

Abstract
Objective:
Rheumatoid arthritis synovial fibroblasts (RASFs) are crucial mediators of synovial inflammation and joint destruction. However, their intrinsic immunoregulatory mechanisms under chronic inflammation remain unclear. Thus, present study was aimed to understand the role of newly identified GTPase, guanylate-binding protein 5 (GBP-5) in RA pathogenesis.
Methods:
The expression of GBP1-GBP7 transcripts was evaluated using qRT-PCR in RA synovial tissues (RASTs) or non-diseased STs (NLSTs). Transient siRNA knockdown and lentiviral overexpression studies in human RASFs examined the regulatory role of GBP-5 on proinflammatory cytokine signaling pathways. Unbiased whole transcriptome RNA-sequencing analysis examined the impact of GBP-5 on RASF molecular functions. These findings were confirmed using in vivo rat model of adjuvant-induced arthritis (AIA).
Results:
Among different GBPs evaluated, GBP-5 was selectively upregulated in RASTs (p<0.05; n=4) and the joints of AIA rats (p<0.05; n=6), and was significantly induced in human RASFs by IL-1β, TNF-α and/or IFN-γ (p<0.05; n=3). Bioinformatics analysis of RNA-sequencing data identified cytokine-cytokine receptor signaling as a major function altered by GBP-5, with IL-6 signaling as a primary target. Knockdown of GBP-5 amplified IL-1β-induced IL-6, IL-8, ENA-78/ CXCL5 by 44%, 54%, 45%, respectively, and MMP-1 production by several-folds, effects which reversed with exogenously delivered GBP-5. Lack of GBP-5 increased IFN-γ-induced proliferation and migration in human RASFs. In vivo GBP-5 knockdown using intra-articular siRNA exacerbated disease onset, severity, synovitis, and bone destruction in AIA.
Conclusion:
Expressed by RASFs in response to cytokine stimulation, GBP-5 has potential to restore cellular homeostasis and blunt inflammation and tissue destruction in RA.
Keywords: Guanylate binding protein 5 (GBP-5), interleukin-1β, synovial fibroblasts, rheumatoid arthritis
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disease of joints in which macrophages, B and T cells, and neutrophils infiltrate the joints and activate synovial fibroblasts (SFs), to cause inflammation and tissue destruction (1, 2). Activated RA synovial fibroblasts (RASFs) play an important role in the perpetuation of local inflammation and tissue destruction (1). Human RASFs produce high levels of IL-6, IL-8, CXC or CC chemokines, and tissue-degrading enzymes such as matrix metalloproteinases (MMPs) in response to inflammatory cytokines such as interleukin-beta (IL-1β) or tumor necrosis factor-alpha (TNF-α) (3–7), making them an attractive target for RA therapies. Since RASFs play a significant role in amplifying inflammation and tissue destruction, there is a paramount therapeutic need to identify endogenous proteins that can regulate RASF functions to limit their contributions to disease pathogenesis. Yet very little is known about the auto-regulatory or anti-inflammatory factors produced by RASFs that play a protective role by an autocrine regulation of this vicious inflammatory cycle. More research is needed to determine whether such factors could restore cellular homeostasis and blunt proinflammatory actions of the cytokines that contribute to RA pathogenesis.
Recent studies provide evidence that a newly-discovered family of IFN-stimulated GTPases (ISGs) called ‘guanylate-binding proteins’ (GBPs) participate centrally in inflammasome activation and host-defense mechanisms (8). The GBP family comprises 7 members (GBP1–7) that are expressed in human cells and are highly responsive to IFN stimulation (8). Besides their function in immunity (9), studies suggest that they regulate cellular proliferation, angiogenesis, and tissue invasion (10–12). Among the GBP family members, GBP-5 has recently been shown to play a crucial role in immunity against infections through NLRP3 inflammasome activation in macrophages (13–15).
Various studies utilizing mass cytometry coupled single-cell transcriptomics and global transcriptome profiling of RASFs have suggested an increase in the expression of different GBPs, including GBP1, GBP2, GBP4, and GBP5 (16, 17), however, their role has never been characterized in RA. In corroboration to these profiling studies, preliminary findings in our lab from RNA sequencing showed GBP-5 to be one of the most highly upregulated genes in RA synovial tissue (RASTs) compared with non-diseased synovial tissues (NLSTs). In the present study, we examined the role GBP-5 in RASF-mediated synovial inflammation and tissue destruction in vitro and in a rat model of adjuvant-induced arthritis (AIA).
MATERIALS AND METHODS
Detailed information about antibodies and reagents, Western immunoblotting, ELISA, and in vitro scratch test are provided in the SI Materials and Methods.
Animals.
Female Lewis rats 6–8 weeks old were purchased from Envigo (East Millstone, NJ). All work was performed at an AAALAC-accredited facility that meets the standards set forth in the Guide for the Care and Use of Laboratory Animals. Rats were housed in pathogen-free conditions on corn cob bedding in plastic microisolator cages on ventilated racks. Animals were maintained on a 12:12h light:dark cycle at 70 ± 2°F and 50 ± 5% relative humidity. Cages were changed weekly. Animals were maintained on a nutritionally complete diet (Teklad/Envigo, Madison, WI) and tap water (in water bottles) ad libitum.
Isolation and culture of SFs from the synovial tissue of human subjects not affected by RA and RA subjects.
The de-identified non-diseased NL and RA synovial tissues (RASTs) were procured under a protocol approved by the Washington State University IRB (IRB#14696) from Cooperative Human Tissue Network (Columbus, OH) and National Disease Research Interchange (Philadelphia, PA). STs from RA patients were obtained from joint surgery or synovectomy according to an IRB protocol and in compliance with the Declaration of Helsinki. NLSTs from were obtained from autopsy or amputation. ST from 6 RA patients were used in the current study (Supplementary (S) table 1, Table S1). RASFs and NLSFs were processed as described earlier (18).
Treatment of RASFs.
Human RASFs were treated with recombinant IL-1 β (10 ng/ml) or TNF-α (20 ng/ml) with or without IFN-γ (10 ng/ml) for 24 h. The expression level of GBP-5 was determined by qRT-PCR and Western immunoblotting methods. Conditioned media was used to determine IL-6, IL-8, MMP-1, and ENA-78/CXCL5 production by ELISA. To understand the signaling mechanism, human RASFs were treated with IL-1β alone or in combination with IFN-γ for 30 min. Whole cell lysates were prepared and used for the analysis of the phosphorylated proteins (p-JNK, p-ERK, p-p38, p-c-Jun, p-NF-κBp65). All blots were re-probed for β-actin to confirm equal loading of proteins. RASFs were seeded in 96-well plates and stimulated with IL-1β or IFN-γ for 24 h to evaluate the effect on cell viability by MTT cell viability assay.
Transient Transfection of siRNA.
To study the effects of GBP-5 knockdown, RASFs were transfected with 120 pmoles of scrambled control (NC) or GBP-5 siRNA using Lipofectamine® 2000 (Life Technologies) for 48 h and then stimulated with IFN-γ and/or IL-1β for 24 h in 6-well format. Conditioned media was used to study IL-6, IL-8, ENA-78/CXCL5, MMP-1, RANTES/CCL5, MIP-1α/CCL2, and MCP-1/CXCL1 production using ELISA method.
RNA extraction and qRT-PCR.
Total RNA was reverse-transcribed using the SuperScript™ First-Strand Synthesis kit (Life Technologies) according to the manufacturer instructions. GBP-5 mRNA transcripts were amplified using the Power SYBR® Green PCR master mix (Life Technologies) and validated primers from the QuantiTect primer assay (Qiagen) (sequence of the primer pairs are provided in Table S2). Quantification of the relative expression of IL-6, IL-8, MMP-1, MMP-3, MMP-12, ENA-78/CXCL5, CCL8, CXCL10, and CCL20 was determined by the ΔΔCt method using GAPDH expression as an endogenous control.
RNA library preparation, sequencing, and bioinformatics analysis.
Detailed method of RNA preparation, sequencing and bioinformatics analysis was performed as described earlier (19) and is provided with experimental details in the SI Materials and Methods.
GBP-5 plasmid transformation and purification.
The validated and sequence confirmed GBP-5 wild-type (Catalog no. HG14977-ANG) plasmid was purchased from Sino biological (Wayne, PA). The bacterial transformation and plasmid purification were performed as per manufacturers’ instructions (Qiagen).
Lentiviral overexpression and transduction protocol.
