

ABSTRACT
Root exudation is one of the primary processes that mediate interactions between plant roots, microorganisms, and the soil matrix, yet the mechanisms by which exudation alters microbial metabolism in soils have been challenging to unravel. Here, utilizing distinct sorghum genotypes, we characterized the chemical heterogeneity between root exudates and the effects of that variability on soil microbial membership and metabolism. Distinct exudate chemical profiles were quantified and used to formulate synthetic root exudate treatments: a high-organic-acid treatment (HOT) and a high-sugar treatment (HST). To parse the response of the soil microbiome to different exudate regimens, laboratory soil reactors were amended with these root exudate treatments as well as a nonexudate control. Amplicon sequencing of the 16S rRNA gene illustrated distinct microbial diversity patterns and membership in response to HST, HOT, or control amendments. Exometabolite changes reflected these microbial community changes, and we observed enrichment of organic and amino acids, as well as possible phytohormones in the HST relative to the HOT and control. Linking the metabolic capacity of metagenome-assembled genomes in the HST to the exometabolite patterns, we identified microorganisms that could produce these phytohormones. Our findings emphasize the tractability of high-resolution multiomics tools to investigate soil microbiomes, opening the possibility of manipulating native microbial communities to improve specific soil microbial functions and enhance crop production.
IMPORTANCE Decrypting the chemical interactions between plant roots and the soil microbiome is a gateway for future manipulation and management of the rhizosphere, a soil compartment critical to promoting plant fitness and yields. Our experimental results demonstrate how soil microbial community and genomic diversity is influenced by root exudates of differing chemical compositions and how changes in this microbiome result in altered production of plant-relevant metabolites. Together, these findings demonstrate the tractability of high-resolution multiomics tools to investigate soil microbiomes and provide new information on plant-soil environments useful for the development of efficient and precise microbiota management strategies in agricultural systems.
KEYWORDS: exometabolomics, metagenomics, rhizodeposition, root exudates, sorghum
INTRODUCTION
The region where plant roots interface with soil, known as the rhizosphere, is one of the most intricate and diverse microbial ecosystems on earth (1). It is widely recognized that exudation of compounds from plant roots is key to assembling these rhizosphere microbial communities (2–4), which in turn can influence plant health and soil biogeochemistry. Plant roots exude up to 40% of their photosynthetically derived carbon, with these compounds ranging from small (e.g., carbohydrates, amino acids, organic acids, and phytohormones) to large (e.g., proteins and mucilage) molecules (5, 6). These exudates provide microbial substrates, as well as signaling molecules, that stimulate microbial activity. For instance, rhizosphere-enriched microorganisms can suppress plant pathogens and provide plant nutrients through nitrogen fixation and metal chelation, among other mechanisms (2, 7–9). Moreover, some members of the rhizosphere can produce plant hormones like indole-3-acetic acid (IAA), gibberellins, cytokinins, and abscisic acid, which modulate plant physiological health (10, 11). The importance of these various contributions highlights the clear need to understand the mechanistic link between plant root exudate chemistry and metabolic changes in the soil microbiome.
Recently there has been an increased focus on plant rhizodeposition and its impact on soil microorganisms. To enhance tractability in a complex system, many of these keystone studies selected model plants (e.g., Brachypodium distachyon and Arabidopsis thaliana) and evaluated their impact on known plant growth-promoting rhizobacteria (PGPR) (e.g., Pseudomonas) (12–15). However, the scaling of these concepts to more complex chemical profiles and soil microbial communities has been constrained by methodological challenges. For example, where exudates were chemically profiled, they were often limited to a small set of known metabolites (16–19), or when microbial communities were analyzed, they often relied on methods that tracked changes in membership (16S rRNA, phospholipid fatty acid profiles), but not metabolic potential over the course of exudation (20, 21). Therefore, there is a current need for studies that employ high-resolution microbial and chemical methods to begin to mechanistically describe the exudate-microbe interactions in agriculturally relevant crop species.
Here, we used a multiomics approach to characterize how root exudation from a model crop plant can drive soil microbial community structure and function. Using three common, but phenotypically diverse, sorghum (Sorghum bicolor (L.) Moench) genotypes, we determined the variation in root exudate chemical composition among these plant varieties. To narrow in on the microbial response to these different exudate profiles, we created plant-free soil microcosms and fed these with two sorghum-informed exudate amendments. We tracked these microcosms over 20 days using exometabolomics, 16S rRNA gene profiling, and genome-resolved metagenomics. Integrating these data, we show that different sorghum exudate treatments led to distinct microbial communities and metabolisms, including the production of three phytohormones. Harnessing this knowledge could support the growing need for sustainable agroecosystems by developing holistic agricultural management strategies that optimize the metabolic capabilities of the soil microbiome.
RESULTS AND DISCUSSION
Sorghum genetics influence root exudation patterns.
Sorghum is one of the most widely produced agricultural crops in the world, serving as a grain, forage, and cover crop for use as human food, livestock feed, and as a biofuel feedstock (22, 23). Due to its diverse agronomic uses, we leveraged three sorghum genotypes known to have distinct aboveground phenotypes that we hypothesized would contribute to distinct root exudate profiles. We selected (i) the grain sorghum BTx623, (ii) the sweet sorghum type Leoti, and (iii) the bioenergy sorghum PI 505735 (23–25). We grew sorghum genotypes in vitro (hydroponically) for 7 days, and soluble exudates were collected in water and analyzed with nontargeted gas chromatography-mass spectrometry (GC-MS).
Metabolites from sorghum seedlings spanned known root exudate metabolite classes ranging from sugars to sugar alcohols, organic acids, and amino acids (Fig. 1A; see Fig. S1 in the supplemental material). Chemical differences were observed between genotypes BTx623 and Leoti, while PI 505735 represented an intermediate root exudate chemical profile (Fig. 1A). Among BTx623 and Leoti, statistical differences in exudate profiles were found (analysis of variance [ANOVA], P < 0.001), with BTx623 exudates enriched in monosaccharides and disaccharides, while Leoti was significantly enriched in organic and amino acids (Fig. 1A; see File S1 in the supplemental material). Given these sorghum genotypes released root exudates with distinct metabolic profiles, we next pursued how these different exudate regimens would alter microbiome membership and metabolic responses.
FIG 1.

Root exudation metabolite profiles from three sorghum genotypes were used to design relevant amendments for soil microcosms. (A) Nonmetric multidimensional scaling (NMDS) of Bray-Curtis distances across the metabolite abundance of Leoti (purple), BTx623 (orange), and PI 505735 (green) sorghum genotypes (stress = 0.06). Ellipses denote the 95% confidence interval for each treatment. Significant metabolite vectors are shown (P < 0.05) and labeled in gray. (B) Butterfly plots show the summed total amount of each compound added across the treatments (Phe, phenylalanine; 5-Oxopro, 5-oxoproline); orange represents the proportion added to high-sugar treatment (HST), and purple represents the proportion added to the high-organic-acid treatment (HOT). For example, of the sucrose added, 95% went to the HST and 5% to the HOT. For actual added concentrations, see Table S3. (C) The microcosm schematic depicts the treatment formulation, where HOT represents Leoti, HST represents BTx623, and Control was buffered medium lacking an exudate treatment. Triplicate microcosms with agricultural soil were maintained for a 20-day experiment, with the sampling schematic denoting when samples were obtained. Days with exudate addition are noted with pipettes, and the day number is in red. Time points with circles represent the samples taken each day, with the total number (n) of samples for each analysis listed. Colors within circles represent the type of sample (HST, orange; HOT, purple; control, teal) taken at that time point.
Laboratory microcosms reveal that exudate treatment structures agricultural soil microbial communities.
We formulated root exudate solutions for laboratory-scale soil reactors (microcosms) using either a high-sugar treatment (HST) representing BTx623 or a high-organic-acid treatment (HOT) representing Leoti. These treatments were run in parallel with exudate-lacking control microcosms. Both HST- and HOT-amended microcosms received the same 11 root exudate compounds (glucose, galactose, fructose, sucrose, trehalose, malic acid, lysine, phenylalanine, 5-oxoproline, citric acid, and shikimic acid) (Table S3), but in various concentrations to model the relative concentrations observed in BTx623 and Leoti exudates (Fig. 1B). The soil microcosms were constructed using soil from semiarid agricultural plots and were amended with exudate treatments daily for the first 5 days and sampled for chemical and microbiological analyses over 20 days (Fig. 1C).
Gene amplicon sequencing of the 16S rRNA gene was used to temporally profile the microbial diversity and membership across the three treatments (HOT, HST, exudate control) (Fig. 1C). We sequenced 81 samples, generating 2,148,831 reads with an average of more than 30,000 reads per sample (File S2). After denoising, a total of 9,818 amplicon sequencing variants (ASVs) were detected, representing 43 phyla (File S2). We first assessed alpha and beta diversity metrics to understand soil microbial community changes in response to exudates.
