

Abstract
Oncolytic virus therapy is a novel cancer treatment modality that has the potential to improve outcomes for patients with currently incurable malignancies. While intravascular delivery of therapeutic viruses provides access to disseminated tumors, this delivery route exposes the virus to opsonizing and inactivating factors in the blood, which limit the effective therapeutic virus dose and contribute to activation of systemic toxicities. When human species C adenovirus HAdv-C5 is delivered intravenously, natural immunoglobulin M antibodies (IgM) and coagulation factor X rapidly opsonize HAdv-C5, leading to virus sequestration in tissue macrophages, and promoting infection of liver cells, triggering hepatotoxicity. Here we show that the hypervariable region 1, HVR1, of the main HAdv-C5 capsid protein, hexon, mediates natural IgM antibody binding to the virus. Using compound targeted mutagenesis of hexon HVR1 loop and other functional sites known to mediate virus-host interactions, we have engineered and obtained a high-resolution cryo-EM structure of an adenovirus vector, Ad5-3M, which resists inactivation by blood factors, avoids sequestration in liver macrophages, and fails to trigger hepatotoxicity after intravenous delivery. Systemic delivery of Ad5-3M to mice with localized and disseminated lung cancer led to viral replication in tumor cells, suppression of tumor growth, and prolonged survival. Thus, compound targeted mutagenesis of functional sites in the virus capsid can be a generalizable approach to tailor virus interactions with the humoral and cellular arms of the immune system, allowing for generation of “designer” viruses with improved therapeutic properties.
One Sentence Summary:
“Designer” viruses for systemic cancer therapy.
Introduction
Oncolytic virotherapy has the potential to improve clinical outcomes in patients who are not responding or have become resistant to immunotherapy or chemotherapy drugs, addressing the need of the vast majority of cancer patents (1). Although direct intra-tumoral injection of the first FDA-approved oncolytic human herpes simplex virus-based drug Imlygic™ showed efficacy in treating patients with loco-regional melanoma lesions (2), disseminated metastatic disease remained largely refractory to Imlygic™ (3), suggesting that systemic delivery of oncolytic viruses will be needed to achieve control of disseminated cancers. While potentially advantageous, intravascular administration renders oncolytic viruses vulnerable to inactivation by humoral components of the innate and adaptive arms of the immune system, which evolved effective mechanisms to limit the spread of bacterial and viral pathogens to vital organs via the bloodstream (4). Natural immunoglobulin M antibodies (IgM) and complement components are among the most abundant proteins in the blood and are the key elements of the host anti-viral defense system that operate by opsonizing, inactivating, and targeting blood-borne viruses for degradation to phagocytic cells of the innate immune system (5). The generation of therapeutic viruses that would resist IgM- and complement-mediated inactivation is notoriously challenging, as natural IgM binding to viruses is thought to occur via “unspecific” low affinity - high avidity interactions with highly regularly structured multifunctional proteins at the virion surface, which are critical for virus entry into host cells. Oncolytic viruses based on vesicular stomatitis virus (VSV), Maraba virus, and Newcastle Disease Virus (NDV), all of which are covered by lipid envelopes, were shown to be sensitive to natural IgM- and complement-mediated inactivation in human serum (6, 7). Several approaches to prevent IgM and complement-dependent virus inactivation have been tested, including substitution of native virus envelope glycoproteins with glycoproteins demonstrating relative resistance to complement fixation (7, 8), generation of oncolytic vector stocks on cell lines with low xenoantigen burden (9), and incorporation of complement inhibitory proteins into the virus particles (6). However, whether any of these approaches will suffice to reduce virus sequestration in immune phagocytic cells or improve therapeutic efficacy after systemic administration to cancer patients remains unclear.
Human adenovirus (HAdv) has a non-enveloped proteinaceous icosahedral capsid of known structure (10, 11). HAdv is highly effective at infecting and killing a variety of cancer cell types (12). While these properties make HAdv attractive for developing novel oncolytic virus drug candidates, systemic delivery of HAdv is known to trigger a potent systemic inflammatory response and hepatotoxicity (13–15), thus limiting clinical utility of this virus platform. Studies involving HAdv vectors based on the most commonly used human species C HAdv-C5 serotype revealed that after intravenous injection, virus particles are rapidly opsonized by coagulation factor X (FX)(16, 17). Although FX binding to HAdv-C5 protects the virus from complement-mediated inactivation (18, 19), FX binding also facilitates highly efficient infection of hepatocytes, triggering hepatotoxicity (16, 17). Furthermore, FX binding to the virus potentiates virus recognition by cells of innate immunity, thus contributing to activation of systemic inflammation (20). After intravenous delivery, HAdv particles are rapidly cleared from the blood by tissue phagocytic cells, most notably by Kupffer cells in the liver (21). When trapped by phagocytic cells, HAdv interacts with cellular β3 integrins via the Arg-Gly-Asp (RGD) amino acid motif in the virus penton base protein(22). This interaction triggers activation of pro-inflammatory IL-1α-dependent signaling, initiating an acute cytokine storm observed after high dose intravenous virus administration (23). Clearly, if a safe and effective oncolytic virus suitable for systemic administration is to be developed, a multipronged approach to tailor virus interactions with humoral factors and tissue phagocytic cells would be required.
In this study, we determined that a large, flexible, and highly negatively charged hyper-variable loop, HVR1, of the HAdv-C5 hexon is the principal site in the virus capsid mediating natural IgM binding to the virus. Using compound targeted mutagenesis of hexon HVR1 loop with mutations abrogating FX binding and re-targeting virus from macrophage β3-integrins to integrin classes expressed on epithelial tumor cells, we have engineered and obtained a high-resolution cryo-EM structure of an HAdv vector, Ad5-3M. This vector resists inactivation by blood factors, avoids sequestration in liver macrophages, and fails to trigger hepatotoxicity after intravenous delivery. Systemic delivery of Ad5-3M to mice with localized and disseminated lung cancer led to viral replication in tumor cells, suppression of tumor growth, and prolonged survival. Our study demonstrates that generation of “designer” viruses that evade innate immune recognition through compound targeted mutagenesis of capsid functional sites is feasible. Ad5-3M represents a platform virus for the development of novel oncolytic drug candidates that can be evaluated in clinical trials where systemic virus administration is warranted to control unresectable or disseminated metastatic disease.
Results
Natural IgMs bind HAdv-C5 via hexon HVR1 loop and mediate virus sequestration in Kupffer cells
Intravascular administration of human adenovirus HAdv-C5 leads to rapid viral sequestration in liver resident macrophages, Kupffer cells (fig. S1A, and (24)). Earlier reports provided evidence that IgM binds directly to HAdv-C5 virus particles (25, 26) and intravenous administration of HAdv-C5 to Rag−/− mice, deficient in immunoglobulin production, resulted in a significant reduction in the ability of Kupffer cells to sequester virus particles from the blood (24, 27). Indeed, we found that liver macrophages in Rag−/− mice or mice deficient in the production of natural IgM and other antibodies (μMT mice) have significantly reduced capacity to sequester HAdv-C5 from the blood after intravenous virus administration (Fig. 1A). Natural IgM binding to viral particles triggers activation of the complement cascade, leading to covalent binding of complement components C3b and C4b to the viral surface (18). We administered virus to mice deficient in complement components C1q, C3, complement receptor CR3 components CD18 and ITGAM, or scavenger receptors CD36, which may sequester IgM-opsonized particles via IgM-associated protein AIM/CD5L (28, 29), and analyzed virus trapping in Kupffer cells 30 minutes later. This analysis showed that only mice deficient in CD36 had reduced capacity to sequester HAdv-C5 from the blood (Fig. 1A and figs. S1, A and B), despite normal levels of IgM in the circulation (fig. S1C). To confirm that liver macrophages utilize CD36 to sequester HAdv-C5 from the blood in WT mice, we injected C57B16 mice with CD36 neutralizing Ab prior to HAdv-C5 administration and analyzed accumulation of virus in Kupffer cells. This analysis confirmed that blocking the CD36 receptor in WT mice leads to a significant reduction in virus accumulation in liver macrophages, compared to mice pre-treated with an isotype control Ab (Fig. 1A, p < 0.0001).
Fig. 1. Natural IgM bind HAdv-C5 through hexon HVR1 loop.
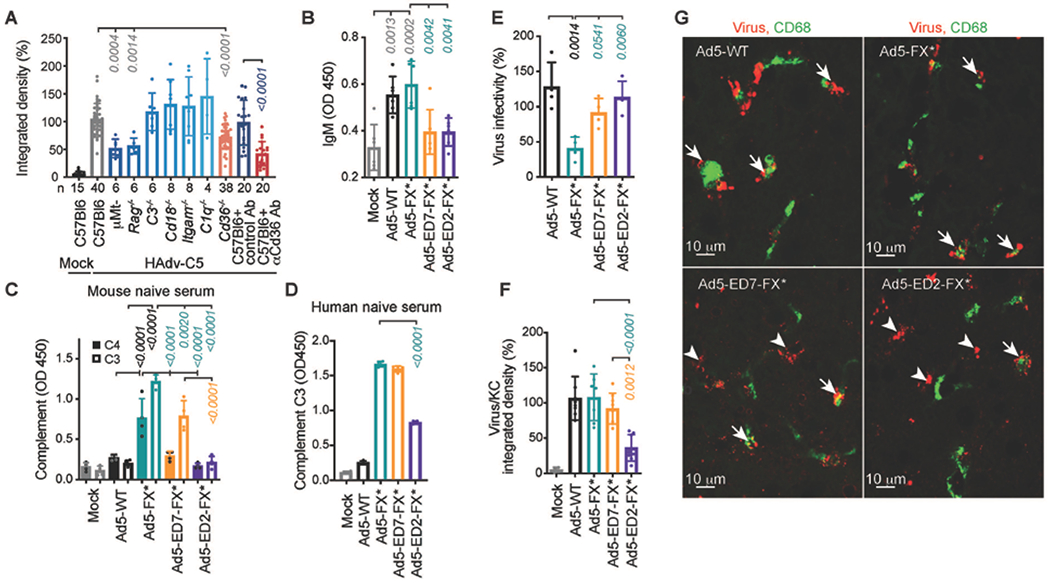
(A) Integrated density of HAdv-C5-positive staining within CD68- and F4/80-positive cells on liver sections from indicated gene-deficient mice or wild-type mice pre-treated with isotype control or anti-CD36 Ab, 30 minutes after intravenous HAdv-C5 administration. Number of replicates (n) are indicated under each bar. (B) ELISA measurement of natural IgM binding to indicated viruses in mouse plasma (n = 6). (C) Detection of complement components C4 and C3 deposition on the surface of the virions for indicated viruses in mouse serum (n = 4). (D) Detection of complement C3 deposition on the surface of the virions for indicated viruses in human naive serum, lacking HAdv-C5-specific neutralizing antibodies (n = 4). (E) Virus infectivity after incubation with 90% of the raw mouse serum normalized to virus infectivity with no serum treatment determined on HEK293 cells (n = 4). (F) Graphical representation and (G) representative images of immunofluorescent staining of sections of livers harvested from WT mice 30 minutes after intravenous injection with indicated viruses. Kupffer cell CD68-specific staining is in green. Staining with adenovirus-specific polyclonal antibodies is in red. Arrows point to virus staining co-localized with Kupffer cells and arrowheads show virus staining that does not co-localize with Kupffer cells (n = 4-8). The p-values of one-way ANOVA with multiple comparison adjustments are shown above the bars. Bars show mean ± SD. The color of the p-value number indicates the partner for multiple comparison tests. The methodology and statistical details are as described in Table S1 and Methods.