Lentivirus containing empty vector and GBP-5 overexpression plasmid were purchased from Origene (Rockville, MD). When 70–80% confluent in 24-well plates, RASFs were transduced with culture media containing GBP-5 LentiORF particle and Polybrene (8 mg/ml). Vector control LentiORF particle (Origene, Cat. No. PS100071V) was used as a negative control. RASFs were infected with 3 multiplicity of infection (MOI) of the lentiviral particles. After 18 h, the new culture media was added. After 48 h, RASFs were serum-starved overnight, followed by stimulation with IL-1β (10 ng/ml) for 24 h. Whole cell lysates were collected to determine GBP-5 expression by the Western immunoblotting method. Conditioned media were collected to quantitate the levels of IL-6, IL-8, MMP-1, and ENA-78/CXCL5.
Rat adjuvant-induced arthritis (AIA).
Rat AIA study was performed as previously described (3, 7, 18). Briefly, female Lewis rats weighing ~130g were injected s.c. at the base of the tail with 300 μl (5 mg/ml stock) of lyophilized Mycobacterium butyricum (Difco Laboratories, Detroit, MI) in sterile mineral oil. The rats were divided into two groups. Group 1 (n=6) received NC siRNA (10 μg in 10 μl atelocollagen) and group 2 (n=6) received GBP-5 siRNA (10 μg in 10 μl atelocollagen) intra-articularly in each ankle on days 7 and 12 (onset of arthritis) and monitored for clinical measurements until day 17. Clinical parameters such as articular index (AI) and ankle circumferences (AC) were measured. AI and AC scores were recorded for each hind joint by a blinded observer and then averaged for each animal described previously (18). Joints and serum samples were collected on day 17. All animal studies were approved by the ethics committee of the Washington State University and conformed to the NIH Guide for the Care the Use of Laboratory Animals (8th edition, 2011).
Micro-computed tomography (μ-CT) Scanning and bone analysis.
Rat ankles fixed in formalin were scanned using the Quantum GX micro-CT Imaging System (Perkin Elmer Waltham, MA). The system acquired the image using 90 kV level and 88 mA with a field of view of 10 mm with a scanning time of 4 min (voxel size 20 μm). The bone marker analysis was performed by standardizing images to QRM-MicroCT-HA phantom (QRM Moehrendorf, Germany). The structural parameters for bone were analyzed with Analyze 12.0 software (AnalyzeDirect, Overland Park, KS, USA). Bone volume, cortex volume, trabecular volume, intra trabecular volume, trabecular tissue volume, and trabecular tissue mean (BMD), were calculated using the region of interest (ROI) tool as described earlier (18).
Histological analysis.
Histological analysis was performed as described previously (20). In brief, harvested joints were decalcified with 10% EDTA for 21 days and then embedded with paraffin. The centerline of the joint was cut sagittally to make 5 μm slices. Sections from NC and GBP-5 siRNA groups were stained with hematoxylin and eosin (H&E). Infiltrating cell and bone destruction were measured with the Leica DM2500 microscope. A pathologist blinded to the study analyzed the slides, inflammatory markers were compared among two groups and presented in Table S3.
Statistical analysis.
A Kruskal-Wallis nonparametric test was used to evaluate the statistical significance of group differences in the protein expression or production from Western blotting or ELISA results in RASFs and different measured parameters obtained from in vivo study. Student’s t-test was performed to calculate statistical differences between variables for bioinformatics analysis. Values shown are Mean ± SE unless stated otherwise. P <0.05 was considered significant.
RESULTS
Significantly enhanced expression of GBP- 5 in RA- affected synovial tissue and induction of GBP- 5 upon cytokine stimulation in human RASFs.
Our RNA-seq data from Illumina identified GBP5 as one of the most highly expressed genes in human RA synovial tissues (RASTs) compared to non-diseased STs (NLSTs) (Figure S1A). Therefore, we profiled the mRNA expression of GBP1–7 in the RASTs and compared to NLST. Among all the hGBPs analyzed, a significant 5-fold increase in GBP5 mRNA expression was observed in RASTs compared to the NLSTs (Figure 1A; p<0.05). Furthermore, densitometric analysis of Western blots showed a similar increase in the protein expression of GBP-5 (Figure 1B, p<0.05). Evaluation of the joint homogenates from naïve and AIA rats also showed ~2-fold increase in GBP-5 expression in AIA joints (Figure 1B, p<0.05). To confirm this in vitro, we stimulated RASFs with IL-1β (10 ng/ml) for up to 48 h to observe a time-dependent increase in GBP-5 expression that peaked around 24 h (Figure 1C; p<0.05 at 24 h). To test if IL-1β is capable of inducing GBP-5 for longer duration and in NLSFs as well, we stimulated RASFs and NLSFs with IL-1β (10 ng/ml) for 24, 48, and 72 h. Analysis of the cell lysates showed that IL-1β induced GBP-5 expression in NLSFs similar to RASFs and the expression levels declined to almost basal values by 72 h of treatment in both groups without affecting the cell viability (Figure S1B–C).
Figure 1:
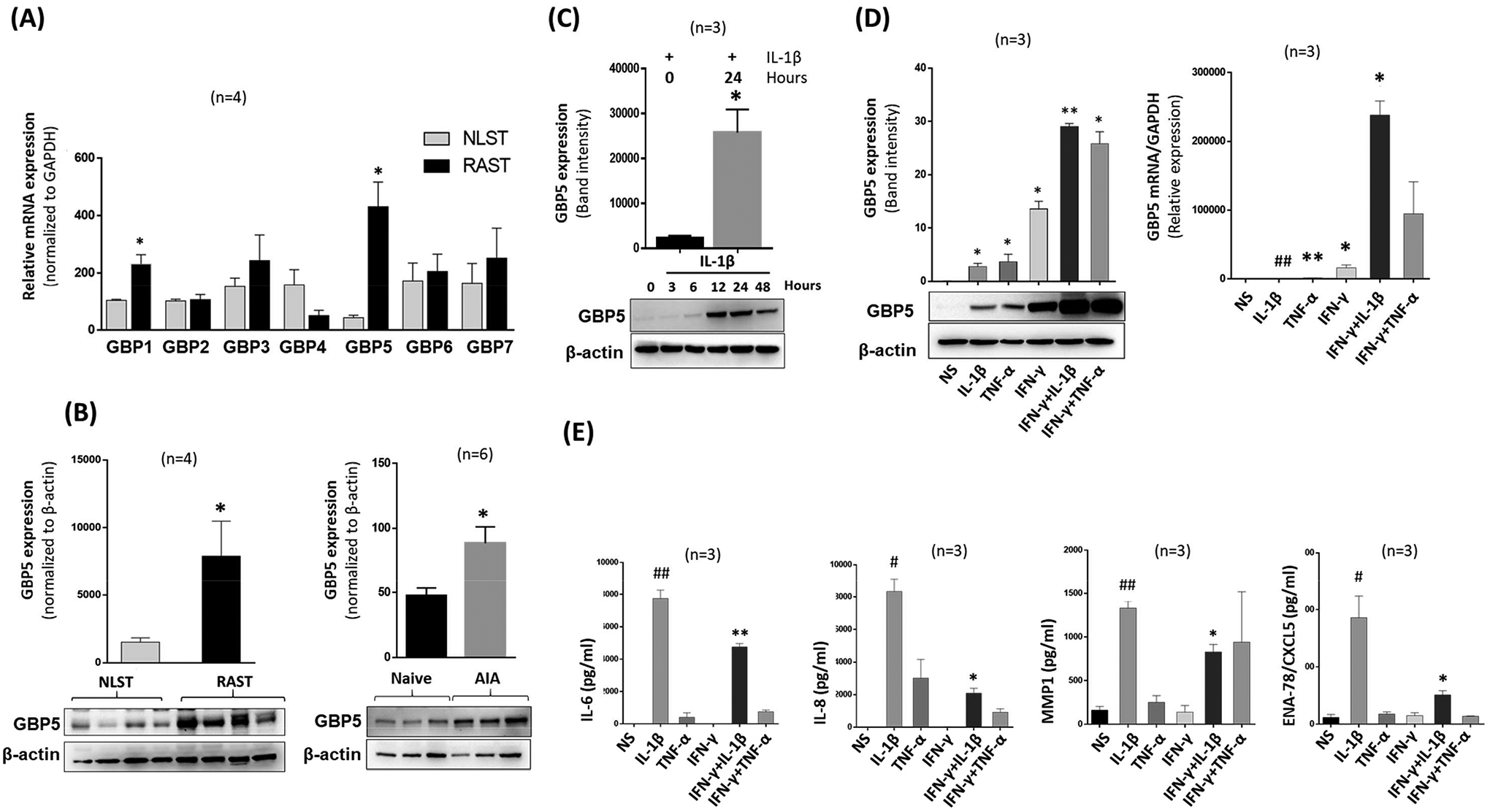
Expression of GBP-5 is significantly higher in RA and IFN-γ preferentially induces a robust GBP-5 expression to suppress IL-1β-induced chemokine production in RASFs. A, qRT-PCR analysis of human RASTs and NLSTs showed a significant increase in GBP5 expression in RASTs compared to NLSTs. B, Expression of GBP-5 protein is observed in RASTs when compared to NLST levels. The expression of GBP-5 at the protein level is upregulated in the joint homogenates of AIA rats compared to naïve rats. C, IL-1β induces time-dependent expression of GBP-5 protein that peaks around 24 h. D, IL-1β (10 ng/ml) or TNF-α (20 ng/ml) induce GBP-5 protein and mRNA expression which is further amplified by IFN-γ. E, IFN-γ significantly downregulates IL-1β-induced IL-6, IL-8, MMP1, and ENA-78/CXCL5 production in RASFs. The results are presented as mean ±SEM. #p<0.05 or ##p<0.01 vs NS; *p<0.05 or **p<0.01 vs IL-1β.