Over time, we observed distinct changes in response to amendments with either HST or HOT (Fig. 2A). For instance, after 1 day of exudate amendment, HST-amended microcosms had significant decreases in ASV richness (P = 0.007), Shannon’s diversity index (P = 0.019), and Pielou’s evenness (P = 0.017). In contrast, we did not detect significant changes in these metrics for HOT or control microcosms in this time period. This suggests HST enriched for select members of the microbial community. In support of this result, we observed an ASV belonging to the genus Pseudomonas (File S2) accounted for 59.4% of the HST community at day 1. Following 5 days of exudate addition, the HST-amended microcosms retained low diversity (P = 0.038), richness (P = 0.042), and evenness (P = 0.017) compared to the HOT and control microcosms (Fig. 2A). By the final day of the experiment, these diversity metrics were no longer significantly different between HST and HOT, highlighting the importance of these amendments in structuring the microbial communities.
FIG 2.
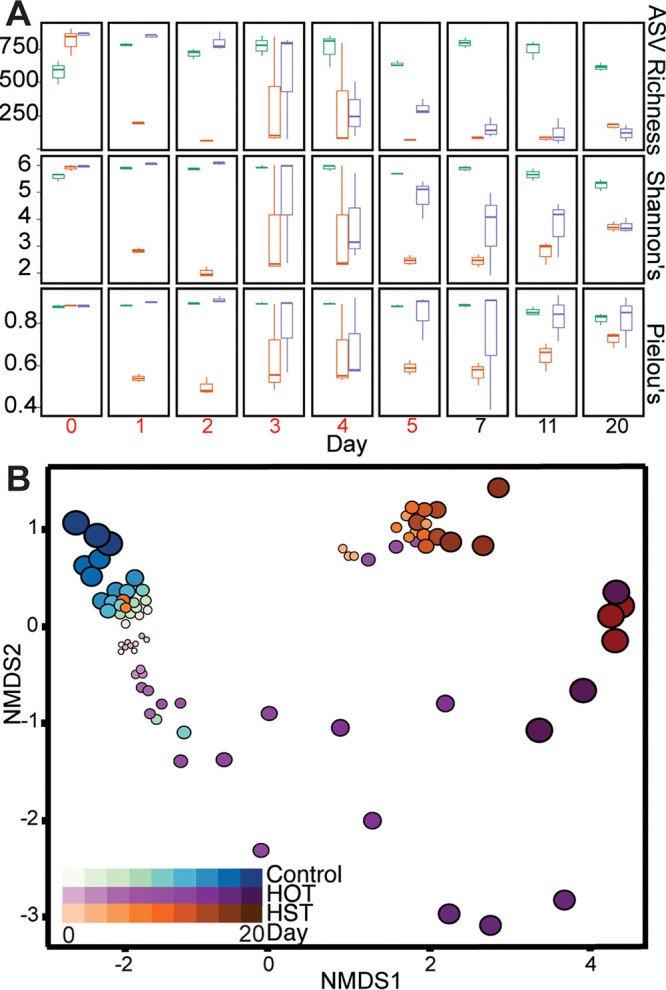
16S rRNA gene diversity metrics and membership changes with exudate amendment. (A) ASV richness (top), Shannon’s diversity index (middle), and Pielou’s evenness for each day highlighting differences in treatment richness, alpha diversity, and community evenness. Coloring corresponds to treatment: control, teal; HST, orange; and HOT, purple. Days where exudates were added are in red (days 0 to 5). (B) Nonmetric multidimensional scaling (NMDS) of Bray-Curtis distances of 16S rRNA amplicon communities showing changes in microbial community structure and membership over time, with colors representing treatment and the size and darkness of circles representing time (stress = 0.09).
Next, changes in the membership and structure of the microbial community across treatments were assessed using the Bray-Curtis dissimilarity matrix and visualized using nonmetric multidimensional scaling (NMDS) (Fig. 2B). We found that HST and HOT amendments significantly shifted microbial community composition and structure during and after periods of synthetic root exudate addition (multiresponse permutation procedure [mrpp] = 0.01, P < 0.05). A permutational multivariate analysis of variance (PERMANOVA) revealed that microbial communities were significantly altered across treatments (P = 0.001) and time (P = 0.001). Building on this, beta dispersion analyses indicated the control exudate communities were the most stable over time, while the HOT had the greatest across-time variation in microbial community membership (Fig. 2B). These microbial community alpha and beta diversity metrics highlight that both treatment and time were factors shaping the soil microbiome in exudate-amended microcosms. Scaling beyond these microcosms, this research highlights the need for understanding the composition and temporal dynamics of natural root exudates.
Exometabolites hint at potential microbial metabolisms stimulated by exudates.
In these soil reactors, we tracked the temporal dynamics of exometabolites (Fig. 1C), which are the extracellular fraction of molecules that are inferred to be produced and/or utilized by soil microorganisms. We classified and assigned the detected exometabolites to three chemical classes: (i) central carbon metabolism, (ii) amino acids and derived compounds, and (iii) phytohormones (Fig. 3; File S1). The exometabolites were coordinated to the microbial communities identified by 16S rRNA gene sequencing, and thus demonstrated differences by treatment and time (Fig. 3; Fig. S2). Relative to the HOT and controls, the HST was enriched in most of the detected exometabolites, likely reflecting a greater metabolic stimulation from the sugar rich treatment (Fig. 3). This finding is not completely unexpected, as prior studies have noted sugar metabolism is more efficient than amino acid metabolism in soil microorganisms (26).
FIG 3.

Exometabolite abundances across treatments. We categorized the detected exometabolites into three chemical classes: (A) central carbon metabolism, (B) amino acids and derivatives, and (C) phytohormones. Within each, heat maps (left) show the relative abundance (represented as a Z-score across samples) of each exometabolite detected. Time increases from left (day 0) to right (day 20) within each treatment, with days where exudate treatments were added in red (days 0 to 5). Ridgeline plots (right) show the relative abundance (0 to 100%) of these exometabolites over time. Coloring corresponds to treatment: control, teal; HST, orange; and HOT, purple.
Metabolites relevant to microbial central carbon metabolism were some of the most enriched exometabolites in HST microcosms (Fig. 3A). We detected six organic acids that changed significantly over time in HST and were significantly enriched in HST compared to the control and HOT. For example, between days 2 and 3, succinic acid and fumaric acid increased 1.5- and 0.9-fold (log2), respectively, in the HST-amended microcosms relative to HOT and the control treatments (Fig. 3A). We also detected increases in itaconic acid (0.7-fold) and oxalic acid (1.6-fold) over time, both of which can be derived from tricarboxylic acid (TCA) cycle intermediates cis-aconitate and oxaloacetate, respectively (27). Furthermore, consumption of malate and citrate was inferred in HST-amended microcosms at the same time points due to their loss over time (Fig. S3). Perhaps indicating metabolite cross-feeding across the community, we also observed a spike in pyruvate at days 2 to 3 in the HST, followed by consumption concomitant with the production of lactic acid across the experiment (Fig. 3A). Collectively, these exometabolite data pointed to HST differentially altering soil carbon pools via the metabolism of the microbiome.
We also detected chemical evidence for production and consumption of amino acid and amino acid-derived compounds by these soil microbial communities. Like the central carbon metabolites, these organic nitrogen exometabolites were most enriched in HST-amended microcosms relative to the other treatments, a somewhat unexpected response given the HOT amendments were initially dosed with these types of compounds (Fig. 1B). Compared to control and HOT-amended microcosms, we observed the production of two nonproteinogenic amino acids (β-alanine and pipecolic acid [PA]) and one aromatic compound (phenyllactic acid [PLA]) in HST-amended microcosms (Fig. 3B). β-Alanine, which was produced over the first 7 days but removed by day 20, is an important amino acid precursor and also necessary for biosynthesis of coenzyme A, a critical cofactor of microbial metabolism in soils (28).
Next, we identified and tracked the production of pipecolic acid (PA), whose production peaks between days 2 and 4 in HST-amended microcosms. This amino acid is thought to be microbially synthesized from lysine (29), a component added to both treatments (Fig. 1B), that was consumed during this period (Fig. S3). PA synthesis has broader ramifications for the entire microbiome, as it is a required intermediate in secondary metabolite production of antibiotics and anthelmintics in many bacteria (29). PA dynamics, which indicate production and subsequent consumption by day 20, suggest its use as a public good (30). Finally, phenyllactic acid (PLA), a phenylalanine derivative, increased 2.6-fold in the HST microcosms over time, peaking at days 11 and 20 (Fig. 3B). Microbes that release amino acid and amino acid-derived compounds could be competitive root colonizers or act as antagonistic agents against target pathogens (31).
