

Abstract
Hyperpolarization-activated cyclic nucleotide–gated (HCN) channels regulate neuronal excitability and represent a possible therapeutic target for major depressive disorder (MDD). These channels are regulated by intracellular cyclic adenosine monophosphate (cAMP). However, the relationship between cAMP signaling and the influence of HCN channels on behavior remains opaque. In this study, we investigated the role of hippocampal cAMP signaling on behavior using chemogenetic technology in mice. Acutely increasing cAMP limited spatial memory and motivated behavior by increasing HCN function. However, chronically elevated cAMP limited surface trafficking of HCN channels by disrupting the interaction between HCN and tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b), an auxiliary subunit. Chronically increased cAMP in the dorsal hippocampus was also sufficient to rescue cognitive deficits induced by chronic stress in mice. These results reveal a behaviorally relevant form of regulation of HCN channel surface expression that has potential as a therapeutic target for cognitive deficits related to chronic stress.
INTRODUCTION
Major depressive disorder (MDD) represents one of the leading causes of disability worldwide (1), in part because of the prominent cognitive dysfunction associated with the disease (2–4). Monoaminergic antidepressants (MAs) are the mainstay of therapy and, in some cases, can be effective in relieving the cognitive manifestations of MDD (5, 6), although many patients remain refractory to treatment (7). Moreover, MA require weeks of treatment to be efficacious, and the mechanism of this delay is poorly understood (8–12). Given these limitations, there is a need for new and synergistic therapies. Hyperpolarization-activated cyclic nucleotide–gated (HCN) channels in the dorsal hippocampus have been identified as a potential therapeutic target for MDD (13–16) and play a role in regulating cognition (17, 18). These voltage-gated channels are regulated by intracellular cyclic nucleotides and influence temporal summation as well as cellular excitability (19, 20). Knockout (21, 22) or knockdown (23–25) of these channels in dorsal CA1 pyramidal neurons leads to a reduction in time spent immobile on the tail suspension and forced swim tasks [described as “antidepressant-like” (23–25) behavior or “motivated” behavior (26)], raising the possibility that antagonizing these channels might be an effective therapeutic strategy for treating MDD. Consistent with this hypothesis, knockout of HCN channels in the basal forebrain has been linked to improved spatial memory performance (17). Conversely, in a rodent model of chronic stress, there is greater expression of HCN channels in the dorsal hippocampus (24), suggesting that these channels may also be involved in the pathophysiology of the stress-related cognitive changes.
There are four genes that encode HCN channels (HCN1 to HCN4), with HCN1 and HCN2 predominant in the hippocampus (27). In vitro, homomeric HCN2 channels exhibit a large increase in opening probability in response to cyclic adenosine monophosphate (cAMP) binding, whereas homomeric HCN1 channels increase by a smaller amount (if at all) (19). in vivo, HCN1 and HCN2 subunits form heterotetrametric channels with intermediate properties, and it is typically not possible to distinguish the contribution of individual isoforms to the electrophysiologic current, Ih. For this reason, we use the phrase “HCN channels” in this report to refer to all isoforms collectively. In CA1 pyramidal neurons, HCN channels are expressed in a characteristic subcellular distribution, with fewer channels expressed in the more proximal stratum radiatum (SR) compared with the distal stratum lacunosum moleculare (SLM) of the apical dendrite (21, 28, 29). This pattern of expression is essential to the synapse-specific regulation of temporal summation by HCN channels, which regulate temporal summation through the temporoammonic (TA) pathway (synapsing onto the distal SLM) more so than the proximal Schaffer collaterals (SCs; synapsing onto the SR) (21, 29, 30). The subcellular distribution of HCN channels is regulated by binding between pore-forming subunits and an auxiliary subunit named tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) (21, 31, 32). TRIP8b-mediated HCN channel trafficking is responsible for both the surface expression and subcellular localization of HCN channels within CA1 pyramidal neurons (21, 29, 33). The interaction of TRIP8b with HCN channels in dorsal CA1 is essential for the channel’s role in regulating motivated behavior (34). TRIP8b knockout (21) and viral rescue of TRIP8b in the dorsal hippocampus are sufficient to bidirectionally regulate both HCN channel function and changes in motivated behavior (34).
TRIP8b binds to HCN at two locations (35–37), and loss of either binding site limits TRIP8b-mediated HCN channel trafficking in vivo (34). TRIP8b competes with cAMP for binding HCN channels, and increasing concentrations of cAMP disrupt TRIP8b binding to HCN in biochemical assays (37–43). In vitro experiments have also shown that cAMP-mediated disruption of TRIP8b binding impedes TRIP8b-mediated HCN surface trafficking (37), although whether or not this occurs in neurons in vivo remains unclear. This question is of particular relevance to understanding the mechanism of action of existing antidepressants, as MA therapies produce substantial elevations in cAMP in both animal models and human patients (44–46) and cAMP has also been suggested to play a role in the therapeutic mechanism of ketamine (47). In this report, we set out to understand how cAMP signaling in the hippocampus affects behavior and whether or not this pathway could rescue the behavioral phenotype of animals subjected to a chronic stress model. We found that acutely increasing cAMP in dCA1 pyramidal neurons increased Ih, the current mediated by HCN channels, and limited both motivated behavior and spatial memory. In contrast, chronically elevated cAMP limited HCN channel surface trafficking and rescued spatial memory after chronic stress. These results have implications both for understanding the mechanism of action of existing MA and for targeting the TRIP8b-HCN interaction as a synergistic therapy for the cognitive sequelae of chronic stress.
RESULTS
Human patients with MDD express more HCN1
Previous work investigating the relationship between HCN channels and MDD demonstrated an increase in hippocampal HCN channels in animal models (24). To determine whether a similar relationship is true for human patients, we performed quantitative reverse transcription polymerase chain reaction (qPCR) using postmortem samples of the CA1 region of the hippocampus from patients with MDD and unaffected controls. We noted an increase in HCN1 expression in human CA1 in both male and female patients with MDD (Fig. 1 and table S1). In contrast, we did not observe changes in other genes whose expression is enriched in the hippocampus [HCN2, PEX5L, KCND2 encoding Kv4.2 channels (48), and DLG4 encoding PSD95]. Cerebellum samples were also available in a subset of patients, but no difference in HCN1 expression was seen in this region, indicating that differences could not be attributed to systematic differences in sample storage or preparation (fig. S1 and table S2).
Fig. 1. HCN1 expression is elevated in human patients with MDD compared to unaffected controls.
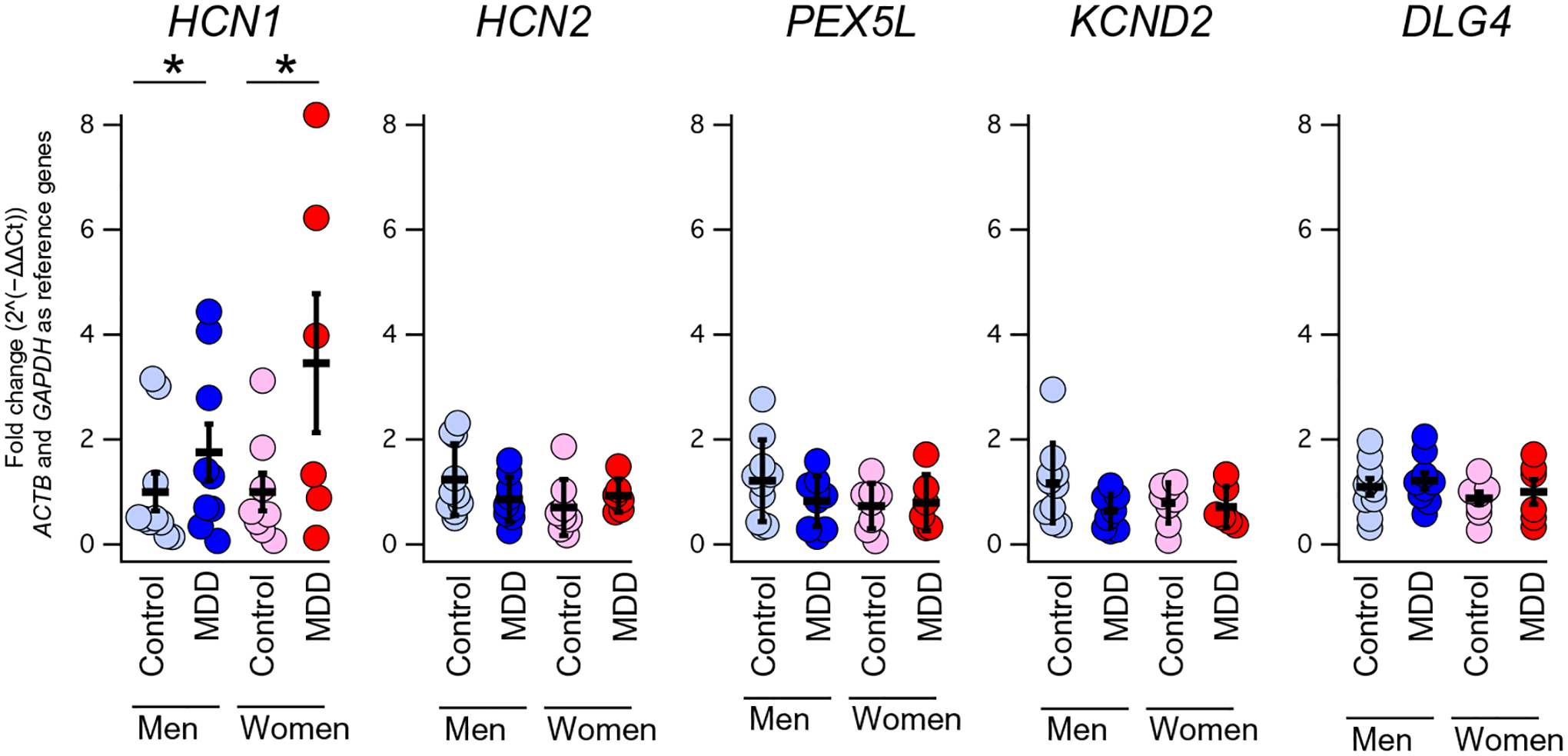
Human hippocampal tissue was obtained and subdissected to isolate CA1. qPCRs were performed to examine the expression of the genes listed above in men and women with and without a diagnosis of MDD. For a list of subjects and statistics, see table S1. *P < 0.05.
