenosine will require nucleobase-protecting groups prior to solid-phase immobilization to prevent unwanted side reactions.
Herein, we have established a new solid-phase synthesis strategy to access peptidouridine-containing small molecules. The compounds were strategically modified to provide maximum chemical diversity in a complex inhibitor library. This approach has provided a streamlined process to access natural product-inspired, nucleoside-containing small molecules. Additionally, in our laboratory this strategy will be employed for other nucleosides, affording potential inhibitors for enzymes that accept non-uridine nucleoside diphosphate sugar substrates.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the National Institute of Health (GM131627 to B.I. and F32GM136023 to C.A.A.) and Massachusetts Institute of Technology Research Support Committee (MIT RSC) is gratefully acknowledged. The authors would like to thank Dr. Leah Seebald (MIT) for providing substantial edits to the manuscript. We also acknowledge the MIT DCIF for NMR analyses and Dr. Mohan Kumar (MIT) with HR-MS assistance.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, full spectroscopic data, and copies of 1H and 13C NMR, and crude HPLC traces for all new compounds (PDF)
REFERENCES
- 1.(a) Madec AGE; Schocker NS; Sanchini S; Myratgeldiyev G; Das D; Imperiali B, Facile Solid-Phase Synthesis and Assessment of Nucleoside Analogs as Inhibitors of Bacterial UDP-Sugar Processing Enzymes. ACS Chem. Biol. 2018, 13 (9), 2542–2550; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Arbour CA; Imperiali B, Uridine natural products: Challenging targets and inspiration for novel small molecule inhibitors. Bioorg. Med. Chem. 2020, 28 (18), 115661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Spork AP; Koppermann S; Schier S; Linder R; Ducho C, Analogues of Muraymycin Nucleoside Antibiotics with Epimeric Uridine-Derived Core Structures. Molecules 2018, 23 (11), 2868; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niro G; Weck SC; Ducho C, Merging Natural Products: Muraymycin–Sansanmycin Hybrid Structures as Novel Scaffolds for Potential Antibacterial Agents. Chem. Eur. J. 2020, 26 (70), 16875–16887; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wiegmann D; Koppermann S; Wirth M; Niro G; Leyerer K; Ducho C, Muraymycin nucleoside-peptide antibiotics: uridine-derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J. Org. Chem. 2016, 12, 769–795; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Leyerer K; Koppermann S; Ducho C, Solid Phase-Supported Synthesis of Muraymycin Analogues. Eur. J. Org. Chem. 2019, 2019 (45), 7420–7431. [Google Scholar]
- 3.Chung BC; Zhao J; Gillespie RA; Kwon D-Y; Guan Z; Hong J; Zhou P; Lee S-Y, Crystal Structure of MraY, an Essential Membrane Enzyme for Bacterial Cell Wall Synthesis. Science 2013, 341 (6149), 1012–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Tran AT; Watson EE; Pujari V; Conroy T; Dowman LJ; Giltrap AM; Pang A; Wong WR; Linington RG; Mahapatra S; Saunders J; Charman SA; West NP; Bugg TDH; Tod J; Dowson CG; Roper DI; Crick DC; Britton WJ; Payne RJ, Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis. Nat. Commun. 2017, 8 (1), 14414; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tran W; Kusay AS; Hawkins PME; Cheung C-Y; Nagalingam G; Pujari V; Ford DJ; Stoye A; Ochoa JL; Audette RE; Hortle E; Oehlers SH; Charman SA; Linington RG; Rubin EJ; Dowson CG; Roper DI; Crick DC; Balle T; Cook GM; Britton WJ; Payne RJ, Synthetic Sansanmycin Analogues as Potent Mycobacterium tuberculosis Translocase I Inhibitors. J. Med. Chem. 2021, 64 (23), 17326–17345. [DOI] [PubMed] [Google Scholar]
- 5.(a) Babič A; Gobec S; Gravier-Pelletier C; Le Merrer Y; Pečar S, Synthesis of 1-C-linked diphosphate analogues of UDP-N-Ac-glucosamine and UDP-N-Ac-muramic acid. Tetrahedron 2008, 64 (38), 9093–9100; [Google Scholar]; (b) Moukha-chafiq O; Reynolds RC, Parallel Solution-Phase Synthesis and General Biological Activity of a Uridine Antibiotic Analog Library. ACS Comb. Sci. 2014, 16 (5), 232–237; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seebald L; Madec AGE; Imperiali B, Deploying Fluorescent Nucleoside Analogues for High-Throughput Inhibitor Screening. ChemBioChem 2020, 21 (1–2), 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozzoli A; Kazmierski W; Kennedy G; Pasquarello A; Pecunioso A, A solid-phase approach to analogues of the antibiotic mureidomycin. Bioorg. Med. Chem. Lett. 2000, 10 (24), 2759–2763. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y; Kurosu M, A new protecting group and linker for uridine ureido nitrogen. Tetrahedron 2012, 68 (24), 4797–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.See Supplementary Information for more details.