To mediate virus sequestration in Kupffer cells, natural pentameric IgMs in the blood must be able to bind HAdv-C5 virions in the presence of FX, which binds HAdv-C5 with near picomolar affinity (16, 17). We hypothesized that a large, highly flexible and negatively-charged hypervariable loop of the major virus capsid protein hexon may protrude outward from the virion surface and mediate natural IgM binding to the virus even in the presence of FX. To experimentally test this hypothesis, we introduced mutations in the HVR1 loop of HAdv-C5 hexon by shortening its length and decreasing the net-negative charge of the loop (virus Ad5-ED7-FX*, fig. S2), or by substituting an entire HVR1 loop with a short stretch of non-charged amino acids GGSG (virus Ad5-ED2-FX*, fig. S2). Both HVR1-mutated viruses had an additional T425A single point mutation in hexon HVR7 loop, ablating virus interaction with FX (20) to avoid FX-mediated shielding of the virus from IgM and complement interactions (18). As positive controls, for our tests we used Ad5-FX* virus (previously reported as Ad-TEA, (20)), ablated for FX binding but possessing unmodified native HAdv-C5 hexon HVR1 loops, as well as fully intact WT HAdv-C5 virus (Ad5-WT, figs. S2 and S3). Incubation of these viruses with mouse EDTA-preserved plasma resulted in IgM binding to Ad5-WT and Ad5-FX* viruses but not to viruses with hexon HVR1 loop mutations (Fig. 1B). Since EDTA inhibits complement activation, we incubated HVR1-loop-mutated and control viruses with complete fresh mouse serum and analyzed deposition of complement components C4 and C3 on the virus surface. These analyses showed that after incubation with serum, complement components C4 and C3 were readily detectable on Ad5-FX* virus (Fig. 1C). In contrast, the introduction of mutations in the hexon HVR1 loop partially prevented complement C4 and C3 deposition on Ad5-ED7-FX* virus, which has shortened but still negatively charged HVR1 loops (Fig. 1C). More dramatic mutation of the HVR1 loop completely prevented deposition of both mouse C4 and C3 complement components on Ad5-ED2-FX*, which has short and net neutral HVR1 loops (Fig. 1C, fig. S2). Incubation of HVR1-mutated viruses with naive human serum, containing low amounts of HAdv-C5-specific neutralizing antibodies, further confirmed that complement C3 deposition on the virus surface is significantly reduced only for Ad5-ED2-FX* virus (Fig. 1D, p < 0.0001), which has the most dramatic HVR1 mutation. Incubation of Ad5-FX* with mouse serum resulted in a significant loss of virus infectivity towards susceptible cells (p = 0.0014); however, Ad5-ED7-FX* and Ad5-ED2-FX* viruses resisted inactivation by mouse serum (Fig. 1E). Importantly, compared to other viruses, Ad5-ED2-FX* accumulated in Kupffer cells in much lower amounts after intravenous delivery (Figs. 1, F and G), consistent with its reduced ability to bind natural IgM and complement.
Re-targeting HAdv-C5 to tumor cell integrins and suppression of penton-mediated activation of pro-inflammatory signaling
The HAdv penton base RGD loop is critical for efficient virus infection as the RGD amino acid motif interacts with cellular integrins of αvβ3 and αvβ5 classes, promoting efficient internalization of virus particles into the cell (22). Furthermore, it was shown that after intravenous administration, the interaction of the virus penton base RGD motif with β3 integrins on tissue macrophages triggers activation of IL-1α-mediated pro-inflammatory signaling that leads to the production of an array of inflammatory cytokines and chemokines in liver and spleen (23). Although potentially advantageous, the deletion of the penton RGD motif negatively affects virus internalization and disassembly kinetics (30). The human laminin-α1 sequence contains the Ser-Ile-Lys-Val-Ala-Val (SIKVAV) amino acid motif, which interacts with integrins of α3β1, α6β1, and α6β4 classes highly expressed on cancer cells of epithelial origin (31, 32). Structural modeling and molecular dynamics simulations of a 48-amino acid sequence from the human laminin-α1 chain, when modeled in place of a native penton RGD loop of HAdv-C5, suggested that the integrin-interacting SIKVAV residues are solvent accessible and presented at the top of flexible α-helixes (fig. S4). This presentation of the SIKVAV residues is similar to that previously modeled for the RGD residues of the HAdv-C5 penton base ((33), fig. S5). To ablate the penton base RGD loop interaction with macrophage β3 integrins, while maintaining virus infectivity towards epithelial tumor cells, we next substituted the entire RGD loop in the HAdv-C5 penton base for a 48-amino acid sequence from the human laminin-α1 chain. We named the HAdv-C5-based virus with substituted penton base RGD loops Ad5-Lam1 (figs. S3 and S6) and analyzed its properties in vitro and in vivo.
The viral uptake into human A549 cells and cell transduction efficiency were similar for Ad5-Lam1 and the RGD-containing Ad5-WT virus (fig. S7, A and B). A549 cells and a panel of human non-small cell lung cancer cell lines express a variety of integrin classes that interact with both RGD and SIKVAV motif-containing ligands (fig. S7, C and D). Incubation of cells with synthetic SIKVAV peptide, but not RGD4C peptide, led to a decrease in cell infection mediated by Ad5-Lam1, while Ad5-WT was insensitive to SIKVAV peptide inhibition (fig. S7B). Importantly, Ad5-Lam1 was insensitive to inhibition with RGD4C peptide, demonstrating that Ad5-Lam1 utilizes non-RGD-dependent integrin binding for tumor cell entry. Re-targeting the virus from RGD – cellular β3 integrin interactions to SIKVAV motif-dependent integrins resulted in a major reduction in activation of inflammatory cytokines, including IL-1β, IL-1β, IL-6 and chemokine CXCL2, after intravenous viral administration (fig. S7E).
High-resolution cryo-EM structure of Ad5-3M and virus infectivity in vitro
Using targeted mutagenesis, we next generated the replication competent virus, Ad5-3M, which comprises an IgM-binding ablating mutation in the hexon HVR1 loop (from Ad-ED2-FX*), the FX-binding ablating mutation T425A in hexon HVR7 loop to reduce FX-dependent hepatocyte infection (20), and the penton-integrin re-targeting mutation to avoid macrophage β3 integrin interactions (from Ad5-Lam1) (figs. S2 and S6). To ascertain the effect of these mutations in Ad5-3M on the viral capsid, we determined a three-dimensional cryo-EM structure at near atomic (3.8-Å) resolution (figs. S8 and S9). The overall structure of the capsid (Fig. 2A) and asymmetric unit (Fig. 2B) closely resemble those of the cryo-EM structure of HAdv-C5 (11) and the x-ray crystallographic structure of the Ad35F vector (10). The key FX interaction site (T425A) is near the top of the hexon and within the central depression of the trimer (Fig. 2C). Filtering the Ad5-3M cryo-EM density map to 5-Å resolution, revealed the location of the Ad5-3M HVR1 loop at the side of the hexon tower (Fig. 2D) and facilitated selection of a reasonable loop model from among a set of models generated by FREAD (34) (Fig. 2E). After cryo-EM guided energy minimization with MDFF (35), there was good agreement between the Ad5-3M and HAdv-C5 hexon coordinates (PDB: 6B1T and 6CGV) indicating that the Ad5-3M hexon mutations did not cause any significant conformational changes in the other HVRs of hexon. Therefore, the combination of the structural analysis and functional binding data strongly suggest that HAdv-C5 HVR1 is the genuine IgM binding site and the lack of IgM binding to Ad5-3M is not a result of gross change in viral capsid folding. Filtering the Ad5-3M cryo-EM density revealed a relatively small protrusion at the top of the penton base that accounts for a small portion of the integrin interacting loop (Figs. 2, F and G). As noted in the local resolution analysis of Ad5-3M (fig. S9), the penton base protrusion is at the lowest resolution (4.5 Å) observed for the capsid indicating flexibility in this region. The most distal amino acids that could be fit into well-defined cryo-EM density are L296 and P381 (Fig. 2H), implying there are 84 residues in the flexible integrin interacting loop. Cryo-EM guided energy minimization with MDFF indicated no significant conformational changes in the penton base other than in the integrin interacting loop.
Fig. 2. High-resolution cryo-EM structure of Ad5-3M virus, possessing compound mutations in hexon HVR1 and HVR7 loops and penton base RGD loop.
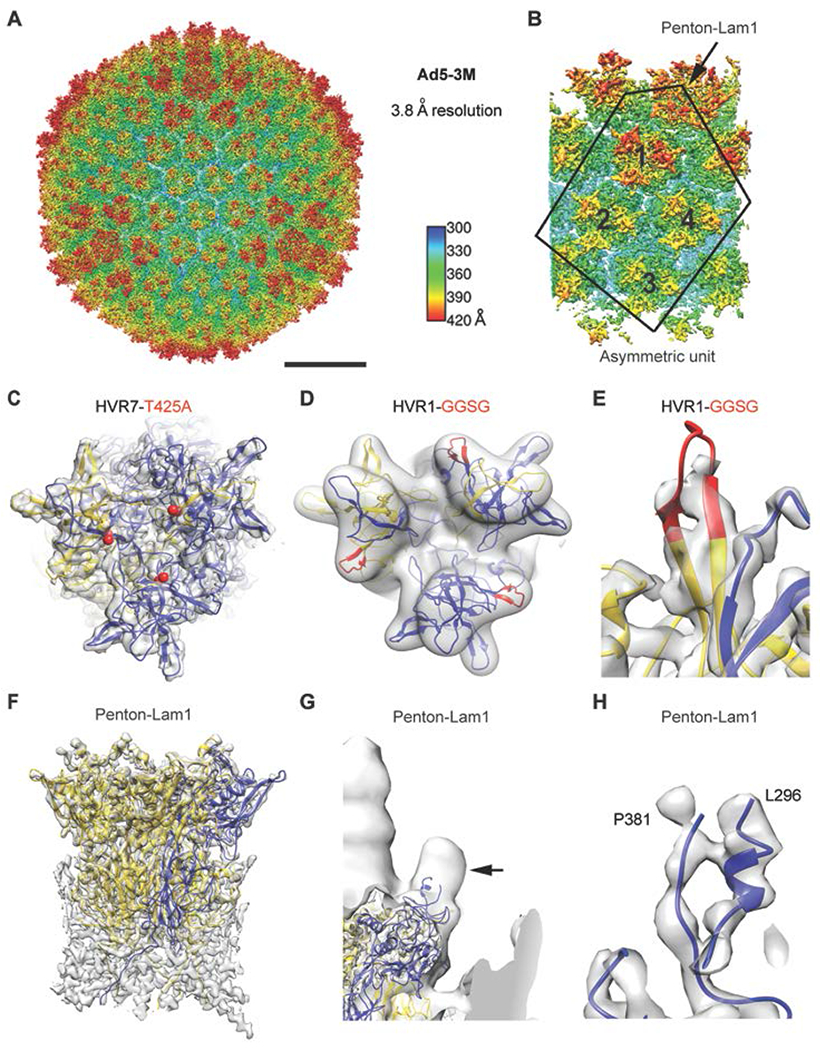
(A) Full Ad5-3M capsid structure at 3.8 Å resolution displayed with radial coloring. Scale bar, 250 Å. (B) Enlarged view of the icosahedral asymmetric unit (outlined), which includes one subunit of penton base with a laminin-α1 derived integrin interacting motif and 4 hexon trimers with mutations in HVR1 and HVR7 loops. (C) Top view of one Ad5-3M hexon trimer with the sites of the Thr-425-Ala (T425A) HVR7 loop point mutation depicted with red spheres. One hexon subunit is in gold, two hexon subunits are in blue, and the cryo-EM density is in transparent gray. (D) Ad5-3M hexon density filtered to 5-Å resolution superimposed with hexon coordinates including the modeled HVR1 loop containing the Gly-Gly-Ser-Gly (GGSG) sequence (red). (E) Enlarged view of Ad5-3M HVR1 loop model shown with unfiltered cryo-EM density. (F) Side view of the Ad5-3M penton base pentamer (one subunit in blue, four subunits in gold). (G) Ad5-3M penton base density filtered to 15-Å resolution reveals a protrusion at the base of integrin interacting loop (arrow). Density for the fiber, which protrudes from the center of the penton base, is observed at the left edge of the panel. (H) Enlarged view of ordered residues at either end of the Ad5-3M penton base integrin interacting loop shown with unfiltered cryo-EM density.