Most notably, we observed a significant increase in the abundance of three phytohormones. Salicylic acid (SA), benzoic acid (BA), and indole-3-acetic acid (IAA) significantly increased (P < 0.01) over the experiment in the HST relative to microcosms treated with HOT or the exudate-lacking control (Fig. 3C). In HST microcosms, BA and SA increased 1-fold (log2), while IAA increased the most, with a 3-fold (log2) increase from day 0 to day 20. In plants, SA, BA, and IAA are vital phytohormones integral to physiological processes like plant defense and development (32). Our detection of these metabolites over time highlights that certain root exudation chemical profiles can stimulate soil microbes to produce phytohormones critical for plant growth and defense.
Curation of a sorghum exudate-responsive microbial genome catalog.
In light of the phytohormone production observed in our HST microcosms, we sought to identify microbial genomes capable of producing these compounds. Towards this goal, we constructed metagenome-assembled genomes (MAGs) representing the suite of soil microbes in the HST and control microcosms. To maximize MAG recovery, we obtained more than 365 Gbp total of sequencing from control and HST microcosms, at three different time points spanning the experiment (control, days 0, 5, and 20, and HST, days 5 and 20, each in triplicate; n = 15 metagenomes) (Fig. 1C). With these data, we reconstructed 371 MAGs that were dereplicated at 99% identity into 243 MAGs, of which 28% were high quality (33) (Fig. 4A; File S2). The genome recovery demonstrated here exceeds those from field-based studies, where assembling and binning are hampered by the vast complexity in agricultural soils (34), further validating the relevance of stimulated laboratory microcosms to increase the microbial genome tractability of soil systems (35, 36).
FIG 4.

Taxonomy of the 243 dereplicated metagenome-assembled genomes in the genome database. (A) Sequential colored rings indicate the most-resolved taxonomic level that could be assigned by GTDB-tk (72). Taxonomic level is denoted in black with a single-letter abbreviation: D, domain; P, phylum; C, class; O, order; F, family; G, genus; and S, species. Ring color corresponds to phylum assignment, with the color legend at right. The treatment condition corresponding to MAG detection is illustrated in the outer ring labeled “Detection”; MAGs detected only in control metagenomes are indicated by teal, MAGs detected only in HST metagenomes are indicated by orange, and MAGs that were detected in both are indicated in brown (see Materials and Methods for detection thresholds). The MAG quality is shown in the outermost ring following the MIMAG standards (33): high quality (HQ; >90% complete and <5% contamination), medium quality (MQ; >50% complete and <10% contamination), and low quality (LQ; here defined as >48% complete and <10% contamination). (B) Stacked bar graph shows the number of dereplicated MAGs recovered that represent novel families, genera, or species according to taxonomy assignments from GTDB-tk. Coloring corresponds to MAG phylum.
Of these MAGs, 49% (n = 119) were detected in HST-amended microcosm samples, while 41% (n = 100) were detected in the control microcosm samples (Fig. 4A; File S2). Notably, only 10% (n = 24) of these MAGs were shared across the two treatments, further supporting our 16S rRNA and exometabolite data showing HST amendments significantly altered the soil microbial community. Our MAG database contains representative soil microbes across 20 phyla, dominated by members of the Actinobacteriota and Proteobacteria (Fig. S4), and includes MAGs belonging to 15 previously unidentified families (6%), 41 previously unidentified genera (17%), and 157 previously unidentified species (65%) (Fig. 4B). We recovered 26 MAGs with partial or full 16S rRNA genes and could directly link 19 of these MAGs to 16S rRNA amplicon sequencing-identified ASVs, providing metabolic blueprints for these taxa. These results highlight the advantage of coupling community profiling (16S rRNA gene) with high-resolution metagenomics to capture the functional potential encoded within the soil microbiome.
Bacterial salicylic acid production is assigned to a potentially new species of Pseudomonas.
We examined which of our MAGs carried the two genes for SA production (pchA, encoding isochorismate synthase, and pchB, encoding isochorismate pyruvate lyase). The first gene, pchA, catalyzes the conversion of chorismate to isochorismate, with the second gene, pchB, catalyzing the subsequent conversion to salicylate (Fig. 5A) (37). Two of our HST-detected MAGs carried a pchA gene, while one of these MAGs also carried pchB. Notably, the MAG that carried both pchA and pchAB likely represents a novel species within the genus Pseudomonas_E (L_E1_T20_B_bin.65) and had two copies of this gene set (Fig. 5B). Consistent with our exometabolite data for SA production (Fig. 3C), this Pseudomonas_E MAG was abundant in HST metagenomes at day 20 (Fig. 5E).
FIG 5.

Diverse MAGs encode biosynthetic potential for salicylic acid, benzoic acid, and indoleacetic acid. (A) Salicylic acid production from chorismate by pchAB. (B) A Pseudomonas MAG carried two biosynthetic gene clusters containing pchAB, including one for the antimicrobial promysalin (left) and another for a predicted siderophore (right). The clusters on the bottom are the reference clusters from antiSMASH. (C) Benzoic acid production from multiple pathways. (D) Indoleacetic acid production pathways. (E) Rank abundance curve of MAGs detected in HST metagenomes at day 20. Bars represent the average relative abundance (n = 3), and error bars represent 1 standard deviation. Bars are colored by MAG phylum, and key phytohormone-producing MAGs are indicated. MAG detection is denoted below abundance bars for potential phytohormone-producing MAGs, with MAGs detected only in HST in orange (n = 18) and MAGs detected in both HST and the control in brown (n = 1). In panels A, C, and D, circles correspond to MAGs carrying each gene in panel E. The circles for the two Mycobacterium MAGs with BA and IAA potential are outlined in pink.
Previous research has shown that bacterial species such as those of the genera Pseudomonas, Bacillus, Mycobacterium, and Azospirillum synthesize this important compound, with pseudomonads, like those enriched in our reactors, exhibiting a propensity for SA production (38). It is recognized that microbially produced SA can act as a plant defense hormone providing pathogen resistance and modulating developmental signaling cascades (39–41). Beyond phytohormone roles, bacterially produced SA can function as a siderophore for metal acquisition from soils (38, 39, 42) or to support antibiotic production (43). Excitingly, further genomic analysis of the Pseudomonas_E MAG revealed one set of pchAB genes, which occurred in a predicted siderophore biosynthetic gene cluster (Fig. 5B). Furthermore, the second pchAB cluster occurred in a 16-kb gene cluster with 100% gene similarity to the promysalin biosynthetic gene cluster from Pseudomonas putida (Fig. 5B). Promysalin is an antibiotic that contains SA and was shown to be selectively antagonistic to other closely related pseudomonads to enable rhizosphere colonization (44). Our data suggest the HST microcosms created conditions that stimulated the Pseudomonas_E MAG to produce SA, which could ultimately aid in its competition for limited resources in the complex soil microbiome. Additionally, the accumulation of SA in the enrichment leaves open the possibility that beyond enhanced microbial metabolism, this exometabolite could also be available to act as a plant defense hormone.
Diverse benzoic acid production pathways encoded by multiple MAGs.
We next investigated BA production potential in our MAGs. Known BA production pathways derive from degradation of aromatic compounds (Fig. 5C) (45). The terminal step of most of these pathways is the oxidation of benzaldehyde to BA by benzaldehyde dehydrogenase (encoded by xylC). In these microcosms, seven MAGs carried xylC, including Stenotrophomonas indicatrix, Arthrobacter koreensis, two MAGs of Rhodococcus erythropolis, and two species of Mycobacterium (Fig. 5C). These MAGs were only detected in the HST microcosms at day 20 (Fig. 5E), matching when BA was observed in exometabolomic data (Fig. 3C).
The S. indicatrix MAG encoded an aryl-alcohol dehydrogenase (EC 1.1.1.90) adjacent to xylC, which would enable reduction of benzyl alcohol to benzaldehyde (Fig. 5C). This is also the most abundant MAG in HST metagenomes at day 20 (Fig. 5E), indicating it may be important to BA production in the HST microcosms. Beyond S. indicatrix, a MAG representing Mycobacterium sp008329585 encoded a benzoylformate decarboxylase (mdlC), which decarboxylates benzoylformic acid to benzaldehyde (Fig. 5C). Finally, an abundant MAG representing a novel species of Sedimentibacter carried a gene for hippurate hydrolase (Fig. 5E). This enzyme cleaves glycine from hippuric acid, producing BA (EC 3.5.1.32) (Fig. 5C). Consistent with the literature, these findings position BA as a central aromatic metabolite in these microcosms.
