

Abstract
Publicly available and validated DNA reference sequences useful for phylogeny estimation and identification of fungal pathogens are an increasingly important resource in the efforts of plant protection organizations to facilitate safe international trade of agricultural commodities. Colletotrichum species are among the most frequently encountered and regulated plant pathogens at U.S. ports-of-entry. The RefSeq Targeted Loci (RTL) project at NCBI (BioProject no. PRJNA177353) contains a database of curated fungal internal transcribed spacer (ITS) sequences that interact extensively with NCBI Taxonomy, resulting in verified name–strain–sequence type associations for >12,000 species. We present a publicly available dataset of verified and curated name–type strain–sequence associations for all available Colletotrichum species. This includes an updated GenBank Taxonomy for 238 species associated with up to 11 protein coding loci and an updated RTL ITS dataset for 226 species. We demonstrate that several marker loci are well suited for phylogenetic inference and identification. We improve understanding of phylogenetic relationships among verified species, verify or improve phylogenetic circumscriptions of 14 species complexes, and reveal that determining relationships among these major clades will require additional data. We present detailed comparisons between phylogenetic and similarity-based approaches to species identification, revealing complex patterns among single marker loci that often lead to misidentification when based on single-locus similarity approaches. We also demonstrate that species-level identification is elusive for a subset of samples regardless of analytical approach, which may be explained by novel species diversity in our dataset and incomplete lineage sorting and lack of accumulated synapomorphies at these loci.
Keywords: Colletotrichum, DNA barcoding, DNA reference sequence, fungi, GenBank, plant protection, plant quarantine, RefSeq, systematics
Global trade of plant products increases the opportunity and incidence of pest establishment to nonnative areas around the world (Chapman et al. 2017; Meyerson and Mooney 2007). The global spread of plant pests (including pathogens) is ostensibly mitigated by the International Standards for Phytosanitary Measures (https://www.ippc.int/en/core-activities/standards-setting/ispms/) of the International Plant Protection Convention (https://www.ippc.int/en/coreactivities/governance/convention-text/) and implemented by National and Regional Plant Protection Organizations (NPPOs and RPPOs, respectively). The identification of pests intercepted during physical inspections of imported plant products is one of the world’s defenses against the movement and establishment of nonnative organisms into new agricultural and natural systems. PPOs rely on pest identifications from specialists around the globe to determine critical information about an organism’s risk to local agriculture and the environment such as ecology, geographic distribution, host range, potential economic impact, etc. Accurate identifications of pests to the lowest possible taxonomic level (i.e., identification specificity) provide critical information for these Pest Risk Assessments, which in turn inform quarantine policy. These activities assist PPOs to strike the best balance between the sometimes-antagonistic priorities of facilitating trade, and protecting agriculture and the environment from pest invasions.
Ascomycota is the largest fungal phylum containing nearly 84,000 described species in the Catalog of Life (James et al. 2020; http://www.catalogueoflife.org/annual-checklist/2019) with another order of magnitude of undescribed species awaiting discovery (Hawksworth and Lücking 2017). The largest share of plant pathogens among fungal phyla are found in this group (Blackwell 2011; Lu et al. 2003). Notable plant pathogens outside of Ascomycota include the rusts and smuts (Basidiomycota), as well as a group of unrelated organisms that superficially resemble Fungi, the Oomycota (e.g., Phytophthora and Pythium). Members of the Ascomycota, specifically members of the subphylum Pezizomycotina that have filamentous growth, represent most intercepted Fungi causing quarantine concerns. The U.S. Department of Agriculture’s (USDA) Animal and Plant Health Inspection Service (APHIS) – Plant Protection and Quarantine (PPQ) serves as the NPPO for the United States of America.
Comparative morphology is the primary method of species recognition used by APHIS-PPQ, and many other NPPOs around the world, to identify Fungi (including Colletotrichum) and make quarantine decisions where time is of the essence. Impediments to accurate and highly specific identifications exist regardless of the technique used (Crous et al. 2016; Inderbitzin et al. 2020; Lücking et al. 2020). Identification of these Fungi using morphology alone may be obscured by certain factors, including a lack of apparent synapomorphies or diagnostic characters during one or more life stages, characters with continuous and overlapping states among related species and genera, and homoplasy at many taxonomic levels (Lücking et al. 2020). Analyses of DNA sequence data have clearly demonstrated that many well-established morphospecies in many commonly intercepted ascomycete genera are actually complexes containing multiple cryptic species (Aung et al. 2020; Damm et al. 2019; Laraba et al. 2021; Norphanphoun et al. 2020; Udayanga et al. 2015; Vaghefi et al. 2021; Wang et al. 2019). This pattern demonstrates that our understanding of species boundaries and delimitations in many of these taxa remain in their early stages (Matute and Sepulveda 2019; Steenkamp et al. 2018) and that recognition of described species may require DNA sequence data (Crous et al. 2016).
The search for a natural classification of life (i.e., one based on evolutionary relatedness determined by patterns of synapomorphies) and stable-diagnostic characters that may be used to recognize its units of diversity led to an incorporation of DNA sequence data and a variety of accompanying analytical methods (Avise and Ball 1990; Barbera et al. 2019; Callahan et al. 2017; Taylor and Hibbett 2013; Taylor et al. 2000; Thiéry et al. 2016). A complicating issue in Fungi is the historical usage of a dual naming system based on the observed sexual or asexual reproductive structures. For a long time, this was the only practical way for mycologists to classify Fungi reliant on the fact that life stages of single species are rarely observed together. The use of DNA sequence comparisons made the abandonment of this system possible, but multiple genera based on sexual or asexual stages need to be treated as synonyms for singular species (Taylor and Hibbett 2013). Much effort will and have been spent toward a fungal taxonomy that fully incorporates DNA sequences (Hibbett et al. 2016; Lücking et al. 2021; Taylor and Hibbett 2013).
The incorporation of molecular characters for species recognition is limited by the availability of reference-quality DNA sequence data and the limitations of single marker analyses (Schoch et al. 2014). These data remain unavailable or unidentified and uncurated in publicly available nucleotide repositories for most described species, and arguably represent the most significant impediment to incorporating DNA sequences as an additional or alternative source of character data (Lücking et al. 2020).
High-quality DNA sequence data that are publicly available, derived from type specimens accessioned into a public biocollection, and associated with valid and community recognized species names (i.e., verified name – type strain – DNA sequence associations) will serve as the best possible references for molecular-based identifications of fungal phytopathogens where they are available. Additionally, best practice is, where possible, not to only rely on data from a single specimen because even the best datasets remain prone to multiple errors. Drawing conclusions on an unknown’s identity, circumscription, and hypothesizing phylogenetic relationships should ideally rest on multiple, reinforcing sources of evidence. Although the utility of these sequence data are limited by use-case, availability, marker selection, taxon sampling, downstream analytical approach, and a priori knowledge of species diversity and boundaries, they nevertheless provide a rich source of character data from the best possible reference source, curated type specimens (Inderbitzin et al. 2020; Schoch et al. 2014). The availability of these data also has advantages for those who rely on identifications of these Fungi by increasing the likelihood that the user community is employing suitable references as their basis for taxon assignments.
Many DNA sequences with identification utility are present in public repositories, such as GenBank. However, many have not been flagged as suitable to serve as a reference, much less verified as such by a taxonomic specialist and curated (Raja et al. 2017; Schoch et al. 2014). Error rates are as high as 20% in some fungal groups (Nilsson et al. 2006); however, rates can be surprisingly low in other groups of Fungi (Leray et al. 2019). Many of these discrepancies can be related to the general confusion on taxonomic identity among submitters of the data, as the public sequence repositories cannot verify the taxonomic accuracy of sequence records with high precision despite recent efforts to improve this (Schäffer et al. 2021). Such a situation, at worst, may lead to downstream misidentifications when the misidentified sequence is used as a reference. This may not be easily remedied in a public repository like GenBank where identifications on public records cannot easily be adjusted. This is also problematic in cases where a species is split into multiple species, but updates to the latest nomenclature on all existing records may not be feasible (Schoch et al. 2020). At best, this situation requires a patient and experienced user to spend a significant amount of time tracking down and verifying sequences that are suitable to serve as references.
The NCBI’s Reference Sequencing (RefSeq) Targeted Loci (RTL) internal transcribed spacer (ITS) database (https://www.ncbi.nlm.nih.gov/refseq/; BioProject PRJNA177353) was created to address the need to identify and separately curate sequences that are suitable to serve as references (Schoch et al. 2014). Each record contains a DNA sequence that is associated with a validated taxonomic name and derived from verified type material using a public BioCollection identifier (https://www.ncbi.nlm.nih.gov/biocollections). A set of verified name–strain–sequence associations curated by NCBI serves as an ideal construct for establishing a publicly available and user-friendly DNA sequence reference set because it interacts directly with NCBI’s separately managed Taxonomy (https://www.ncbi.nlm.nih.gov/taxonomy; Schoch et al. 2020) and nucleotide (https://www.ncbi.nlm.nih.gov/nucleotide/) databases and is searchable using NCBI’s WebBLAST tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi; Sayers et al. 2021). Although designated as a “universal” barcode for Fungi (Schoch et al. 2012), limitations exist when using any single DNA sequence to characterize an organism in any way. However, the RefSeq ITS sequence records may serve as a primer for species identification and a source for finding additional sequence markers that could serve as references to support identification efforts. The Fungi RefSeq ITS dataset contains reference-quality sequence records for >12,000 species, including >190 orders and 2,700 genera.