Acute increases in hippocampal cAMP limit motivated behavior and spatial memory
MA increases hippocampal cAMP signaling (44, 45), and cAMP increases HCN channel function (19). Given that increasing HCN channel function limits motivated behavior (34), these observations raise the possibility that HCN channels could limit the therapeutic efficacy of MA. To isolate the effect of cAMP from the other pathways activated by MA, we used the rM3D(Gs) DREADD (designer receptors exclusively activated by designer drugs) construct, which leads to the production of cAMP in the presence of clozapine-N-oxide (CNO) (49, 50). To confirm that rM3D(Gs) increases cAMP signaling and augments Ih, we performed in vitro experiments using human embryonic kidney (HEK) cells expressing HCN2 and rM3D(Gs). We observed an increase in HCN channel function in response to bath application of CNO (fig. S2); hence, we next generated an adeno- associated virus (AAV) expressing rM3D(Gs) regulated by a CaMKII (calcium/calmodulin-dependent protein kinase II) promoter (referred to as AAV-DREADD; fig. S3). Expression of AAV-DREADD in the dorsal hippocampus of adult male mice led to the production of cAMP after intraperitoneal injection of CNO (Fig. 2A and table S3). Similar to HEK experiments, whole-cell recordings from CA1 pyramidal neurons infected with AAV-DREADD showed an acute increase in the sag ratio (a current clamp measurement of the current mediated by HCN channels) in response to bath application of CNO (Fig. 2, B and C) that was not observed in uninfected cells (table S3). These results indicate that AAV-DREADD–mediated increases in cAMP lead to an increase in HCN channel function.
Fig. 2. AAV-DREADD increases hippocampal cAMP and influences behavior.
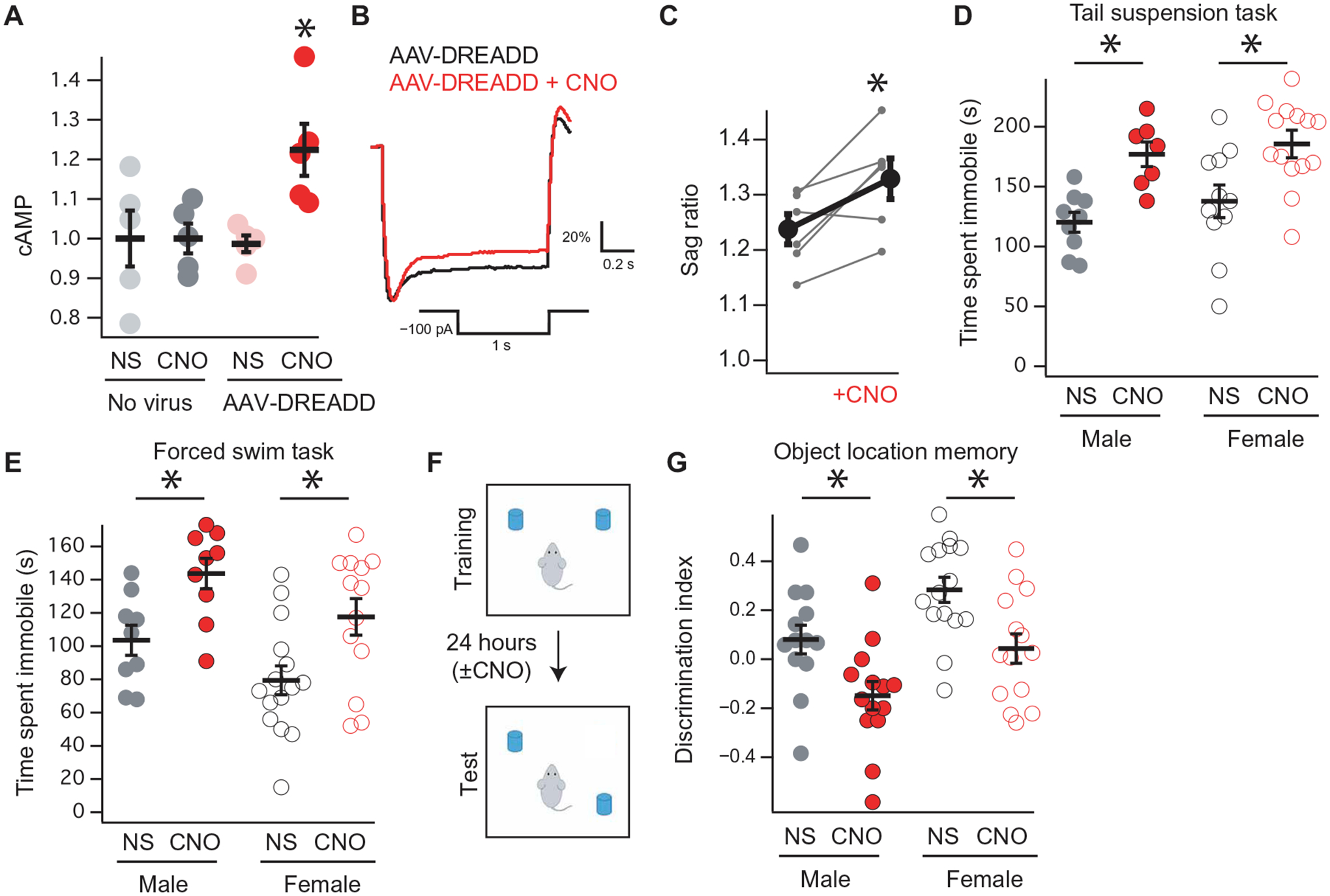
(A) Hippocampal cAMP production in animals injected with hippocampal AAV-DREADD and intraperitoneal CNO. A 2 × 2 ANOVA showed an interaction between virus (none/AAV-DREADD) and intraperitoneal injection (saline/CNO): F1,16 = 5.0492, P = 0.0391, n = 5, 5, 5, and 5. (B) Representative current clamp recording from a GFP+ CA1 pyramidal neuron at a holding potential of −70 mV in the presence or absence of CNO. Quantified in (C) (pre-CNO: 1.24 ± 0.02; post-CNO: 1.32 ± 0.03; paired t test, t6 = −3.26, P = 0.017, n = 7, N = 5). (D) Wild-type male and female mice were bilaterally injected with AAV-DREADD and received either intraperitoneal CNO or saline (NS) before TST [(D) male NS: 120.2 ± 8.3 s, n = 9; male CNO: 177.0 ± 10.2 s, n = 7; female NS: 137.7 ± 13.6 s, n = 11; female CNO: 187.4 ± 11.5 s, n = 11; means ± SEM] and FST [(E) male NS: 103.5 ± 9.0 s, n = 9; male CNO: 143.6 ± 9.1 s, n = 9; female NS: 79.4 ± 8.6 s, n = 15; female CNO: 117.6 ± 11.0 s, n = 13]. (F) Schematic for the object location memory (OLM) task, adapted from Maroso et al. (18). Either intraperitoneal CNO or intraperitoneal saline was administered immediately after training. (G) OLM performance from male and female mice bilaterally injected with AAV-DREADD after intraperitoneal saline or CNO is administered (male NS: 0.08 ± 0.05, n = 14; male CNO: −0.14 ± 0.05, n = 13; female NS: 0.28 ± 0.05, n = 15; female CNO: 0.04 ± 0.06, n = 14). *P < 0.05 for (D), (E), and (G). Asterisk denotes main effect of “NS/CNO”; see supplementary tables for details.
Previous work has shown that knockdown of HCN reduced immobility time in tests of motivated behavior (23–25); hence, we tested whether acutely increasing HCN channel function with a rise in cAMP would alter these behaviors. Toward this end, we bilaterally injected the dorsal hippocampi of adult male and female mice with AAV-DREADD. The animals were then administered either intraperitoneal saline [normal saline (NS)] or CNO and subjected to the tail suspension test (TST), forced swim test (FST), or open-field test (OFT) (Fig. 2, D and E, and table S3). We observed that intraperitoneal injection of CNO led to more time immobile in TST and FST without affecting OFT (Fig. 2, D and E, and fig. S4). No difference in any of the three assays was seen in response to intraperitoneal injection of CNO in animals that had received a control virus [AAV–green fluorescent protein (GFP)] or no virus at all (fig. S5 and table S4).
HCN channel function in the dorsal hippocampus has also been implicated in spatial memory (17), and previous work has suggested that cannabinoid signaling interferes with spatial memory in the object location memory (OLM) task, in part, by augmenting hippocampal Ih function (18). We next bilaterally injected AAV-DREADD into the dorsal hippocampi of adult male and female mice and then intraperitoneally injected either saline or CNO after the training session of the OLM task (Fig. 2, F and G). Mice that received CNO performed worse on day 2 of the OLM task. These results indicate that acute increases in hippocampal cAMP increase HCN channel function and limit both motivated behavior and spatial memory.
Hcn2R/R mice reveal that hippocampal cAMP produces changes in behavior through HCN channels
On the basis of our electrophysiology data showing an increase in Ih, we reasoned that cAMP binding to HCN channels was involved in the change in behavior. HCN pore-forming subunits homo- and heterotetramerize in vivo to produce channels with intermediate properties, although HCN2 subunits are highly sensitive to cAMP (51). To isolate the effect of increasing HCN channel function from the other signaling pathways regulated by cAMP, we generated a knock-in mouse line expressing a mutant HCN2 subunit that is insensitive to cAMP [HCN2(R591E), Hcn2R/R] (fig. S6). This mutation has been characterized in vitro and functions similarly to wild-type channels, with the exception that its gating does not shift in the presence of cAMP (51, 52). A recent report also investigated the effects of a series of HCN2 mutations including R591E and found no difference in heterotetramerization of the mutant channel with wild-type channels, nor was there a difference in surface expression of the channel in thalamic nuclei (52).
Hcn2R/R animals were born in a Mendelian ratio and were indistinguishable from wild-type littermates by eye. Immunohistochemical and Western blot analyses of the hippocampus revealed no changes in HCN1, HCN2, or TRIP8b (Fig. 3, A and B; fig. S7; and table S5), and immunohistochemistry revealed no difference in HCN2 subcellular distribution (Fig. 3, C and D). Whole-cell recordings from CA1 pyramidal neurons of Hcn2R/R mice revealed reduced sag ratio (Fig. 3, E and F) and a hyperpolarized V50 (the membrane potential at which half of the channels are open) in the presence of 8-Br-cAMP (8-bromoadenosine-cAMP), a cell-permeable cAMP analog (Fig. 3G) (38). 8-Br-cAMP had no effect on the membrane resistance of wild-type cells but increased the membrane resistance of Hcn2R/R cells and their excitability (Fig. 3, H and I). This change in membrane resistance (Rm) was similar to that previously seen in response to protein kinase A (PKA) activation, leading to removal of Kv channels from the cell surface (48), and indicates that cAMP normally reduces the membrane resistance (Rm) by increasing Ih and, in parallel, increases Rm through PKA (48). Given the reduction in Ih in Hcn2R/R mice, we next investigated the TA pathway, which is strongly regulated by Ih (21, 28, 29). Whole-cell recordings were made from CA1 pyramidal neurons with a bipolar stimulating electrode placed over the TA pathway (Fig. 3J). To isolate the contribution of HCN channels, recordings were made before and after application of ZD7288, an HCN channel antagonist (53). ZD7288 increased temporal summation in Hcn2+/+ cells, whereas it did not affect temporal summation in Hcn2R/R cells (Fig. 3, K and L). These results indicate less dendritic Ih and greater TA pathway temporal summation in the Hcn2R/R animals. Given the reduction in hippocampal Ih, we sought to test whether the Hcn2R/R animals would show more motivated behavior (15, 16, 21, 24). Consistent with this prediction, we observed that male and female Hcn2R/R animals showed reduced immobility time on TST (Fig. 4A) and FST (Fig. 4B and table S6) without a change in OFT (fig. S8A and table S7).