GBP-5 is an ISG robustly induced in response to IFN-γ in different cell types (11, 13, 14). However, its expression in RASFs activated by proinflammatory cytokines in RA pathogenesis has remained untested. Activation of RASFs with IL-1β (10 ng/ml) or TNF-α (20 ng/ml) alone or in combination with IFN-γ (10 ng/ml) for 24 h confirmed an upregulation in the expression of GBP-5. The combination of IFN-γ with IL-1β or TNF-α further amplified the expression of GBP-5 protein compared to IFN-γ alone (Figure 1D, p<0.01) and produced a 10-fold increase in mRNA level (Figure 1D, p<0.05). However, anti-inflammatory cytokines such as IL-10 (1–100 ng/ml) or IL-35 (1–100 ng/ml) failed to induce GBP-5 expression on RASFs (Figure S1E). Evaluation of the conditioned media from RASFs treated with IL-1β or TNF-α alone or in combination with IFN-γ for 24 h showed that IFN-γ was able to significantly reduce the levels of IL-1β-induced IL-6, IL-8, MMP-1, and ENA-78/CXCL5 production by 38%, 74%, 37%, and 72%, respectively (Figures 1E, p<0.05).
Identification by RNA- Seq analysis of a novel anti-inflammatory role of GBP- 5 mediated through modulation of cytokine- cytokine receptor interactions.
To determine the role of GBP-5 in IL-1β-induced inflammation, we used a loss-of-function model by performing RNA-seq analysis on IL-1β-stimulated RASFs with/without GBP-5 transiently knocked down. Among the panel of 20,803 genes, the expression of 3,315 genes was significantly modulated ≥ 2.5-fold upon IL-1β stimulation in RASFs, as depicted in the heatmap (Figure 2A). Further analysis of the data showed that of these modulated genes, 1353 genes were significantly downregulated, and 825 genes were significantly upregulated with GBP-5 knockdown (Figure 2A).
Figure 2:
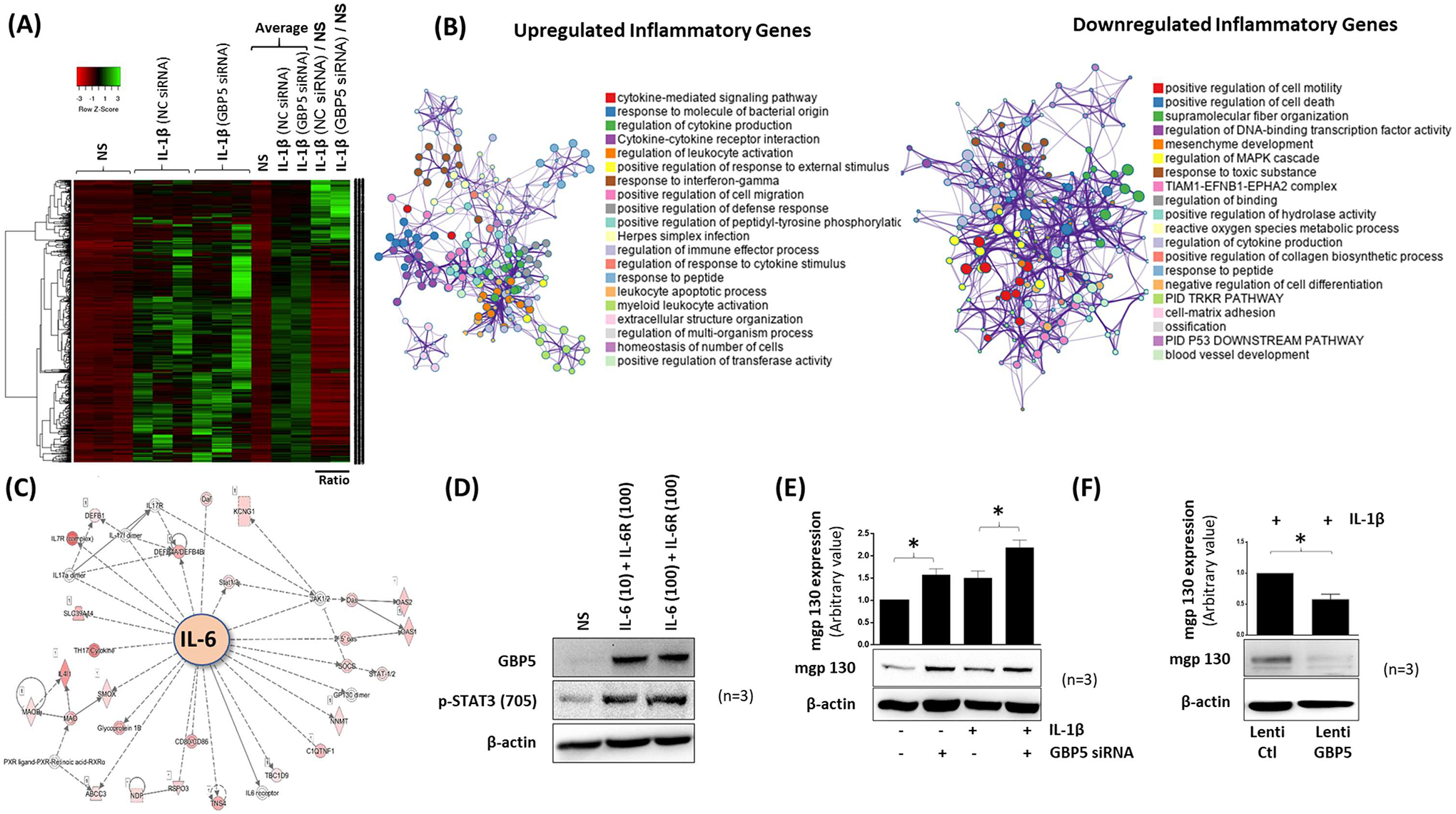
Loss of GBP-5 exacerbates IL-1β-induced inflammatory and tissue destructive genes in human RAST. A, A heat map of 3315 differentially modulated genes is presented based on t-test qualification. Red represents lower expression; green represents higher expression. B, Network of enriched GO terms for upregulated and downregulated genes where the node sizes represent the proportion of the number of the gene into a specific term. Different colors were selected for cluster ID. C, IPA deciphers the role of GBP-5 in IL-6 signaling pathway. D, Co-stimulation of human RASFs with IL-6 (10 or 100 ng/ml) and IL-6R (100 ng/ml) upregulates the expression of GBP-5 and p-STAT3 (Tyr705). E, mgp130 is upregulated by IL-1β stimulation for 24 h and further augmented upon GBP-5 knockdown in both constitutive and IL-β-treated RASFs. F, Overexpression of GBP-5 was achieved by lentiviral delivery method followed by IL-1β stimulation for 24 h. Exogenous GBP-5 reduced the expression of mgp130 in human RASFs. The values presented in the graphs are mean ±SEM of three independent experiments. *p<0.05 NC versus siRNA treatment.