While there are limited reports today that microbial BA production can have a direct benefit to plants, it is well known that plant BA and derivatives are precursors to the production of secondary metabolites and hormones (46). BA is also known to improve plant stress tolerance and contribute to growth regulation (46). In support, exogenous application of BA has been associated with improved growth and yield in quinoa and drought-stressed soybean (47, 48). As such, BA synthesis by the soil microbiome and its relationship with plant health are an area of research that warrants more investigation.
Bacterial indoleacetic acid production encoded by diverse MAGs.
Tryptophan (Trp) is the primary precursor for IAA biosynthesis in microorganisms (49), with five known biosynthetic pathways characterized in bacteria (50). We surveyed our MAG database for these genes and found 14 HST-detected MAGs capable of IAA production using two different pathways (File S3), consistent with reports that auxin biosynthesis is widely encoded in plants (43, 49). Nine MAGs carried the indole-pyruvate decarboxylase (ipdC) gene, the key gene for converting Trp to IAA via indole-3-pyruvate (Fig. 5D). These MAGs spanned four phyla, with six belonging to Proteobacteria, covering several of the most abundant MAGs at day 20 in the HST metagenomes (Fig. 5E). Beyond this pathway, three MAGs carried iaaH for IAA production via indoleacetamide (Fig. 5D). One of these MAGs, a novel species of Xanthobacteraceae, carried both iaaH and ipdC. This fits with previous observations of single organisms encoding redundant IAA biosynthesis pathways (51). Of note, two mycobacterial MAGs with IAA production potential also had BA production potential. One potentially novel Mycobacterium species carried ipdC and xylC, while Mycobacterium sp008329585 carried iaaH in addition to mdlC and xylC, indicating some soil microbes can produce multiple phytohormones.
IAA is a phytohormone in the auxin family that is necessary for proper plant development and can be used to stimulate root biomass growth (51, 52). From a microbial perspective, IAA production offers a competitive colonization strategy over non-IAA-producing strains (53, 54), and it has been shown that microbially produced IAA in the rhizosphere can elicit positive plant growth promotion and disease suppression effects (18, 53–55). However, these effects are dose dependent, as high concentrations of IAA can also inhibit plant growth (56–58). The reduced complexity of our laboratory microcosm system allowed us to demonstrate that IAA can accumulate in soil microbiomes, and its production is redundantly encoded across 4 different phyla, offering new perspectives on the microbial contributions to soil IAA pools.
It is important to note that the metagenomic results presented here only represent the genomic capabilities of a hypothesized genome; thus, additional experimental evaluation of gene expression would be required to definitively confirm specific bacterial synthesis of SA, BA, and IAA and their roles as potential species of plant growth-promoting rhizobacteria (PGPR). Furthermore, gene expression could inform whether exometabolite production was a direct microbial response to exudates versus an indirect response via the priming effect (59). Yet, we find these MAG results, coupled with exometabolite evidence for these compounds through time, to be an exciting platform for targeted studies aimed at harnessing the power of microbial metabolisms for improving agroecosystems.
Conclusion.
Coupling a reduced-complexity, tractable laboratory soil system with high-resolution multiomics approaches afforded new perspectives on the complex interactions between plant root exudation and the soil microbiome. We first showed that root exudate amendments modeled after two different sorghum genotypes drastically impacted overall soil microbial community diversity and membership. Second, shifts in microbial community structure mirrored differences in exometabolites. Finally, we linked interesting plant-relevant metabolites to bacterial genomes, highlighting new roles for specific microorganisms in phytohormone biosynthesis. We have provided a publicly available, genome-resolved microbial database for researchers interested in the sorghum rhizosphere. Our findings suggest root exudate composition influenced microbial production of possible phytohormones that could impact host plant physiology, as well as govern metal acquisition and antibiotic production from soils. Future studies can scale these hypotheses to the field to analyze the impact of plant genotype, root exudates, and associated microbial communities on overall plant performance. In summary, the results of this study represent an important step toward decoding the complex chemical language between plants and their rhizosphere microbial communities, a translation required to optimize these interactions for enhanced agricultural management and production.
MATERIALS AND METHODS
Sorghum root exudate collections.
Mature seeds from three sorghum genotypes (Leoti, BTx623, and PI 505735) were first sterilized by placing seeds in sterile 50-mL conical tubes with 45 mL 95% ethanol. Tubes were vortexed, ethanol was removed, and 45 mL of Captan fungicide solution (0.2 g Captan fungicide in 45 mL sterile water) was added to remove any fungal components on the seeds, and the mixture was vortexed for 3 h. After removal of the Captan fungicide, 45 mL of 100% Clorox bleach (8.25% sodium hypochlorite active ingredient) and 3 droplets of dish soap (Ajax Triple Action Orange) were added for the final sterilization step, and seeds were shaken for 20 min. In a sterile tissue culture hood, the bleach was removed using sterile techniques, and the seeds were rinsed five times with sterile water. Seeds were blotted dry and placed on germination paper inside a clean 600-mL beaker with a solution of 1 mM CaCl2. After 7 days of growth, seedlings were removed from the paper and pooled into a 250-mL glass bottle filled with 80 mL of ultrapure water. Root exudates were collected for each genotype in separate bottles. The bottles were covered with aluminum foil to protect roots from the light and were placed on a rotary shaker. After 2 h, the roots were removed, blotted dry, and weighed. The root exudate suspensions containing the root exudates were filtered through a 0.2-μm-pore filter membrane to remove root detritus and microbial cells. Samples were frozen at −80°C. We recognize that the use of hydroponic growth chambers does not exactly replicate soil growth conditions of sorghum (60); however, this system provided a highly controllable, tractable, and sterile environment that eliminated confounding microbial or soil influences for more accurate downstream analytical detection. Samples were lyophilized completely before resuspension in 15 mL sterile high-performance liquid chromatography (HPLC)-grade water. Resuspended exudates were vortexed thoroughly to remove all residue from the bottle, transferred to a clean 50-mL Falcon tube, and dried completely under nitrogen gas. One milliliter of liquid chromatography-mass spectrometry (LC-MS)-grade 80% acetonitrile was added to the Falcon tube with dried exudates and vortexed until thoroughly resuspended. A 0.5-mL portion was transferred to a clean 2-mL glass vial, and 5 μg of glucose-13C6, d-arabinose-13C5, sucrose-13C12, galactose-13C6, and fructose-13C6 was added to each vial and dried completely under nitrogen prior to derivatization.
Sample derivatization for sorghum root exudates.
Dried samples were resuspended in 75 μL of 0.2 M methoxyamine hydrochloride in pyridine (Sigma), incubated at 60°C for 45 min, vortexed, sonicated for 10 min, incubated a second time at 60°C for 45 min, and allowed to cool to ambient temperature. Thirty microliters of each sample was then transferred to a separate 2-mL glass vial for MSTFA (N-methyl-N-trimethylsilyltrifluoroacetamide) derivatization and the other 30 μL for acetic anhydride (AA) derivatization. Ninety microliters of 100% AA was added to AA vials, and the mixture was vortexed for 30 s, incubated at 60°C for 60 min, and cooled for 10 min at ambient temperature. Thirty microliters of MSTFA plus 1% trimethylchlorosilane (TMCS) (Thermo Scientific) was added to the MSTFA vials, and the mixture was vortexed for 30 s, incubated at 60°C for 35 min, and cooled to ambient temperature before transferring to vial inserts. AA samples were dried completely under nitrogen, resuspended in 40 μL of 100% ethyl acetate, and then loaded into glass inserts for analysis.
Nontargeted and targeted GC-MS analyses for sorghum root exudates.
Metabolites were analyzed and detected using a Trace 1310 gas chromatograph (Thermo) coupled to an ISQ mass spectrometer (Thermo). Samples (1 μL) were injected into an injection port at 285°C and a 1:10 split ratio. Separation was accomplished with a 30-m TG-5MS column (Thermo Scientific [0.25-mm inside diameter i.d., 0.25-μm film thickness]) and a helium gas at a flow rat of 1.2 mL/min. The oven temperature program started at 80°C for 30 s, was ramped to 330°C at 15°C/min, and then was held at the final temperature for 8 min. The transfer line and ion source were maintained at 300°C and 260°C, respectively. Masses between 50 and 650 m/z were scanned at 5 scans/s after electron impact ionization. A pooled quality control (QC) sample (made by combining an equal volume of all samples) was injected after every 6 samples to monitor instrument stability throughout the analysis.
Synthetic exudate preparation.