Fig. 3. Hcn2R/R mice have less Ih and more temporal summation in the TA pathway.
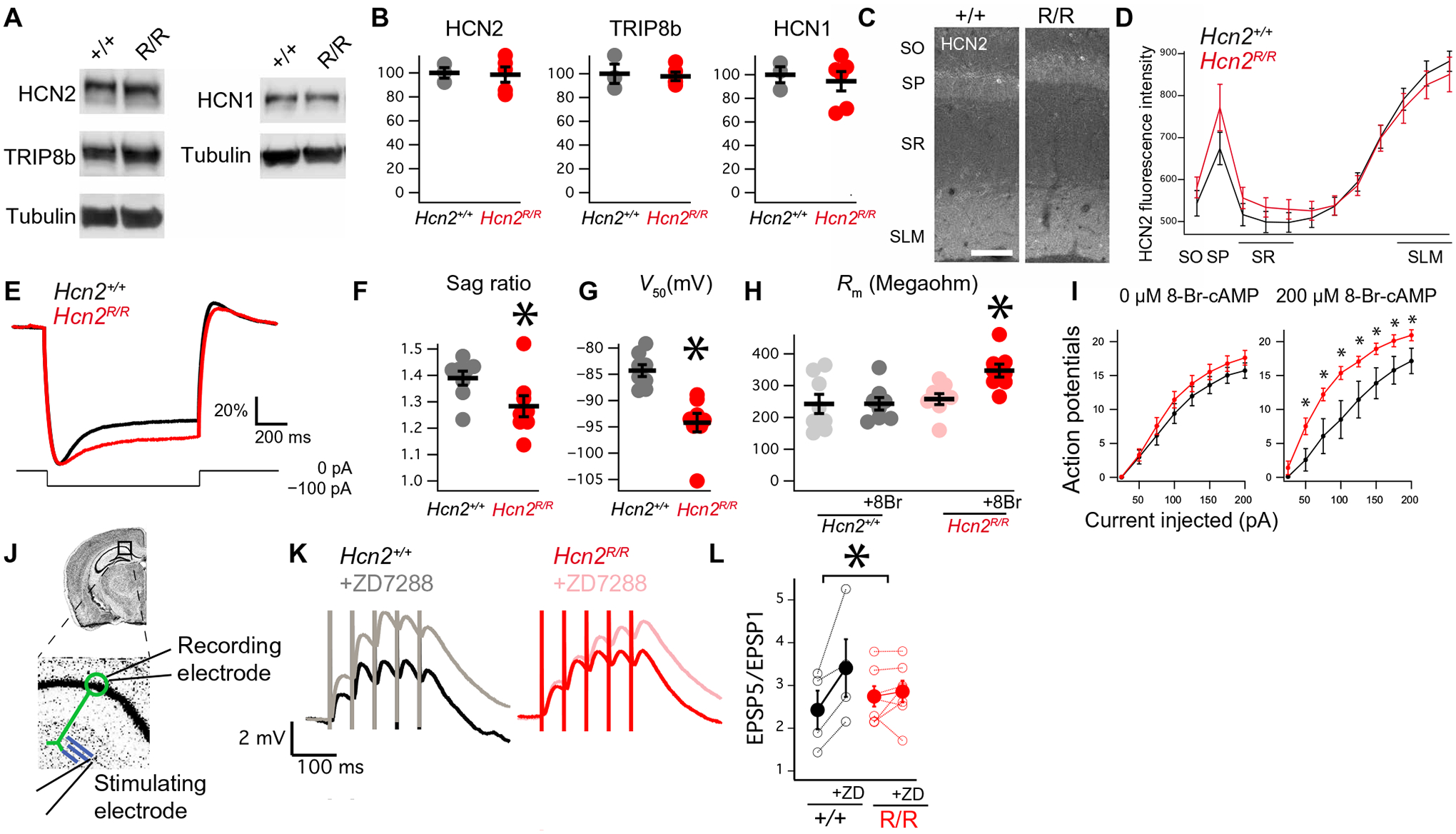
(A) Representative Western blots of HCN1, HCN2, or TRIP8b, quantified in (B) (HCN1 Hcn2+/+: 100.0 ± 6.7; Hcn2R/R: 94.4 ± 8.2, n = 3 and 6; HCN2 Hcn2+/+: 100.0 ± 4.4; Hcn2R/R: 98.5 ± 6.4, n = 3 and 5; TRIP8b Hcn2+/+: 100.0 ± 8.1; Hcn2R/R: 97.8 ± 3.5, n = 3 and 5). Uncropped blots are provided in fig. S7. (C) Representative images of CA1 stained with anti-HCN2 antibody. Scale bar, 100 μm. (D) Quantification of HCN2 staining [Hcn2+/+: 881.8 ± 24.1 arbitrary units (AU); Hcn2R/R: 850.5 ± 41.1 AU, n = 4 and 4]. (E) Representative current clamp recordings showing response to −100-pA current injection from a −70-mV potential. Note that the y axis is scaled to maximal deflection to facilitate comparison (see Materials and Methods). (F) Quantification of sag ratio (Hcn2+/+: 1.38 ± 0.02; Hcn2R/R: 1.28 ± 0.03, n = 8 and 8, N = 5 and 4). (G) Half activation potential for Ih in the presence of 8-Br-cAMP (Hcn2+/+: −84.3 ± 1.1 mV; Hcn2R/R: −94.2 ± 1.7 mV, n = 8 and 8, N = 5 and 4). (H) Membrane resistance at −60 mV in the presence or absence of 8-Br-cAMP (Hcn2+/+, pre–8-Br-cAMP: 242.5 ± 30.2 megohms; Hcn2+/+, post–8-Br-cAMP: 242.0 ± 19.6 megohms; Hcn2R/R, pre–8-Br-cAMP: 257.6 ± 17.2 megohms; Hcn2R/R, post–8-Br-cAMP: 347.2 ± 20.4 megohms; n = 8 and 8, N = 5 and 4). (I) Action potentials produced in response to current injection in the absence of 8-Br-cAMP (left) or the presence of 8-Br-cAMP (right). (J) Recording configuration for examining the TA pathway. Whole-cell recording was performed from CA1 cell body (green), whereas bipolar simulating electrode was placed on the TA pathway (in blue). (K) Representative traces obtained from the configuration in (J) showing temporal summation before and after application of ZD7288. (L) Quantification of the change in temporal summation (change in EPSP5/EPSP1 with ZD7288 Hcn2+/+: 0.97 ± 0.34 mV; Hcn2R/R: 0.11 ± 0.16 mV, n = 4 and 7, N = 4 and 5). *P < 0.05. SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; SLM, stratum lacunosum moleculare.
Fig. 4. Hippocampal HCN channels limit motivated behavior and spatial memory.
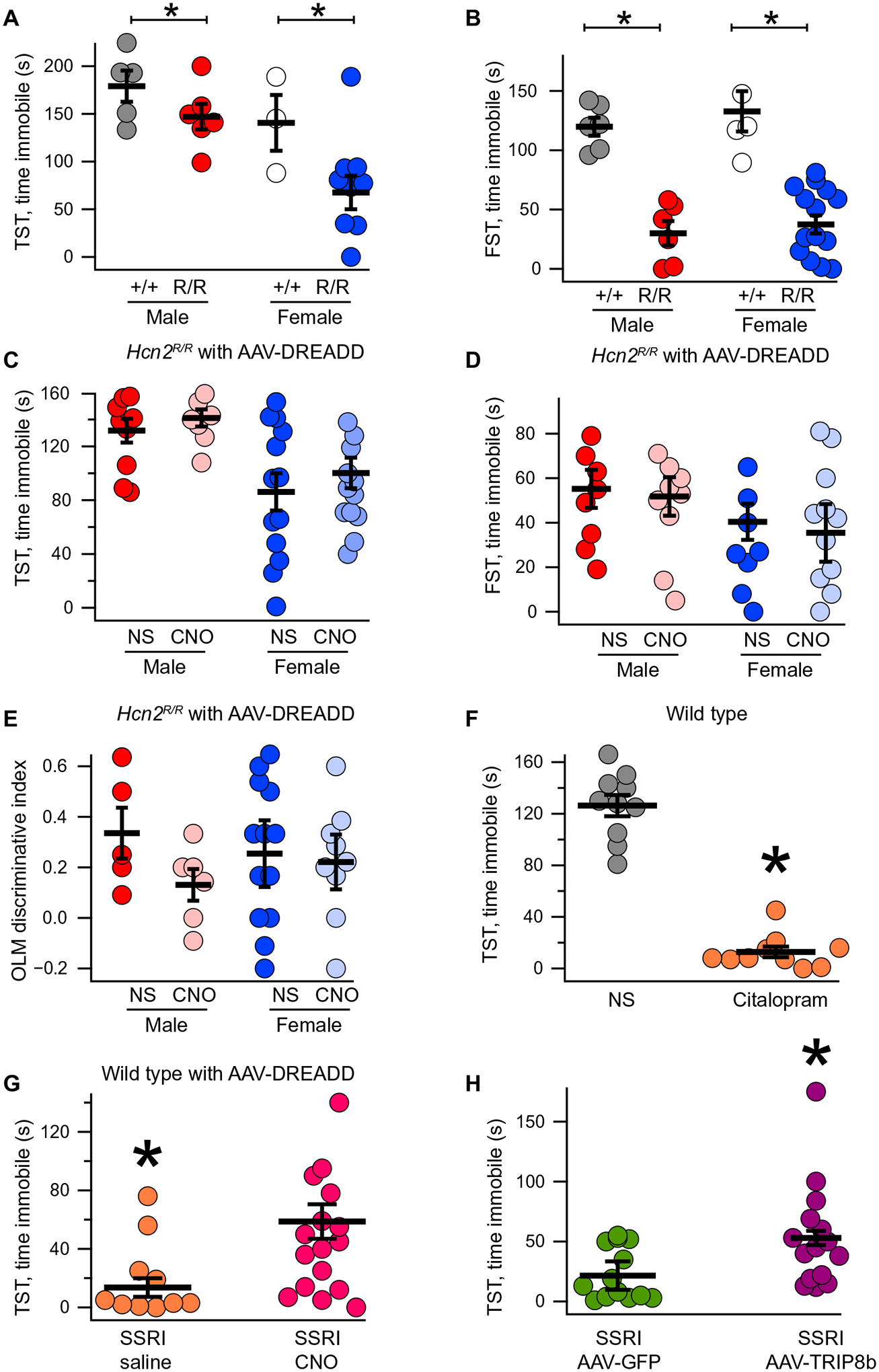
Male and female Hcn2+/+ and Hcn2R/R mice were subjected to TST [(A) male +/+: 179.1 ± 16.3 s; male R/R: 147.0 ± 13.44 s; female +/+: 140.6 ± 29.2 s; female R/R: 67.6 ± 17.5 s] and FST [(B) male +/+: 119.8 ± 7.6 s; male R/R: 30 ± 10.2 s; female +/+: 132.8 ± 16.9 s; female R/R: 37.4 ± 7.6 s, n = 6, 6, 5, and 15]. (C) Male and female Hcn2R/R mice were bilaterally injected with AAV-DREADD and then received intraperitoneal saline or CNO before TST (male R/R saline: 131.7 ± 8.88 s; male R/R CNO: 141.1 ± 6.36 s; female R/R saline: 86.15 ± 13.82 s; female R/R CNO: 100.21 ± 11.36 s, n = 10, 8, 13, and 14). (D) An identical setup was used for FST (male R/R saline: 55.2 ± 8.54 s; male R/R CNO: 51.8 ± 8.65 s; female R/R saline: 40.1 ± 12.9 s; female R/R CNO: 35.4 ± 8.11 s; n = 9, 10, 12, and 12). (E) Hcn2R/R males and females were bilaterally injected in the dorsal CA1 with AAV-DREADD and then subjected to the OLM task. After the training day, mice received either intraperitoneal saline or intraperitoneal CNO (discriminative index: male R/R saline: 0.33 ± 0.10; male R/R CNO: 0.13 ± 0.06; female R/R saline: 0.34 ± 0.13; female R/R CNO: 0.26 ± 0.10; n = 5, 6, 13, and 9). (F) Wild-type male mice were intraperitoneally injected with either saline or citalopram (SSRI) before TST (saline: 126.3 ± 8.23 s; SSRI: 12.8 ± 4.13 s; n = 10 and 10). (G) Wild-type male mice were bilaterally injected with AAV-DREADD and then received SSRI along with either saline or CNO before TST (SSRI/saline: 13.57 ± 6.36 s; SSRI/CNO: 58.66 ± 11.76 s; n = 14 and 18). (H) Wild-type male mice were bilaterally injected with either AAV-GFP or AAV-TRIP8b and then received intraperitoneal citalopram (AAV-GFP/SSRI: 21.57 ± 5.93 s; AAV-TRIP8b/SSRI: 53.00 ± 11.05 s; n = 14 and 15). Please note that between-sex differences are not reported above (see table S6 for statistics). *P < 0.05.