GO analysis of the Differentially Expressed Genes (DEGs) using Metascape showed that knockdown of GBP-5 primarily modulates cytokine-mediated signaling pathways (Figure S2A). In corroboration with this observation, the list of top upregulated inflammatory genes confirmed the previous results (Figure 2B). GO studies using ToppGene Suite analysis of the upregulated genes predicted the role of GBP-5 in regulating top RASF molecular functions such as cytokine/chemokine activity and their signaling receptor binding (Figure S2B) and affected pathways such as cytokine signaling in the immune system, interferon signaling, and cytokine-cytokine receptor signaling (Figure S2C). Functional network and canonical pathway analysis of upregulated genes predicted the top biological function affected by GBP-5 to include response to cytokine, defense response, immune response, response to biotic stimulus, and cellular response to cytokine stimulus (Figure S2D). The top affected cellular component function included extracellular space, cell surface, extracellular matrix, IκB/NF-κB complex, and integral component of the plasma membrane (Figure S2E). Notably, GO analysis of the downregulated genes showed regulation of cell motility as an important cellular function affected (Figure 2B).
Among top IL-1β-activated genes that further amplified in the absence of GBP-5 included those that govern inflammation (IL-6 and IL-8), chemotaxis (CXCL1, CXCL2, CXCL3, CXCL5, CXCL10, CCL7, CCL8, CCL20, and CCL3), and tissue remodeling (MMP-1, MMP-3, and MMP-12) (Figure S2F). Further validation of the RNA-seq data by qRT-PCR showed a significant increase in IL-1β-induced MMP3, MMP12, CCL8, CXCL10, and CCL20 mRNA expression with the knockdown of GBP-5 compared to the NC group (Figure S2G, p<0.05 for all).
Interestingly, the interactome generated from Ingenuity Pathway Analysis (IPA) showed IL-6 signaling to be centrally affected by GBP-5 knockdown in IL-1β-stimulated RASFs (Figure 2C). To validate this IPA finding, we stimulated RASFs with IL-6/IL-6R for 24 h. Western blotting results showed that IL-6/IL-6R induced GBP-5 expression along with p-STAT3(Tyr705) expression in RASFs (Figure 2D). Interestingly, knockdown of GBP-5 in RASFs upregulated both the constitutive as well as IL-1β-induced expression of membrane-bound gp130 (mgp130), a key transmembrane receptor in IL-6 signaling (3) (Figure 2E, p<0.05). Furthermore, overexpression of GBP-5 using a lentiviral delivery method significantly reduced IL-1β-induced mgp130 expression in RASFs (Figure 2F, p<0.05).
Significantly higher levels of soluble mediators of inflammation and cartilage destruction induced by IL- 1β in the absence of GBP- 5.
We knocked down GBP-5 before activating RASFs with IL-1β, TNF- α alone or in combination with IFN-γ for 24 h. The results of ELISAs showed that, compared to NC siRNA, GBP-5 knockdown significantly reduced the ability of IFN-γ to inhibit IL-1β-induced inflammation, as GBP-5 knockdown further amplified IL-6 and IL-8 production by 84% and 39%, respectively (Figures 3A, p<0.05 for IL-6). This suggests that IFN-γ primarily relies on GBP-5 to inhibit IL-1-induced inflammatory mediators. Strikingly, GBP-5 knockdown in IL-1β-stimulated RASFs resulted in ~8-fold increase in MMP-1 and ~43% increase in ENA-78/CXCL5 production compared to the levels in NC siRNA group (Figures 3B, p<0.05 and p<0.01).
Figure 3:
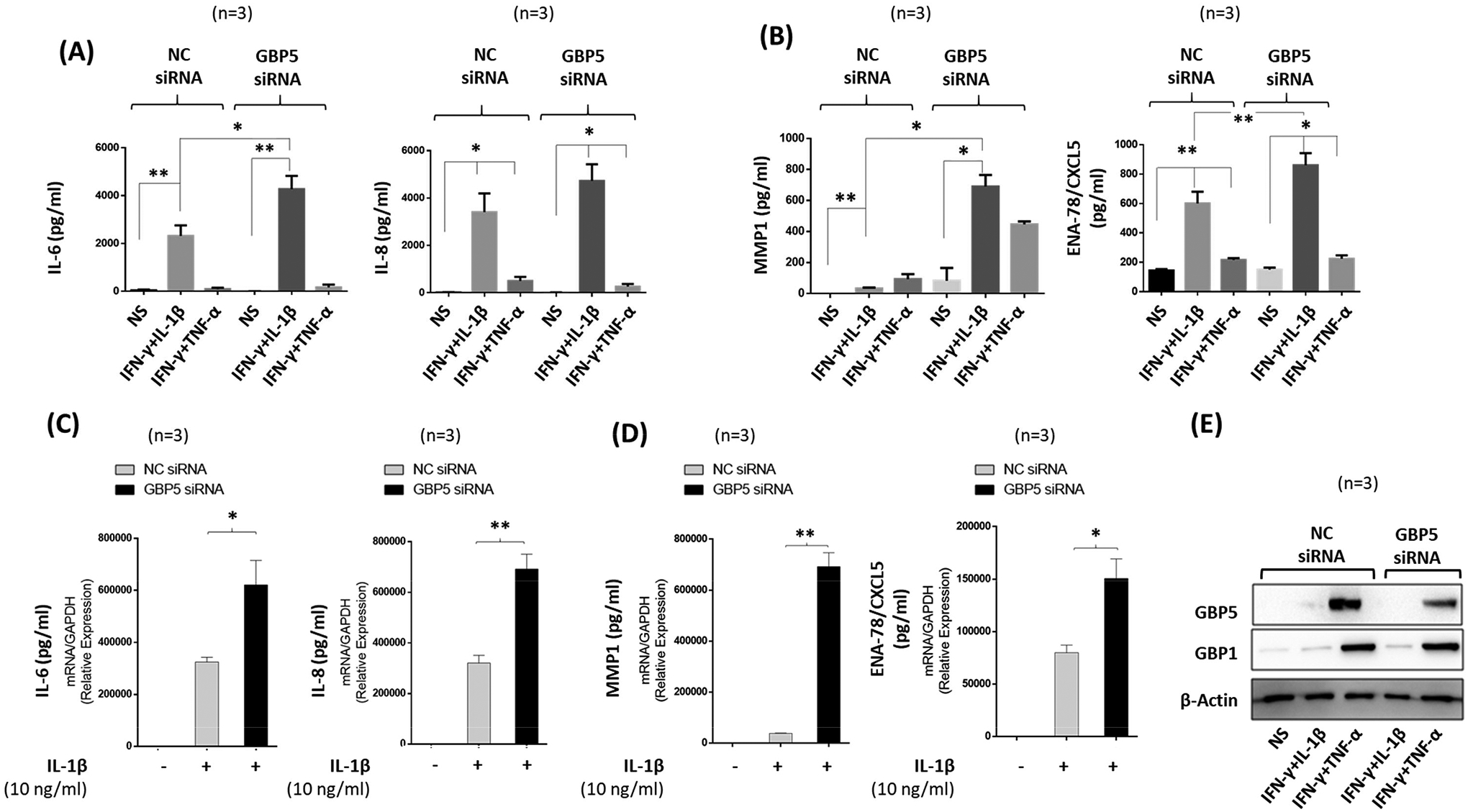
Knockdown of GBP-5 by transient transfection causes loss of IFN-γ protection and further amplifies IL-1β-induced inflammation. A-B, Human RASFs were transfected with scrambled (NC) or GBP-5 siRNA (120 pM) for 48 h followed by serum starvation and stimulation with IFN-γ (10 ng/ml) and IL-1β (10 ng/ml) or TNF-α (20 ng/ml) for 24 h. Knockdown of GBP-5 nullified the effect of IFN-γ and upregulated IL-1β–induced IL-6, IL-8, MMP-1, and ENA-78/CXCL5 production. C-D, RASFs stimulated with IL-1β alone also showed significant increase of IL-6, IL-8, MMP-1, and ENA-78/ CXCL5 mRNA expression in GBP-5 knockdown samples compared to NC samples. E, Western immunoblotting showing the efficiency of GBP-5 knockdown in human RASFs. The values presented in the graphs are mean ±SEM of three independent experiments. *p<0.05 or **p<0.01 vs NS; *p<0.05 or **p<0.01 for NC versus siRNA treatment.