- 9.Boas U; Brask J; Jensen KJ, Backbone Amide Linker in Solid-Phase Synthesis. Chem. Rev. 2009, 109 (5), 2092–2118. [DOI] [PubMed] [Google Scholar]
- 10.Carbajo D; El-Faham A; Royo M; Albericio F, Optimized Stepwise Synthesis of the API Liraglutide Using BAL Resin and Pseudoprolines. ACS Omega 2019, 4 (5), 8674–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shannon SK; Barany G, Colorimetric Monitoring of Solid-Phase Aldehydes Using 2,4-Dinitrophenylhydrazine. J. Comb. Chem. 2004, 6 (2), 165–170. [DOI] [PubMed] [Google Scholar]
- 12.(a) Lukose V; Walvoort MT; Imperiali B, Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface. Glycobiology 2017, 27 (9), 820–833; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karlyshev AV; Everest P; Linton D; Cawthraw S; Newell DG; Wren BW, The Campylobacter jejuni general glycosylation system is important for attachment to human epithelial cells and in the colonization of chicks. Microbiology (Reading) 2004, 150 (Pt 6), 1957–1964. [DOI] [PubMed] [Google Scholar]
- 13.Walvoort MTC; Lukose V; Imperiali B, A Modular Approach to Phosphoglycosyltransferase Inhibitors Inspired by Nucleoside Antibiotics. Chem. Eur. J. 2016, 22 (11), 3856–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaakoush NO; Mitchell HM, Campylobacter concisus - A new player in intestinal disease. Front Cell Infect Microbiol 2012, 2, 4–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro V; Rodríguez H; Albericio F, CuAAC: An Efficient Click Chemistry Reaction on Solid Phase. ACS Comb. Sci. 2016, 18 (1), 1–14. [DOI] [PubMed] [Google Scholar]
- 16.Hudson KL; Bartlett GJ; Diehl RC; Agirre J; Gallagher T; Kiessling LL; Woolfson DN, Carbohydrate–Aromatic Interactions in Proteins. J. Am. Chem. Soc. 2015, 137 (48), 15152–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castro V; Blanco-Canosa JB; Rodriguez H; Albericio F, Imidazole-1-sulfonyl Azide-Based Diazo-Transfer Reaction for the Preparation of Azido Solid Supports for Solid-Phase Synthesis. ACS Comb. Sci. 2013, 15 (7), 331–334. [DOI] [PubMed] [Google Scholar]
- 18.Grieco P; Gitu PM; Hruby VJ, Preparation of ‘side-chain-to-side-chain’ cyclic peptides by Allyl and Alloc strategy: potential for library synthesis. J. Pept. Res. 2001, 57 (3), 250–256. [DOI] [PubMed] [Google Scholar]
- 19.Oliver M; Le Corre L; Poinsot M; Corio A; Madegard L; Bosco M; Amoroso A; Joris B; Auger R; Touzé T; Bouhss A; Calvet-Vitale S; Gravier-Pelletier C, Synthesis, biological evaluation and molecular modeling of urea-containing MraY inhibitors. Org. Biomol. Chem. 2021, 19 (26), 5844–5866. [DOI] [PubMed] [Google Scholar]
- 20.Jampilek J, Heterocycles in Medicinal Chemistry. Molecules 2019, 24 (21), 3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46 (1), 3–26. [DOI] [PubMed] [Google Scholar]
- 22.Feng BY; Shoichet BK, A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1 (2), 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGovern SL; Caselli E; Grigorieff N; Shoichet BK, A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45 (8), 1712–1722. [DOI] [PubMed] [Google Scholar]
- 24.Wang X-K; Jia Y-M; Li Y-X; Yu C-Y, Total Synthesis of Pseudouridimycin. Org. Lett. 2022. [DOI] [PubMed] [Google Scholar]
- 25.Moukha-Chafiq O; Reynolds RC; Wilson JC; Snowden TS, Parallel Solution Phase Synthesis and Preliminary Biological Activity of a 5′-Substituted Cytidine Analog Library. ACS Comb. Sci 2019, 21 (9), 628–634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.