We next investigated whether or not cAMP binding to HCN channels was responsible for the reduction in motivated behavior that we observed in response to acute increases in cAMP in wild-type mice (Fig. 2, D to G), so we bilaterally injected the dorsal hippocampi of male and female Hcn2R/R animals with AAV-DREADD. Unlike in wild-type mice, intraperitoneal injection of CNO in these animals did not produce a change in TST (Fig. 4C), FST (Fig. 4D), or OFT (fig. S8B). We also investigated whether or not hippocampal HCN2 channels were responsible for the effect of acute cAMP on spatial memory. Unlike the wild-type mice, acutely increasing hippocampal cAMP in male and female Hcn2R/R mice had no effect on OLM performance (Fig. 4E). These results indicate that cAMP signaling through HCN channels is responsible for the acute reduction in motivated behavior and impaired spatial memory observed in wild-type animals (Fig. 2, D to F).
Hippocampal HCN channels limit the behavioral response to antidepressants
On the basis of our observation that acutely increasing cAMP in CA1 limited motivated behavior, we next speculated that HCN channels in dorsal CA1 might limit the changes in behavior seen with acute MA administration. To test this hypothesis, we used citalopram, a selective serotonin reuptake inhibitor (SSRI) that produces an increase in motivated behavior after acute intraperitoneal administration (Fig. 4F) (54). We bilaterally injected the dorsal hippocampus of wild-type mice with AAV-DREADD, and after 3 weeks (to allow for viral expression), the animals received either intraperitoneal saline or intraperitoneal CNO 40 min before the behavioral assay and intraperitoneal citalopram 30 min prior (Fig. 4G). Mice receiving both CNO and citalopram spent more time immobile compared with saline and citalopram, indicating that acutely increasing hippocampal cAMP limits the behavioral response to citalopram.
We next asked whether directly increasing hippocampal HCN channel expression would have a similar effect. To avoid altering the composition of HCN1 and HCN2 channels or the subcellular distribution of the channels (21), we overexpressed the most abundant TRIP8b isoform in the dorsal hippocampus [AAV-TRIP8b; see Materials and Methods (34)]. We have previously characterized the effect of AAV-TRIP8b on CA1 HCN channels and shown that overexpression of the virus leads to an increase in HCN channel surface trafficking (34). Thirty minutes before the behavioral experiment, mice received an intraperitoneal injection of either saline or citalopram (Fig. 4H and table S6). Although citalopram promoted motivated behavior, overexpression of TRIP8b limited this effect. These results indicate that increasing hippocampal HCN function limits the behavioral response to antidepressants.
Chronically elevated hippocampal cAMP promotes motivated behavior and spatial memory
On the basis of previous in vitro work showing that cAMP disrupts the interaction between TRIP8b and HCN (36, 37, 43), we predicted that chronically elevated cAMP could produce a reduction in surface HCN in vivo. This hypothesis suggests that chronically elevated cAMP in the hippocampus should have the opposite behavioral effect from acutely elevated cAMP, as the TRIP8b-HCN interaction is disrupted and HCN channels are lost from the cell surface. To test this hypothesis, we bilaterally injected the dorsal CA1 of wild-type male mice with AAV-DREADD or an AAV-GFP control virus and allowed several weeks for the viruses to express before administering either CNO or saline chronically in the animal’s drinking water for 3 weeks. Two hours before testing, all mice were switched to tap water to minimize the acute effects of DREADD activation (49). We noted that the AAV-DREADD–injected animals receiving CNO in their drinking water spent less time immobile in both the TST (Fig. 5A) and FST tasks (Fig. 5B and table S8) without a change in OFT (fig. S9 and table S10). Moreover, a separate cohort of male mice that were bilaterally injected with AAV-DREADD but received just 24 hours of CNO in the drinking water did not exhibit a change in motivated behavior (fig. S10 and table S11), indicating that this change in behavior is specifically the result of chronically elevated cAMP signaling. We next investigated whether or not spatial memory would similarly be altered in the chronically elevated cAMP condition. Wild-type male and female mice were bilaterally injected into the dCA1 with AAV-DREADD followed by 3 weeks of either saline or CNO in the drinking water. Consistent with the results for motivated behavior, chronically elevated cAMP improved performance on the OLM task (Fig. 5C and table S8). Combined, these results indicate that acute and chronic changes in cAMP have opposing effects on behavior through bidirectional regulation of HCN channel function.
Fig. 5. Chronically elevated cAMP signaling promotes motivated behavior and spatial memory.
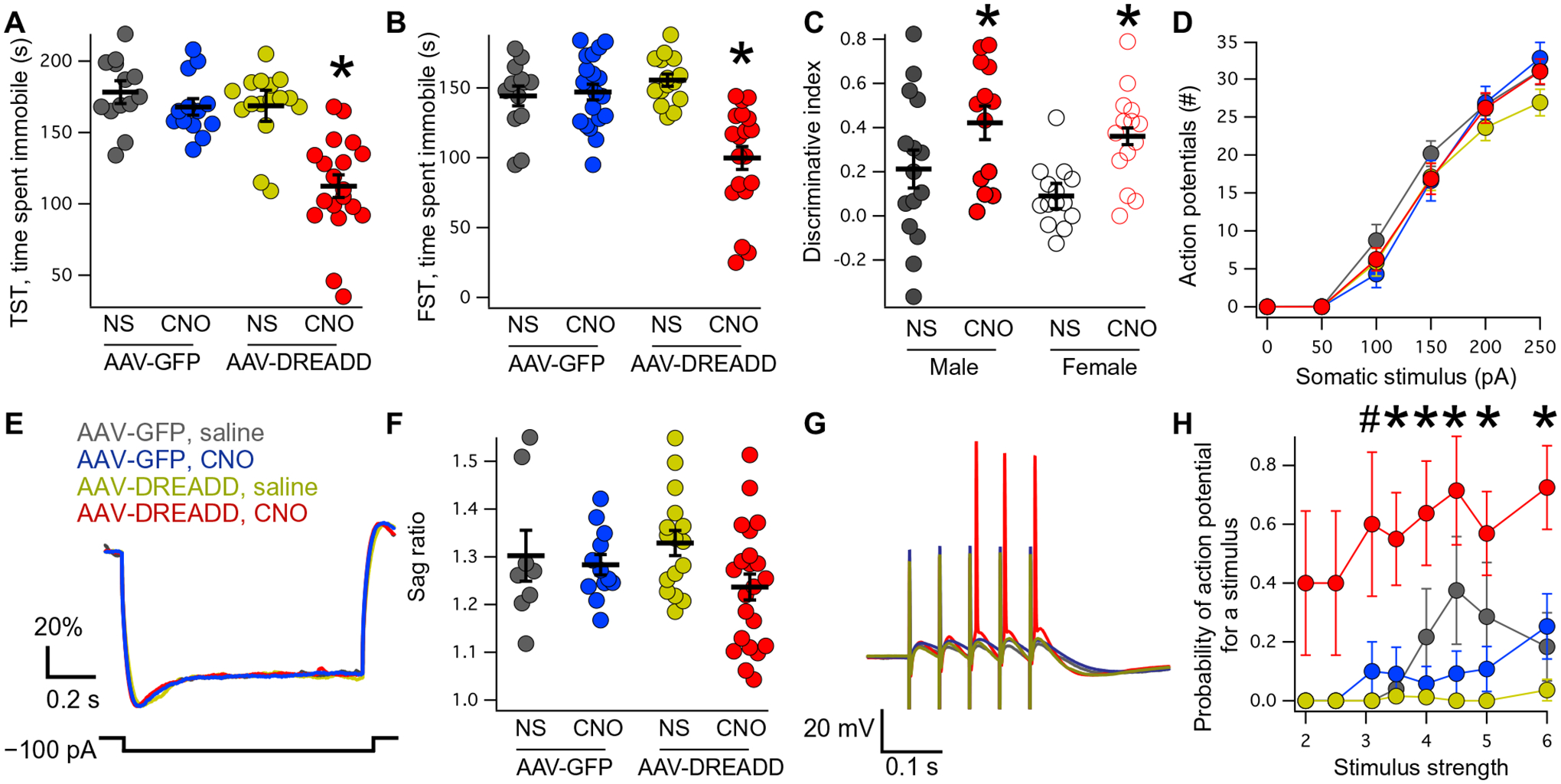
(A) Male mice were bilaterally injected with AAV-GFP or AAV-DREADD into the dorsal CA1 and then received 3 weeks of either saline (NS) or CNO before TST (AAV-GFP/NS: 178.2 ± 8.0 s; AAV-GFP/CNO: 167.8 ± 5.8 s; AAV-DREADD/NS: 168.6 ± 10.8 s; AAV-DREADD/CNO: 112.3 ± 7.9 s). An identical setup was used before FST [(B) AAV-GFP/NS: 144.2 ± 7.0 s; AAV-GFP/CNO: 147 ± 5.6 s; AAV-DREADD/NS: 155.5 ± 4.3 s; AAV-DREADD/CNO: 99.2 ± 8.2 s]. (C) Male and female mice were bilaterally injected with AAV-DREADD into dorsal CA1 and then received either NS or CNO in their drinking water for 3 weeks before OLM testing (male AAV-DREADD/NS: 0.21 ± 0.08; male AAV-DREADD/CNO: 0.42 ± 0.07; female AAV-DREADD/NS: 0.08 ± 0.03; female AAV-DREADD/CNO: 0.36 ± 0.05; n = 15, 13, 14, and 14). (D) Number of action potentials fired in each condition as a function of somatic current injection (AAV-GFP/NS: n = 7; AAV-GFP/CNO: n = 11; AAV-DREADD/NS: n = 16; AAV-DREADD/CNO: n = 21; tables S8 and S9). (E) Representative current clamp traces from all four conditions in response to hyperpolarizing 100-pA current injection. The y axis is scaled as in previous figures. (F) Quantification of somatic sag ratio (AAV-GFP/NS: 1.30 ± 0.05; AAV-GFP/CNO: 1.28 ± 0.02; AAV-DREADD/NS: 1.32 ± 0.02; AAV-DREADD/CNO: 1.23 ± 0.02). (G) Representative current clamp traces obtained during stimulation of the TA pathway. Note that colors represent the same conditions as in (A), (D), and (E). (H) Quantification of the proportion of stimuli producing action potentials as a function of TA stimulating electrode intensity (see tables S8 and S9 for statistics). For all panels, *P < 0.05. For (H), #P < 0.05 for effect of drinking water, *P < 0.05 for interaction of drinking water and virus.