In addition, our results at the transcriptional level showed that the lack of GBP-5 further increased IL-1β-induced IL-6 and IL-8 gene expression by 40–50% (Figures 3C, p<0.05 and p<0.01) We also found a 4-fold increase in IL-1β-induced MMP-1 and ~2-fold increase in ENA-78/CXCL5 production in GBP-5 siRNA-treated samples compared to the NC siRNA group (Figures 3D, p<0.05 and p<0.01). Several additional inflammatory genes identified in IPA were confirmed by qRT-PCR method (Figure S2F). Successful knockdown of GBP-5 was confirmed by Western immunoblotting (Figure 3E). Furthermore, knockdown of GBP-5 in human dermal fibroblasts also resulted in a significant upregulation of IL-1β-induced IL-6 and IL-8 production (Figure S3A–B, p<0.05, p<0.01), suggesting that GBP-5 elicits its anti-inflammatory actions in fibroblasts of other origin as well.
Suppression of inflammation and amplification of the function of IFNγ.
To evaluate if preconditionting of RASFs with GBP-5 blunts inflammatory pathways, cells were transfected with control or lentiviral GBP-5 overexpression vector, followed by IL-1β (10 ng/ml) stimulation for 24 h. The transduction success was confirmed using Western immunoblotting (Figure 4A). Overexpression of GBP-5 resulted in 40%, 43%, and 17% reduction in IL-1β-induced IL-6, IL-8, and ENA-78/CXCL5 production, respectively (Figure 4B, p<0.01), with no significant modulation in RANTES/CCL5, MCP-1/CXCL1, and MIP-1α/CCL2 production (Figure S4A–C). Since MAPK signaling is important in IL-1β-driven RA pathogenesis (21) , we tested the impact of GBP-5 overexpression on IL-1β-induced MAPKs in RASFs. Western immunoblot analysis of lysates treated with IL-1β after GBP-5 overexpression revealed marked inhibition of IL-1β-induced phosphorylation of p38 and JNK, thereby downregulating the activation of transcription factors p-NF-κBp65 and p-c-Jun (Figure 4C). To further test the potential combinatorial effects of GBP-5 and IFN-γ, we transiently overexpressed GBP-5 and treated RASFs with IFN-γ (0.5–10 μg/ml) followed by IL-1β stimulation for 24 h. Intriguingly, in the presence of GBP-5, the ability of IFN-γ to inhibit IL-1β-induced IL-6, IL-8, and ENA-78/CXCL5 production was enhanced when compared to IFN-γ treatment alone at the respective concentrations (Figure 4 D–F, p<0.05). These results suggest that GBP-5 is not only able to reduce inflammatory mediators, but also adds a combinational effect to IFN-γ’s function in human RASFs.
Figure 4:
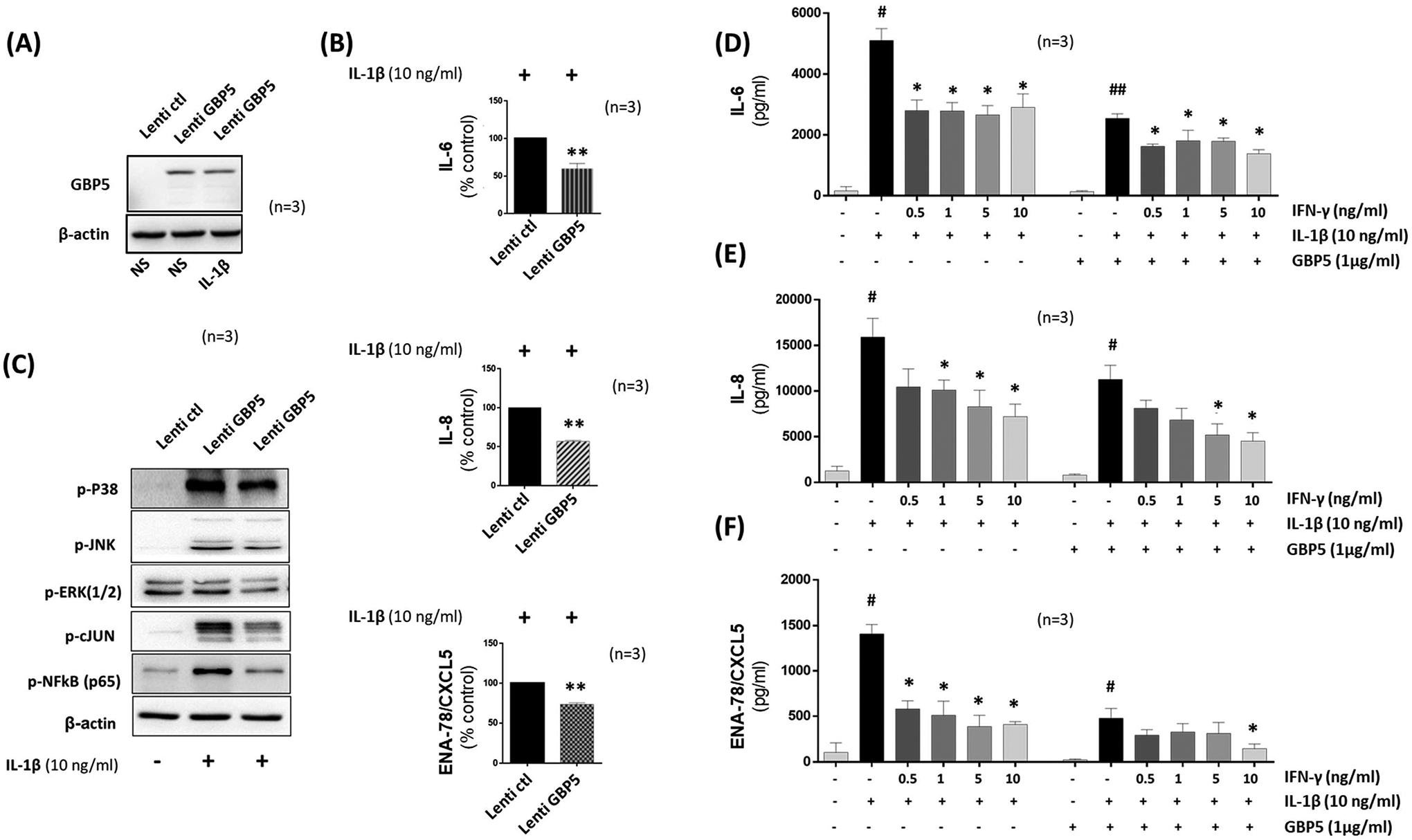
Exogenously overexpressed GBP-5 abrogates IL-1β-induced chemokine production and adds to the protective action of IFN-γ in human RASFs. Human RASFs were transduced with control (NC) and GBP-5 lentiviral particles at 3MOI for overnight. After 48 h, cells were treated with IL-1β or IFN-γ for 24 h. Whole Cell extracts were collected to determine the expression of GBP-5. A, Western immunoblotting showing the exogenous expression of GBP-5 overexpression. B, The ELISA result showed significant reduction in IL-1β-induced IL-6, IL-8, and ENA-78/CXCL5 production. C, Human RASFs were transduced as described in (A) and treated with IL-1β for 30 min. Western immunoblotting analysis showed a marked decreased in the expression of IL-1β-induced level of p-c-Jun, p-NF-κBp65, p-JNK, p-ERK1/2, and p-P38. β-actin was tested as a loading control. D-F, Human RASFs were transfected with empty vector or GBP-5 plasmid (1μg/ml) in 12-well plates for 48 h. Cells were treated with IL-1β (10 ng/ml) or IL-1β and IFN-γ at the indicated concentration for 24 h. Conditioned media was collected to determine the level of IL-6, and IL-8, and ENA-78/CXCL5 production. *p<0.05 or **p<0.01 vs NC; #p<0.05 or ##p<0.01 vs NS; *p<0.05 or **p<0.01 vs IL-1β.
Propagation of migration, inflammation, and tissue destruction following GBP- 5 knockdown in RASFs.
Since GBP-5 is classified as an ISG (22), it is crucial to understand its broader regulatory role in RASFs when both IFN-γ and IL-1β pathways are activated. First, we performed RNA-seq on IL-1β and/or IFN-γ treated RASFs to confirm that IFN-γ suppresses IL-1β-induced inflammatory pathways. Our results presented as a heatmap in Figure S5A showed a marked regulation by IFN-γ of 1350 t-test qualified genes activated by IL-1β. IPA data revealed key pathways affected by IFN-γ (Figure S5B). Next, we knocked down GBP-5 in IL-1β+IFN-γ treated RASFs and compared with NC siRNA control. RNA-seq analysis after t-test qualification identified 269 genes differentially regulated by GBP-5 siRNA compared to the NC siRNA group (Figure 5A). Follow-up IPA using these differentially regulated genes identified pathways, including IL-1 signaling, PI3K/AKT signaling, and osteoarthritis pathway among top 10 pathways affected by GBP-5 knockdown (Figure 5B). Importantly, some of the top genes altered (Figure S5C) matched those most affected when GBP-5 was knocked down with IL-1β alone (Figure S2E), suggesting that GBP-5 acts as an anti-inflammatory agent in response to IL-1β stimulation.