Incubation of Ad5-3M with mouse plasma and serum demonstrated that natural IgM antibodies and complement components C4 and C3 were unable to bind to this virus (Fig. 3, A to C), and human C3 complement deposition on the virus was also significantly reduced (Fig. 3D; p < 0001). Importantly, as a result of three simultaneous mutations in the virus capsid, Ad5-3M was resistant to inactivation by both mouse and human sera containing low neutralizing antibody titer (Fig. 3, E and F).
Fig. 3. Ad5-3M virus is resistant to natural IgM- and complement-mediated opsonization and inactivation in mouse and human sera.
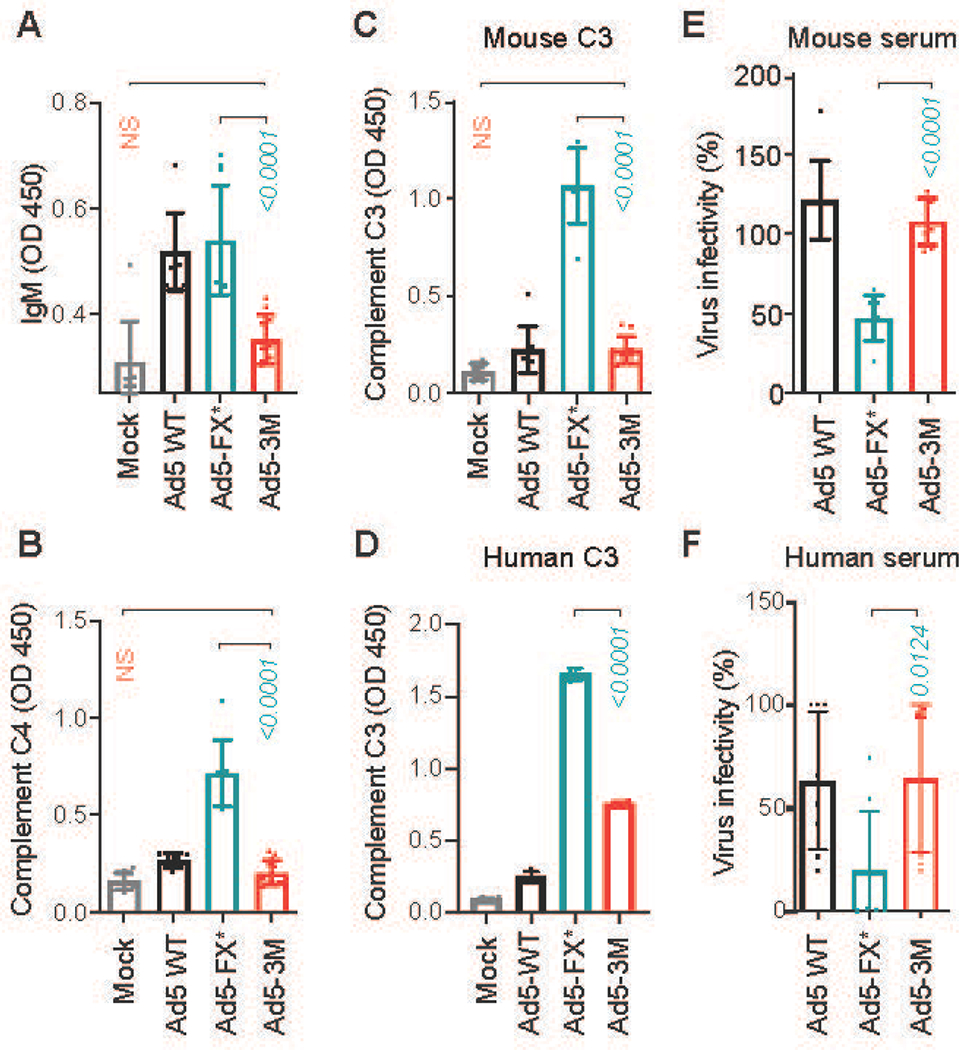
(A) ELISA measurement of natural IgM binding to the indicated viruses in mouse plasma. Detection of deposition of complement components C4 (B) and C3 (C) on the surface of the virions for indicated viruses in mouse serum using ELISA (n = 8-12). (D) Detection of deposition of human complement C3 on the surface of the virions for indicated viruses in human serum using ELISA (n = 4). (E and F) Virus inactivation in raw serum. Indicated viruses were incubated with 90% raw mouse serum (E) (n = 8), and human serum (F) containing low amounts of neutralizing antibodies (n = 8). Virus infectivity after incubation with serum was normalized to virus infectivity with no serum treatment. The p-values of one-way ANOVA with multiple comparison adjustments are shown above the bars. Bars show mean ± SD. The color of the p-value number indicates the partner for multiple comparison tests. N. S. – not significant; p > 0.05. The methodology and statistical details are as described in Table S1 and Methods.
To test whether this combination of three mutations has any detrimental effect on virus transduction of tumor cells in vitro, we analyzed Ad5-3M transduction efficacy in a panel of eight human non-small cell lung adenocarcinoma cell lines. Comparative analysis of Ad5-3M infectivity to the parental HAdv-C5-based vector showed that there was no difference in tumor cell transduction efficacy between the two viruses (fig. S10A). The viral growth kinetics and virus-mediated cell death were also comparable in all tumor cell lines tested (fig. S10B).
Ad5-3M avoids sequestration in liver tissue and poorly activates inflammatory cytokines after intravenous delivery
Since natural IgMs demonstrated reduced binding to Ad5-3M (Fig. 3A), we next examined whether this property would translate into reduced viral sequestration in liver resident macrophages, Kupffer cells, after intravenous virus administration to mice. Using immunofluorescent staining we found that while unmodified HAdv-C5 was readily trapped in Kupffer cells (fig. S11), co-localization of Ad5-3M particles with liver macrophages was significantly reduced after intravenous virus administration to WT mice (p < 0.0001; Ad5-WT vs Ad5-3M. Fig. 4A and fig. S11). Next, we used quantitative PCR to analyze the amount of viral genomes present in Kupffer cells, isolated from mice 30 minutes after viral injection. This analysis showed a 5-fold reduction in the amount of viral genomic DNA present in purified Kupffer cells after administration of mice with Ad5-3M, compared to HAdv-C5 (Fig. 4B). Adenovirus sequestration in Kupffer cells triggers necrotic cell death (36, 37), which is not unique to the species C adenovirus HAdv-C5 as other HAdv currently being developed for human gene therapy applications, including HAdv-B11 (38) and HAdv-B35 (39), are also highly potent at triggering necrotic death of Kupffer cells after intravenous virus administration (Figs. 4, C and D). In contrast, intravenous administration of Ad5-3M failed to trigger necrotic death of Kupffer cells, consistent with its ability to escape sequestration in these liver macrophages after intravenous administration (Fig. 4, C and D). The analysis of the amount of virus present in whole livers after intravenous virus delivery showed that liver tissue sequestered large quantities of HAdv-C5, HAdv-B11, and to a lesser degree HAdv-B35 (Fig. 4E). In contrast, only 15.66% of the administered viral dose of Ad5-3M was associated with liver tissue 1 hour after intravenous injection (Fig. 4E), while more Ad5-3M was accumulated in the spleen, compared to Ad5-WT virus (fig. S12, A and B). Because hepatocytes in mice support HAdv-C5 genomic DNA replication, and both HAdv-C5 and Ad5-3M are replication-competent viruses, we analyzed the amount of virus genomic DNA in the liver 24 and 48 hours after virus administration. At the 48 hour time point this analysis showed 172-fold higher amounts of viral genomic DNA as well as 37-fold higher amounts of virus-encoded luciferase expression in the livers of mice administered with HAdv-C5, compared to Ad5-3M virus (Fig. 4F). The profoundly efficient replication of HAdv-C5 in the liver triggers severe hepatotoxicity (Fig. 4, G and H) and mice were moribund and had to be sacrificed within 72 hours post administration of the wild-type HAdv-C5 virus. However, no hepatotoxicity was observed when the same dose of Ad5-3M was injected intravenously to mice (Fig. 4, G and H) and all animals survived systemic virus delivery. Even though the spleen accumulated higher amounts of Ad5-3M compared to HAdv-C5 (fig. S12A), Ad5-3M failed to trigger activation of the inflammatory cytokines IL-1α, IL-1β, IL-6, TNF-α and chemokines CXCL1 and CXCL2, which were readily activated in the spleen after intravenous injection of HAdv-C5 (Fig. 4I and fig. S12C). Incubation of primary human macrophages with Ad5-3M triggered reduced secretion of inflammatory cytokines TNF-α and IL-6, compared to the amounts of these cytokines secreted from primary macrophages after their incubation with unmodified Ad5-WT virus (Fig. 4J).
Fig. 4. Ad5-3M virus escapes sequestration in liver macrophages and fails to trigger hepatotoxicity and inflammatory cytokine activation after systemic delivery.