Previous attempts at focused curation at NCBI Taxonomy included the genus Trichoderma and its sexual morph synonym Hypocrea (Robbertse et al. 2017). It resulted in several improvements for the public sequence records for the genus and set the stage for making improvements to the taxonomic quality of the new fungal genome data submissions gathering pace in the public repositories. However, it proved to be a manually intensive process with few areas for automation, and was difficult to scale to other genera. Despite comprehensive taxonomic treatments, challenges to accurate presentation of sequence data related to recent taxonomic practice remain for this genus (Cai and Druzhinina 2021) and several others. Here we continue previous work and extend it in a collaboration between NCBI curators and USDA–APHIS–PPQ National Identification Services to improve public records for Colletotrichum, another large and well-studied genus with important implications for plant quarantine.
Colletotrichum Corda species are among the most frequently encountered plant-associated Fungi worldwide and considered one of the world’s most important groups of fungal pathogens (Dean et al. 2012). These ascomycetes interact with a wide range of host plants in a wide range of ecological associations, including as pathogens of economically important plant groups such as Proteaceae (Lubbe et al. 2004), Agavaceae (Farr et al. 2006), Camellia (Wang et al. 2016), Vitis (Yan et al. 2015), Citrus (You et al. 2007), Persea (Yakoby et al. 2000), Capsicum (Sangdee et al. 2011), Phaseolus (Kamfwa et al. 2021), Fragaria (Soares et al. 2021), Vaccinium (Liu et al. 2020), Malus (Khodadadi et al. 2020), and many others (Cannon et al. 2012). Colletotrichum is the fourth most frequently intercepted fungal genus at U.S. ports-of-entry for which quarantine action is taken on shipments of plants and plant products, making them responsible for interruptions to trade more often than nearly any other genus containing phytopathogens (PPQ internal data, 2010 to 2020). Suites of morphological characters commonly used to recognize Colletotrichum species are often not diagnostic within the species complexes, and can mislead identifications even to the level of species complex (Crouch 2014; Damm et al. 2009, 2012a, b, 2019; Weir et al. 2012). Colletotrichum is perhaps distinguishable from other commonly intercepted genera with species that are difficult to identify using morphology in that its species diversity and evolutionary relationships have been more thoroughly studied and characterized in recent years (Bhunjun et al. 2021; Cannon et al. 2012; Crouch 2014; Damm et al. 2009, 2012a, b, 2019; Weir et al. 2012). Colletotrichum therefore stands out as a candidate to prioritize for exhaustive inclusion in RefSeq at NCBI.
The primary goals of this article are to identify and improve the public availability of reference quality DNA sequence data for all 200+ species of Colletotrichum; determine whether the marker loci commonly used in the literature to identify Colletotrichum species and to study their molecular systematics are suitable (e.g., phylogeny reconstruction); to evaluate commonly used strategies for identifying Colletotrichum species; and to provide strategic guidance to the plant health community for conducting DNA sequence-based identifications of unknown Colletotrichum isolates or those intercepted in international or domestic trade of agricultural commodities. We expect that resulting insights will assist the plant health community in their efforts to protect domestic agriculture from incursions of Colletotrichum pathogens and assist the Colletotrichum research community with their biodiversity research efforts that reciprocally inform plant protection efforts. However, to properly accomplish these goals, we additionally explore other basic questions regarding the suitability of these data for species identification and molecular systematics research.
Materials and Methods
Steps taken to evaluate and curate the Colletotrichum names, classification, and molecular sequence data are briefly summarized below. Each associated taxon, strain, and sequence are hereafter referred to as “verified,” as a type of shorthand. More details are found in Supplementary Text S1.
Colletotrichum-type strains nomenclature and classification.
A list of Colletotrichum names and their known species–complex associations was compiled from recent phylogeny-based revisionary work (Crouch 2014; Damm et al. 2009, 2012a, b, 2013, 2014, 2019; Liu et al. 2014; Weir et al. 2012), Index Fungorum (http://www.indexfungorum.org/names/names.asp), and MycoBank (https://www.mycobank.org/; Table 1). Orthographic variants were resolved using the Index Fungorum. Each name and its type material associations were verified by reviewing the original descriptions of each species. Each name was evaluated for proper description, designation of a nomenclatural type, and deposition of a type voucher and associated cultures (ex-type) into a public biorepository.
Table 1.
List of all Colletotrichum species names and corresponding publicly available reference sequences that were verified during this study; all internal transcribed spacer (ITS) sequence accessions beginning with “NR” are curated in the Fungi RefSeq Targeted Loci (RTL) ITS database at NCBI (BioProject PRJNA177353), whereas the others are not and did not pass quality standards for RTL inclusion; however, all sequences listed here (ITS included) were incorporated into our phylogenetic analyses as described in the Materials and Methods (see main article); epithets are sorted based on species complex membership and alphabetically therein; an asterisk (*) indicates a member of Clade Graminicola that has been recognized as member of the C. caudatum species complex
In this article we follow the conventions of the International Code of Nomenclature for Algae, Fungi, and Plants (https://www.iapt-taxon.org/nomen/main.php; Turland et al. 2018) for italicizing all formal scientific names regardless of rank and capitalizing all taxa above species level. We follow the PhyloCode (http://phylonames.org/code/; Cantino and de Queiroz 2020) and use the prefix “clade” (e.g., clade Acutatum) when referring to species complex names of Colletotrichum when they are recovered by our analyses as monophyletic and well supported (Thines et al. 2020).
Colletotrichum reference marker selection.
Colletotrichum DNA sequences from GenBank’s core nucleotide database representing the ITS and 11 other nuclear encoded marker loci (gene name, Saccharomyces cerevisiae gene symbol in brackets for cross-reference), ACT (actin, ACT1), Apn2 (DNA-lyase 2, APN2), ApMat intergenic region (ApMat), CAL (calmodulin, CMD1), CHS (chitin synthase class I, CHS1), GAPDH (glyceraldehyde-3-phosphate dehydrogenase, TDH3), GS (glutamine synthetase, GLN1), HIS (histone H3, HHT1), Mat1-Apn1, SOD (superoxide dismutase, SOD2), and TUB2 (beta-tubulin, TUB2) were selected after verifying their association with each type strain. Quality of the ITS sequence data was evaluated and then promoted to the RTL project for Fungi ITS (BioProject PRJNA177353). These data were compiled to complete a database of verified name – type strain – DNA sequence associations (Table 1).
Colletotrichum RTL ITS sequences were compared with archival GenBank ITS sequences to evaluate the range of Colletotrichum ITS sequences in the current GenBank and confirm their origins. Six marker loci from our reference dataset (ITS, ACT, CHS, GAPDH, HIS, and TUB2) that are commonly used in phylogenetic studies and DNA-based identifications of Colletotrichum were mined from all 117 Colletotrichum genome assemblies in GenBank at the time (end of 2020; Supplementary Table S1). Each locus was examined to determine copy number in Colletotrichum genomes and variation among copies and primer sites in an effort to evaluate the suitability of sequence data produced from expected PCR products for phylogeny reconstruction and species identification. For reference, in the RefSeq genome assembly of C. fructicola, these partial marker sequences are present in genes with the following locus tag identifiers: CGMCC3_g6665 (ACT), CGMCC3_g835 (CHS), CGMCC3_g7976 (GAPDH), CGMCC3_g10906 (HIS), and CGMCC3_g2839 (TUB).
Evaluation of methods for identification of Colletotrichum sequences.
The evolutionary relationships among all Colletotrichum reference sequences and sequences mined from genomes of GenBank were evaluated using phylogenetic analyses with Monilochaetes infuscans (CBS 869.96; JQ005843, JQ005801, JX546612, JQ005822, JQ005780, and JQ005864) as an outgroup. Multiple sequence alignments were created separately for ACT, CAL, CHS, GAPDH, HIS, ITS, and TUB2 using the program MAFFT v7.450 (https://mafft.cbrc.jp/alignment/software/; Katoh et al. 2002; Katoh and Standley 2013) within the program Geneious Prime 2020.0.5 (https://www.geneious.com), and then all seven were concatenated to create a combined matrix, henceforth referred to as the “concatenated alignment” (or CA). The remaining regions (ApMat, Apn2, GS, Mat1-Apn1, and SOD) were not aligned and analyzed because of low levels of taxon coverage across the genus for each (Table 2). The resulting phylogenies were used to test the monophyly and species composition of each major clade and species complex, and to test the accuracy and specificity of each respective submitter-designated genome identity in GenBank. Sequence similarity measures (SID1, SID2, and SID3), which are defined in Supplementary Text S1, were evaluated for their utility to determine clade- and species-level assignments of unknowns relative to phylogeny-based assignments.
Table 2.