Given the extended time course required for changes in behavior, we speculated that changes in transcription could be responsible. cAMP signaling is coupled to transcription through several transcription factors including cAMP response element–binding protein (CREB) (55–57). We next investigated whether or not CREB overexpression was sufficient to reproduce these changes in behavior given that previous work has emphasized a possible role for CREB signaling in the effects of MA (45, 58). We generated a virus expressing a constitutively active form of CREB (59) but saw no difference in TST, FST, or OFT after expression in the dorsal hippocampus (fig. S11 and table S12).
Chronically elevated hippocampal cAMP disrupts the TRIP8b-HCN interaction
To determine the molecular mechanism responsible for the changes in behavior in response to chronically elevated cAMP, we performed whole-cell recordings from GFP+ cells in male mice injected with either AAV-DREADD or AAV-GFP and then receiving CNO or saline in their drinking water for 3 weeks. Somatic recordings revealed no change in membrane potential, membrane resistance, excitability, or sag ratio (Fig. 5, D and F, and tables S8, S9, and S13). Although we did not observe a change in the sag ratio measured at the soma, we reasoned that because most of the HCN channels that are regulated by TRIP8b are expressed in the dendrites (20), they are unlikely to be electrically accessible using somatic current injections. Given that dendritic HCN channels regulate the TA pathway synapsing onto the distal SLM, we next investigated the strength of this pathway using a bipolar stimulating electrode while recording from the soma (as in Fig. 3J) (21, 29, 30). Across the four conditions, we noted that only cells from mice in the chronically elevated cAMP condition (AAV-DREADD with chronic CNO) produced more action potentials in response to TA pathway stimulation (Fig. 5, G and H, and tables S9, S13, and S14). Stimulating the SC pathway that synapses onto the more proximal SR [and is less regulated by HCN channels (29, 30)] did not reveal a difference (fig. S12 and tables S15 and S16).
We reasoned that the increase in strength of the TA pathway could be related to a loss of HCN channels on the surface of the dendrites in response to chronically elevated cAMP (36, 37, 43). To test this hypothesis, we examined temporal summation of the TA pathway in the presence or absence of ZD7288. Cells from AAV-DREADD–injected mice that received saline (vehicle) in their drinking water exhibited a large increase in TA pathway temporal summation after bath application of ZD7288, consistent with a previous report (30) (Fig. 6, A and B, and table S18). However, cells from mice that received CNO for 3 weeks showed little change in TA pathway temporal summation after application of ZD7288, indicating that there was little contribution of dendritic Ih to begin with. These results indicate a loss of surface HCN channels after chronically elevated cAMP.
Fig. 6. Chronically elevated cAMP disrupts the TRIP8b-HCN interaction.
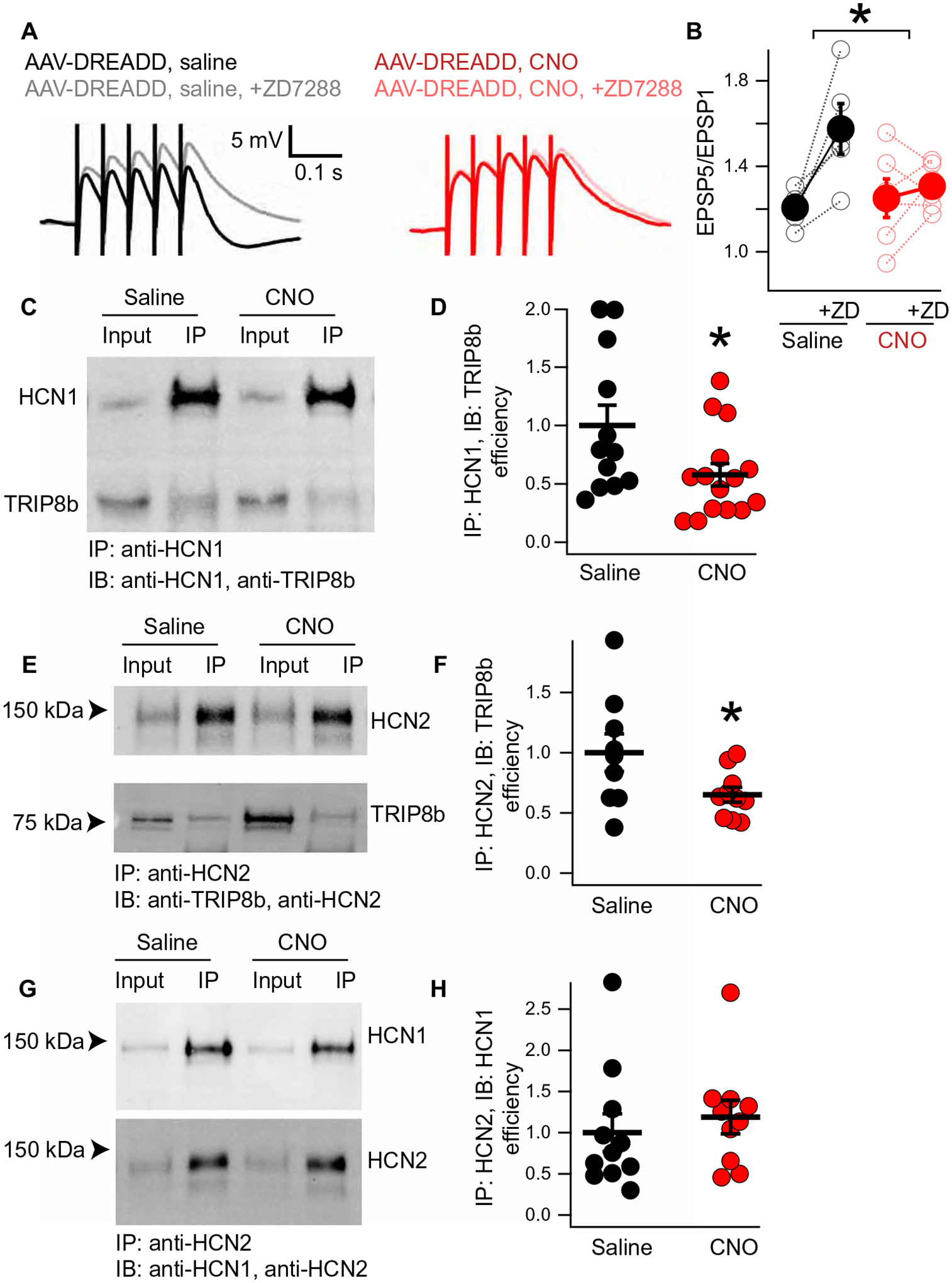
(A) Whole-cell recordings from GFP+ CA1 pyramidal neurons were performed from mice injected with AAV-DREADD and receiving either saline (left) or CNO (right) in their drinking water for 3 weeks. Quantification in (B) (temporal summation after ZD7288 − before ZD7288; AAV-DREADD/NS: 0.36 ± 0.11; AAV-DREADD/CNO: 0.05 ± 0.07; n = 5 and 6). (C) Representative coimmunoprecipitation reaction was performed using anti-HCN1 antibodies in hippocampal lysate from mice injected with AAV-DREADD and receiving either saline or CNO in their drinking water. IP, immunoprecipitation; IB, immunoblotting. (D) Quantification of the results in (C) (AAV-DREADD/NS: 1.00 ± 0.17; AAV-DREADD/CNO: 0.58 ± 0.10; n = 12 and 15). (E and F) A similar experiment was performed using anti-HCN2 antibodies to immunoprecipitate TRIP8b in complex with HCN2 from mice hippocampi after AAV-DREADD injection and chronic NS or CNO (AAV-DREADD/NS: 1.00 ± 0.15; AAV-DREADD/CNO: 0.65 ± 0.06; n = 9 and 10). (G and H) Using an identical setup to that described for (E) and (F), we examined the amount of HCN1 bound to HCN2 by immunoprecipitating with an anti-HCN2 antibody (AAV-DREADD/NS: 1.00 ± 0.22; AAV-DREADD/CNO: 1.18 ± 0.20; n = 11 and 10) (see table S18 for statistics). *P < 0.05
We next investigated the mechanism responsible for the change in HCN channel surface expression in response to chronically elevated cAMP. qPCR showed no difference in the expression of Hcn1 or Pex5l (the gene encoding TRIP8b; fig. S13 and table S17), so we investigated mechanisms of posttranslational regulation. We performed coimmunoprecipitation reactions using an anti-HCN1 antibody and the hippocampi of mice injected with AAV-DREADD and receiving either CNO or saline in their drinking water for 3 weeks (Fig. 6, C and D; fig. S14; and table S18). We noted that less TRIP8b bound to HCN1 in the hippocampi of mice receiving CNO in their drinking water compared with mice receiving saline in their drinking water. To extend these results, we subsequently performed immunoprecipitation using an anti-HCN2 antibody. As with the anti-HCN1 antibody, we immunoprecipitated less TRIP8b from mice receiving chronically elevated cAMP when using the anti-HCN2 antibody (Fig. 6, E and F, and fig. S15). To ensure that these results were not the results of changes in the stoichiometry of HCN subunits, we also examined HCN1 subunits bound to HCN2 by immunoprecipitating with an anti-HCN2 antibody and blotting for HCN1. Here, we noticed no difference in the HCN1 subunits bound to HCN2 after chronically elevated cAMP treatment (Fig. 6, G and H, and fig. S15). These results indicate that chronically elevated cAMP disrupts the TRIP8b-HCN interaction and ultimately limits surface trafficking of HCN channels.