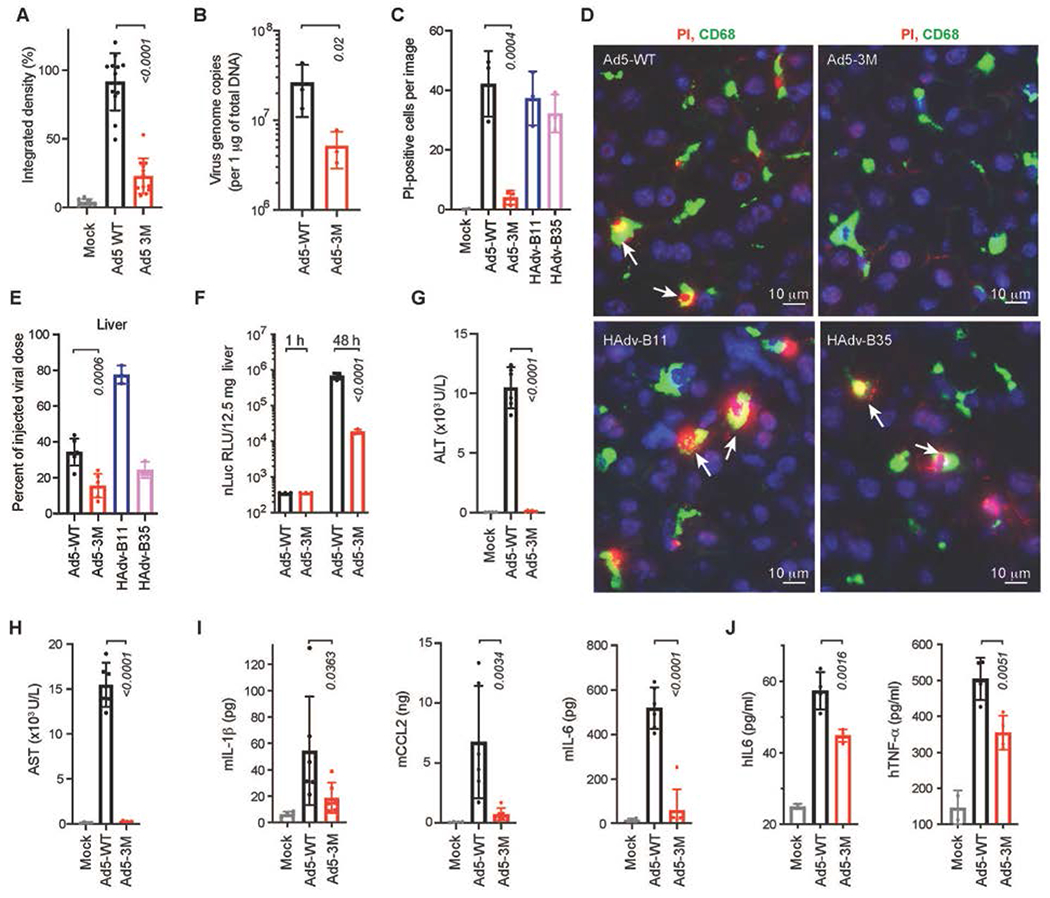
(A) Integrated density of virus-positive staining within CD68- and F4/80-positive cells for indicated viruses on liver sections at 1 h post virus injection, normalized to the median integrated density of Ad5-WT virus staining (n = 8-12). (B) Comparison of virus DNA copies in Kupffer cells by qPCR analysis for indicated viruses 30 min post intravenous virus injection (n = 3). (C) Graphical representation and (D) immunofluorescent staining of liver sections showing in vivo necrosis (PI permeability, red) of CD68-positive Kupffer cells (green) after administration of mice with indicated viruses. DAPI staining of the nuclei is in blue. Arrows point to the PI-positive necrotic Kupffer cells (n = 2-3). (E) Accumulation of viral DNA in livers of mice 1 hour after intravenous virus administration, when compared to the total injected dose for indicated viruses (n = 3-6). (F) Activity of virus-encoded nano-luciferase in liver lysates at the indicated time points (n = 3). Liver function was evaluated by measuring alanine aminotransferase (ALT) (G), and aspartate aminotransferase (AST) (H) in mouse serum 48 h post virus injection (n = 4-6). (I) Comparison of cytokine and chemokine concentration in the spleens of mice (pg or ng per 130mg of spleen tissue) at 1 h post intravenous virus injection (n = 4-6). (J) Amounts of human TNF-α and IL-6 released from primary human macrophages 72 h after their incubation with indicated viruses (n = 4). Bars show mean ± SD. The p-values of one-way ANOVA with multiple comparison tests are shown above the bars, the color of the p-value number indicates the comparison partner. The methodology for individual settings are as described in Table S1 and Methods.
Ad5-3M extends survival of mice with localized and disseminated cancer
To analyze whether Ad5-3M is able to infect and replicate in human tumors after intravenous injection, we first grafted human lung adenocarcinoma A549 cells into the flank of NSG mice subcutaneously. When the tumors reached between ~300 mm3 in size, mice were injected with Ad5-3M virus intravenously. The expression of virus-encoded nano-luciferase, tumor growth kinetics, and mouse survival was monitored over time and compared to a control group of tumor-bearing mice treated with unmodified Ad-WT virus or buffer. These analyses showed that 12 days after beginning treatment, virus-encoded nano-luciferase expression was localized exclusively to tumor sites (Fig. 5A). Longitudinal analysis of nano-luciferase expression in tumors showed that transgene luminescence reached its peak at 12 days post virus administration. However, transgene luminescence remained high for up to 80 days after virus treatment (Fig. 5B). In addition, Ad5-3M virus DNA was readily detectible in tumors at this late time point after virus treatment (Fig. 5C), suggesting long-lasting viral persistence in tumors for this model of localized disease. This finding is consistent with suppression of tumor growth after Ad5-3M administration (Fig. 5D). All mice treated with unmodified Ad5-WT virus succumbed within 5 days due to hepatotoxicity. The median survival of mice treated with buffer was 33 days and all mice in this group succumbed to tumor progression by 43 days post treatment (Fig. 5E). In contrast, none of the tumor-bearing mice treated with Ad5-3M had to be sacrificed for the duration of the study (Fig. 5E).
Fig. 5. Ad5-3M transduces tumors, suppresses tumor growth, and extends survival of mice with localized and disseminated tumors after systemic delivery.
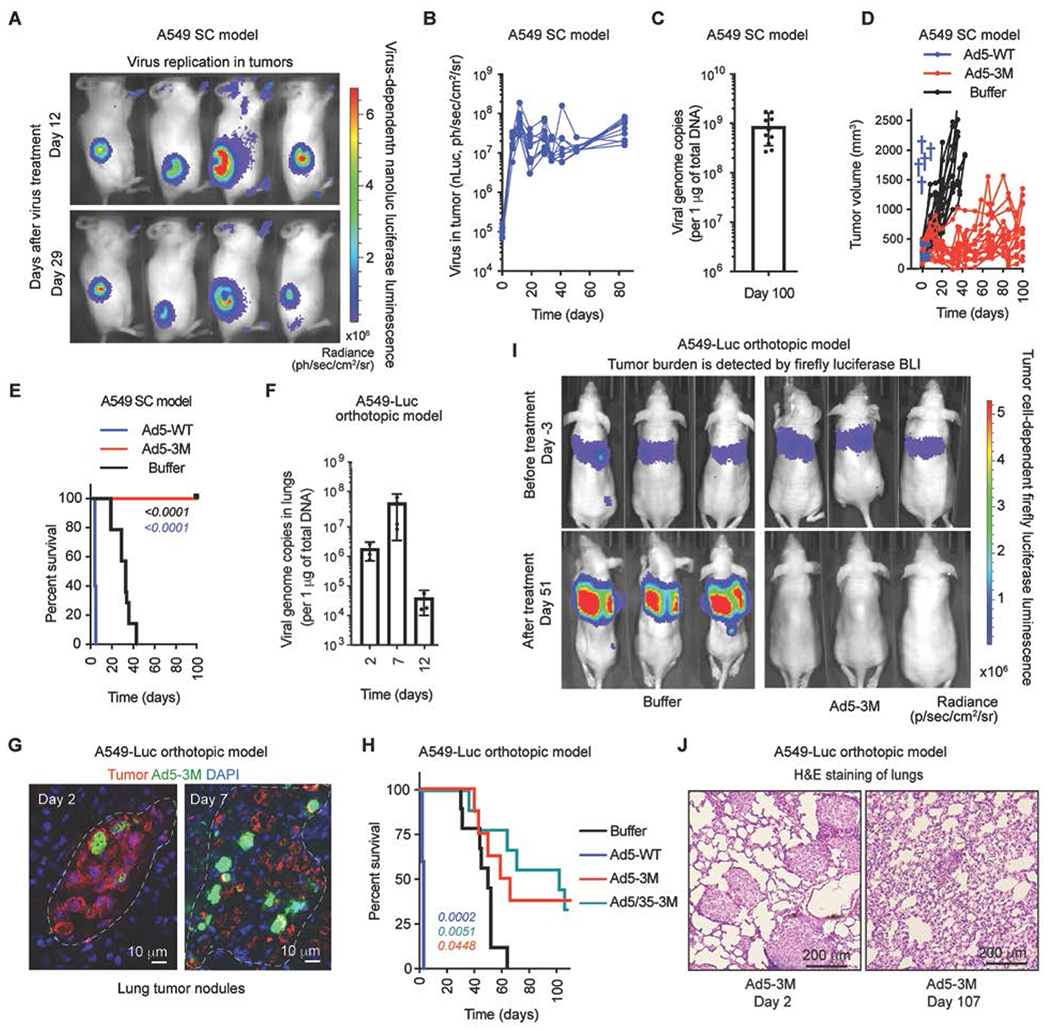
(A) In vivo bioluminescence imaging (BLI) of subcutaneous tumor-bearing mice at days 12 and 29 post Ad5-3M treatment. The color indicates bioluminescence intensity of virus-encoded nano-luciferase. SC – subcutaneous. (B) Activity of virus-encoded nano-luciferase in tumor-bearing mice treated with Ad5-3M (n = 10). (C) The amounts of viral genomic DNA in the tumors on day 100 after Ad5-3M treatment (n = 10). (D) Kinetics of subcutaneous tumor growth in mice treated with Ad5-WT (n = 5), Ad5-3M (n = 15), or Buffer (n = 14). Blue crosses indicate deaths of animals treated with Ad5-WT. (E) Log-rank survival plot of subcutaneous tumor-bearing mice over time treated with Ad5-WT (n = 5), Ad5-3M (n = 15), or Buffer (n = 14). (F) Viral genome copies in the lungs of orthotopic tumor-bearing mice at the indicated time points post Ad5-3M treatment, measured by qPCR (n = 3-4). (G) Immunofluorescent staining of lung sections from disseminated orthotopic lung tumor-bearing mice at days 2 and 7 post Ad5-3M injection. Staining with human mitochondria-specific antibodies, recognizing human-derived tumor cells, is in red. Staining with adenovirus-specific polyclonal antibodies is in green. Nuclei-specific DAPI staining is in blue. The anatomical boundaries of tumor nodules are depicted with dotted lines. (H) Log-rank survival plot of mice with disseminated orthotopic lung tumors after treatment with Ad5-WT (n = 5), Ad5-3M (n = 8), Ad5/35-3M (n = 9), or Buffer (n = 9). The methodology and statistical details are as described in Table S1 and Methods. The color of the p-value number indicates the virus-treated comparison partner to the Buffer group. (I) In vivo whole body bioluminescent imaging of disseminated orthotopic lung tumor-bearing mice 3 days before (upper panels) and 51 days after (lower panels) indicated treatments. Representative images of tumor burden in mice on day 3 and on day 51 after treatment with buffer are shown. Selected images on day 51 after treatment with Ad5-3M are shown for 3 out of 8 mice that showed complete disappearance of tumor luminescence. The color indicates the intensity of luminescence, reflecting tumor burden in the lungs. (J) Hematoxylin and eosin staining of sections of lungs harvested from mice with disseminated lung tumors on days 2 and 107 after Ad5-3M treatment. Representative images are shown.