Summary of the distribution of verified species “name–strain–sequence” associations by species complex/clade and distribution of available DNA sequences by marker locus as determined during this study
| Clade/complex | No. of species | ACT |
Apn2 |
ApMat |
CAL |
CHS-1 |
GAPDH |
GS |
HIS |
ITS |
Mat1Apn2 |
SOD |
TUB2 |
Total |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | b | #a | xc | ||
|
| |||||||||||||||||||||||||||
| Acutatum | 41 | 39 | 0.95 | 0 | 0.00 | 0 | 0.00 | 1 | 0.02 | 37 | 0.90 | 40 | 0.98 | 0 | 0.00 | 33 | 0.80 | 41 | 1.00 | 0 | 0.00 | 0 | 0.00 | 41 | 1.00 | 232 | 5.7 |
| Agaves | 4 | 4 | 1.00 | 0 | 0.00 | 0 | 0.00 | 1 | 1.00 | 2 | 0.50 | 3 | 0.75 | 0 | 0.00 | 4 | 1.00 | 4 | 1.00 | 0 | 0.00 | 0 | 0.00 | 3 | 0.75 | 21 | 5.25 |
| Boninense | 25 | 24 | 0.96 | 0 | 0.00 | 0 | 0.00 | 19 | 0.76 | 21 | 0.84 | 24 | 0.96 | 0 | 0.00 | 19 | 0.76 | 24 | 0.96 | 0 | 0.00 | 0 | 0.00 | 25 | 1.00 | 156 | 6.24 |
| caudatum | 5 | 0 | 0.00 | 4 | 0.80 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 5 | 1.00 | 4 | 0.80 | 3 | 0.60 | 0 | 0.00 | 16 | 3.2 |
| dematium | 17 | 17 | 1.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 15 | 0.88 | 17 | 1.00 | 0 | 0.00 | 7 | 0.41 | 17 | 1.00 | 0 | 0.00 | 0 | 0.00 | 16 | 0.94 | 89 | 5.2 |
| Destructivum | 16 | 16 | 1.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 16 | 1.00 | 16 | 1.00 | 0 | 0.00 | 13 | 0.81 | 16 | 1.00 | 0 | 0.00 | 0 | 0.00 | 16 | 1.00 | 93 | 5.8 |
| dracaenophilum | 8 | 7 | 0.88 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 3 | 0.38 | 7 | 0.88 | 0 | 0.00 | 3 | 0.38 | 8 | 1.00 | 0 | 0.00 | 0 | 0.00 | 8 | 1.00 | 36 | 4.5 |
| Gigasporum | 7 | 6 | 0.86 | 0 | 0.00 | 0 | 0.00 | 4 | 0.57 | 6 | 0.86 | 7 | 1.00 | 0 | 0.00 | 6 | 0.86 | 7 | 1.00 | 0 | 0.00 | 0 | 0.00 | 7 | 1.00 | 43 | 6.1 |
| Gloeosporioides | 50 | 48 | 0.96 | 0 | 0.00 | 26 | 0.52 | 35 | 0.70 | 39 | 0.78 | 47 | 0.94 | 13 | 0.26 | 6 | 0.12 | 49 | 0.98 | 0 | 0.00 | 14 | 0.28 | 47 | 0.94 | 324 | 6.5 |
| Graminicola | 18 | 14 | 0.78 | 2 | 0.11 | 0 | 0.00 | 0 | 0.00 | 10 | 0.56 | 4 | 0.22 | 0 | 0.00 | 5 | 0.28 | 17 | 0.94 | 2 | 0.11 | 3 | 0.17 | 14 | 0.78 | 71 | 3.9 |
| Magnum | 8 | 8 | 1.00 | 0 | 0.00 | 0 | 0.00 | 1 | 0.13 | 7 | 0.88 | 8 | 1.00 | 0 | 0.00 | 5 | 0.63 | 7 | 0.88 | 0 | 0.00 | 0 | 0.00 | 8 | 1.00 | 44 | 5.5 |
| Orbiculare | 9 | 9 | 1.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 8 | 0.89 | 9 | 1.00 | 1 | 0.11 | 8 | 0.89 | 9 | 1.00 | 0 | 0.00 | 0 | 0.00 | 9 | 1.00 | 53 | 5.9 |
| Orchidearum | 8 | 8 | 1.00 | 0 | 0.00 | 0 | 0.00 | 1 | 0.13 | 8 | 1.00 | 8 | 1.00 | 0 | 0.00 | 8 | 1.00 | 8 | 1.00 | 0 | 0.00 | 0 | 0.00 | 8 | 1.00 | 49 | 6.1 |
| Spaethianum | 7 | 7 | 1.00 | 0 | 0.00 | 0 | 0.00 | 1 | 0.14 | 4 | 0.57 | 7 | 1.00 | 0 | 0.00 | 4 | 0.57 | 7 | 1.00 | 0 | 0.00 | 0 | 0.00 | 7 | 1.00 | 37 | 5.3 |
| Truncatum | 6 | 6 | 1.00 | 0 | 0.00 | 0 | 0.00 | 1 | 0.17 | 5 | 0.83 | 6 | 1.00 | 1 | 0.17 | 2 | 0.33 | 6 | 1.00 | 0 | 0.00 | 0 | 0.00 | 5 | 0.83 | 32 | 5.3 |
| unassociated | 9 | 9 | 1.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 9 | 1.00 | 8 | 0.89 | 0 | 0.00 | 7 | 0.78 | 9 | 1.00 | 0 | 0.00 | 0 | 0.00 | 8 | 0.89 | 50 | 5.5 |
| Total | 238 | 222 | 0.90 | 6 | 0.06 | 26 | 0.03 | 64 | 0.23 | 190 | 0.74 | 211 | 0.85 | 15 | 0.03 | 130 | 0.60 | 234 | 0.98 | 6 | 0.06 | 20 | 0.07 | 222 | 0.88 | 1,346 | 5.7 |
Number of species with an available sequence that was verified.
Average proportion of verified sequences per species.
Average number of markers per taxon.
NCBI RefSeq’s genome-assembly–derived multiprotein distance tree.
The RefSeq group at NCBI makes use of a protein distance interactive tree tool to assist during the curation of fungal genomes. The tree has been built incrementally using a matrix of dissimilarities and the tree optimized so that the dissimilarities between genomes should equal the tree distances as much as possible by using the least-squares method. After the tree has been built, the taxonomic names are mapped on the tree nodes by solving the maximum-parsimony (MP) problem for each taxon in the NCBI Taxonomy, allowing one to find genomes whose NCBI taxonomic assignment does not match their position in the tree.
Results
Evaluation of the Colletotrichum names, classification and molecular sequence data are summarized below. More detailed results are available in Supplementary Text S1.
Colletotrichum-type strains nomenclature and classification in NCBI’s taxonomy.
In September 2021, the NCBI Taxonomy contained 297 species (Fig. 1) names in the genus Colletotrichum (248 with sequences obtained from type material) with new names being added constantly. These are classified in 13 species complex nodes at NCBI Taxonomy, with 48 species not placed in any species complex. As part of this specific project, we compiled 238 Colletotrichum species with verified name – type strain – DNA sequence associations (Table 1) and verified species complex membership for each species by phylogeny (discussed below; Table 1). This information was then used to update the NCBI Taxonomy database (Schoch et al. 2020). During the process, several names listed as unpublished and labeled with temporary labels that were not updated by their original submitters after publication could be adjusted. Updates also included the additions of type material information for >35 species names. Additionally, NCBI Taxonomy changed at least five orthographic variants with alternate spelling and could update 12 species names to invalid status (“nom. inval.”) because procedures for declaring a correct new name (Aime et al. 2021) were not followed according to the International Code of Nomenclature for Algae, Fungi, and Plants (https://www.iapt-taxon.org/nomen/main.php; Turland et al. 2018).
Fig. 1.
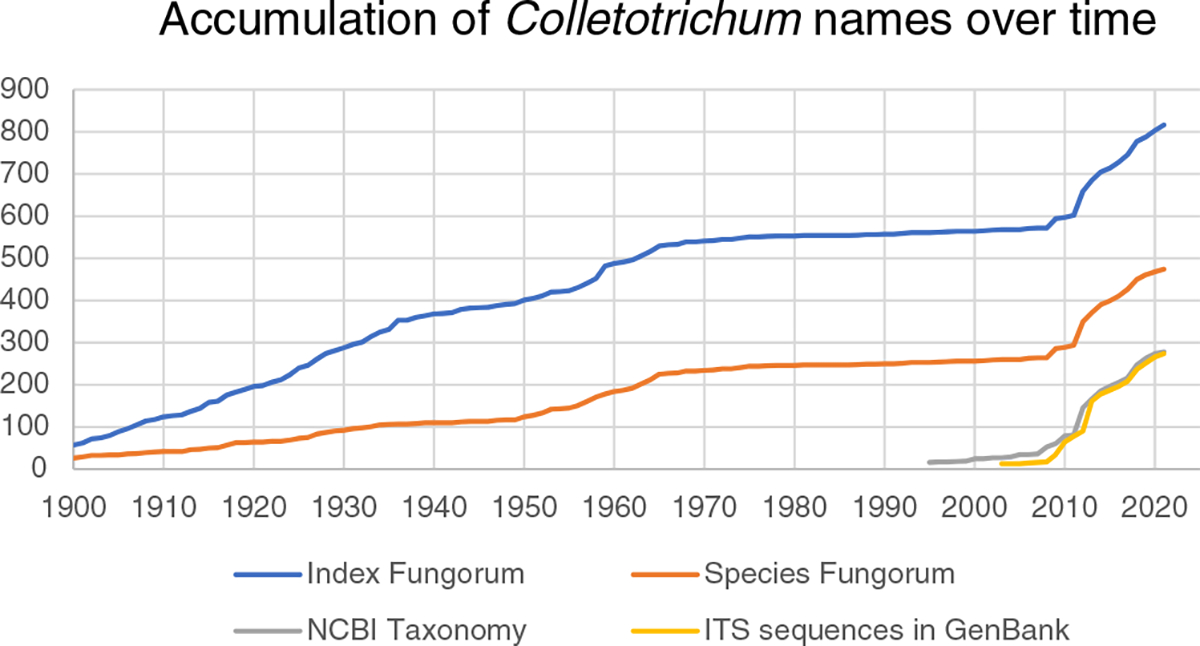
The accumulation per year of all new species names and combinations in Colletotrichum, from 1900 to present. Names and new combinations in the nomenclature resource Index Fungorum (http://www.indexfungorum.org) are indicated, with accepted species names in the taxonomy resource Species Fungorum (http://www.speciesfungorum.org) as well as the release dates of accepted names in NCBI Taxonomy (https://www.ncbi.nlm.nih.gov/taxonomy). An additional line indicates the first instances of internal transcribed spacer sequence records submitted to GenBank (https://www.ncbi.nlm.nih.gov/nuccore) for species names in Colletotrichum.