Exudate treatments were formulated based on the results of the exudate profiles for the two most divergent sorghum genotypes (Leoti and BTx623). The high-sugar treatment (HST) was formulated to mimic BTx623, with higher sugar and lower organic acid composition, and the high-organic-acid treatment (HOT) was formulated to mimic the Leoti exudate profiles, with higher organic acid and lower sugar composition (see Table S3 and Files S5 and S6 in the supplemental material). The 11 synthetic root exudate compounds were chosen based on practical considerations and availability of standards. Organic acid root exudate relative quantification values were normalized to fructose peak area and scaled to the absolute quantification of fructose in that sample. Treatment calculations can be found in Files S5 and S6. The exudate-lacking medium control treatment (control) was a 10 mM phosphate buffer, consisting of ammonium chloride, disodium phosphate, and sodium dihydrogen at a pH of 6.5 (Table S1). Exudates found in each genotype were weighed out to their respective masses (Table S3) to supply equal exudates for 6 days of exudate addition (Fig. 1C, days 0 to 5) to the microcosms, homogenized, suspended in 10 mL 10 mM phosphate buffer, vortexed, aliquoted into 6 tubes for each day of exudate addition, and frozen at −80°C until use. Aliquots were removed 30 min before use, incubated at 24°C until thawed, and added to microcosms at the respective time point.
Soil samples.
The soil (microbial inoculum) was collected from agricultural fields at the Colorado State University Agricultural Research and Education Center (CSU-ARDEC) near Fort Collins, CO, on 4 October 2019. The climate at the site is semiarid, with a mean annual precipitation of 408 mm and a mean annual temperature of 10.2°C (average from 1981 to 2010 [https://usclimatedata.com/]). The soil is classified as an Aridic Haplustalf. Three 2-cm-diameter soil core samples to a depth of approximately 15 cm were collected from each of seven different plots. The soil was stored at −20°C until microcosm construction. Twenty grams of soil from each replicate/plot was pooled and homogenized to create the representative bulk soil repository used in the following microcosms experiment.
Microcosm experimental setup.
Microcosms were established and sampled as previously described (35, 60). Briefly, 5 g of homogenized soil and 35 mL of phosphate-buffered (pH 6.5) minimal medium (Table S1) were added to sterile 50-mL conical tubes to construct each microcosm. Microcosms were vortexed and allowed to settle for 5 min. Then, day 0 samples were taken by removing 1 mL of soil slurry for exometabolomics analysis and 1 mL of soil slurry for DNA extraction. After this initial sampling, 2 mL of exudate treatment was added to each microcosm and vortexed, and a second 1-mL aliquot was immediately taken for exometabolomics analysis. At this point, for each microcosm the bottle caps were removed and replaced with a sterile foam stopper for the rest of the experiment to maintain oxic conditions and prevent colonization by contaminating microbes. Microcosms were incubated in an orbital shaker set at 200 rpm at 24°C for 20 days. Each exudate treatment (i.e., HST, HOT, and control) was conducted in triplicate, and treatments were added (2 mL) to microcosms on days 0, 1, 2, 3, 4, and 5. After day 5, no additional exudate treatments were applied, but microcosms were maintained until day 20, which afforded additional samples taken at days 7, 11, and 20 (Fig. 1C). Samples were collected at roughly the same time each day and collected with aseptic techniques to ensure no additional microbial influence was introduced. All collected samples were immediately frozen at −80°C until processing.
Sample preparation and extraction for exometabolomics analysis.
Samples were thawed at 4°C overnight and centrifuged for 20 min at 18,000 × g, and supernatant was transferred to a preweighed 1.5-dram vial and refrozen at −80°C for subsequent lyophilization. Samples were lyophilized for 24 h until all water was sublimated. Samples were weighed to calculate total exometabolite mass and then were resuspended in 4-mL sterile HPLC-grade water. A volume equivalent to 0.50 mg was transferred to a new, preweighed 2-mL glass vial and dried under N2. Finally, each sample was resuspended in 500 μL of sterile HPLC-grade water, vortexed for 1 min, and sonicated for 15 min. Two 250-μL aliquots were transferred to new 2-mL vials, respectively, and samples were dried under N2. This yielded two 0.25-mg subsamples for analysis by GC-MS and ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) as described below.
Targeted UPLC-MS/MS for phytohormone analysis of exometabolites.
Subsamples of 0.25 mg were extracted in 75 μL of a spiked methanol solution containing 100% methanol with 65.2 ng/mL absciscic acid-d6 (ABA-d6), 62.5 ng/mL salicylic acid-d6, and 90.0 ng/mL jasmonic acid-d5 (Sigma). After solvent addition, samples were placed on a shaker plate for 1 h at the highest speed setting, centrifuged at 3,500 × g at 4°C for 5 min, and transferred to glass inserts. A final centrifugation step at 3,500 × g for 15 min at 4°C was completed to ensure any precipitate was in the bulb of the vial insert. Five microliters of exometabolite samples was injected onto a PerkinElmer UPLC-MS/MS system, equipped with a PerkinElmer QSight LX50 solvent delivery module. An Acquity UPLC T3 column (1 by 100 mm, 1.8 μm; Waters Corporation) was used for chromatographic separation. Mobile phase A consisted of LC‐MS-grade water with 0.1% formic acid, and mobile phase B consisted of 100% LC‐MS grade acetonitrile. The elution gradient was initially set at 0.1% B for 1 min, which was increased to 55.0% B at 12 min and further increased to 97.0% B at 15 min, then decreased to 0.1% B at 15.5 min. The column was reequilibrated for 4.5 min for a total run time of 20 min. The flow rate was set to 200 μL/min, and the column temperature was maintained at 45°C. Samples were held at 4°C in the autosampler. Detection was performed on a PerkinElmer QSight 220 triple-quadrupole mass spectrometer in selected reaction monitoring (SRM) mode. The transitions monitored for each phytohormone compound can be found in Table S2. The MS was operated with an electrospray ionization (ESI) voltage of 4,500 V in positive mode and −3,500 V in negative mode. Nebulizer gas flow was set at 350 arbitrary units, and drying gas was set to 120 arbitrary units. The source temperature was 315°C, and the hot-surface induced desolvation (HSID) temperature was set to 200°C.
Nontargeted GC-MS analysis of exometabolites.
Sample preparation was conducted as previously described (23, 61) and as described above for sorghum root exudates. Briefly, dried samples were resuspended in 50 μL of pyridine containing 25 mg/mL methoxyamine hydrochloride (Sigma), centrifuged, incubated at 60°C for 45 min, vortexed for 30 s, sonicated for 10 min, centrifuged briefly, and incubated a second time for 45 min at 60°C. Samples were cooled to room temperature and centrifuged for 2 min. Then, 50 μL of MSTFA plus 1% TMCS (Thermo Fisher) was added, and samples were vortexed for 30 s, centrifuged, and incubated a third time at 60°C for 35 min. Samples were cooled to room temperature and centrifuged, and 80 μL of supernatant was transferred to glass vial inserts within glass vials. Samples were centrifuged a final time for 10 min before analysis. Metabolites were separated with a 30-m TG-5MS column (Thermo Scientific [0.25-mm i.d., 0.25-μm film thickness]) and detected using a PerkinElmer Clarus 690 gas chromatograph coupled to a Clarus SQ 8S mass spectrometer (PerkinElmer). Samples (1 μL) were injected at a 10:1 split ratio onto the column with a 1.0-mL/min helium gas flow rate. The gas chromatography inlet was held at 285°C, the transfer line was held at 300°C, and the source temperature was held at 260°C. The GC oven program started at 80°C for 30s, followed by a ramp of 15°C/min to 330°C, followed by an 8‐min hold. Masses between 50 and 620 m/z were scanned at 4 scans/s under electron impact ionization. Injection of QCs was analyzed after every 6th sample to ensure proper instrument function and to detect any analytical variation.
GC-MS and LC-MS data analysis.
Nontargeted GC-MS data (both sorghum exudates and exometabolites) were processed within the R statistical software (62) using methods previously described (63). For GC-MS samples, .cdf files were processed through the following workflow: (i) XCMS software was used for preprocessing to identify molecular features (64), (ii) features were further normalized to total ion current (TIC), and (iii) the package RAMClustR (65) was used for clustering features into spectra and prepared for subsequent identification of spectra in RAMSearch (66) using external databases, including Golm (http://gmd.mpimp-golm.mpg.de/) and NIST (http://www.nist.gov). For exometabolite data, prior to TIC normalization, features were normalized by linearly regressing run order versus QC feature intensities to account for instrument signal intensity drift. For root exudate data, relative quantitation was also normalized by root weight. Targeted quantification of sugars from the GC-MS data was performed using Chromeleon 7.2 (Thermo Fisher). The integrated peak area for each sugar was normalized to its corresponding internal standard, with the exceptions that arabinose-13C5 was used for xylose, glucose-13C6 was used for mannose, and sucrose-13C12 was used for trehalose and maltose. Quantification was determined using a linear regression of an 8-point standard curve for each sugar. Final concentrations were normalized to root weight. For phytohormone analysis of exometabolites, LC-MS data were processed using Simplicity 3Q (v1.5) (PerkinElmer) bioinformatics software for sample processing. Briefly, the peak area for each phytohormone compound was normalized to internal standard peak area, and quantification was assessed using a linear regression against an external calibration curve. Exudate and exometabolite data are provided in File S1.