NSG mice are advantageous for evaluating the oncolytic potency of viral vectors due to their high efficiency grafting of tumor cells of human origin. However, this mouse model has profound hyposplenia and lacks circulating IgM in the blood, thus artificially limiting the impact of these factors on bio-distribution of the virus after intravenous delivery. To analyze the oncolytic potency of Ad5-3M in a more physiologically relevant mouse model, we established disseminated tumors in the lungs of NCr/Nude mice. These mice lack functional T cells, but they have normal sized spleens and wild-type amounts of IgM antibodies in the blood, as serum from mice of this strain effectively inactivates HAdv vectors unable to bind FX (18). Intravenous administration of A549-Luc cells into NCr/Nude mice leads to the formation of numerous disseminated tumor sites anatomically resembling late-stage metastatic disease (fig. S13). As A549-Luc cells constitutively express firefly luciferase, the efficacy of tumor grafting in the lungs, as well as the change in tumor burden over time, can be monitored via non-invasive imaging methodology. Analysis of the amount of Ad5-3M viral DNA in the lungs of tumor-bearing mice demonstrated a 35-fold increase in viral DNA between 2 to 7 days post intravenous virus injection (Fig. 5F). Histological analysis of tumor transduction by Ad5-3M further showed that while only sparse individual tumor cells were transduced with the virus at day 2 post virus administration, extensive foci of virally-transduced cells were detectable on sections of lung tumors harvested 7 days after viral treatment (Fig. 5G). All tumor-bearing mice treated with unmodified Ad5-WT succumbed within 3 days due to hepatotoxicity (p = 0.0002; Ad5-WT vs. Buffer; Fig. 5H). The median survival of mice sham-treated with buffer was 49 days and for the mice treated with Ad5-3M, the median survival was extended to 61.5 days (p = 0.0448; Ad5-3M vs. Buffer; Fig. 5H). Remarkably 3 out of 8 mice treated with Ad5-3M exhibited a complete response, as measured by the lack of tumor-encoded luciferase luminescence in the lungs on day 51 (Fig. 5I) and survival up to 107 days post virus treatment (Fig. 5H, termination of experiment). The additional targeting of Ad5-3M to tumors via human CD46 receptor by substituting HAdv-C5 receptor-interacting fiber knob domain for that of HAdv-B35 in the Ad5/35-3M vector further increased anti-tumor potency. Administration of Ad5/35-3M into tumor-bearing mice extended median survival of this virus-treated group to 101 days (p = 0.0051; Ad5/35-3M vs. Buffer; Fig. 5H). Importantly, systemic delivery of Ad5/35-3M triggered significantly lower systemic toxicity, compared to the parental Ad5-WT virus (fig. S14). Histological analysis of lungs harvested from complete responder mice on day 107 post treatment showed that tumor sites were converted into collagen-rich scar-like structures sparsely distributed through the lung parenchyma (Fig. 5J). Collectively, these data show that intravenous treatment with Ad5-3M extends the survival of mice with localized and disseminated cancer.
Ad5-3M extends survival of mice grafted with human non-small cell lung cancer patient-derived xenografts
To analyze whether systemic therapy with Ad5-3M would improve survival of animals grafted with primary tumor cells derived from NSCLC patients, we first tested the sensitivity of a panel of human patient-derived xenograft (PDX) samples to Ad5-3M infection in vitro. To do that, four NSCLC PDX tumor models were obtained from Jackson laboratories and freshly-harvested tumor tissues were cultured in media with the addition of Ad5-3M. Fluorescence microscopy analysis for Ad5-3M-encoded GFP gene expression showed that all four of the tested PDX samples had numerous GFP-positive cells 72-120 hours after addition of the virus (Fig. 6A), demonstrating that Ad5-3M can infect and replicate in primary human tumor cells. Next we grafted two of these PDX tumors on NSG mice and 2-3 weeks later, the mice were randomized and treated intravenously with unmodified Ad5-WT virus, buffer, or Ad5-3M. The analysis of viral-encoded luciferase expression demonstrated that Ad5-3M efficiently infected PDX tumors in vivo after intravenous virus delivery (Fig. 6, B and C) and that the amounts of viral DNA and nano-luciferase expression in PDX tumors increased over 500-fold between 2 to 21 days post treatment (Fig. 6, D and E). Furthermore, the virus infection of tumor cells triggered their necrotic death in vivo (Fig. 6F). Similar to the localized A549 model, all mice treated with unmodified Ad5-WT succumbed within 5 days after the virus administration due to hepatotoxicity (Fig. 6, G to J). Although primary human PDX tumors grew extremely aggressively in these mouse models, intravenous administration of Ad5-3M delayed tumor progression and significantly extended animal survival (Fig. 6, H and J; p < 0.0001).
Fig. 6. Ad5-3M transduces primary NSCLC tumors, suppresses tumor growth, and extends survival of PDX tumor-bearing mice after systemic administration.
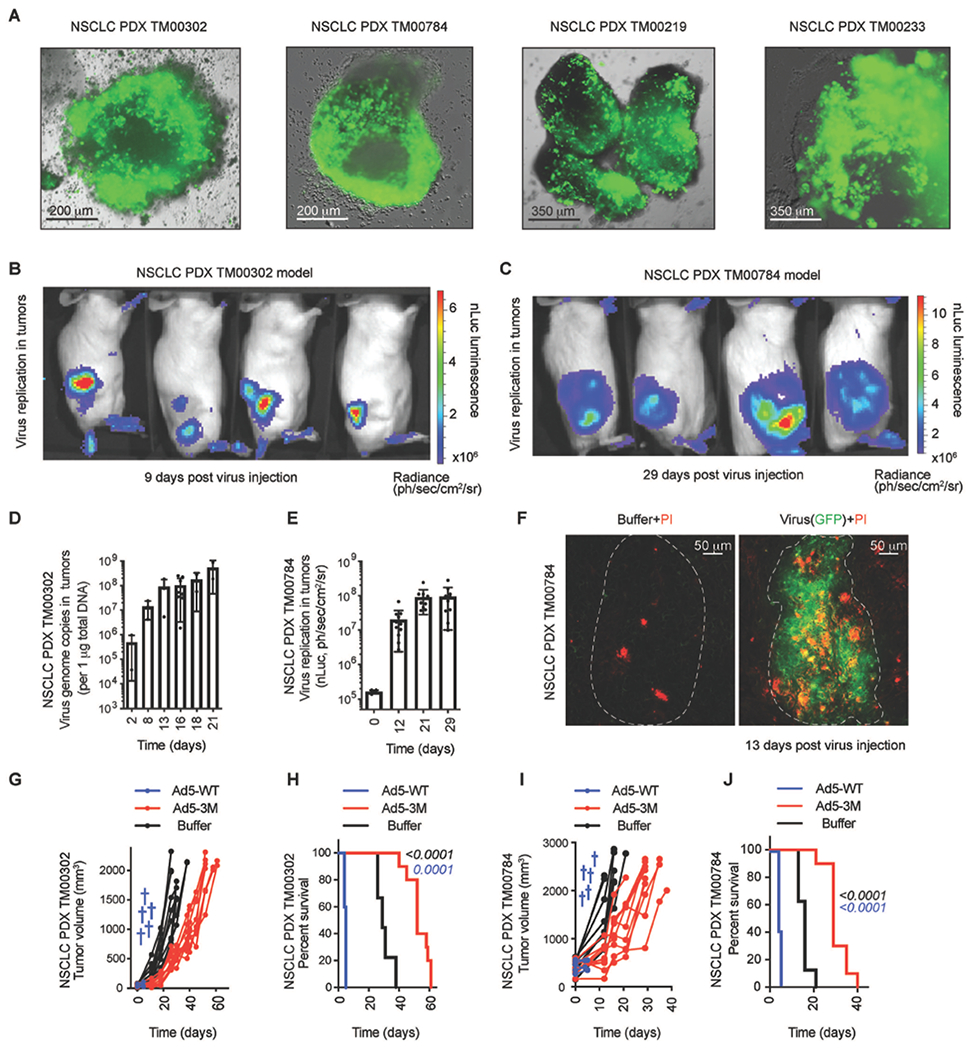
(A) Fluorescent images of virus-driven GFP expression in primary NSCLC PDX tumor explants 3-5 days after Ad5-3M infection. Representative images of virus transduction for four individual PDX models are shown. In vivo BLI of NSCLC PDX models TM00302 (B) and TM00784 (C) subcutaneously grafted to NSG mice and treated with Ad5-3M intravenously. The color indicates intensity of luminescence of virus-encoded nano-luciferase. (D) Amounts of viral genomic DNA in TM00302-derived PDX tumors at the indicated times post intravenous Ad5-3M administration (n = 2-8). (E) Activity of virus-encoded nano-luciferase in subcutaneous TM00784 PDX tumors at the indicated time points after intravenous Ad5-3M administration (n = 4-12). (F) Fluorescent images of sections of PDX-tumors harvested from mice treated with Ad5-3M or buffer. Virus-driven GFP expression is in green and necrotic propidium iodide (PI)-positive cells are in red. The anatomical boundaries of tumor nodules are depicted with dotted lines. (G) Kinetics of tumor growth and (H) Log-rank survival plot of TM00302 PDX-tumor-bearing mice treated with Ad5-WT (n = 5), Ad5-3M (n = 10), or Buffer (n = 9). (I) Kinetics of tumor growth and (J) log-rank survival plot of TM00784 PDX-tumor-bearing mice treated with Ad5-WT (n = 5), Ad5-3M (n = 10), or buffer (n = 8). The blue crosses indicated death of the animals treated with Ad5-WT. The color of the p-value number indicates the comparison partner to Buffer group. The methodology and statistical details are as described in Table S1 and Methods.
Discussion
Systemic delivery of therapeutic viruses is a highly desirable approach in cancer therapy due to its potential to provide virus access to disseminated tumors and reverse cancer progression at late, metastatic stages of disease. However, this delivery route exposes therapeutic viruses to humoral factors and cells of the innate and adaptive arms of the immune system, which together act as a key barrier to the safety and efficacy of this approach. Previous studies of various oncolytic virus platforms, adopted for systemic administration, demonstrated that exposure of therapeutic vectors to human serum leads to dramatic reduction in virus infectivity, mediated primarily by natural IgM- and complement-dependent mechanisms. Specifically, when VSV- and Maraba virus-based vectors were exposed to normal human serum, virus infectivity was reduced 2-orders of magnitude for Maraba virus and up to 6-orders of magnitude for VSV (7). This potent virus inactivation by human sera was also observed for oncolytic NDV virus, which loses infectivity even when normal human serum is diluted by a factor of 20 (6). The development of oncolytic vectors that would resist IgM- and complement-mediated inactivation is challenging, as IgM binding to the virus surface is thought to occur via “non-specific” low affinity-high avidity interactions with multifunctional structural virus proteins that are critical for productive infection of host cells. Natural IgM binding promotes highly efficient virus sequestration in phagocytic cells of the innate immune system, most notably in resident macrophages of liver and spleen (24, 27, 40, 41). Innate phagocytic cells sense therapeutic viruses as genuine pathogens and, as a component of the host’s default antiviral defense program, they activate both local and systemic inflammation, leading to dose-limiting toxicities. It is certain that to realize their true potential as novel therapeutics for disseminated cancer, oncolytic viruses must be able to avoid deleterious interactions with multiple factors of innate and adaptive immunity without compromising safety and therapeutic efficacy.