Colletotrichum reference marker selection.
Availability of verified sequences from each of the 12 evaluated marker loci was highly variable among species. ACT was available for 223 (94%) species, Apn2 for six (2.5%), ApMat for 27 (11.3%), CAL for 64 (26.9%), CHS-1 for 191 (80.3%), GADPH for 212 (89.1%), GS for 15 (6.3%), HIS for 131 (55.0%), ITS for 235 (98.7%), MatApn1 for 6 (2.5%), and TUB2 for 223 (93.7%; Table 2). Availability of sequences from some marker loci varied among clades. For example, Apn2 and Mat1Apn2 are only available for species of the C. caudatum species complex and clade Graminicola, and ApMat is only available for species within clade Gloeosporioides; but ACT, CHS-1, GAPDH, HIS, ITS, and TUB2 are available at a high frequency within most clades (Table 2). The number of available verified sequences per species within each of the major clades varied from 6.5 in clade Gloeosporioides to 3.2 in the clade Caudatum (Table 2).
Colletotrichum ITS in NCBI’s RefSeq targeted loci project.
A total of 226 Colletotrichum taxa have an ITS record in the RTL database. Their sequence lengths ranged from 454 to 612 bases with some including small subunit- or/and large subunit-flanking regions. During this focused review of Colletotrichum, some RTL records were suppressed for the following reasons: type material from subjective synonym; source material not from type or annotated as reference material in NCBI Taxonomy; invalid organism name; ITS coverage not enough; sequence quality issues or replaced by another more complete sequence record. Three records are partial for both the ITS1 and ITS2, 184 records are complete for both the ITS1 and ITS2, nine records are complete for ITS1 only, and 29 records are complete for ITS2 only (Fig. 2).
Fig. 2.
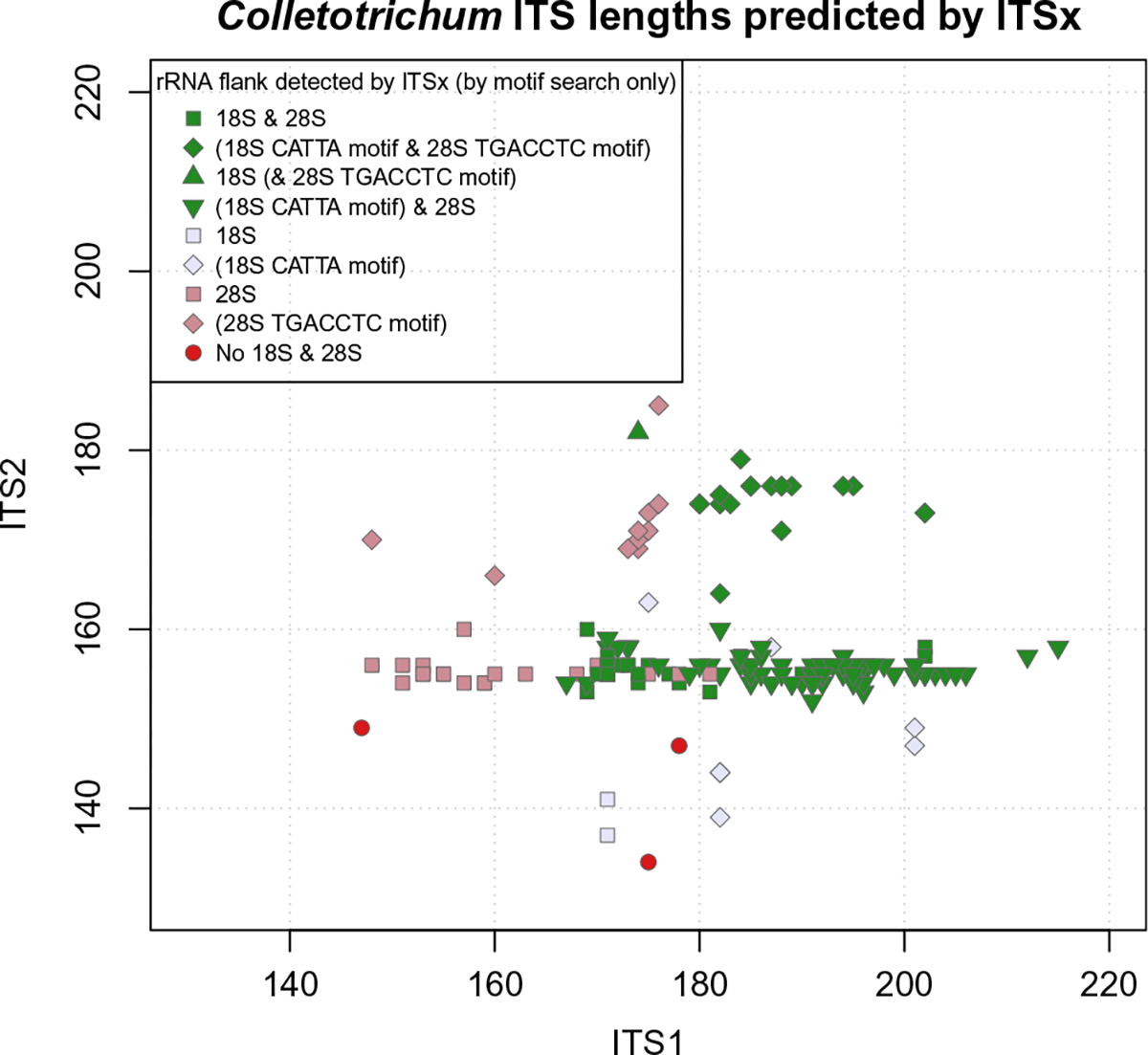
Graphical display of internal transcribed spacer (ITS) ITS1 × ITS2 lengths from Colletotrichum RefSeq Targeted Loci sequence records according to ITSx prediction, with colors indicating the presence of rRNA gene flanks from ITSx and other detection methods. Green markers indicate the presence of partial rRNA genes on both ends, thus implying a complete ITS region. Pink markers indicate the presence of a partial 28S gene flank (complete ITS2 region) and lavender markers indicate the presence of a partial 18S gene flank (complete ITS1 region).
Most Colletotrichum RTL ITS sequences (from 169 taxa) contained the same 5.8S gene variant as seen in record NR_111190.1. The remaining Colletotrichum sequences in RTL contained three 5.8S gene variants seen in 40, six, and three other Colletotrichum sequences and a few unique variants in seven different sequences (Fig. 3). Most of the RTL ITS sequences represent discrete species from 13 species complexes/major clades. The remaining species (8%) are not associated with a complex. A comparison of megaBLAST alignments between taxa from different Colletotrichum species complexes and between complexes and unassigned Colletotrichum taxa produced a median identity of 90.7%, and observed identities varied between 85.7 and 98.7% (Fig. 4). A 100% identity over a 440-base megaBLAST alignment for various RTL ITS sequences was observed among 59 Colletotrichum taxa, and frequently between taxa within clade Gloeosporioides and within clade Acutatum. ITS sequences of Colletotrichum species generally contain insufficient information for distinguishing different species within species complexes. It is therefore recommended that ITS not be used alone or as an independent locus to determine species-level identification of unknown specimens.
Fig. 3.

An alignment showing identities to first sequence with a dot, and the variation of 5.8S gene sequences in the Colletotrichum RefSeq Targeted Loci internal transcribed spacer dataset with variants of the highest frequency ordered from the top (most [75%] having the same variant as seen in record NR_111190.1) to bottom (last seven being unique to each sequence record).
Fig. 4.