Figure 5:
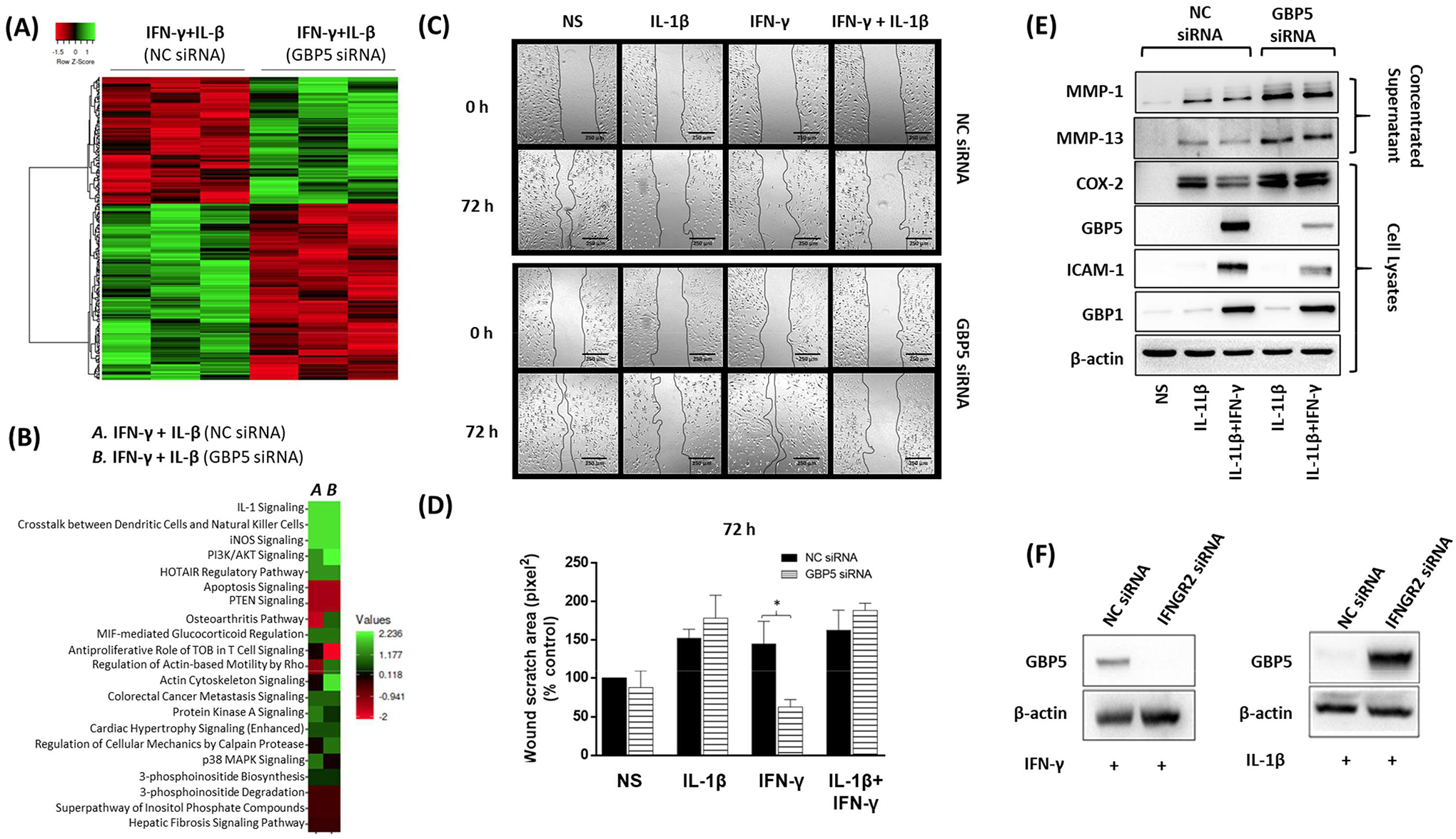
Loss of GBP-5 enhanced the migration, inflammation, and tissue destructive properties of human RASFs. A, A heat map of 269 differentially regulated genes is presented based on t-test qualification. B, Pathways identified to be affected by GBP-5 siRNA in IL-1β+IFN-γ stimulation. C, The represented images of wound-healing assay of RASFs transfected with scrambled control (NC) or GBP-5 siRNA for 48 h followed by stimulation with IL-1β/ and or IFN-γ for 72 h. After scratching (0 h) and after 72 h, the pictures from the same area were acquired. D, The width of the scratched area was measured at 72 h and compared with 0 h width and presented as a percentage. E, Cells were treated with IL-1β (10 ng/ml), IFN-γ (10 ng/ml) or in combination with GBP-5 siRNA for 24 h to study MMP-1, MMP-13, COX-2, ICAM-1, GBP-5 and GBP1 expression normalized with β-actin F, RASFs were transfected with IFNGR2 or NC siRNA (120 pmol) for 48 h, followed by stimulation with IFN-γ (10 ng/ml) or IL-1β (10 ng/ml) for 24 h to study GBP-5 expression. *p < 0.05.
Next, we evaluated the effect of GBP-5 knockdown on RASF functions. In an in vitro scratch test, we observed that GBP-5 knockdown had no effect on migration in IL-1β presence, but it significantly increased the invasiveness of RASFs in IFN-γ treated samples (Figures 5C–D; p<0.05). Western blot analysis of the concentrated supernatants from a similar treatment showed a marked increase in IL-β- or IL-1β+IFN-γ-induced MMP-1 and MMP-13 production with GBP-5 siRNA (Figure 5E). Analysis of cell lysates showed a marked increase in COX-2 expression, with no effect on ICAM-1 expression in GBP-5 siRNA group (Figure 5E).
Evaluation of the MAPK signaling pathway showed that GBP-5 knockdown significantly enhanced the basal and IL-1β+IFN-γ stimulated expression of p-JNK (Figures S5D–E). Since JNK-activated transcription factor AP-1 plays a central role in upregulating proteins that cause inflammation (COX-2) and tissue destruction (MMP-1 and MMP-13) in RA, these findings underline the anti-inflammatory and potential tissue protective actions of GBP-5 in RA.
To understand if GBP-5 expression can be induced independent of IFN-γ signaling, we knocked down IFN-γ receptor 2 (IFNGR2) using siRNA in human RASFs and stimulated cells with IFN-γ or IL-1β for 24 h. As expected, the inhibition of IFNGR2 nullified the expression of IFN-γ-induced GBP-5 (Figure 5F). Notably, IL-1β-induced GBP-5 expression was not altered by IFNGR2 knockdown, suggesting that IL-1β-induced GBP-5 expression in human RASFs is independent of IFN-γ signaling pathway.
Exacerbation of synovial inflammation and bone destruction in rat AIA with intraarticular administration of GBP-5 siRNA.
To characterize the role of GBP in vivo, we administered NC siRNA or GBP-5 siRNA into the hind ankles on day 7 and day 12 (from the onset of the disease). AI score and AC were measured on day 0, day 7, and from day 11 through 16 with termination on day 17. When compared to AIA rats given NC siRNA, the administration of GBP-5 siRNA exacerbated the disease severity in AIA rats (Figure 6A, p<0.05). The AI score of GBP-5 siRNA group was ~25% higher compared to NC group at day 16 (Figure 6A, p<0.05). The efficiency and selectivity of GBP-5 knockdown in the ankles was confirmed by Western immunoblotting (Figure 6B).