In this study, we demonstrate that interactions of HAdv-C5 virus with humoral factors and phagocytic cells of innate immunity can be tailored via compound targeted mutagenesis of viral capsid proteins. Using targeted mutagenesis we determined that natural IgMs bind to the HAdv-C5 virion surface via a large, flexible, and highly negatively charged HVR1 loop of the main capsid protein hexon. Importantly, we showed that virus variants with mutated hexon HVR1 loops were resistant to complement-mediated inactivation after incubation with undiluted neat mouse and human sera, an experimental condition resembling exposure of therapeutic vectors to humoral factors after intravenous administration in clinical settings. In contrast to the established paradigm, our data suggests that there is a significant degree of specificity for natural IgM binding to a virus surface. Thus, finding and mutating key moieties in other oncolytic viruses could be a generalizable approach for the development of therapeutic vectors that would resist IgM- and complement-mediated inactivation after systemic administration. We further found that after intravenous delivery, HAdv variants with mutated hexon HVR1 loops escape sequestration in liver resident macrophages, Kupffer cells. Previous studies suggested that HAdv-C5 sequestration in Kupffer cells is mediated by natural IgMs (24, 27). We confirmed and extended those findings and demonstrated that Kupffer cells in antibody-deficient μMT mice have a significantly reduced capacity to sequester unmodified wild-type HAdv-C5 after systemic virus delivery. Upon analysis of macrophage receptors that mediate trapping of IgM-opsonized virus particles, we found that a scavenger receptor, CD36, is required for sequestering HAdv-C5 from the blood. A previous study by Stichling et al, demonstrated that scavenger receptor SR-A6 (MARCO) on murine alveolar macrophages can mediate direct uptake of HAdv-C5 particles via the hexon HVR1 loop (42). Because virus sequestration in Kupffer cells in CD36−/− and antibody-deficient mice is significantly reduced, compared to wild-type animals, our data indicates that opsonization of virus by natural IgMs may be a dominant mechanism, mediating virus sequestration by liver macrophages after systemic virus delivery. It is noteworthy that although unmodified wild-type HAdv-C5 is resistant to complement-mediated inactivation, natural IgMs still mediate its sequestration in Kupffer cells. This data suggests that oncolytic virus resistance to complement-dependent inactivation does not necessarily indicate the lack of natural IgM binding to the virus. As Kupffer cells represent the largest pool of macrophages in the human body, IgM binding to the virus even without subsequent complement-mediated inactivation can lead to virus sequestration in the liver, thereby reducing the virus dose that can reach disseminated tumors and compromising efficacy.
Intravenous injection of high doses of HAdv-C5 leads to severe hepatotoxicity and systemic inflammation (13, 15, 41). Our previous studies showed that when trapped by tissue immune phagocytic cells, HAdv-C5 interaction with β3 integrins triggers activation of IL-1α and production of an array of inflammatory cytokines and chemokines in an IL-1R-dependent manner (23). The HAdv-C5 interaction with β3 integrins promotes virus internalization into the cell and is mediated by the RGD amino acids located in the virus capsid protein penton base (43). Using structural modeling and targeted mutagenesis, we introduced a mutation into the HAdv-C5 penton, which ablated virus interaction with β3 integrins and instead re-targeted the virus to α3β1, α6β1, and α6β4 integrins expressed on human epithelial tumor cells. Re-targeting the virus from macrophage β3 integrins resulted in greatly reduced activation of inflammatory cytokines after systemic administration.
Using these insights, we next constructed a replication-competent virus, Ad5-3M, which incorporates three capsid mutations simultaneously: in the hexon HVR1 loop, to avoid natural IgM binding to the virus; a FX binding-ablating mutation T425A in the hexon HVR7 loop, to reduce virus infection of liver cells (20); and in the penton base to prevent virus interaction with macrophage β3 integrins and to re-target virus infection to tumor cells. We performed a high-resolution cryo-EM structural analysis of Ad5-3M, which showed that these engineered mutations had no material impact on the overall structure of the capsid. We showed that Ad5-3M was resistant to IgM- and complement-mediated inactivation by human serum, and after intravenous administration, it had reduced sequestration in Kupffer cells and liver tissue as a whole. Unlike parental unmodified HAdv-C5 and species B adenoviruses HAdv-B11 and HAdv-B35, Ad5-3M also failed to trigger necrotic death of Kupffer cells in vivo. Systemic delivery of Ad5-3M resulted in greatly reduced activation of inflammatory cytokines in the spleen and failed to trigger hepatotoxicity. Taken together, Ad5-3M represents a platform virus configuration, engineered to resist inactivation by humoral factors and to avoid exuberant activation of innate immunity after systemic administration.
A limitation of our work is that Ad5-3M retained sensitivity to neutralization by human sera with high amounts of HAdv-C5-specific neutralizing antibodies, which neutralize the virus in a complement-independent manner (fig. S15). Previous studies showed that HAdv-C5 can be made resistant to pre-existing anti-viral immunity by mutating all hexon HVRs (44). A meta-analysis of published data revealed that 30 to 70 % of adults in the US lack HAdv-C5-specific neutralizing antibodies (45), indicating that the Ad5-3M platform can be immediately used in patients that lack virus-specific neutralizing immunity.
To demonstrate the anti-tumor efficacy of Ad5-3M after systemic administration, in this study we have utilized subcutaneous and disseminated orthotopic lung cancer models of transplanted human lung adenocarcinoma cells into immune-deficient mice, as well as subcutaneous PDX models of xenografts established from primary tumor samples harvested from NSCLC patients. NSG and other strains of immune-deficient mice, such as NOD/SCID and CB17/Icr-SCID, are routinely used to analyze the anti-tumor activity of oncolytic viruses after intravenous delivery, as reported for the oncolytic HAdv-B11-based virus enadenotucirev (46, 47) and an oncolytic VSV-based virus with LCMV envelope, VSV-GP (8). However, NSG mice lack circulating natural IgM antibodies and have profound hyposplenia, thus artificially limiting the impact of these factors on viral bio-distribution and virus sequestration in liver Kupffer cells. Nevertheless, studies with NSG mice can provide valuable information on the oncolytic potency of novel viral drug candidates. Our studies with NSG mice showed that after intravenous administration of Ad5-3M, the virus infected tumor nodules, suppressed tumor growth, and prolonged survival of mice with subcutaneous adenocarcinoma and primary NCSLC PDX tumors. To demonstrate anti-tumor activity of Ad5-3M in a model more closely recapitulating virus-host interactions in the human system, we also utilized NCr/Nude mice with an orthotopic disseminated lung cancer model. NCr/Nude mice have an intact and fully functional innate immune system, they do not exhibit hyposplenia, and they have normal levels of circulating natural IgM antibodies and complement, which are capable of inactivating FX-binding ablated adenovirus vectors (18). All tumor-bearing mice treated with unmodified Ad5-WT virus succumbed within 3 days post virus administration due to severe hepatotoxicity. Intravenous administration of Ad5-3M significantly extended survival of mice with disseminated orthotopic lung tumors (61.5 days vs 49 days for sham-treated controls, p = 0.0448). Additional targeting of Ad5-3M to CD46 receptor overexpressed on human tumor cells by modifying the receptor-interacting domain of the virus fiber protein in Ad5/35-3M virus, extended median survival of mice with disseminated tumors to 101 days post treatment (p = 0.0051). Importantly, after systemic therapy with Ad5-3M, thirty seven percent of mice enrolled in the study completely cleared disseminated tumors from the lungs and converted tumor nodules into collagen-rich scar-like structures. It is certain that evaluation of Ad5-3M-based vectors in clinical trials will ultimately be needed to assess the therapeutic potential of this vector platform at controlling disseminated tumors after systemic virus administration in cancer patients.
Taken together, our data demonstrate that the safety and efficacy of therapeutic viruses can be improved by compounding targeted modifications in virus capsid proteins that mediate virus-host interactions. Our studies show that structural analysis and modeling is a powerful and complementary tool when designing novel virus-based drug candidates. Engineering viruses to evade innate immunity offers a generalizable approach forward in the development of systemically delivered virus therapies for human cancers that are resistant to current treatment modalities.
Materials and Methods
Study design.
The objective of this study was to analyze the feasibility of developing replication-competent HAdv-C5-based virus variants that would be safe for systemic administration at such doses that would demonstrate therapeutic efficacy in mouse models of localized and disseminated cancers. We used structural modeling and targeted mutagenesis approaches to ablate interactions of Ad5-3M virus with coagulation FX, natural IgM, and macrophage integrins of β3 class, which allowed the virus to avoid sequestration in the liver and triggered greatly reduced inflammation after systemic administration. We further used structural modifications to additionally target virus to tumors via enabling its interactions with integrin classes and CD46 receptor expressed on tumor cells. We used grafting of a human adenocarcinoma line and primary NSCLC PDX tumors onto immune-deficient NSG mice as models of localized disease. We grafted a human adenocarcinoma line into NCr/Nude mice to generate an orthotopic model of disseminated lung cancer. In all models tested, we used systemic delivery of Ad5-3M and evaluated therapeutic efficacy. The experiment duration, inclusion and endpoint criteria were set prior to the experiments. For each experiment the group size and statistical details are described in the figure legends or Table S1. Primary data for the main figures of the study are reported in Data file S1.
Mice.
All animal studies were carried out with the approval of the Institutional Animal Care and Use Committee of the Emory University, Atlanta, USA. C57Bl/6, Rag−/−, μMT, C3−/−, CD36−/−, CD18−/−, and Itgam−/− mice were purchased from The Jackson Laboratory, C1q−/− mice were kindly provided by M. Diamond (Washington University, St. Louis, MO). All mice were on C57Bl/6 genetic background, matched by age and housed in specific-pathogen free facilities. NCr/Nude athymic female nude mice were purchased from Taconic Biosciences, NOD/SCID/IL2rγnull (NSG) mice were purchased from The Jackson Laboratory. More details and genotypes are in Table S2.
Cryo-EM structural analysis of Ad5-3M.