Soil DNA extraction and library preparation.
Total genomic DNA was extracted from the microcosms using the Zymo Quick-DNA fecal/soil microbe microprep kit. 16S rRNA gene amplicon sequencing was performed on the Illumina MiSeq using 251-bp paired-end reads and the Earth Microbiome Project primers 515F/806R (1), for an average of more than 30,000 reads per sample (see File S2 for individual sample data). The 16S rRNA partial gene reads were analyzed, and reads were demultiplexed using QIIME2 (2019.10) (67). Using DADA2 (68), demultiplexed reads were denoised to produce an amplicon sequence variant (ASV) table and filtered to remove noisy sequences, chimeras, and singletons. Feature classification was completed by comparing the ASV table against the trained full-length SILVA classified (silva132.250) database for taxonomic classification. The ASV table was filtered to contain ASVs that were observed in at least 2 samples, and the output files were visualized in QIIME2. Samples L_E1_T5_A (HST day 5 rep A), L_E2_T11_A, and L_E2_T20_A (HOT days 11 and 20 rep A) yielded insufficient sequencing results and were excluded from subsequent analyses. The ASV feature table is provided in File S2.
Metagenomics analysis.
Metagenomic DNA from day 0 (control) and day 5 (control and HST) metagenomes (n = 9) was sequenced at the Genomics Shared Resource at the University of Colorado Cancer Center using the NovaSeq6000 platform. Metagenomic DNA from day 20 (control and HST, n = 6) was prepared for metagenomic sequencing using the Nextera XT low-input Illumina library creation kit, and samples were sequenced at the Department of Energy Joint Genome Institute on the Illumina NovaSeq 6000. FastQ files were trimmed using Sickle (v1.33) (69). Day 0 and day 5 reads were concatenated within each of the time points/treatments (i.e., triplicate controls at day 5) for coassembly. Day 0 and day 5 (both control and HST) coassemblies and the day 20 individual assemblies were assembled with IDBA-UD (70). Within each assembly, scaffolds greater than 2.5 kb were binned into metagenome-assembled genomes (MAGs) using MetaBAT2 (v2.12.1) (71). MAGs were assessed for completion and contamination using checkM (72). A MAG was retained if it was >48% complete with <10% contamination and was assigned quality following MIMAG guidelines (33). Using dRep (73), MAGs were dereplicated to 99% identity. MAG taxonomy was assigned using GTDB-tk (v1.5.0, R06-RS202), and taxonomic novelty was defined as the first unnamed taxonomy level (74). To obtain MAG abundance, trimmed metagenomic reads from individual samples were mapped to the dereplicated MAG set using bbmap (75) (v38.70) at minid = 95 and output as sam files, which were converted to sorted bam files using samtools (76) (v1.9). CoverM (v0.3.2) was used to determine MAG relative abundance as described by McGivern et al. (35). MAGs were annotated using DRAM (77). Biosynthetic gene clusters were detected using the antiSMASH webserver with default parameters (v6.0) (78). See File S2 for MAG quality, taxonomy, and mapping, File S3 for DRAM annotations, and see https://doi.org/10.5281/zenodo.5639650 for raw annotations.
Statistical analyses.
Sorghum genotype ordinations were generated using metaMDS function from the vegan (79) package in R (v4.0.2) (62) and visualized with ggplot2 (80). Metabolite loadings were calculated with the envfit function in vegan. Alpha diversity of microbial communities was calculated using the diversity function from the vegan package (79) in R (v4.0.2) (62) with Shannon’s (H), Pielou’s, and richness indices. To estimate beta diversity across treatments, we utilized the Bray-Curtis dissimilarity matrix, which was visualized by nonmetric multidimensional scaling (NMDS) in R with the ggplot2 package with stress of the nonparametric fit for the goodness of fit for the model. Significance of compositional differences across treatments was quantified using mrpp and the betadisper commands from the vegan package, with an ANOVA model in R. Significance between communities across time and treatment was quantified using beta-group-significance commands in QIIME2 (2019.10) (66) with a PERMANOVA model. Fold changes for exometabolite dynamics were calculated and converted to log2 abundances via the log function. Two-way ANOVA and pairwise comparisons were completed in GraphPad Prism (v8.2.1) using grouped analyses with Sidak’s multiple comparison testing with alpha = 0.05.
Data availability.
The MAGs, metagenomes, and 16S rRNA amplicon sequencing reads from this data set have been deposited in National Center for Biotechnology Information under BioProject accession no. PRJNA725542 (for biosample accession numbers, see File S2). MAGs are also provided in archive (https://doi.org/10.5281/zenodo.5142207). Metabolomics (exometabolites and root exudates) data have been deposited in the EMBL-EBI MetaboLights database with the identifier MTBLS3164 (81). The complete data set can be accessed at https://www.ebi.ac.uk/metabolights/MTBLS3164.
ACKNOWLEDGMENTS
B.B.M. and K.C.W. were fully or partially supported by an Early Career Award to K.C.W. from the National Science Foundation under award no. 1750189. R.A.D. was supported by an Early Career Award from the U.S. Department of Energy to K.C.W., Office of Science, Office of Biological and Environmental Research under award no. DE-SC0019746. A portion of this work was conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, which is supported by the Office of the U.S. Department of Energy under contract no. DE-AC02-05CH11231 (proposal ID 2049, Wrighton). A portion of this work was performed by the University of Colorado Anschutz's Genomics Shared Resource supported by the Cancer Center Support Grant (P30CA046934). Sorghum root exudate analyses were conducted under DOE DER award DE-SC0014395.
We thank Linxing Yao in the ARC-BIO at Colorado State University for assistance in analysis of the sorghum root exudates.
Footnotes
Supplemental material is available online only.
Contributor Information
Jessica E. Prenni, Email: Jessica.prenni@colostate.edu.
Gladys Alexandre, University of Tennessee at Knoxville.