Hippocampal cAMP signaling rescues spatial memory deficits after chronic social defeat
Our results outlined above raise the possibility that chronically elevated cAMP in the hippocampus could improve cognition. To investigate this possibility, we used the chronic social defeat (CSD) model to examine the effect of chronic stress on lasting behavioral changes in mice (60, 61). Social interaction testing after CSD is used to define a group that is “resilient” to CSD (and continues to interact with an unfamiliar mice) and a second group that is “susceptible” to CSD (less likely to interact with an unfamiliar mouse) (60). Previous reports have shown that CSD leads to deficits in cognition in susceptible mice, including impaired spatial memory (62, 63). To test this hypothesis, we bilaterally injected the dorsal CA1 of male C57Bl/6J mice with AAV-DREADD. These animals were then subjected to CSD (see Materials and Methods) followed by social interaction testing to identify susceptible and resilient mice (Fig. 7, A and B). Half of the susceptible mice were randomly assigned to receive CNO in their drinking water for 3 weeks, whereas the remainder of the mice received saline (vehicle control). Afterward, susceptible mice that received CNO in their drinking water (chronically elevated cAMP) performed better in the OLM task compared with susceptible mice receiving only saline in their drinking water (Fig. 7C and table S19). Susceptible mice receiving CNO in their drinking water also spent less time immobile in the FST (Fig. 7D and table S19). These results establish that chronically elevated cAMP signaling in the dorsal hippocampus rescues both motivated behavior and spatial memory after chronic stress.
Fig. 7. Chronically elevated cAMP rescues motivated behavior and spatial memory after social defeat.
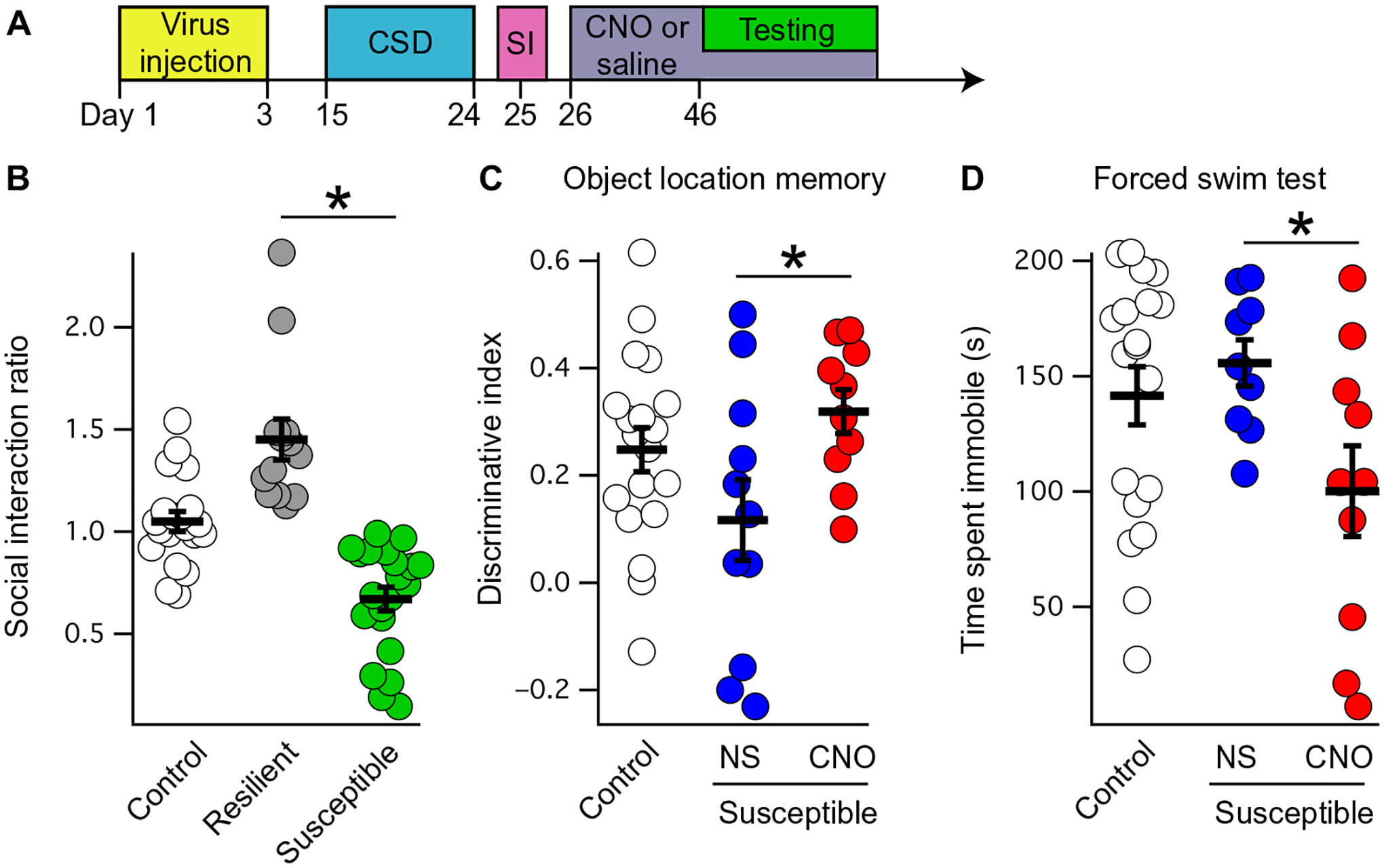
(A) Schematic of the experimental paradigm, with AAV-DREADD injected (yellow box) into the bilateral dCA1 of male mice before CSD. After CSD (blue box), susceptible mice were identified on social interaction (SI) testing (pink box), followed by 3 weeks of either saline or CNO in the drinking water (purple box). During behavioral testing (green box), all mice received tap water for 2 hours before testing but afterward were returned to their designated water condition afterward. (B) CSD leads to mice that are defined as either resilient (continue to interact with a novel mouse; gray circles) or susceptible (interact less with a novel mouse; green circles). Mice that are naïve to CSD are labeled “Control” (social interaction ratio for control: 1.04 ± 0.04; resilient: 1.45 ± 0.10; susceptible: 0.67 ± 0.05; n = 20, 13, and 21; means ± SEM). (C) Susceptible mice received either saline (NS) or CNO in their drinking water for 3 weeks before OLM testing (discriminative index for NS: 0.11 ± 0.07; CNO: 0.31 ± 0.04; n = 11 and 10; two-tailed t test, t19 = −2.29, P = 0.033). (D) Susceptible mice that received saline or CNO after CSD were also subjected to forced swim testing (time immobile for NS: 155.66 ± 10.06 s; CNO: 100.13 ± 19.68 s; n = 9 and 10; two-tailed t test, t17 = 2.42, P = 0.026). *P < 0.05 (see table S19 for additional details).
DISCUSSION
In this study, we identified a form of cAMP-dependent regulation of HCN channel trafficking that can influence motivated behavior and spatial memory. Our findings have translational relevance given that we observed an increase in the expression of HCN1 in the CA1 region of patients with MDD, providing human evidence linking HCN channels to MDD. We also noted that increased hippocampal HCN channel function in mice was sufficient to limit the behavioral response to citalopram, an MA. In light of the increased HCN channel expression in patients with MDD, our results raise the possibility that the therapeutic effects of MA are initially constrained by the presence of hippocampal HCN channels. In this framework, the beneficial effects of MA on motivated behavior and spatial memory come about in response to chronically elevated cAMP, a result of chronic MA therapy (44, 45, 64, 65), and this mechanism could be an explanation for the delayed therapeutic effect of MA (8, 66). Previous work on the role of cAMP in MDD has emphasized the CREB signaling pathway (67), which is thought to contribute to the therapeutic effect of MA in hippocampal regions outside of CA1 (45, 58). Consistent with this idea, we found that CREB overexpression in dorsal CA1 was not sufficient to mediate changes in behavior, and a likely possibility is that MAs exert their effects on behavior through distinct pathways in various brain regions (45, 58).
To our knowledge, there are no animal models that exhibit the biphasic change in behavior that we observed in response to cAMP signaling in dorsal CA1 pyramidal neurons (1, 8, 68). Although acute increases in cAMP increase HCN channel function and limit both motivated behavior and spatial memory, chronically elevated cAMP produced the opposite behavioral changes through a newly discovered pathway whereby cAMP disrupts HCN surface trafficking (fig. S17). This is consistent with the improvement in spatial memory noted in animal models with reduced forebrain expression of HCN1 (17) as well as other work showing impaired spatial memory with increasing Ih in dorsal CA1 (18). TRIP8b binds to HCN channels at two sites, and one of these binding sites directly competes with cAMP (36, 37, 43). Our previous in vivo work established that both TRIP8b-HCN interaction sites are required for dendritic enrichment of HCN channels (34), and in this report, we provide evidence that elevated concentrations of cAMP play a role in regulating HCN channel trafficking in vivo. This form of HCN channel regulation could serve as a form of homeostasis to match HCN channel function to cAMP signaling, with transient elevations in cAMP increasing Ih but sustained increases in cAMP limiting the density of HCN channels at the neuronal membrane. It remains to be seen whether this mechanism occurs at physiologic concentrations of cAMP signaling and whether or not other signaling factors are involved.
Our results with the Hcn2R/R mice lead to several interesting predictions about the role of HCN1 and HCN2 in vivo in the hippo-campus. HCN1 is the predominant subunit in the dorsal hippocampus (27), although HCN1 and HCN2 are thought to form both homotetramers and heterotetramers in vivo (51). However, we noted no difference in the subcellular distribution of HCN2 on Western blot or immunohistochemistry in Hcn2R/R mice. This is unexpected given that we have previously noted that the R591E mutation is sufficient to disrupt binding of TRIP8b to HCN2 (37) and observed that loss of TRIP8b binding to HCN channels leads to loss of the dendritic enrichment of HCN as well as a reduction in the total quantity of HCN protein (34, 69). The most likely explanation for the intact gradient of HCN2 in the Hcn2R/R mice is that there are relatively few HCN2 homomers and that TRIP8b binding to the HCN1 subunit of HCN1/HCN2(R591E) heterotetramers is sufficient to maintain HCN2 dendritic trafficking (fig. S17). Previous reports have established that HCN1 channel opening is minimally affected by cAMP binding, whereas HCN2 exhibits a large depolarization in V50 in response to cAMP (70). Given that we noted that Hcn2R/R pyramidal neurons are less sensitive to cAMP, this suggests that HCN2 is primarily responsible for conferring cAMP sensitivity to heterotetramers, although HCN1 makes up the bulk of Ih current.