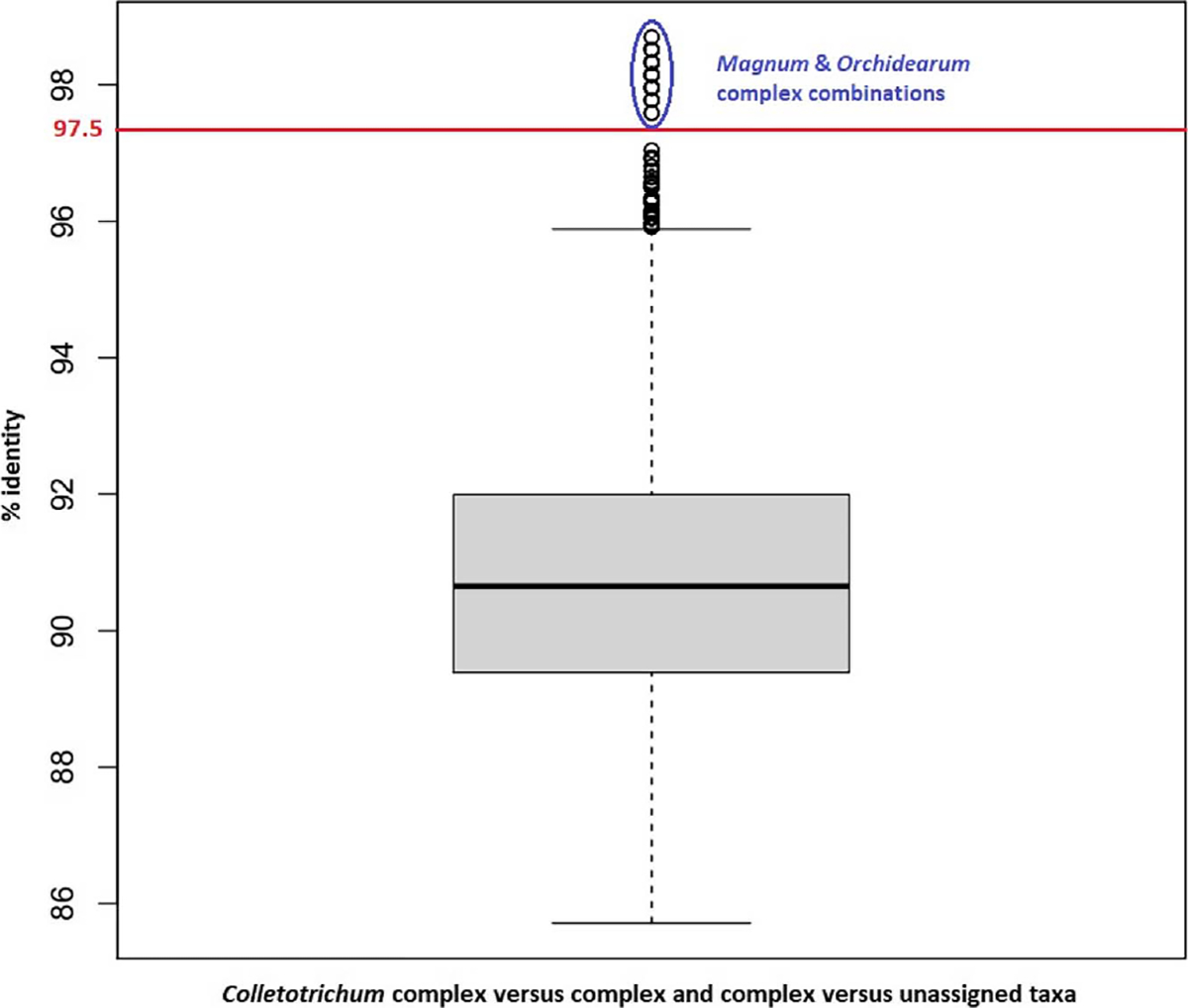
A boxplot displaying the distribution of % identities (from pairwise RefSeq Targeted Loci internal transcribed spacer megaBLAST alignments) between taxa from different Colletotrichum species complexes and between complexes and unassigned Colletotrichum taxa. The gray box demarks the interquartile range (IQR = 25th percentile [Q1] to 75th percentile [Q3]) with the bottom and top black lines indicating the minimum observed identity and Q3 + 1.5 * IQR, respectively.
The placement of ITS records in a strict consensus MP tree (from 320 trees) showed that the ITS sequences of 224 species tend to be placed together with their own species complex members or separate if not belonging to a complex (Fig. 5). Only two ITS sequences, one from C. riograndense and one from C. orchidis, did not follow this pattern. Instead, the ITS placement for C. riograndense was some distance away from clade Spaethianum and among species that do not belong to any species complex. The ITS placement for C. orchidis was in clade Destructivum, instead of the C. dematium species complex.
Fig. 5.
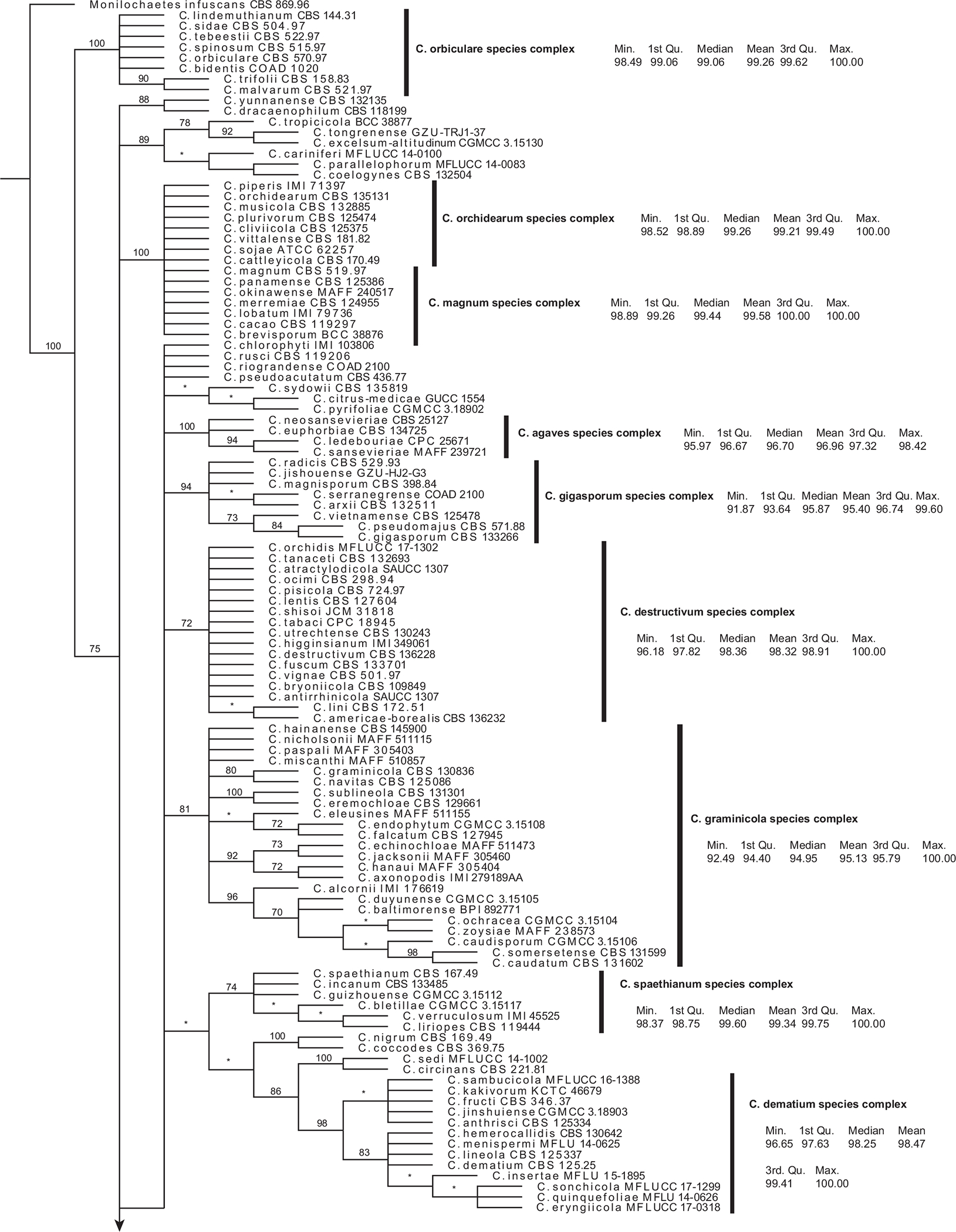
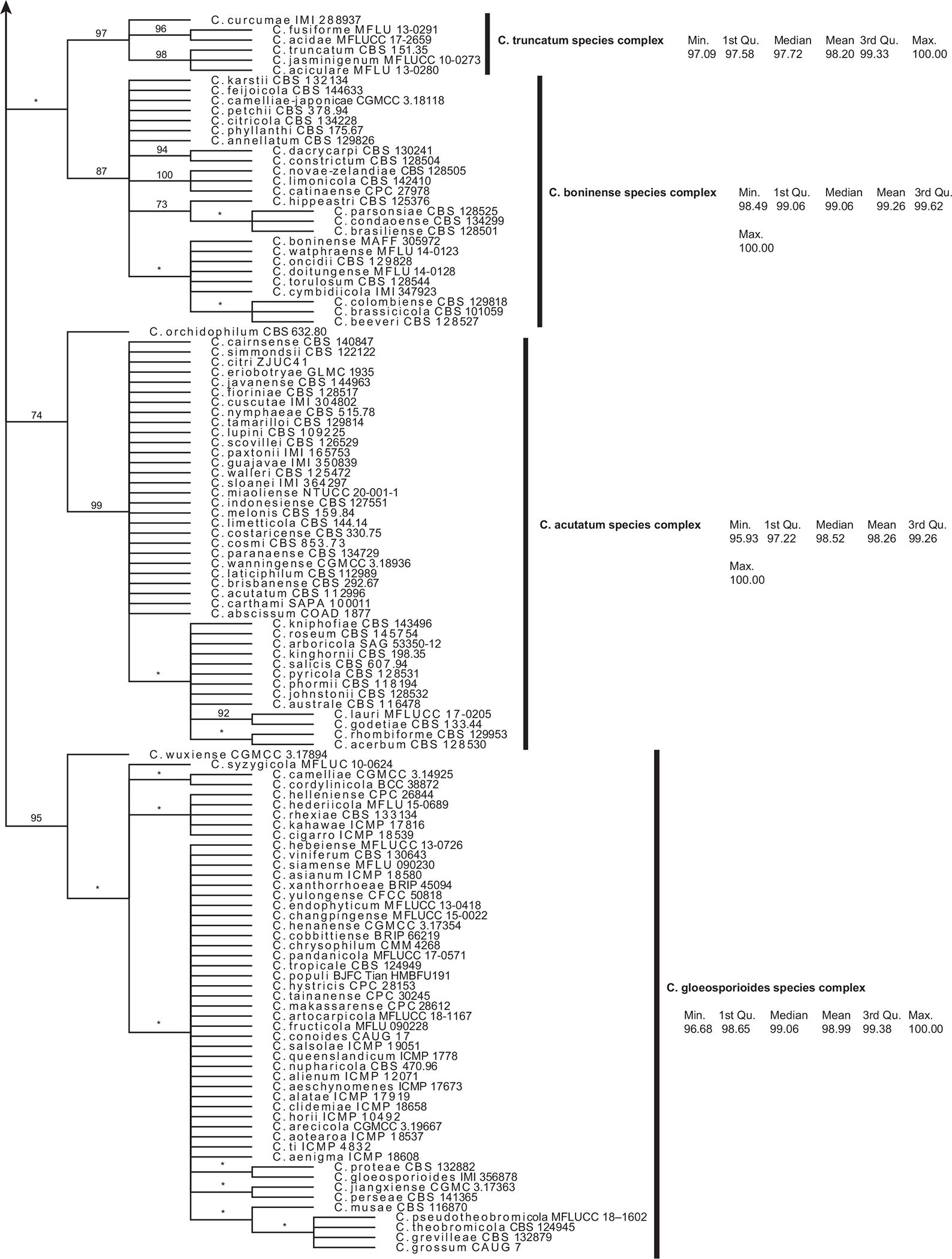
A strict consensus phylogeny of 320 most parsimonious trees resulting from analysis of all Colletotrichum ITS sequence data. Jackknife support values above 70% are shown at clade nodes. Clades with less than 70% jackknife support are represented by an asterisk and considered unsupported. Clades are annotated by species complex and descriptive statistics of the % identities (from pairwise RTL ITS megaBLAST alignments) within each species complex.