REFERENCES
- 1.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, Rivera JLA, Al-Moosawi L, Alverdy J, Amato KR, Andras J, Angenent LT, Antonopoulos DA, Apprill A, Earth Microbiome Project Consortium, et al. 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457–463. 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM. 2006. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266. 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- 3.Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loqué D, Bowen BP, Firestone MK, Northen TR, Brodie EL. 2018. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3:470–480. 10.1038/s41564-018-0129-3. [DOI] [PubMed] [Google Scholar]
- 4.Korenblum E, Dong Y, Szymanski J, Panda S, Jozwiak A, Massalha H, Meir S, Rogachev I, Aharoni A. 2020. Rhizosphere microbiome mediates systemic root metabolite exudation by root-to-root signaling. Proc Natl Acad Sci USA 117:3874–3883. 10.1073/pnas.1912130117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg G, Smalla K. 2009. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68:1–13. 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- 6.Micallef SA, Channer S, Shiaris MP, Colón-Carmona A. 2009. Plant age and genotype impact the progression of bacterial community succession in the Arabidopsis rhizosphere. Plant Signal Behav 4:777–780. 10.1093/jxb/erp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiao X, Takishita Y, Zhou G, Smith DL. 2021. Plant associated rhizobacteria for biocontrol and plant growth enhancement. Front Plant Sci 12:634796. 10.3389/fpls.2021.634796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh RK, Singh P, Li H-B, Song Q-Q, Guo D-J, Solanki MK, Verma KK, Malviya MK, Song X-P, Lakshmanan P, Yang L-T, Li Y-R. 2020. Diversity of nitrogen-fixing rhizobacteria associated with sugarcane: a comprehensive study of plant-microbe interactions for growth enhancement in Saccharum spp. BMC Plant Biol 20:220. 10.1186/s12870-020-02400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manoj SR, Karthik C, Kadirvelu K, Arulselvi PI, Shanmugasundaram T, Bruno B, Rajkumar M. 2020. Understanding the molecular mechanisms for the enhanced phytoremediation of heavy metals through plant growth promoting rhizobacteria: a review. J Environ Manage 254:109779. 10.1016/j.jenvman.2019.109779. [DOI] [PubMed] [Google Scholar]
- 10.Boiero L, Perrig D, Masciarelli O, Penna C, Cassán F, Luna V. 2007. Phytohormone production by three strains of Bradyrhizobium japonicum and possible physiological and technological implications. Appl Microbiol Biotechnol 74:874–880. 10.1007/s00253-006-0731-9. [DOI] [PubMed] [Google Scholar]
- 11.Spaepen S, Vanderleyden J, Remans R. 2007. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol Rev 31:425–448. 10.1111/j.1574-6976.2007.00072.x. [DOI] [PubMed] [Google Scholar]
- 12.Chaparro JM, Badri DV, Vivanco JM. 2014. Rhizosphere microbiome assemblage is affected by plant development. ISME J 8:790–803. 10.1038/ismej.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaparro J, Badri D, Bakker M, Sugiyama A, Manter D, Vivanco J. 2013. Root exudation of phytochemicals in Arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS One 8:e55731. 10.1371/journal.pone.0055731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Micallef SA, Shiaris MP, Colón-Carmona A. 2009. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot 60:1729–1742. 10.1093/jxb/erp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawasaki A, Donn S, Ryan P, Mathesius U, Devilla R, Jones A, Watt M. 2016. Microbiome and exudates of the root and rhizosphere of Brachypodium distachyon, a model for wheat. PLoS One 11:e0164533. 10.1371/journal.pone.0164533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassan S, Mathesius U. 2012. The role of flavonoids in root-rhizosphere signalling: opportunities and challenges for improving plant–microbe interactions. J Exp Bot 63:3429–3444. 10.1093/jxb/err430. [DOI] [PubMed] [Google Scholar]
- 17.Cai T, Cai W, Zhang J, Zheng H, Tsou AM, Xiao L, Zhong Z, Zhu J. 2009. Host legume-exuded antimetabolites optimize the symbiotic rhizosphere. Mol Microbiol 73:507–517. 10.1111/j.1365-2958.2009.06790.x. [DOI] [PubMed] [Google Scholar]
- 18.Khan AL, Halo BA, Elyassi A, Ali S, Al-Hosni K, Hussain J, Al-Harrasi A, Lee I-J. 2016. Indole acetic acid and ACC deaminase from endophytic bacteria improves the growth of Solanum lycopersicum. Electron J Biotechnol 21:58–64. 10.1016/j.ejbt.2016.02.001. [DOI] [Google Scholar]
- 19.Maki Y, Soejima H, Kitamura T, Sugiyama T, Sato T, Watahiki MK, Yamaguchi J. 2021. 3-Phenyllactic acid, a root-promoting substance isolated from Bokashi fertilizer, exhibits synergistic effects with tryptophan. Plant Biotechnol (Tokyo) 38:9–16. 10.5511/plantbiotechnology.20.0727a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finney DM, Buyer JS, Kaye JP. 2017. Living cover crops have immediate impacts on soil microbial community structure and function. J Soil Water Conserv 72:361–373. 10.2489/jswc.72.4.361. [DOI] [Google Scholar]
- 21.Hu L, Robert CAM, Cadot S, Zhang X, Ye M, Li B, Manzo D, Chervet N, Steinger T, van der Heijden MGA, Schlaeppi K, Erb M. 2018. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat Commun 9:2738. 10.1038/s41467-018-05122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.SARE. 2012. Managing cover crops profitably, 3rd ed. Sustainable Agriculture Research and Education (SARE) program. https://www.sare.org/wp-content/uploads/Managing-Cover-Crops-Profitably.pdf.
- 23.Sheflin AM, Chiniquy D, Yuan C, Goren E, Kumar I, Braud M, Brutnell T, Eveland AL, Tringe S, Liu P, Kresovich S, Marsh EL, Schachtman DP, Prenni JE. 2019. Metabolomics of sorghum roots during nitrogen stress reveals compromised metabolic capacity for salicylic acid biosynthesis. Plant Direct 3:e00122. 10.1002/pld3.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper EA, Brenton ZW, Flinn BS, Jenkins J, Shu S, Flowers D, Luo F, Wang Y, Xia P, Barry K, Daum C, Lipzen A, Yoshinaga Y, Schmutz J, Saski C, Vermerris W, Kresovich S. 2019. A new reference genome for Sorghum bicolor reveals high levels of sequence similarity between sweet and grain genotypes: implications for the genetics of sugar metabolism. BMC Genomics 20:420–420. 10.1186/s12864-019-5734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ratnavathi CV, Chavan UD. 2016. Sorghum syrup and other by products, p 253–310. In Ratnavathi CV, Patil JV, Chavan UD (ed), Sorghum biochemistry. Academic Press, San Diego, CA. [Google Scholar]
- 26.Manzoni S, Taylor P, Richter A, Porporato A, Ågren GI. 2012. Environmental and stoichiometric controls on microbial carbon-use efficiency in soils. New Phytol 196:79–91. 10.1111/j.1469-8137.2012.04225.x. [DOI] [PubMed] [Google Scholar]
- 27.Steiger MG, Blumhoff ML, Mattanovich D, Sauer M. 2013. Biochemistry of microbial itaconic acid production. Front Microbiol 4:23. 10.3389/fmicb.2013.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leonardi R, Jackowski S. 13 August 2007, posting date. Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus 2007. 10.1128/ecosalplus.3.6.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He M. 2006. Pipecolic acid in microbes: biosynthetic routes and enzymes. J Ind Microbiol Biotechnol 33:401–407. 10.1007/s10295-006-0078-3. [DOI] [PubMed] [Google Scholar]
- 30.Cavaliere M, Feng S, Soyer OS, Jiménez JI. 2017. Cooperation in microbial communities and their biotechnological applications. Environ Microbiol 19:2949–2963. 10.1111/1462-2920.13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whipps JM. 2001. Microbial interactions and biocontrol in the rhizosphere. J Exp Bot 52:487–511. 10.1093/jexbot/52.suppl_1.487. [DOI] [PubMed] [Google Scholar]
- 32.Parray JA, Shameem N. 2020. Signalome: communication between crops and microbiomes, p 137–179. In Parray JA, Shameem N (ed), Sustainable agriculture. Academic Press, San Diego, CA. [Google Scholar]
- 33.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, The Genome Standards Consortium, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. 2014. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci USA 111:4904–4909. 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGivern BB, Tfaily MM, Borton MA, Kosina SM, Daly RA, Nicora CD, Purvine SO, Wong AR, Lipton MS, Hoyt DW, Northen TR, Hagerman AE, Wrighton KC. 2021. Decrypting bacterial polyphenol metabolism in an anoxic wetland soil. Nat Commun 12:2466. 10.1038/s41467-021-22765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narrowe AB, Borton MA, Hoyt DW, Smith GJ, Daly RA, Angle JC, Eder EK, Wong AR, Wolfe RA, Pappas A, Bohrer G, Miller CS, Wrighton KC, Orsi W. 2019. Uncovering the diversity and activity of methylotrophic methanogens in freshwater wetland soils. mSystems 4:e00320-19. 10.1128/mSystems.00320-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaille C, Reimmann C, Haas D. 2003. Isochorismate synthase (PchA), the first and rate-limiting enzyme in salicylate biosynthesis of Pseudomonas aeruginosa. J Biol Chem 278:16893–16898. 10.1074/jbc.M212324200. [DOI] [PubMed] [Google Scholar]
- 38.Mishra AK, Baek K-H. 2021. Salicylic acid biosynthesis and metabolism: a divergent pathway for plants and bacteria. Biomolecules 11:705. 10.3390/biom11050705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakker PAHM, Ran L, Mercado-Blanco J. 2014. Rhizobacterial salicylate production provokes headaches! Plant Soil 382:1–16. 10.1007/s11104-014-2102-0. [DOI] [Google Scholar]