The excitability of CA1 pyramidal neurons is regulated by many currents, including Kv and Ih. cAMP regulates PKA and leads to internalization of A-type K channels to increase CA1 excitability (48) as well as modulates Ih to reduce excitability. We observed that bath application of 8-Br-cAMP led to no change in membrane resistance in the wild-type cells, although there was an increase in both excitability and membrane resistance in the Hcn2R/R cells. This suggests that, at baseline, there is some degree of balance between the pathways through which cAMP produces an excitatory or inhibitory influence on CA1 pyramidal neurons and that loss of HCN2’s cAMP sensitivity leads to the excitatory pathways [such as internalization of A-type K channels (48)] playing a larger role.
There is an increasing recognition of the importance of excitatory neurotransmission in MDD and antidepressant efficacy (1, 71, 72). In particular, the TA pathway has been shown to be strengthened by chronic antidepressant treatment and weakened in animal models relevant to MDD (73, 74). HCN channels play an essential role in limiting temporal summation through the TA pathway (21, 29, 30); hence, this synapse is a natural focal point for unifying the burgeoning literature on the role of hippocampal HCN channels in MDD with existing theories of MDD and its treatment. Although it is challenging to extrapolate from animal studies, we suspect that dorsal hippocampal HCN channels limit the behavioral effects of MA and may contribute to the delayed therapeutic effect of MA. Existing theories for the delayed therapeutic effect of MA have focused on desensitization of midbrain serotonin receptors (75), although many subtypes of serotonin receptors are expressed in the hippocampus (76) and the possibility exists that there are multiple mechanisms responsible for the delayed therapeutic action. Acute administration of SSRI is known to increase hippocampal serotonergic signaling in the rodent, and serotonergic signaling increases Ih in CA1 pyramidal neurons (77, 78). We observed that increasing hippocampal HCN channel function (either via TRIP8b overexpression or by increasing hippocampal cAMP signaling) limits the behavioral response to acute SSRI administration. These results suggest that the acute production of cAMP in the dorsal hippocampus by SSRI may, at least initially, counteract the desired behavioral changes that are elicited through other mechanisms and signaling pathways.
We speculate that compounds that limit HCN channel function in the dorsal hippocampus could be used as antidepressants by specifically treating the cognitive symptoms associated with chronic stress (15) based on the observation that chronically elevated cAMP was sufficient to rescue spatial memory and motivated behavior after CSD. HCN channels in brain regions outside of CA1 have often been shown to produce opposing effects on behavior from those reported here (16, 22, 79–81), and combined with concerns about limiting HCN channel function in the heart, it is unlikely that directly targeting HCN channels will lead to a clinically useful therapy. However, TRIP8b is not thought to be expressed in these other regions and cell types (31, 81), and targeting the TRIP8b-HCN interaction may prove to be more effective as a therapy while avoiding off-target effects from antagonizing HCN channels in cells outside of dorsal CA1 (82).
It is important to note that there are several limitations of our study. First, our human data are from an extremely small sample, and the limitations associated with the dataset prevent comparing treatment status and disease severity. Ideally, future work would more formally test the hypothesis that there are changes in HCN channels in patients with MDD and would specifically examine changes in protein expression with a larger sample size. A second limitation is in our use of DREADD technology to influence cAMP concentrations. Although we have attempted to control for the influence of CNO and DREADD expression independent of DREADD activation by CNO, the possibility remains that there are unanticipated effects of these manipulations. It would be worthwhile to validate our results using alternative methods of increasing cAMP, ideally only in the dendritic compartment of CA1 pyramidal neurons. Similarly, having a method to rapidly dissociate TRIP8b from HCN and then observe the effects on behavior would permit a more direct examination of the role of the TRIP8b-HCN interaction than the one we provide here. As new molecular tools are developed, it will be interesting to reexamine the question of how posttranslational modifications in the hippocampus translate into changes in behavior.
MATERIALS AND METHODS
Study design
The rationale for this study was to investigate the molecular biology of HCN channel trafficking and specifically to investigate whether cAMP influences channel trafficking. All animal subjects were randomized by cage to different experimental conditions using a random number generator. All experimentalists were blinded to experimental condition for scoring animal behavior and recording electrophysiological data. For behavioral data, qPCR, Western blotting, coimmunoprecipitation, and immunohistochemistry, each n represents a distinct animal or human sample. For electrophysiology, each n represents one cell and animal number (described with N) is reported either in the figure caption or in the corresponding supplementary table. The data that support the findings of this study are available from the corresponding author upon reasonable request. No unpublished analysis routines were used in the completion of this study. All comparisons were planned before beginning experimentation, and only two-sided comparisons were used (with α = 0.05). No formal power analyses were conducted before experimentation.
Statistical analysis
All data are presented as means ± SEM in figure captions and in graphs displayed in figures. All data were subjected to tests of normality (Shapiro-Wilk) and homoscedasticity (Levene’s) as described else-where (83). Data that did not meet these assumptions were compared using nonparametric methods: Wilcoxon rank sum test in place of t tests, Kruskal-Wallis test in place of one-way analysis of variance (ANOVA), and 2 × 2 ANOVA of an aligned rank transformation of the data (84) in place of a 2 × 2 ANOVA of the raw data. Otherwise, t tests and ANOVA analyses were performed as described in the article.
Supplementary Material
Funding:
This work was supported by NIH grant R01-NS059934 (to D.M.C.), R01MH106511 (to D.M.C.), R21MH113262 (to D.M.C.), R21MH104471 (to D.M.C.), R21MH126272 (to D.M.C.), Brain and Behavior Research Foundation NARSAD 25138 (to Y.H.), R01-NS 107424-01 (to C.Z.), and Vanderbilt Institute for Clinical and Translational Research VR52450 and VR53895 (to Y.H.). Immunofluorescence images were performed at the Vanderbilt Cell Imaging Shared Resource supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126. Part of behavioral experiments were carried out at Vanderbilt Neurobehavioral Core supported by Vanderbilt Kennedy Center and the EKS NICHD of the NIH under award no. 1P50HD103537-01.
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests:
The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Duman RS, Shinohara R, Fogaça MV, Hare B, Neurobiology of rapid-acting antidepressants: Convergent effects on GluA1-synaptic function. Mol. Psychiatry 24, 1816–1832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abas MA, Sahakian BJ, Levy R, Neuropsychological deficits and CT scan changes in elderly depressives. Psychol. Med 20, 507–520 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Gould NF, Holmes MK, Fantie BD, Luckenbaugh DA, Pine DS, Gould TD, Burgess N, Manji HK, Zarate CA Jr., Performance on a virtual reality spatial memory navigation task in depressed patients. Am. J. Psychiatry 164, 516–519 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Elliott R, Sahakian BJ, McKay AP, Herrod JJ, Robbins TW, Paykel ES, Neuropsychological impairments in unipolar depression: The influence of perceived failure on subsequent performance. Psychol. Med 26, 975–989 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Burt DB, Niederehe G, Zembar MJ, Depression and memory impairment: A meta-analysis of the association, its pattern, and specificity. Psychol. Bull 117, 285–305 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Rosenblat JD, Kakar R, McIntyre RS, The cognitive effects of antidepressants in major depressive disorder: A systematic review and meta-analysis of randomized clinical trials. Int. J. Neuropsychopharmacol 19, pyv082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kessler RC, Bromet EJ, The epidemiology of depression across cultures. Annu. Rev. Public Health 34, 119–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yun S, Reynolds RP, Masiulis I, Eisch AJ, Re-evaluating the link between neuropsychiatric disorders and dysregulated adult neurogenesis. Nat. Med 22, 1239–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, Chang EF, Gutierrez AJ, Kriegstein AR, Mathern GW, Oldham MC, Huang EJ, Garcia-Verdugo JM, Yang Z, Alvarez-Buylla A, Human hippocampal neurogenesis drops sharply in children to undetectable levelsin adults. Nature 555, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, Hen R, Mann JJ, Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 22, 589–599.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagi Y, Medrihan L, George K, Barney M, McCabe KA, Greengard P, Emergenceof 5-HT5A signaling in parvalbumin neurons mediates delayed antidepressant action. Mol. Psychiatry 25, 1191–1201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, Enkavi G, Biojone C, Cannarozzo C, Sahu MP, Kaurinkoski K, Brunello CA, Steinzeig A, Winkel F, Patil S, Vestring S, Serchov T, Diniz CRAF, Laukkanen L, Cardon I, Antila H, Rog T, Piepponen TP, Bramham CR, Normann C, Lauri SE, Saarma M, Vattulainen I, Castrén E, Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313.e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao JL, Covington HE, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han MH, Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J. Neurosci 30, 16453–16458 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah MM, HCN1 channels: A new therapeutic target for depressive disorders? Sci. Signal 5, pe44 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Lyman KA, Han Y, Chetkovich DM, Animal models suggest the TRIP8b-HCN interaction is a therapeutic target for major depressive disorder. Expert Opin. Ther. Targets 21, 235–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ku SM, Han M-H, HCN channel targets for novel antidepressant treatment. Neurotherapeutics 14, 698–715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A, A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119, 719–732 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Maroso M, Szabo GG, Kim HK, Alexander A, Bui AD, Lee S-H, Lutz B, Soltesz I, Cannabinoid control of learning and memory through HCN channels. Neuron 89, 1059–1073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wahl-Schott C, Biel M, HCN channels: Structure, cellular regulation and physiological function. Cell. Mol. Life Sci 66, 470–494 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee C-H, MacKinnon R, Structures of the human HCN1 hyperpolarization-activated channel. Cell 168, 111–120.