Colletotrichum ITS sequences in the GenBank archive.
The “Entrez” query described in the “Methods” section of Supplementary Text S1 produced 21,808 records from the Nucleotide GenBank database and potentially contained an ITS region from a Colletotrichum species. However, 299 of these sequences had quality issues and were removed from further identity evaluation. Of the GenBank sequences, 20,629 out of 21,509 sequences likely contained the ITS region of a Colletotrichum species, because these sequences produced the expected alignment lengths, identities, and sequence-end coverage when possible. Ultimately, ~97% of the Colletotrichum-labeled GenBank records could be verified as originating from Colletotrichum, but the rest had either sequence-quality issues or better alignment and identity to other Fungi.
The Colletotrichum ITS dataset in RTL was also used to search for records in GenBank not labeled as Colletotrichum. A total of 1,553 records in the BLAST Nt database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch) were found to have good alignment and identity to one or more Colletotrichum RTL ITS sequences. Almost all (99%) of these sequence records were associated with an unspecified name (e.g., “fungal endophyte” <taxon name> sp., uncultured <taxon name>) and a few records (23) were associated with specified binomial names from other genera.
Genomic evaluation of Colletotrichum marker loci.
Using 11 representative 5.8S gene sequences (Fig. 3) representing the variation in the 226 RTL sequences as the query in a BLAST search against 117 Colletotrichum WGS assemblies produced hits with alignment lengths longer than 100 bases in 80 assemblies. A cmsearch (https://manpages.ubuntu.com/manpages/xenial/man1/cmsearch.1.html) with the 5.8S RFAM model against WGS contigs containing hits, identified 79 assemblies containing complete 5.8S genes. The majority (87%) of these complete 5.8S genes found in the WGS assemblies were identical to gene variants in the RTL ITS sequences or at most had a two-nucleotide base difference. The rest of the 5.8S gene sequences in the WGS assemblies were significantly different. These copies were either exposed to repeat-induced–point (RIP) mutation or originated from a contaminating source. However, in a few cases, sequence quality issues contributed to the difference. Four assemblies contained 5.8S genes from a contaminating source (Penicillium, Curvularia, Aspergillus, Thozetella, and Trichosporon species), and 10 assemblies from five taxa (C. siamense, C. lindemuthianum, C. tanaceti, C. trifolii, and Colletotrichum sp. COLG25) contained 22 5.8S copies with a RIP signature (e.g., Supplementary Fig. S1) as determined by the web-based tool The RIPper (https://bio.tools/RIPper; van Wyk et al. 2019).
The number of ITS copies included in the 117 submitted genome assemblies varied widely. ITS copies were not detected in 39 assemblies, whereas one or more copies (partial or complete) were detected in 78 assemblies. Fifty-one assemblies contained at least one copy and 27 assemblies contained between two and 41 copies per assembly. One-hundred-and-four assemblies were from species that are represented in the RTL ITS dataset and of those assemblies that did include ITS sequences, 50 contained ITS copies for which the RTL ITS sequence of the same taxon had >99.4% identity (two or less mismatches and one or less gap). ITS copies that did vary more significantly, either originated from a contaminating source, were misassembled (chimeric; e.g.: PUHP01001126.1 6,079 to 6,888), or were of low sequencing quality.
Amplified regions of all five protein coding marker genes were detected in 106 of the 117 Colletotrichum genome assemblies (Supplementary Table S1). Not all five loci (continuous region of ~260 to 600 marker bases) were detected in a few (11) assemblies for the following reasons: the expected sequence region was not represented in the assembly (five cases); only a very short piece of the amplified gene region aligned (one case); the locus was split over two contigs (two cases); and mismatches toward the ends of the megaBLASTn alignment caused the query coverage to not extend past 98% (four cases).
Evaluation of identification methods for Colletotrichum.
Summaries of single and multilocus alignment statistics may be found in Table 3. The program Guidance2 (http://guidance.tau.ac.il/) significantly reduced the number of aligned base positions from 430 to 132 for GAPDH, indicating much of the original alignment is homoplasious at the genus level. A continuous 78-bp span in the HIS alignment (positions 193 to 270) was removed by hand because of alignment difficulty at the genus level. The MP heuristic search found a total of the 80 most parsimonious trees with lengths of 10,672 during eight of the 1,000 TBR replicates. The number of nodes with at least 70% jackknife (JK) support in the MP consensus tree was 173 (Table 3). The Incongruence Length-Difference Test (Farris et al. 1994) indicated no significant difference between any single locus of the CA and the remaining loci, suggesting that no underlying genus-wide incongruence exists among the involved marker loci. However, topological comparisons among single marker trees indicated variation in the phylogenetic utility of these markers for clade assignment, relationships among clades, and species identification (Supplementary Fig. S2).
Table 3.
Numerical descriptions of each single marker alignment, the concatenated alignment (CA), the corresponding most optimal maximum-likelihood trees, and the maximum-parsimony strict consensus tree resulting from analysis of the CAa
| Item description | ACT | CAL | CHS | GAPDHg2 | HIS | ITS | TUB2 | CA |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Number of taxa | 320 | 66 | 291 | 309 | 229 | 238 | 324 | 339 |
| Number of aligned base positions | 354 | 869 | 300 | 132 | 365 | 651 | 849 | 3,521 |
| Number of variable characters | 245 | 467 | 134 | 43 | 111 | 275 | 563 | 1,839 |
| Number of PI characters | 204 | 387 | 111 | 20 | 96 | 187 | 477 | 1,485 |
| % PI characters | 57.63 | 44.53 | 37.00 | 15.15 | 26.30 | 28.73 | 56.18 | 42.18 |
| Nodes with at least 70% bootstrap support | 109 | 37 | 59 | 2 | 61 | 53 | 154 | 200 |
| Nodes with at least 70% jackknife support | NA | NA | NA | NA | NA | NA | NA | 173 |
| MPT length | NA | NA | NA | NA | NA | NA | NA | 10,672 |
| Model of DNA sequence evolution | GTR+I+G | GTR+I+G | GTR+I+G | GTR+I+G | GTR+I+G | GTR+I+G | GTR+I+G | GTR+I+G |
PI, Parsimony informative; MPT, most parsimonious tree.
Multilocus phylogenetic analysis of the CA under maximum-likelihood (ML) and MP yielded phylogenies with highly similar topologies and clade support (Fig. 6). These phylogenies indicated strong support (defined here as presence of a clade in the ML and MP consensus trees and JK or bootstrap [BS] support >70%) for 12 of the 14 recognized species complexes as clades. Support values are reported here in the following format: MP JK%/ML BS% and “*” indicates support <70%. Clades Boninense (95/76) and Orbiculare (100/100) were among these 12; however, these analyses agreed that C. feijoicola is a well-supported member of clade Boninense, which is in contrast with its previously published placement in clade Orbiculare (Bhunjun et al. 2021). Clade Graminicola was also among these 12; however, members of the C. caudatum species complex (sensu Crouch 2014) plus three C. graminicola species complex members (C. ochracea, C. duyuense, and C. caudisporum) comprised a well-supported clade (98/99) nested within another containing all remaining C. graminicola species complex members (93/99), so we refer here to this monophyletic unit collectively as “clade Graminicola.”
Fig. 6.