Purified Ad5-3M particles were frozen on Quantifoil grids. Cryo-EM movies (5,812 in total) were collected using a ThermoFisher Titan Krios transmission electron microscope equipped with a Gatan imaging filter (GIF) and Gatan K2 Summit direct detector operated in super-resolution mode (University of California, Los Angeles). Motion correction was performed with MotionCorr (48), contrast transfer function parameters estimated with GCTF (49), and particle picking performed with EMAN2 in Swarm mode (50). A dataset of 5,817 particle images was processed with RELION 2.0 (51) (pixel size 2.14 Å, box size 540 pixels) and cisTEM (52) (pixel size 1.07 Å, box size 1080 pixels). The final structure included information from 2,545 particle images. Docking of the HAdv-C5 asymmetric unit (PDB: 6B1T) enabled optimization of the Ad5-3M pixel size (1.04 Å). The resolution of the Ad5-3M structure, as determined by cisTEM gold-standard Fourier Shell Coefficient (FSC), is 3.8 Å at the 0.143 threshold (fig. S8). Local resolution assessment of Ad5-3M was performed with MonoRes (53) (fig. S9).
Analysis of virus accumulation in Kupffer cells.
Virus associated with Kupffer cells was calculated using a published protocol (54) with the following modifications: OCT-preserved frozen liver sections were stained with anti-CD68-AF647 and anti F4/80-AF647 antibody to define Kupffer cells. Virus was stained with polyclonal anti-adenovirus rabbit antibody and anti-rabbit-AF594 secondary antibody. The images were analyzed using ImageJ/Fiji software (NIH). The integrated intensity for virus-specific staining within the boundaries of Kupffer cells in different mouse strains (and with different viruses) was normalized to the median of the integrated intensity of Ad5-WT in the C57Bl6 mouse strain.
Analysis of virus-induced Kupffer cell death in vivo.
To test the amount of Kupffer cell death after virus injection, mice were injected with different viruses (3x1010 vp/mouse). Fifty minutes after virus injection 50 μg of PI was injected in 200 μl of PBS, and 10 minutes after PI injection mice were sacrificed. Livers were harvested and preserved in OCT by freezing. Liver slices were cut and images of PI-positive cells were taken while marking the exact areas for alignment of PI-positive images with images stained for Kupffer cells. Later the slides were fixed and stained with anti-CD68-AF647 antibodies for Kupffer cells.
Assessment of transgene expression in livers.
Livers from virus injected animals were flash frozen in liquid nitrogen and at the time of measurement were thawed, weighed, and disrupted using bullet blender tissue homogenizer (Next advance). Nano-luciferase activity was measured using Nano-Glo luciferase assay (Promega), according to the manufacturer’s instructions. The luminescence was measured on a Synergy HTX plate reader (BioTek).
Analysis of inflammatory cytokine and chemokine production.
Mice were intravenously injected via tail vein with different viruses (3x1010 vp/mouse). One hour later mice were sacrificed and spleens were harvested and flash frozen in liquid nitrogen. Before cytokine measurement the spleens were thawed, weighed, and lysed using Bio-Plex cell lysis kit (Bio-Rad) according to manufacturer’s instructions. For determining cytokines concentration in the blood, the plasma was collected at 6 h post virus injection. Cytokine concentrations were measured using Bio-Plex Pro mouse cytokine 23-Plex assay (Bio-Rad) on a Bio-Plex Magpix multiplex reader (Bio-Rad) using Bio-Plex manager 6.1 software (Bio-Rad) following manufacturer’s recommendations. The final data is calculated as pg/ng per 130 mg of total spleen tissue or pg/ml.
Statistical analyses.
All statistical tests were performed in Prism 8 (GraphPad Software). For each experiment the appropriate test was determined based on data that was analyzed. The statistical tests, results of null hypothesis testing, and n numbers for each experiment are listed in Table S1. The p-values are shown on the figures. All experiments were performed in replicates with each measurement shown on the figures. Sample sizes were determined using G*Power 3.1 software (55) based on the previously determined/estimated standard deviations and desired/estimated effect size (0.4 to 2). In more detail, for estimation of the number of animals for testing the oncolytic effect in tumor models, we performed a pilot study with a total of 10 NSG mice injected with A549 tumor cells. Tumor-bearing mice were randomized in two cohorts and treated with Ad5-3M virus or buffer. On day 19 post treatment the buffer-treated mice had mean tumor measurement of 11 mm by 17 mm (width x length). We estimated that we would consider oncolytic virotherapy effective if the mean of one dimension measurement of tumor size would differ by at least 3 mm between virus- and buffer-treated groups. The pooled standard deviation of the one dimension measurement of tumor size on day 19 in the pilot experiment was 2.04 mm. Thus the minimum effect size that we will consider effective is 1.47. Using G*Power 3.1 software and parameters of f=1.47, α=0.05, 1-β=0.95, df=10, number of group=2, and “a priori” set up, the G*Power program calculated a total sample size of n=19. We performed the experiments with similar numbers of mice also adding five mice for injection with Ad-WT to confirm Ad-WT virus toxicity in tumor-bearing mouse models.
Supplementary Material
Supplementary Materials and Methods
Fig. S1. Efficient HAdv-C5 virus sequestration in liver macrophages requires natural IgM antibodies and scavenger receptor CD36.
Fig. S2. Hexon HVR1 sequence comparison for viruses used in the study.
Fig. S3. Schematic diagrams of the genomic structure of viral vectors used in this study and specific mutations introduced into the hexon and penton base capsid proteins of each adenovirus variant.
Fig. S4. Structural modeling and MD simulations of the penton base containing the Lam1 insertion.
Fig. S5. Modeling of penton base loop in HAdv-C5.
Fig. S6. Penton base loop sequence comparison for viruses used in the study.
Fig. S7. HAdv-C5 de-targeting from β3-integrins reduces inflammation.
Fig. S8. Resolution assessment of Ad5-3M capsid.
Fig. S9. Local resolution assessment of Ad5-3M.
Fig. S10. Ad5-3M effectively infects a variety of non-small cell lung cancer (NSCLC) cell lines.
Fig. S11. Ad5-3M escapes Kupffer cell sequestration.
Fig. S12. Ad5-3M bio-distribution after intravenous delivery.
Fig. S13. A549-luc-C8 cells form tumor nodules in lungs after intravenous injection.
Fig. S14. Intravenous administration replication competent Ad5/35-3M virus does not trigger systemic toxicity.
Fig. S15. Human sera with high HAdv-C5-specific neutralizing antibody titers neutralize Ad5-WT and capsid-modified adenoviruses.
Table S1. (Microsoft Excel format). Statistical summary report.
Table S2. Mouse strains used in the study.
Table S3. List of antibodies used in the study.
Table S4. Sequences of the qPCR primers.
Date file S1. (Microsoft Excel format). Primary data for main figures of the paper.
Data file S2. (Microsoft Excel format). Primary data for figures shown in Supplementary Materials.
Acknowledgements:
We are thankful to Eric Irons (Buffalo University) for analyzing in vitro properties of Ad5-Lam1 virus and Neetu M. Gulati for assistance with preparing Ad5-3M cryo-EM grids.
Funding:
The authors acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057, 1S10OD018111, and 1U24GM116792), NSF (DBI-1338135) and CNSI at UCLA. We thank the Case Western Reserve University High Performance Computing staff for their assistance with campus computing resources. Preliminary image processing was performed using the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. This work used the XSEDE Large Memory Nodes (Bridges Large) at the Pittsburgh Supercomputing Center through allocation MCB170163. This work was supported by NIH grant AI107960 to D.M.S and P.L.S. and by NIH grant AI065429, David C. Lowance Endowment Fund, and Children’s Healthcare of Atlanta Research Trust to D.M.S. C.C.E. acknowledges support from the NIH T32 GM008803 training grant. The analysis of Ad5/35-3M was supported by funding from AdCure Bio to D.M.S.
Footnotes
Competing interests: D.M.S. has equity interest and is a chief scientific officer of AdCure Bio, which develops adenovirus technologies for therapeutic use. D.M.S. is an inventor on issued US patents No. 9,982,276, Penton-mutated, integrin-retargeted adenovirus vectors with reduced toxicity and their use; and No.10,376,549, Detargeted adenovirus variants and related methods; and pending US patent application 16/460,160 and European patent application 16740545.5, Detargeted adenovirus variants and related methods, submitted by AdCure Bio. All other authors declare that they have no competing interests.
Data and materials availability:
The cryo-EM density map of Ad5-3M was deposited in the Electron Microscopy Data Bank (EMDB) under accession number EMD-21049. Requests for Ad5/35-3M virus should be directed to AdCure Bio (www.adcurebio.com). All raw data collected during this study is tabulated and available in Data files S1 and S2.