Figure 6:
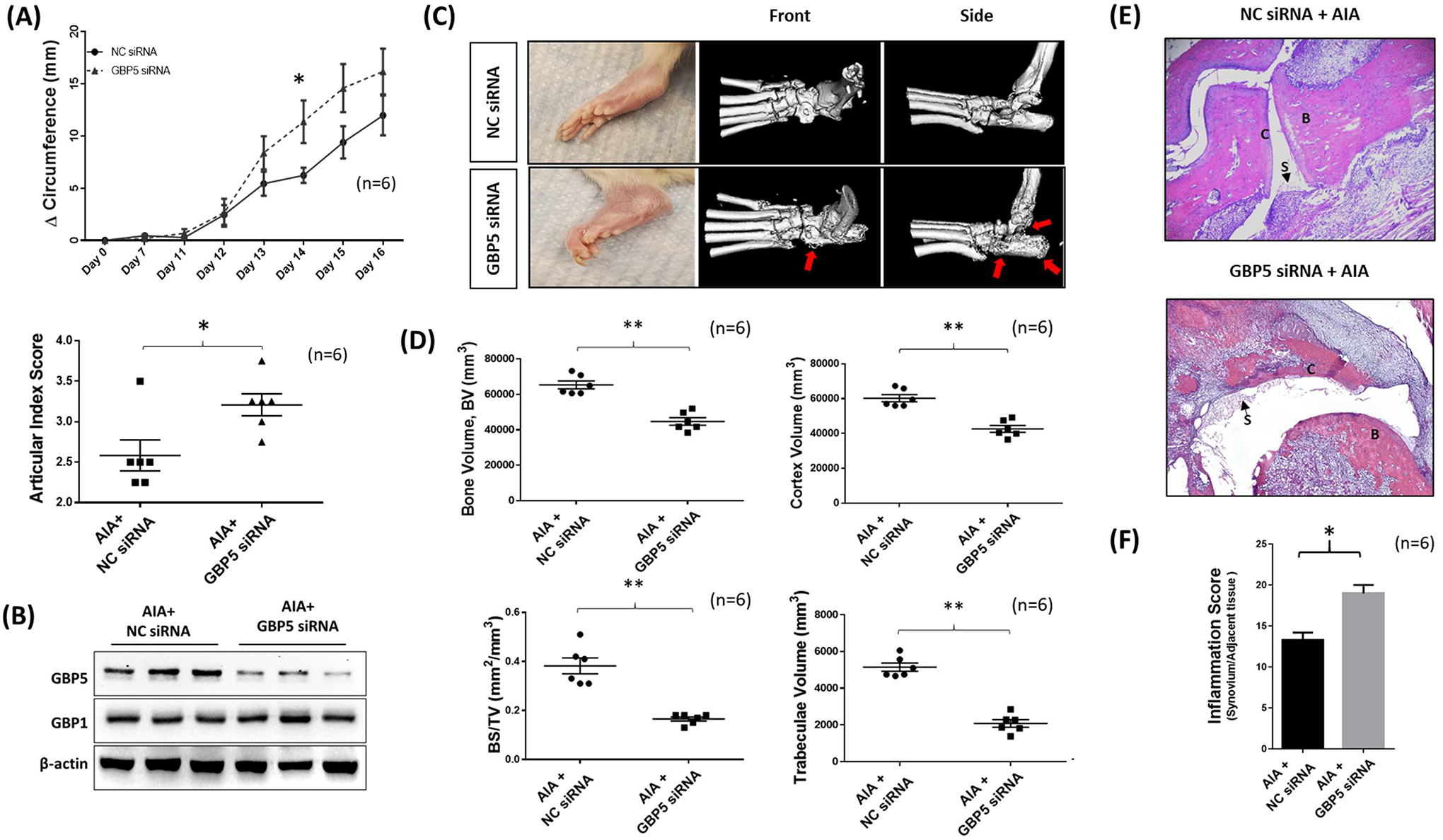
Lack of GBP-5 exacerbates inflammation and bone destruction in AIA rats. A, GBP-5siRNA and NC siRNA were injected with atelocollagen (10 μg in 10 μl atelocollagen) on day 7 and day 12 and articular index score, normalized to day 0 were averaged comparisons and final values represented as the mean of “n” number of animals per group. Mean ± SEM is shown for ankle Δ circumferences (in millimeters) were determined. Ankle articular index scores (range 0–4) on day 16 of the group was averaged. B, Western immunoblotting showed the effective knockdown of GBP-5 in rat ankle without affecting other GBPs such as GBP1. C, μ-CT analysis was performed after the termination of the study. Red arrow indicates the area of bone destruction. D, Bone marker analysis was conducted using the ROI tool in Analyze direct software using phantom standard to calibrate. E, Joints after decalcification were sliced (5 μm) and stained for H&E to identify bone (B) destruction, cartilage (C) degradation, and synovitis (S). F, Histological analysis of H&E slides for synovial inflammation. **p < 0.01; *p < 0.05.
Next, we analyzed the impact of GBP-5 on bone remodeling by using μCT-imaging of ankles. We found that the GBP-5 siRNA group elicited severe bone damage compared to the NC siRNA arthritis group (Figure 6C). In-depth bone analysis (Figure S6A–E) showed that rat ankles from the GBP-5 siRNA group had significantly less bone volume, lower cortex volume, lower bone surface density, and decreased trabeculae volume (Figure 6D). Histopathological evaluation of H&E stained joints and periarticular connective tissue showed significant accentuation of acute and chronic inflammation of joints and periarticular connective tissue as well as associated sequelae including synovitis (S), erosion of articular cartilage (C), subchondral bone loss (B) and remodeling, pannus formation and fibrous ankylosis in AIA rats treated with GBP-5 siRNA compared to NC siRNA (Figure 6E). The inflammatory score (Table S3) showed higher inflammatory parameters in GBP-5 siRNA group compared to NC siRNA group (Figure 6F, p<0.05).
DISCUSSION
In the present study, we identified GBP-5 as a unique endogenous anti-inflammatory protein expressed by RASFs in response to inflammation, that has the ability to inhibit IL-1β-induced synovial inflammation and tissue destruction in RA. Importantly, we identified that GBP-5 not only directly blunts cytokine-mediated inflammation, but also centrally contributes to the anti-inflammatory function of IFN-γ. We hypothesize that GBP-5 is upregulated in the inflamed joint as a negative feedback response to restore cellular homeostasis in human RASFs and blunt the actions of proinflammatory cytokines that contribute to RA pathogenesis. These findings may present an opportunity for therapeutic approaches harnessing GBP-5 to suppress synovial inflammation and bone/cartilage destruction, a primary therapeutic need for effective management and treatment of RA.
The GBP family belongs to the ISGs that are active in host cell defense (23). Although the anti-viral activity of the GBP family is less potent than that of Mx family proteins (9), each GBP protein elicits a unique role in host defense mechanisms and displays clear associations with specific disease pathogenesis. A study by Hu et al. showed a significantly higher expression of GBP2 in the saliva of patients with Sjogren’s syndrome compared to healthy controls (24). Similarly, an increased expression of GBP1 was observed in skin lesions of cutaneous lupus erythematosus and in islets of type I diabetes patients (25). Why GBPs are upregulated in these diseased conditions and whether they serve as autocrine or paracrine regulators in the pathogenesis remains elusive.
While IFN-γ was initially characterized as proinflammatory due to its ability to upregulate IL-12, TNF-α, and IP-10 in diseases such as autoimmune thyroid disease (26), insulin-dependent diabetes mellitus (27) and lupus (28), it has also displayed anti-inflammatory activity by inhibiting Th17 differentiation, resulting in the reduced levels of IL-17 in the collagen-induced arthritis (CIA) model (29). A knockdown of the IFN-γ gene or blockade of IFN-γ signaling using an anti-IFN-γ antibody or IFN-γ receptor knockout resulted in aggravated joint destruction in CIA mice, further confirming that IFN-γ inhibits one or more crucial inflammatory pathways in RA (30, 31). However, it is unclear which protein(s) are critical specifically in mediating the anti-inflammatory effects of IFN-γ in RA. Our finding that the inhibition of IL-1β-induced inflammation by IFN-γ was completely lost in the absence of GBP-5 addresses that gap in understanding of IFN-γ biology. These findings, coupled with GBP-5’s anti-inflammatory activity in human dermal fibroblasts, provide evidence of a broader impact that could be exploited for IL-1β-driven diseases.
The ability of GBP-5 to suppress the detrimental effects of IL-1β in activated RASFs independent of IFN-γ brings its therapeutic potential to the forefront. Despite circumstantial evidence of protective action, IFN-γ has not been used as a therapeutic option due to lack of safety. While our results are open to interpretation, we hypothesize that testing a combinatorial use of IFN-γ at a suboptimal dose with an engineered GBP-5 protein in pre-clinical models of RA may shed light on two synergistic approaches in controlling the disease severity and progression. This is timely and clinically relevant given the fact that adverse events such as opportunistic infections from JAK inhibitors are partly due to their ability to inhibit IFN-γ and its functions (32–34).