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis AS, Vaidya SP, Blaiss CA, Liu Z, Stoub TR, Brager DH, Chen X, Bender RA, Estep CM, Popov AB, Kang CE, Van Veldhoven PP, Bayliss DA, Nicholson DA, Powell CM, Johnston D, Chetkovich DM, Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J. Neurosci 31, 7424–7440 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yun S, Reynolds RP, Petrof I, White A, Rivera PD, Segev A, Gibson AD, Suarez M, DeSalle MJ, Ito N, Mukherjee S, Richardson DR, Kang CE, Ahrens-Nicklas RC, Soler I, Chetkovich DM, Kourrich SXD, Coulter DA, Eisch AJ, Stimulationof entorhinal cortex–dentate gyrus circuitry is antidepressive. Nat. Med 24, 658–666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim CS, Chang PY, Johnston D, Enhancement of dorsal hippocampal activity by knockdown of HCN1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron 75, 503–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CS, Brager DH, Johnston D, Perisomatic changes in h-channels regulate depressive behaviors following chronic unpredictable stress. Mol. Psychiatry 18, 892–903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher DW, Han Y, Lyman KA, Heuermann RJ, Bean LA, Ybarra N, Foote KM, Dong H, Nicholson DA, Chetkovich DM, HCN channels in the hippocampus regulate active coping behavior. J. Neurochem 146, 753–766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai H-C, Finkelstein J, Kim S-Y, Adhikari A, Thompson KR, Andalman AS, Gunaydin LA, Witten IB, Deisseroth K, Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493, 537–541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Notomi T, Shigemoto R, Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J. Comp. Neurol 471, 241–276 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z, Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat. Neurosci 5, 1185–1193 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Piskorowski R, Santoro B, Siegelbaum SA, TRIP8b splice forms act in concert to regulate the localization and expression of HCN1 channels in CA1 pyramidal neurons. Neuron 70, 495–509 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magee JC, Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat. Neurosci 2, 848 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Santoro B, Wainger BJ, Siegelbaum SA, Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J. Neurosci 24, 10750–10762 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bankston JR, Camp SS, DiMaio F, Lewis AS, Chetkovich DM, Zagotta WN, Structure and stoichiometry of an accessory subunit TRIP8b interactionwith hyperpolarization-activated cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. U.S.A 109, 7899–7904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA, TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channelsin the brain. Neuron 62, 802–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han Y, Heuermann RJ, Lyman KA, Fisher D, Ismail QA, Chetkovich DM, HCN-channel dendritic targeting requires bipartite interaction with TRIP8b and regulates antidepressant-like behavioral effects. Mol. Psychiatry 22, 458–465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, Surmeier DJ, Baram TZ, Macdonald RL, Chetkovich DM, Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J. Neurosci 29, 6250–6265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santoro B, Hu L, Liu H, Saponaro A, Pian P, Piskorowski RA, Moroni A, Siegelbaum SA, TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J. Neurosci 31, 4074–4086 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han Y, Noam Y, Lewis AS, Gallagher JJ, Wadman WJ, Baram TZ, Chetkovich DM, Trafficking and gating of hyperpolarization-activated cyclic nucleotide-gated channels are regulated by interaction with tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) and cyclic AMP at distinct sites. J. Biol. Chem 286, 20823–20834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Müller CS, Thumfart J-O, Vlachos A, Deller T, Pfeifer A, Fleischmann BK, Roeper J, Fakler B, Klöcker N, Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron 62, 814–825 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Hu L, Santoro B, Saponaro A, Liu H, Moroni A, Siegelbaum SA, Binding of the auxiliary subunit TRIP8b to HCN channels shifts the mode of action of cAMP. J. Gen. Physiol 142, 599–612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saponaro A, Pauleta SR, Cantini F, Matzapetakis M, Hammann C, Donadoni C, Hu L, Thiel G, Banci L, Santoro B, Moroni A, Structural basis for the mutual antagonismof cAMP and TRIP8b in regulating HCN channel function. Proc. Natl. Acad. Sci. U.S.A 111, 14577–14582 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyman KA, Han Y, Heuermann RJ, Cheng X, Kurz JE, Lyman RE, Van Veldhoven PP, Chetkovich DM, Allostery between two binding sites in the ion channel subunit TRIP8b confers binding specificity to HCN channels. J. Biol. Chem 292, 17718–17730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bankston JR, DeBerg HA, Stoll S, Zagotta WN, Mechanism for the inhibitionof the cAMP dependence of HCN ion channels by the auxiliary subunit TRIP8b. J. Biol. Chem 292, 17794–17803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saponaro A, Cantini F, Porro A, Bucchi A, DiFrancesco D, Maione V, Donadoni C,Introini B, Mesirca P, Mangoni ME, Thiel G, Banci L, Santoro B, Moroni A, A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. eLife 7, e35743 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nibuya M, Nestler EJ, Duman RS, Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci 16, 2365–2372 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blendy JA, The role of CREB in depression and antidepressant treatment. Biol. Psychiatry 59, 1144–1150 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Donati RJ, Dwivedi Y, Roberts RC, Conley RR, Pandey GN, Rasenick MM, Postmortem brain tissue of depressed suicides reveals increased Gs localization in lipid raft domains where it is less likely to activate adenylyl cyclase. J. Neurosci 28, 3042–3050 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM, NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 24, 1833–1843 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoffman DA, Johnston D, Downregulation of transient K+ channels in dendritesof hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J. Neurosci 18, 3521–3528 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guettier J-M, Gautam D, Scarselli M, de Azua IR, Li JH, Rosemond E, Ma X,Gonzalez FJ, Armbruster BN, Lu H, Roth BL, Wess J, A chemical-genetic approach to study G protein regulation of β cell function in vivo. Proc. Natl. Acad. Sci. U.S.A 106, 19197–19202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roth BL, DREADDs for neuroscientists. Neuron 89, 683–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen S, Wang J, Siegelbaum SA, Properties of hyperpolarization-activated pacemaker current defined by coassembly of Hcn1 and Hcn2 subunits and basal modulation by cyclic nucleotide. J. Gen. Physiol 117, 491–504 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammelmann V, Stieglitz MS, Hülle H, Le Meur K, Kass J, Brümmer M, Gruner C, Rötzer RD, Fenske S, Hartmann J, Zott B, Lüthi A, Spahn S, Moser M, Isbrandt D, Ludwig A, Konnerth A, Wahl-Schott C, Biel M, Abolishing cAMP sensitivity in HCN2 pacemaker channels induces generalized seizures. JCI Insight 4, e126418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I, Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancementof inhibition to hyperexcitability. Nat. Med 7, 331–337 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowley JJ, Blendy JA, Lucki I, Strain-dependent antidepressant-like effectsof citalopram in the mouse tail suspension test. Psychopharmacology 183, 257–264 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Lonze BE, Ginty DD, Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Carlezon W Jr., R. Duman, E. Nestler, The many faces of CREB. Trends Neurosci 28, 436–445 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Molteni R, Calabrese F, Cattaneo A, Mancini M, Gennarelli M, Racagni G, Riva MA, Acute stress responsiveness of the neurotrophin BDNF in the rat hippocampus is modulated by chronic treatment with the antidepressant duloxetine. Neuropsychopharmacology 34, 1523–1532 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Chen A, Shirayama Y, Shin KH, Neve RL, Duman RS, Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol. Psychiatry 49, 753–762 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Barco A, Alarcon JM, Kandel ER, Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108, 689–703 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, Russo SJ, LaPlant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ, Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Golden SA, Covington HE, Berton O, Russo SJ, A standardized protocol for repeated social defeat stress in mice. Nat. Protoc 6, 1183–1191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X-D, Chen Y, Wolf M, Wagner KV, Liebl C, Scharf SH, Harbich D, Mayer B, Wurst W, Holsboer F, Deussing JM, Baram TZ, Muller MB, Schmidt MV, Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol. Dis 42, 300–310 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP, Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J. Neurosci 36, 2590–2604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Czysz AH, Schappi JM, Rasenick MM, Lateral diffusion of Gαs in the plasma membrane is decreased after chronic but not acute antidepressant treatment: Role of lipid raft and non-raft membrane microdomains. Neuropsychopharmacology 40, 766–773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujita M, Richards EM, Niciu MJ, Ionescu DF, Zoghbi SS, Hong J, Telu S, Hines CS, Pike VW, Zarate CA, Innis RB, cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol. Psychiatry 22, 754–759 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belmaker RH, Agam G, Major depressive disorder. N. Engl. J. Med 358, 55–68 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Senese NB, Rasenick MM, Traynor JR, The role of G-proteins and G-protein regulating proteins in depressive disorders. Front. Pharmacol 9, 1289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nestler EJ, Hyman SE, Animal models of neuropsychiatric disorders. Nat. Neurosci 13, 1161–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han Y, Lyman KA, Foote KM, Chetkovich DM, The structure and function of TRIP8b, an auxiliary subunit of hyperpolarization-activated cyclic-nucleotide gated channels. Channels 14, 110–122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson RB, Siegelbaum SA, Hyperpolarization-activated cation currents: From molecules to physiological function. Annu. Rev. Physiol 65, 453–480 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Duman RS, Aghajanian GK, Synaptic dysfunction in depression: Potential therapeutic targets. Science 338, 68–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X, An excitatory synapse hypothesis of depression. Trends Neurosci 38, 279–294 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kallarackal AJ, Kvarta MD, Cammarata E, Jaberi L, Cai X, Bailey AM, Thompson SM, Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J. Neurosci 33, 15669–15674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kvarta MD, Bradbrook KE, Dantrassy HM, Bailey AM, Thompson SM, Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol 114, 1713–1724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Racagni G, Popoli M, Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin. Neurosci 10, 385–400 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berumen LC, Rodríguez A, Miledi R, García-Alcocer G, Serotonin receptors in hippocampus. ScientificWorldJournal 2012, 1–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gasparini S, DiFrancesco D, Action of serotonin on the hyperpolarization-activated cation current (Ih) in rat CA1 hippocampal neurons. Eur. J. Neurosci 11, 3093–3100 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Bickmeyer U, Heine M, Manzke T, Richter DW, Differential modulation of Ih by 5-HT receptors in mouse CA1 hippocampal neurons. Eur. J. Neurosci 16, 209–218 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, Li X, Dietz DM, Pan N, Vialou VF, Neve RL, Yue Z, Han M-H, Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science 18, 313–319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhong P, Vickstrom C, Liu X, Hu Y, Yu L, Yu H-G, Liu Q-S, HCN2 channels in the ventral tegmental area regulate behavioral responses to chronic stress. eLife 7, e32420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng J, Umschweif G, Leung J, Sagi Y, Greengard P, HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron 101, 662–672.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Han Y, Lyman K, Clutter M, Schiltz GE, Ismail QA, Prados DB, Luan CH, Chetkovich DM, Identification of small-molecule inhibitors of hyperpolarization-activated cyclic nucleotide-gated channels. J. Biomol. Screen 20, 1124–1131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lovett-Barron M, Andalman AS, Allen WE, Vesuna S, Kauvar I, Burns VM, Deisseroth K, Ancestral circuits for the coordinated modulation of brain state. Cell 171, 1411–1423.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wobbrock JO, Findlater L, Gergle D, Higgins JJ, The aligned rank transformfor nonparametric factorial analyses using only ANOVA procedures. Proc. SIGCHI Conf. Hum. Factors Comput. Syst 11, 143–146 (2011). [Google Scholar]
- 85.Vogel-Ciernia A, Wood MA, Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci 69, 8.31.1–8.31.17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou C, Huang Z, Ding L, Deel ME, Arain FM, Murray CR, Patel RS, Flanagan CD, Gallagher MJ, Altered cortical GABAA receptor composition, physiology,and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J. Biol. Chem 288, 21458–21472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou C, Ding L, Deel ME, Ferrick EA, Emeson RB, Gallagher MJ, Altered intrathalamic GABAA neurotransmission in a mouse model of a human genetic absence epilepsy syndrome. Neurobiol. Dis 73, 407–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shin M, Simkin D, Suyeoka GM, Chetkovich DM, Evaluation of HCN2 abnormalitiesas a cause of juvenile audiogenic seizures in Black Swiss mice. Brain Res 1083, 14–20 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Shin M, Chetkovich DM, Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J. Biol. Chem 282, 33168–33180 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chung WK, Shin M, Jaramillo TC, Leibel RL, LeDuc CA, Fischer SG, Tzilianos E, Gheith AA, Lewis AS, Chetkovich DM, Absence epilepsy in apathetic, a spontaneous mutant mouse lacking the h channel subunit, HCN2. Neurobiol. Dis 33, 499–508 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spandidos A, Wang X, Wang H, Seed B, PrimerBank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38, D792–D799 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X, Spandidos A, Wang H, Seed B, PrimerBank: A PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–D1149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.