References and Notes:
- 1.Senior M, Checkpoint inhibitors go viral. Nat Biotechnol 37, 12–17 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH Jr., Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS, Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33, 2780–2788 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Andtbacka RH, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, Amatruda T, Zager JS, Cranmer L, Hsueh E, Chen L, Shilkrut M, Kaufman HL, Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Annals of surgical oncology 23, 4169–4177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, Zinkernagel RM, Control of early viral and bacterial distribution and disease by natural antibodies. Science 286, 2156–2159 (1999); published online EpubDec 10 ( 10.1126/science.286.5447.2156). [DOI] [PubMed] [Google Scholar]
- 5.Maloney BE, Perera KD, Saunders DRD, Shadipeni N, Fleming SD, Interactions of viruses and the humoral innate immune response. Clin Immunol 212, 108351 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rangaswamy US, Cotter CR, Cheng X, Jin H, Chen Z, CD55 is a key complement regulatory protein that counteracts complement-mediated inactivation of Newcastle Disease Virus. The Journal of general virology 97, 1765–1770 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Tesfay MZ, Ammayappan A, Federspiel MJ, Barber GN, Stojdl D, Peng KW, Russell SJ, Vesiculovirus neutralization by natural IgM and complement. J Virol 88, 6148–6157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muik A, Stubbert LJ, Jahedi RZ, Geibeta Y, Kimpel J, Dold C, Tober R, Volk A, Klein S, Dietrich U, Yadollahi B, Falls T, Miletic H, Stojdl D, Bell JC, von Laer D, Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res 74, 3567–3578 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Pipperger L, Koske I, Wild N, Mullauer B, Krenn D, Stoiber H, Wollmann G, Kimpel J, von Laer D, Banki Z, Xenoantigen-Dependent Complement-Mediated Neutralization of Lymphocytic Choriomeningitis Virus Glycoprotein-Pseudotyped Vesicular Stomatitis Virus in Human Serum. J Virol 93, e00567–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kundhavai Natchiar S, Venkataraman S, Mullen TM, Nemerow GR, Reddy VS, Revised Crystal Structure of Human Adenovirus Reveals the Limits on Protein IX Quasi-Equivalence and on Analyzing Large Macromolecular Complexes. J Mol Biol 430, 4132–4141 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Dai X, Wu L, Sun R, Zhou ZH, Atomic Structures of Minor Proteins VI and VII in Human Adenovirus. J Virol 91, e00850–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, Zhang W, Ehrhardt A, Expanding the Spectrum of Adenoviral Vectors for Cancer Therapy. Cancers (Basel) 12, 1139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML, Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80, 148–158 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Morral N, O’Neal WK, Rice K, Leland MM, Piedra PA, Aguilar-Cordova E, Carey KD, Beaudet AL, Langston C, Lethal toxicity, severe endothelial injury, and a threshold effect with high doses of an adenoviral vector in baboons. Hum Gene Ther 13, 143–154. (2002). [DOI] [PubMed] [Google Scholar]
- 15.Brunetti-Pierri N, Palmer DJ, Beaudet AL, Carey KD, Finegold M, Ng P, Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum Gene Ther 15, 35–46 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov DM, Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci, U S A 105, 5483–5488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waddington SN, Mcvey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SMK, Greig JA, Denby L, Custers J, Morita T, Francischetti IMB, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Hlavenga MJE, Nicklin SA, Baker AH, Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132, 397–409 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Qiu Q, Tian J, Smith JS, Conenello GM, Morita T, Byrnes AP, Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat Med 19, 452–457 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Bottermann M, Foss S, Caddy SL, Clift D, van Tienen LM, Vaysburd M, Cruickshank J, O’Connell K, Clark J, Mayes K, Higginson K, Lode HE, McAdam MB, Sandlie I, Andersen JT, James LC, Complement C4 Prevents Viral Infection through Capsid Inactivation. Cell Host Microbe 25, 617–629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doronin K, Flatt JW, Di Paolo NC, Khare R, Kalyuzhniy O, Acchione M, Sumida JP, Ohto U, Shimizu T, Akashi-Takamura S, Miyake K, MacDonald JW, Bammler TK, Beyer RP, Farin FM, Stewart PL, Shayakhmetov DM, Coagulation Factor X Activates Innate Immunity to Human Species C Adenovirus. Science 338, 795–798 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B, Kay MA, The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol 71, 8798–8807. (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR, Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73, 309–319 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Di Paolo NC, Miao EA, Iwakura Y, Murali-Krishna K, Aderem A, Flavell RA, Papayannopoulou T, Shayakhmetov DM, Virus Binding to a Plasma Membrane Receptor Triggers Interleukin-1 alpha-Mediated Proinflammatory Macrophage Response In Vivo. Immunity 31, 110–121 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khare R, Hillestad ML, Xu Z, Byrnes AP, Barry MA, Circulating antibodies and macrophages as modulators of adenovirus pharmacology. J Virol 87, 3678–3686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doszpoly A, de la Cuesta F, Lopez-Gordo E, Benezech C, Nicklin SA, Baker AH, Human Adenovirus Serotype 5 Is Sensitive to IgM-Independent Neutralization In Vitro and In Vivo. Viruses 11, 616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy MR, Doszpoly A, Turner G, Nicklin SA, Baker AH, The relevance of coagulation factor X protection of adenoviruses in human sera. Gene Ther 23, 592–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian J, Xu Z, Smith JS, Byrnes AP, Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol 82, 11705–11713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiramoto E, Tsutsumi A, Suzuki R, Matsuoka S, Arai S, Kikkawa M, Miyazaki T, The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci Adv 4, eaau1199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurokawa J, Arai S, Nakashima K, Nagano H, Nishijima A, Miyata K, Ose R, Mori M, Kubota N, Kadowaki T, Oike Y, Koga H, Febbraio M, Iwanaga T, Miyazaki T, Macrophage-derived AIM is endocytosed into adipocytes and decreases lipid droplets via inhibition of fatty acid synthase activity. Cell Metab 11, 479–492 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Shayakhmetov DM, Eberly AL, Li ZY, Lieber A, Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosorne escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J Virol 79, 4553–4553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kibbey MC, Grant DS, Kleinman HK, Role of the SIKVAV site of laminin in promotion of angiogenesis and tumor growth: an in vivo Matrigel model. Journal of the National Cancer Institute 84, 1633–1638 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Giannelli G, Astigiano S, Antonaci S, Morini M, Barbieri O, Noonan DM, Albini A, Role of the alpha3beta1 and alpha6beta4 integrins in tumor invasion. Clin Exp Metastasis 19, 217–223 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Flatt JW, Kim R, Smith JG, Nemerow GR, Stewart PL, An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS One 8, e61571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi Y, Deane CM, FREAD revisited: Accurate loop structure prediction using a database search algorithm. Proteins 78, 1431–1440 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K, Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Paolo NC, Doronin K, Baldwin LK, Papayannopoulou T, Shayakhmetov DM, The transcription factor IRF3 triggers “defensive suicide” necrosis in response to viral and bacterial pathogens. Cell reports 3, 1840–1846 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, Byrnes AP, Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Molecular Therapy 13, 108–117 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Machiels JP, Salazar R, Rottey S, Duran I, Dirix L, Geboes K, Wilkinson-Blanc C, Pover G, Alvis S, Champion B, Fisher K, McElwaine-Johnn H, Beadle J, Calvo E, A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE). Journal for immunotherapy of cancer 7, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakurai F, Mizuguchi H, Yamaguchi T, Hayakawa T, Characterization of in vitro and in vivo gene transfer properties of adenovirus serotype 35 vector. Mol Ther 8, 813–821 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Ferguson MS, Lemoine NR, Wang Y, Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol 2012, 805629 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Chirmule N, Gao GP, Qian R, Croyle M, Joshi B, Tazelaar J, Wilson JM, Acute cytokine response to systemic adenoviral vectors in mice is mediated by dendritic cells and macrophages. Molecular Therapy 3, 697–707 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Stichling N, Suomalainen M, Flatt JW, Schmid M, Pacesa M, Hemmi S, Jungraithmayr W, Maler MD, Freudenberg MA, Pluckthun A, May T, Koster M, Fejer G, Greber UF, Lung macrophage scavenger receptor SR-A6 (MARCO) is an adenovirus type-specific virus entry receptor. PLoS Pathog 14, e1006914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nemerow GR, Cell receptors involved in adenovirus entry. Virology 274, 1–4 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Roberts DM, Nanda A, Havenga MJE, Abbink P, Lynch DM, Ewald BA, Liu J, Thorner AR, Swanson PE, Gorgone DA, Lifton MA, Lemckert AAC, Holterman L, Chen B, Dilraj A, Carville A, Mansfield KG, Goudsmit J, Barouch DH, Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 441, 239–243 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Mennechet FJD, Paris O, Ouoba AR, Salazar Arenas S, Sirima SB, Takoudjou Dzomo GR, Diarra A, Traore IT, Kania D, Eichholz K, Weaver EA, Tuaillon E, Kremer EJ, A review of 65 years of human adenovirus seroprevalence. Expert Rev Vaccines 18, 597–613 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Illingworth S, Di Y, Bauzon M, Lei J, Duffy MR, Alvis S, Champion B, Lieber A, Hermiston T, Seymour LW, Beadle J, Fisher K, Preclinical Safety Studies of Enadenotucirev, a Chimeric Group B Human-Specific Oncolytic Adenovirus. Molecular therapy oncolytics 5, 62–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, Thorne S, Reid T, Ni S, Lieber A, Fisher K, Seymour L, Rubanyi GM, Harkins RN, Hermiston TW, Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One 3, e2409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng YF, Agard DA, MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang K, Gctf: Real-time CTF determination and correction. Journal of Structural Biology 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ, EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Scheres SH, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grant T, Rohou A, Grigorieff N, cisTEM, user-friendly software for single-particle image processing. Elife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilas JL, Gomez-Blanco J, Conesa P, Melero R, de la Rosa-Trevin JM, Oton J, Cuenca J, Marabini R, Carazo JM, Vargas J, Sorzano COS, MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure 26, 337-+ (2018). [DOI] [PubMed] [Google Scholar]
- 54.Smith JS, Xu ZL, Byrnes AP, A quantitative assay for measuring clearance of adenovirus vectors by Kupffer cells. Journal of Virological Methods 147, 54–60 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Faul F, Erdfelder E, Buchner A, Lang AG, Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav Res Methods 41, 1149–1160 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Chan KY, Trabuco LG, Schreiner E, Schulten K, Cryo-electron microscopy modeling by the molecular dynamics flexible fitting method. Biopolymers 97, 678–686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang J, Zhang Y, I-TASSER server: new development for protein structure and function predictions. Nucleic acids research 43, W174–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K, Scalable molecular dynamics with NAMD. Journal of Computational Chemistry 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphrey W, Dalke A, Schulten K, VMD: Visual molecular dynamics. J Mol Graph Model 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE, UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muck-Hausl M, Solanki M, Zhang W, Ruzsics Z, Ehrhardt A, Ad 2.0: a novel recombineering platform for high-throughput generation of tailored adenoviruses. Nucleic acids research 43, e50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mittereder N, March KL, Trapnell BC, Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol 70, 7498–7509. (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zarif JC, Hernandez JR, Verdone JE, Campbell SP, Drake CG, Pienta KJ, A phased strategy to differentiate human CD14+monocytes into classically and alternatively activated macrophages and dendritic cells. Biotechniques 61, 33–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jarvi J TM, Wang M, Cheng M, Creamer-Hente M, Refining the PDX Model: Development of a less invasive and disposable tumor engraftment process. Laboratory Animal Science Professional 3, 47–51 (2016). [Google Scholar]
- 66.Alcoser SY, Kimmel DJ, Borgel SD, Carter JP, Dougherty KM, Hollingshead MG, Real-time PCR-based assay to quantify the relative amount of human and mouse tissue present in tumor xenografts. Bmc Biotechnol 11, 124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods
Fig. S1. Efficient HAdv-C5 virus sequestration in liver macrophages requires natural IgM antibodies and scavenger receptor CD36.
Fig. S2. Hexon HVR1 sequence comparison for viruses used in the study.
Fig. S3. Schematic diagrams of the genomic structure of viral vectors used in this study and specific mutations introduced into the hexon and penton base capsid proteins of each adenovirus variant.
Fig. S4. Structural modeling and MD simulations of the penton base containing the Lam1 insertion.
Fig. S5. Modeling of penton base loop in HAdv-C5.
Fig. S6. Penton base loop sequence comparison for viruses used in the study.
Fig. S7. HAdv-C5 de-targeting from β3-integrins reduces inflammation.
Fig. S8. Resolution assessment of Ad5-3M capsid.
Fig. S9. Local resolution assessment of Ad5-3M.
Fig. S10. Ad5-3M effectively infects a variety of non-small cell lung cancer (NSCLC) cell lines.
Fig. S11. Ad5-3M escapes Kupffer cell sequestration.
Fig. S12. Ad5-3M bio-distribution after intravenous delivery.
Fig. S13. A549-luc-C8 cells form tumor nodules in lungs after intravenous injection.
Fig. S14. Intravenous administration replication competent Ad5/35-3M virus does not trigger systemic toxicity.
Fig. S15. Human sera with high HAdv-C5-specific neutralizing antibody titers neutralize Ad5-WT and capsid-modified adenoviruses.
Table S1. (Microsoft Excel format). Statistical summary report.
Table S2. Mouse strains used in the study.
Table S3. List of antibodies used in the study.
Table S4. Sequences of the qPCR primers.
Date file S1. (Microsoft Excel format). Primary data for main figures of the paper.
Data file S2. (Microsoft Excel format). Primary data for figures shown in Supplementary Materials.
Data Availability Statement
The cryo-EM density map of Ad5-3M was deposited in the Electron Microscopy Data Bank (EMDB) under accession number EMD-21049. Requests for Ad5/35-3M virus should be directed to AdCure Bio (www.adcurebio.com). All raw data collected during this study is tabulated and available in Data files S1 and S2.