Activated RASFs become hyperproliferative and secrete factors that promote inflammation, neovascularization, and cartilage degradation (35). In response to IL-1β stimulation, RASFs produce IL-6, IL-8, ENA-78/CXCL5, and IL-23, factors known to promote Th17 differentiation. We have shown that IL-1β relies on MAPK and NF-κB pathways to propagate inflammatory signaling in RASFs (3, 4). GBP-5 overexpression through the lentiviral method markedly reduced IL-1β-induced p-P38, p-JNK, and p-ERK1/2 activation, and the downstream transcription factors p-c-Jun and NF-κBp65 in the signaling cascade. While these findings are of great interest and open to further interpretation, several unique actions of GBP-5 remain to be elucidated. These include a possible interaction and modulation of proteins that are proximal to IL-1 receptor (IL-1R), activation of anti-inflammatory proteins by GBP-5 that serve as negative feedback control of IL-1R mediated signaling, or cross-talk with key serine/threonine kinases upstream of MAPK or NF-κB pathways. To support this concept, TNFAIP3 (A20) is one such protein produced by SFs that has been shown to suppress inflammatory responses and bone destruction in vitro and in pre-clinical models of RA (36–38).
Though we characterized the role of GBP-5 in the context of inflammation and tissue destruction (GBPs reviewed in ref. (12)), the study has several limitations that open the door to future investigation. Firstly, further mechanistic studies are needed to identify which pathways are exploited by IL-1β to induce GBP-5 expression, given that IFNGR2 knockdown did not inhibit IL-1β-induced GBP-5 expression in RASFs. Secondly, beyond RASFs, the impact of GBP-5 on other cell types relevant to RA pathogenesis warrants further examination. Finally, complementary to our loss-of-function study in vivo, studies testing the efficacy of overexpression or exogenous delivery of GBP-5 protein in amelioration of the disease will further validate these initial findings.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Disease Research Interchange and the Cooperative Human Tissue Network for providing RA synovial tissue. Authors thank Dr. David A Fox, University of Michigan, Ann Arbor for providing some RASF cell lines generated in his lab. Authors also thank Ms. Ruby J. Siegel for critical reading of the manuscript.
FUNDING
This study was supported by the NIH R01 AR072615 grant and the funds from Washington State University.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
REFERENCES
- 1.Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol. 2005;115(2):118–28. [DOI] [PubMed] [Google Scholar]
- 2.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed S, Marotte H, Kwan K, Ruth JH, Campbell PL, Rabquer BJ, et al. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc Natl Acad Sci USA. 2008;105(38):14692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed S, Pakozdi A, Koch AE. Regulation of interleukin-1beta-induced chemokine production and matrix metalloproteinase 2 activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2006;54(8):2393–401. [DOI] [PubMed] [Google Scholar]
- 5.Akhtar N, Singh AK, Ahmed S. MicroRNA-17 Suppresses TNF-α Signaling by Interfering with TRAF2 and cIAP2 Association in Rheumatoid Arthritis Synovial Fibroblasts. J Immunol. 2016;197(6):2219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones BA, Riegsecker S, Rahman A, Beamer M, Aboualaiwi W, Khuder SA, et al. Role of ADAM-17, p38 MAPK, cathepsins, and the proteasome pathway in the synthesis and shedding of fractalkine/CX₃ CL1 in rheumatoid arthritis. Arthritis Rheum. 2013;65(11):2814–25. [DOI] [PubMed] [Google Scholar]
- 7.Singh AK, Umar S, Riegsecker S, Chourasia M, Ahmed S. Regulation of Transforming Growth Factor β-Activated Kinase Activation by Epigallocatechin-3-Gallate in Rheumatoid Arthritis Synovial Fibroblasts: Suppression of K(63)-Linked Autoubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6. Arthritis Rheumatol. 2016;68(2):347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol. 2016;17(5):481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol. 2016;428(17):3495–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guenzi E, Topolt K, Cornali E, Lubeseder-Martellato C, Jorg A, Matzen K, et al. The helical domain of GBP-1 mediates the inhibition of endothelial cell proliferation by inflammatory cytokines. EMBO J. 2001;20(20):5568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guenzi E, Topolt K, Lubeseder-Martellato C, Jorg A, Naschberger E, Benelli R, et al. The guanylate binding protein-1 GTPase controls the invasive and angiogenic capability of endothelial cells through inhibition of MMP-1 expression. EMBO J. 2003;22(15):3772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haque M, Siegel RJ, Fox DA, Ahmed S. Interferon-stimulated GTPases in Autoimmune and Inflammatory Diseases: Promising role of Guanylate binding protein (GBP) family. Rheumatology 2020. [ 10.1093/rheumatology/keaa609]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP-5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336(6080):481–5. [DOI] [PubMed] [Google Scholar]
- 14.Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Sturzel CM, et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe. 2016;19(4):504–14. [DOI] [PubMed] [Google Scholar]
- 15.Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Rühl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol. 2015;16(5):476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You S, Yoo SA, Choi S, Kim JY, Park SJ, Ji JD, et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc Natl Acad Sci USA. 2014;111(1):550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fechtner S, Singh AK, Srivastava I, Szlenk CT, Muench TR, Natesan S, et al. Cannabinoid Receptor 2 Agonist JWH-015 Inhibits Interleukin-1β-Induced Inflammation in Rheumatoid Arthritis Synovial Fibroblasts and in Adjuvant Induced Arthritis Rat via Glucocorticoid Receptor. Front Immunol. 2019;10:1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh AK, Haque M, O’Sullivan K, Chourasia M, Ouseph MM, Ahmed S. Suppression of monosodium urate crystal-induced inflammation by inhibiting TGF-β-activated kinase 1-dependent signaling: role of the ubiquitin proteasome system. Cell Mol Immunol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67(7):909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tretina K, Park ES, Maminska A, MacMicking JD. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J Exp Med. 2019;216(3):482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 2004;25(11):601–9. [DOI] [PubMed] [Google Scholar]
- 24.Hu S, Gao K, Pollard R, Arellano-Garcia M, Zhou H, Zhang L, et al. Preclinical validation of salivary biomarkers for primary Sjögren’s syndrome. Arthritis Care Res. 2010;62(11):1633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundberg M, Krogvold L, Kuric E, Dahl-Jørgensen K, Skog O. Expression of Interferon-Stimulated Genes in Insulitic Pancreatic Islets of Patients Recently Diagnosed With Type 1 Diabetes. Diabetes. 2016;65(10):3104–10. [DOI] [PubMed] [Google Scholar]
- 26.Caturegli P, Hejazi M, Suzuki K, Dohan O, Carrasco N, Kohn LD, et al. Hypothyroidism in transgenic mice expressing IFN-gamma in the thyroid. Proc Natl Acad Sci USA. 2000;97(4):1719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell IL, Kay TW, Oxbrow L, Harrison LC. Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice. J Clin Invest. 1991;87(2):739–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-γ and systemic autoimmunity. Discov Med. 2013;16(87):123–31. [PMC free article] [PubMed] [Google Scholar]
- 29.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56(4):1145–51. [DOI] [PubMed] [Google Scholar]
- 30.Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997;158(11):5501–6. [PubMed] [Google Scholar]
- 31.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. 1997;158(11):5507–13. [PubMed] [Google Scholar]
- 32.Kato M New insights into IFN-gamma in rheumatoid arthritis: role in the era of JAK inhibitors. Immunol Med. 2020;43(2):72–8. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Yuan L, Yang J, Lei Y, Zhang H, Xia L, et al. Changes in Serum Cytokines May Predict Therapeutic Efficacy of Tofacitinib in Rheumatoid Arthritis. Mediators Inflamm. 2019;2019:5617431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21(1):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gantier MP, Stunden HJ, McCoy CE, Behlke MA, Wang D, Kaparakis-Liaskos M, et al. A miR-19 regulon that controls NF-κB signaling. Nucleic Acids Res. 2012;40(16):8048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hah YS, Lee YR, Jun JS, Lim HS, Kim HO, Jeong YG, et al. A20 suppresses inflammatory responses and bone destruction in human fibroblast-like synoviocytes and in mice with collagen-induced arthritis. Arthritis Rheum. 2010;62(8):2313–21. [DOI] [PubMed] [Google Scholar]
- 38.Yoon HK, Byun HS, Lee H, Jeon J, Lee Y, Li Y, et al. Intron-derived aberrant splicing of A20 transcript in rheumatoid arthritis. Rheumatology. 2013;52(3):427–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.