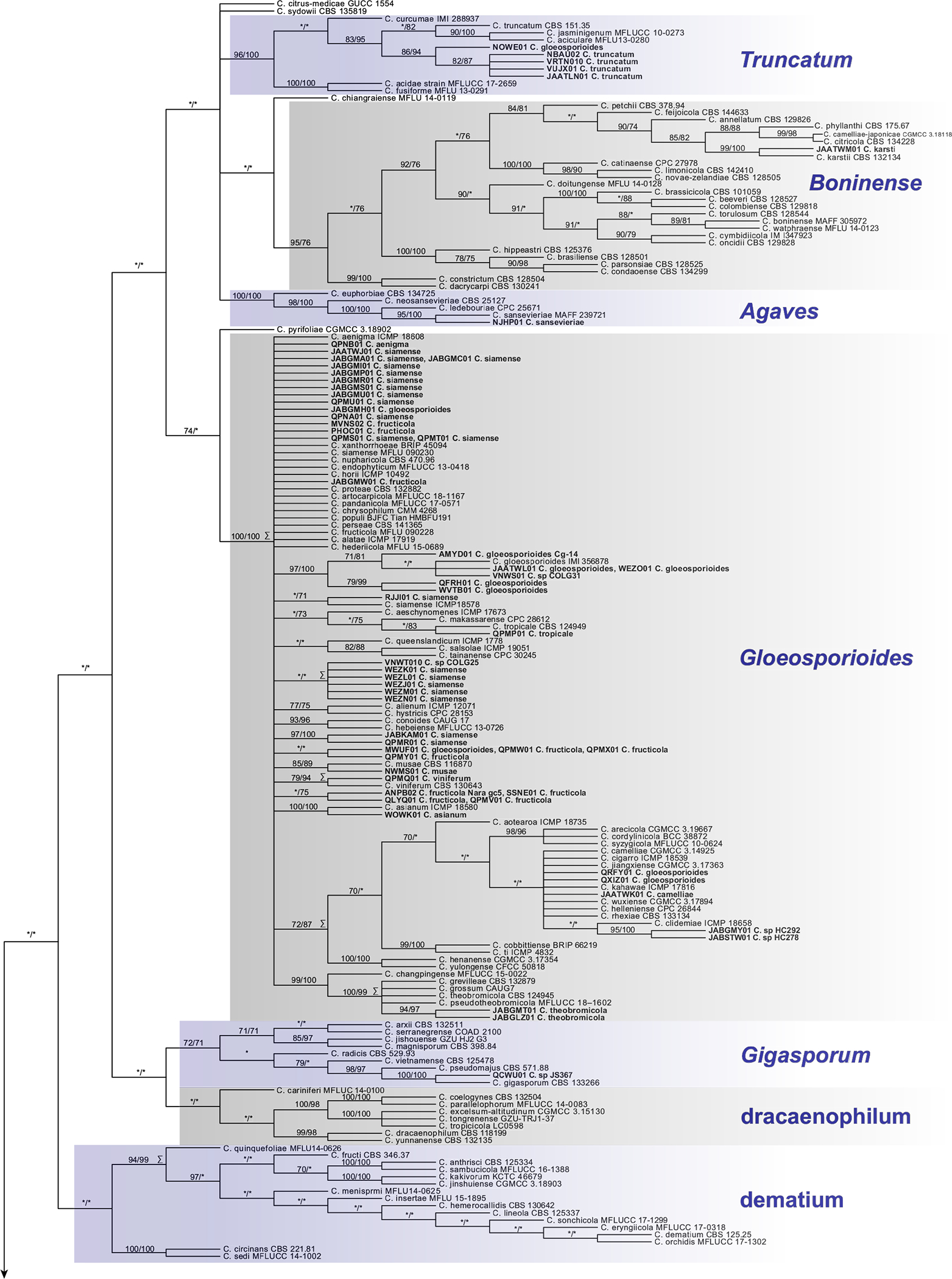
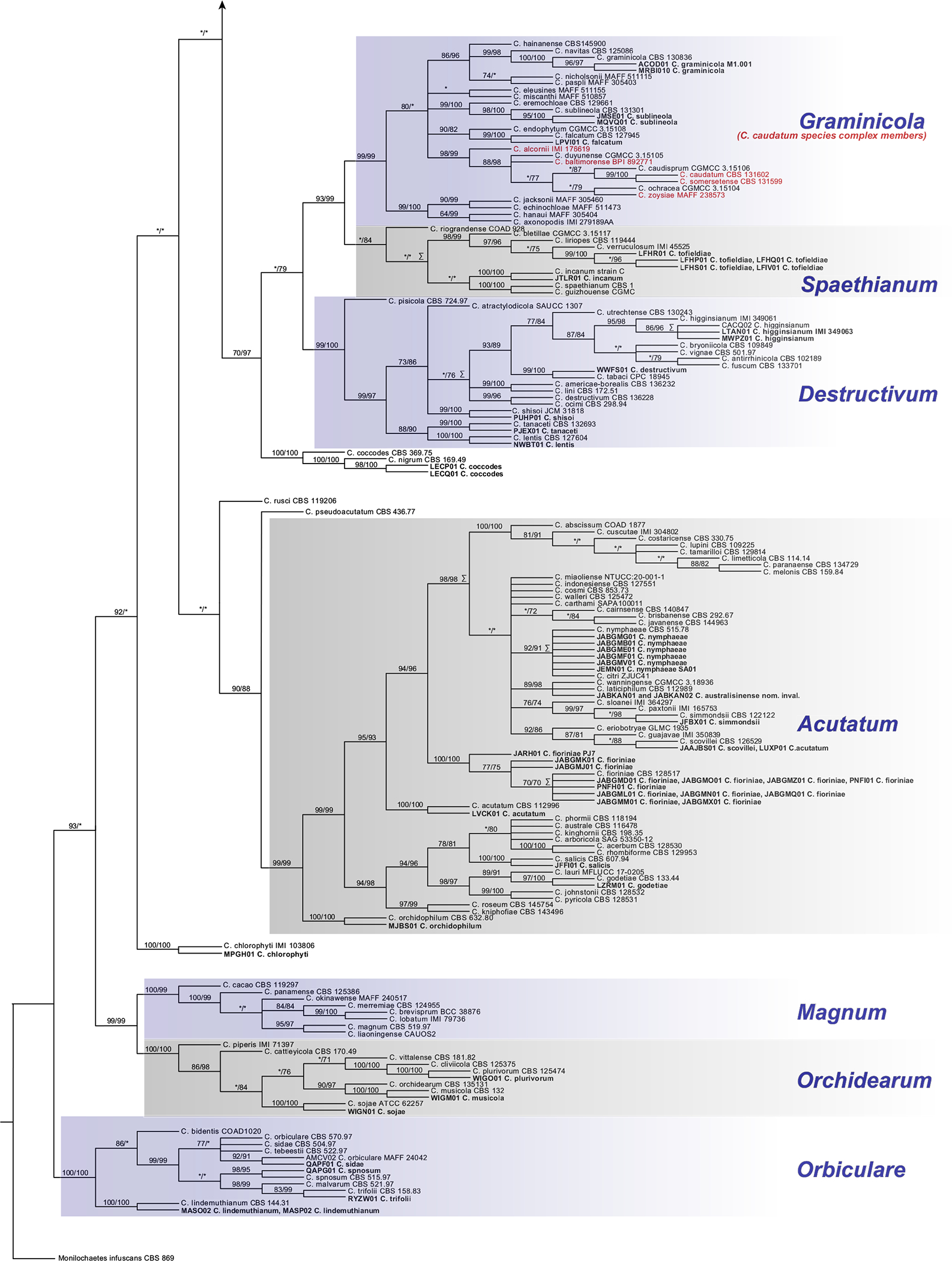
The strict consensus (SC) phylogeny representing a summary of all equally maximum-parsimony (MP) trees resulting from MP analysis of the concatenated alignment (ACT, CAL, CHS, GAPDH, HIS, ITS, and TUB2; see main article for definitions). Names of clades corresponding with previously recognized species complexes that are well supported by jackknife (JK) or bootstrap (BS) are capitalized and italicized. These support values (JK/BS) are shown only on nodes that were also recovered in the most optimal maximum-likelihood (ML) tree and only when ≥70%. Nodes with an asterisk indicate JK or BS support of <70%. Taxa in bold represent the GenBank genome assemblies. Multiple taxa at a terminal indicate that their concatenated sequences were identical and represented only once in the analyzed alignment. The MP SC tree and the most optimal ML tree were highly congruent with respect to topology. Nodes with incongruity between these trees are highlighted with the symbol “Σ.”
Neither MP JK or ML BS support were recovered for all members of the C. dematium species complex or the C. dracaenophilum species complex. All members of the C. dematium species complex were recovered (94/99), except for C. circinans and C. sedi, which were well supported as sister species (100/100) to the remaining members of the complex in the MP strict consensus tree. However, this relationship between the core members of the C. dematium species complex and C. circinans and C. sedi is not supported by either MP JK or ML BS. All members of the C. dracaenophilum species complex were recovered as monophyletic in the MP strict consensus tree, but this clade is similarly unsupported by MP JK and ML BS. Monophyly of five of its members, however, was well supported (100/98).
Little well-supported resolution was found for relationships among basal portions of the MP and ML phylogenies, including relationships among species complexes, except for a clade (99/99) containing clades Magnum and Orchidearum, and a clade (70/97) containing clades Graminicola, Spaethianum, Destructivum, and a clade (100/100) containing C. coccodes and C. nigrum. This resulted in a large polytomy at the base of the Colletotrichum phylogeny containing lineages that correspond with known species complexes, their unsupported constituents, and several other lineages with one or a few described species, including: C. chlorophyte; C. pyrifoliae (which is sister to clade Gloeosporioides [74/*]); C. rusci; C. chiangraiense; C. sydowii; and C. citrus-medicae. Finally, C. orchidophilum was a well-supported sister to the clade Acutatum (99/99) and C. pseudoacutatum was a well-supported sister to the clade containing C. orchidophilum plus clade Acutatum (90/88).
Comparisons of our phylogeny-based identifications with the corresponding submitter’s identifications indicated that both were identical for 56 (47.9%) of the 117 genome accessions (Table 4). Fifty-eight (49.6%) were either well supported for an alternative identification at the species level relative to the submitter-provided name (four cases with different species-level identifications: LECP01, LECQ01, LUXP01, and WWFS01); or no or poor support for the accession’s submitted identity was found (52 cases, e.g., QPNB01 was identified as C. aenigma by the submitter, but we identified it by phylogeny as Colletotrichum sp.); or genomes with only genus level names were identified to species level (two cases: QCWU01 and VNWS01).
Table 4.