- 40.Islam MN, Ali MS, Choi S-J, Park Y-I, Baek K-H. 2019. Salicylic acid-producing endophytic bacteria increase nicotine accumulation and resistance against wildfire disease in tobacco plants. Microorganisms 8:31. 10.3390/microorganisms8010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tufail A, Arfan M, Gurmani AR, Khan A, Bano A. 2013. Salicylic acid induced salinity tolerance in maize (Zea mays). Pak J Bot 45:75–82. [Google Scholar]
- 42.Meyer JM, Azelvandre P, Georges C. 1992. Iron metabolism in Pseudomonas: salicylic acid, a siderophore of Pseudomonas fluorescens CHAO. Biofactors 4:23–27. [PubMed] [Google Scholar]
- 43.Khalid A, Arshad M, Zahir Z. 2004. Screening plant growth-promoting rhizobacteria for improving growth and yield of wheat. J Appl Microbiol 96:473–480. 10.1046/j.1365-2672.2003.02161.x. [DOI] [PubMed] [Google Scholar]
- 44.Li W, Estrada-de los Santos P, Matthijs S, Xie G-L, Busson R, Cornelis P, Rozenski J, De Mot R. 2011. Promysalin, a salicylate-containing Pseudomonas putida antibiotic, promotes surface colonization and selectively targets other Pseudomonas. Chem Biol 18:1320–1330. 10.1016/j.chembiol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 45.Noda S, Kitazono E, Tanaka T, Ogino C, Kondo A. 2012. Benzoic acid fermentation from starch and cellulose via a plant-like β-oxidation pathway in Streptomyces maritimus. Microb Cell Fact 11:49. 10.1186/1475-2859-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Widhalm JR, Dudareva N. 2015. A familiar ring to it: biosynthesis of plant benzoic acids. Mol Plant 8:83–97. 10.1016/j.molp.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Allah MMS, El-Bassiouny HMS, Elewa TAE, El Sebai T. 2015. Effect of salicylic acid and benzoic acid on growth, yield and some biochemical aspects of quinoa plant grown in sandy soil. International Journal of ChemTech Research 8:216–225. https://sphinxsai.com/2015/ch_vol8_no12/1/(216-225)V8N12CT.pdf. [Google Scholar]
- 48.Anjum S, Ehsanullah Xue L, Wang L, Saleem MF, Huang CJ. 2013. Exogenous benzoic acid (BZA) treatment can induce drought tolerance in soybean plants by improving gas-exchange and chlorophyll contents. Austr J Crop Sci 7:555–560. [Google Scholar]
- 49.Patten CL, Glick BR. 1996. Bacterial biosynthesis of indole-3-acetic acid. Can J Microbiol 42:207–220. 10.1139/m96-032. [DOI] [PubMed] [Google Scholar]
- 50.Carreño-Lopez R, Campos-Reales N, Elmerich C, Baca BE. 2000. Physiological evidence for differently regulated tryptophan-dependent pathways for indole-3-acetic acid synthesis in Azospirillum brasilense. Mol Gen Genet 264:521–530. 10.1007/s004380000340. [DOI] [PubMed] [Google Scholar]
- 51.Spaepen S, Vanderleyden J. 2011. Auxin and plant-microbe interactions. Cold Spring Harb Perspect Biol 3:a001438. 10.1101/cshperspect.a001438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luziatelli F, Ficca AG, Bonini P, Muleo R, Gatti L, Meneghini M, Tronati M, Melini F, Ruzzi M. 2020. A genetic and metabolomic perspective on the production of indole-3-acetic acid by Pantoea agglomerans and use of their metabolites as biostimulants in plant nurseries. Front Microbiol 11:1475. 10.3389/fmicb.2020.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu SF, Wei JY, Chen HW, Liu YY, Lu HY, Chou JY. 2015. Indole-3-acetic acid: a widespread physiological code in interactions of fungi with other organisms. Plant Signal Behav 10:e1048052. 10.1080/15592324.2015.1048052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lebrazi S, Fadil M, Chraibi M, Fikri-Benbrahim K. 2020. Screening and optimization of indole-3-acetic acid production by Rhizobium sp. strain using response surface methodology. J Genet Eng Biotechnol 18:21. 10.1186/s43141-020-00035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Islam S, Akanda AM, Prova A, Islam MT, Hossain MM. 2015. Isolation and identification of plant growth promoting rhizobacteria from cucumber rhizosphere and their effect on plant growth promotion and disease suppression. Front Microbiol 6:1360. 10.3389/fmicb.2015.01360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan L, Chen J, Ren S, Shen H, Rong B, Liu W, Yang Z. 2020. Complete genome sequence of Mycobacterium Mya-zh01, an endophytic bacterium, promotes plant growth and seed germination isolated from flower stalk of Doritaenopsis. Arch Microbiol 202:1965–1976. 10.1007/s00203-020-01924-w. [DOI] [PubMed] [Google Scholar]
- 57.Xie H, Pasternak JJ, Glick BR. 1996. Isolation and characterization of mutants of the plant growth-promoting rhizobacterium Pseudomonas putida GR12-2 that overproduce indoleacetic acid. Curr Microbiol 32:67–71. 10.1007/s002849900012. [DOI] [Google Scholar]
- 58.Bunsangiam S, Thongpae N, Limtong S, Srisuk N. 2021. Large scale production of indole-3-acetic acid and evaluation of the inhibitory effect of indole-3-acetic acid on weed growth. Sci Rep 11:13094. 10.1038/s41598-021-92305-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fontaine S, Mariotti A, Abbadie L. 2003. The priming effect of organic matter: a question of microbial competition? Soil Biol Biochem 35:837–843. 10.1016/S0038-0717(03)00123-8. [DOI] [Google Scholar]
- 60.Pantigoso HA, He Y, DiLegge MJ, Vivanco JM. 2021. Methods for root exudate collection and analysis, p 291–303. In Carvalhais LC, Dennis PG (ed), The plant microbiome: methods and protocols. Springer US, New York, NY. [DOI] [PubMed] [Google Scholar]
- 61.Borton MA, Hoyt DW, Roux S, Daly RA, Welch SA, Nicora CD, Purvine S, Eder EK, Hanson AJ, Sheets JM, Morgan DM, Wolfe RA, Sharma S, Carr TR, Cole DR, Mouser PJ, Lipton MS, Wilkins MJ, Wrighton KC. 2018. Coupled laboratory and field investigations resolve microbial interactions that underpin persistence in hydraulically fractured shales. Proc Natl Acad Sci USA 115:E6585–E6594. 10.1073/pnas.1800155115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
- 63.Chaparro JM, Holm DG, Broeckling CD, Prenni JE, Heuberger AL. 2018. Metabolomics and ionomics of potato tuber reveals an influence of cultivar and market class on human nutrients and bioactive compounds. Front Nutr 5:36. 10.3389/fnut.2018.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. 2006. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78:779–787. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 65.Broeckling CD, Afsar FA, Neumann S, Ben-Hur A, Prenni JE. 2014. RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal Chem 86:6812–6817. 10.1021/ac501530d. [DOI] [PubMed] [Google Scholar]
- 66.Broeckling CD, Ganna A, Layer M, Brown K, Sutton B, Ingelsson E, Peers G, Prenni JE. 2016. Enabling efficient and confident annotation of LC-MS metabolomics data through MS1 spectrum and time prediction. Anal Chem 88:9226–9234. 10.1021/acs.analchem.6b02479. [DOI] [PubMed] [Google Scholar]
- 67.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joshi NF. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33). https://github.com/najoshi/sickle.
- 70.Peng Y, Leung HCM, Yiu SM, Chin FYL. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 71.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olm MR, Brown CT, Brooks B, Banfield JF. 2017. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11:2864–2868. 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36:1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bushnell B. 2014. BBMap: a fast, accurate, splice-aware aligner. https://www.osti.gov/biblio/1241166-bbmap-fast-accurate-splice-aware-aligner?msclkid=97b474b4c33111ec8dab200dda4bca44.
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, Gazitúa MC, Daly RA, Smith GJ, Vik DR, Pope PB, Sullivan MB, Roux S, Wrighton KC. 2020. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res 48:8883–8900. 10.1093/nar/gkaa621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, Weber T. 2021. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res 49:W29–W35. 10.1093/nar/gkab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin P, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2020. vegan: community ecology package, vR package version 2.5-7. https://CRAN.R-project.org/package=vegan.
- 80.Wickham H. 2016. ggplot2: elegant graphics for data analysis. https://ggplot2.tidyverse.org.
- 81.Haug K, Cochrane K, Nainala VC, Williams M, Chang J, Jayaseelan KV, O’Donovan C. 2020. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res 48:D440–D444. 10.1093/nar/gkz1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Download aem.00226-22-s0001.xlsx, XLSX file, 0.1 MB (53.8KB, xlsx)
File S2. Download aem.00226-22-s0002.xlsx, XLSX file, 2.7 MB (2.8MB, xlsx)
File S3. Download aem.00226-22-s0003.xlsx, XLSX file, 2.8 MB (2.9MB, xlsx)
Fig. S1 to S4 and Tables S1 to S3. Download aem.00226-22-s0004.pdf, PDF file, 0.3 MB (367.5KB, pdf)
File S5. Download aem.00226-22-s0005.xlsx, XLSX file, 0.1 MB (71.2KB, xlsx)
File S6. Download aem.00226-22-s0006.xlsx, XLSX file, 0.2 MB (204KB, xlsx)
Data Availability Statement
The MAGs, metagenomes, and 16S rRNA amplicon sequencing reads from this data set have been deposited in National Center for Biotechnology Information under BioProject accession no. PRJNA725542 (for biosample accession numbers, see File S2). MAGs are also provided in archive (https://doi.org/10.5281/zenodo.5142207). Metabolomics (exometabolites and root exudates) data have been deposited in the EMBL-EBI MetaboLights database with the identifier MTBLS3164 (81). The complete data set can be accessed at https://www.ebi.ac.uk/metabolights/MTBLS3164.