A list of the 117 Colletotrichum genome assemblies accessioned into GenBank with corresponding submitter identification, phylogeny-based identification, similarity-based identifications resulting from analysis using the Classify Sequences Plugin in Geneious Prime v.2021.2 for each of three sets of criteria (SID1, SID2, and SID3), and summary of correspondence between identification methods, and among marker loci based on similarity statistics
| GenBank accession | Accession ID |
Phylogeny ID |
IDs same? | Similarity ID |
%I1 − %I1i | Phylogeny ID and similarity species-level ID identical? |
Similarity ID |
Incongruent locim | Corroborated?n | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Species | Complexa | Speciesb | Complexc | Most similar match (%I)d | %I with acc. IDe | SID1: speciesf | SID2: speciesg | SID3: speciesh | SID1 | SID2 | SID3 | Taxa with I ≥ 99%j | Loci matchedk | Proportion congruentl | |||||
|
| ||||||||||||||||||||
| ACOD01 | Graminicola | graminicola | Graminicola/caudatum | graminicola | Yes | Graminicola | graminicola (99.79) | – | graminicola | graminicola | graminicola | 5.18 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| AMCV02 | Orbiculare | orbiculare | Orbiculare | sp. | No | Orbiculare | orbiculare (99.58) | – | sp. | sp. | sp. | 0.18 | Yes | Yes | Yes | 2 | 5 of 5 | 5 of 5 | 0 | N/A |
| AMYD01 | Gloeosporioides | gloeosporioides | Gloeosporioides | gloeosporioides | Yes | Gloeosporioides | gloeosporioides (99.88) | – | gloeosporioides | gloeosporioides | gloeosporioides | 3.21 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 3 of 4 | ACT = 0.35 | No |
| ANPB02 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.43) | – | sp. | sp. | sp. | 0.13 | Yes | Yes | Yes | 3 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.24 | No |
| CACQ02 | Destructivum | higginsianum | Destructivum | higginsianum | Yes | Destructivum | higginsianum (99.86) | – | higginsianum | sp. | higginsianum | 0.49 | Yes | No | Yes | 5 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| JAAJBS01 | Acutatum | scovillei | Acutatum | scovillei | Yes | Acutatum | scovillei (99.88) | – | scovillei | sp. | scovillei | 0.55 | Yes | No | Yes | 3 | 5 of 5 | 5 of 5 | 0 | N/A |
| JAATLN01 | Truncatum | truncatum | Truncatum | sp. | No | Truncatum | aciculare (99.10) | 99.10 | aciculare | sp. | aciculare | 0.51 | No | Yes | No | 2 | GAPDH | 1 of 1 | 0 | N/A |
| JAATWJ01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.15) | – | sp. | sp. | sp. | 0.02 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 0 of 4 | ACT = 0.35, CHS = 0.41, GAPDH = 0.54, TUB2 = 0.44 | No |
| JAATWK01 | Gloeosporioides | camelliae | Gloeosporioides | sp. | No | Gloeosporioides | camelliae (99.68) | – | sp. | sp. | sp. | 0.14 | Yes | Yes | Yes | 2 | ACT, GAPDH, TUB2 | 3 of 3 | 0 | N/A |
| JAATWL01 | Gloeosporioides | gloeosporioides | Gloeosporioides | gloeosporioides | Yes | Gloeosporioides | gloeosporioides (99.47) | – | gloeosporioides | gloeosporioides | gloeosporioides | 2.19 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JAATWM01 | Boninense | karsti | Boninense | karsti | Yes | Boninense | karsti (99.19) | – | karsti | karsti | karsti | 1.09 | Yes | Yes | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 4 of 4 | 0 | N/A |
| JABGLZ01 | Gloeosporioides | theobromicola | Gloeosporioides | sp. | No | Gloeosporioides | theobromicola (99.27) | – | theobromicola | sp. | sp. | 0.25 | No | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 4 of 4 | 0 | No |
| JABGMA01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.22) | 99.09 | sp. | sp. | sp. | 0.13 | Yes | Yes | Yes | 3 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | ACT = 0.87, GAPDH = 1.07 | No |
| JABGMB01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.39) | – | nymphaeae | nymphaea | nymphaea | 0.68 | No | No | No | 1 | 5 of 5 | 4 of 5 | CHS = 1.06 | No |
| JABGMC01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.22) | 99.09 | sp. | sp. | sp. | 0.13 | No | No | No | 3 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | ACT = 0.87, GAPDH = 1.07 | No |
| JABGMD01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGME01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.39) | – | nymphaeae | nymphaeae | nymphaeae | 0.68 | No | No | No | 1 | 5 of 5 | 3 of 5 | ACT = 0.01, CHS = 0.35 | No |
| JABGMF01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.09) | – | nymphaeae | nymphaeae | nymphaeae | 0.67 | No | No | No | 1 | 5 of 5 | 3 of 5 | ACT = 0.01, HIS = 1.05 | No |
| JABGMG01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.09) | – | nymphaeae | nymphaeae | nymphaeae | 0.67 | No | No | No | 1 | 5 of 5 | 3 of 5 | ACT = 0.01, HIS = 1.05 | No |
| JABGMH01 | Gloeosporioides | gloeosporioides | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.53) | 96.12 | sp. | sp. | sp. | 0.09 | Yes | Yes | Yes | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.95 | No |
| JABGMI01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.22) | – | sp. | sp. | sp. | 0.19 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.35, CHS = 0.41, TUB2 = 0.44 | No |
| JABGMJ01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.7) | – | fioriniae | fioriniae | fioriniae | 3.44 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMK01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.7) | – | fioriniae | fioriniae | fioriniae | 3.44 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGML01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMM01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMN01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMO01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMP01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.13) | 99.09 | sp. | sp. | sp. | 0.04 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | ACT = 0.87, CHS = 0.41 | No |
| JABGMQ01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMR01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.22) | – | sp. | sp. | sp. | 0.19 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.35, CHS = 0.41, TUB2 = 0.44 | No |
| JABGMS01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.22) | – | sp. | sp. | sp. | 0.19 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.35, CHS = 0.41, TUB2 = 0.44 | No |
| JABGMT01 | Gloeosporioides | theobromicola | Gloeosporioides | sp. | No | Gloeosporioides | grevilleae (99.33) | 99.08 | grevilleae | sp. | sp. | 0.25 | No | Yes | Yes | 2 | 5 of 5 | 2 of 5 | CHS = 0.34, GAPDH = 0.73, TUB2 = 0.15 | No |
| JABGMU01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (98.96) | – | sp. | sp. | sp. | 0.12 | Yes | Yes | Yes | 0 | ACT, CHS, GAPDH, TUB2 | 0 of 4 | ACT = 0.35, CHS = 0.41, GAPDH = 0.01, TUB2 = 0.59 | No |
| JABGMV01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.03) | – | nymphaeae | nymphaeae | nymphaeae | 0.46 | No | No | No | 1 | 5 of 5 | 3 of 5 | ACT = 0.01, HIS = 1.05 | No |
| JABGMW01 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.53) | – | fructicola | sp. | sp. | 0.23 | No | Yes | Yes | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.10 | No |
| JABGMX01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABGMY01 | N/A | sp. | Gloeosporioides | sp. | N/A | Gloeosporioides | rhexiae (99.85) | N/A | rhexiae | sp. | sp. | 0.41 | No | Yes | Yes | 2 | TUB2 | 1 of 1 | 0 | No |
| JABGMZ01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JABKAM01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.15) | – | sp. | sp. | sp. | 0.12 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.64, CHS = 1.01, TUB2 = 0.44 | No |
| JABKAN01 | Acutatum | australisinense (nom inval) | Acutatum | sp. | No | Acutatum | wanningense (99.77) | N/A | wanningense | wanningense | wanningense | 1.35 | No | No | No | 1 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.01 | No |
| JABSTW01 | N/A | sp. | Gloeosporioides | sp. | N/A | Gloeosporioides | rhexiae (99.85) | N/A | rhexiae | sp. | sp. | 0.41 | No | Yes | Yes | 2 | TUB2 | 1 of 1 | 0 | No |
| JARH01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (98.97) | 98.97 | sp. | sp. | sp. | 2.71 | No | No | No | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| JEMN01 | Acutatum | nymphaeae | Acutatum | sp. | No | Acutatum | nymphaeae (99.09) | – | nymphaeae | nymphaeae | nymphaeae | 0.67 | No | No | No | 1 | 5 of 5 | 3 of 5 | ACT = 0.01, HIS = 1.05 | No |
| JFBX01 | Acutatum | simmondsii | Acutatum | simmondsii | Yes | Acutatum | simmondsii (99.82) | – | simmondsii | simmondsii | simmondsii | 1.03 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JFFI01 | Acutatum | salicis | Acutatum | salicis | Yes | Acutatum | salicis (99.82) | – | salicis | salicis | salicis | 2.36 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| JMSE01 | Graminicola | sublineola | Graminicola/Caudatum | sublineola | Yes | Graminicola | sublineola (99.79) | – | sublineola | sublineola | sublineola | 0.96 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| JTLR01 | Spaethianum | incanum | Spaethianum | incanum | Yes | Spaethianum | incanum (99.86) | – | incanum | incanum | incanum | 6.89 | Yes | Yes | Yes | 1 | ACT, GAPDH, HIS, TUB2 | 4 of 4 | 0 | N/A |
| LECP01 | Unassociated | coccodes | Unassociated | nigrum | No | Unassociated | nigrum (99.65) | 97.87 | nigrum | nigrum | nigrum | 1.78 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| LECQ01 | Unassociated | coccodes | Unassociated | nigrum | No | Unassociated | nigrum (99.60) | 97.81 | nigrum | nigrum | nigrum | 1.79 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFHP01 | Spaethianum | tofieldiae | Spaethianum | sp. | No | Spaethianum | liriopes (97.5) | N/A | sp. | sp. | sp. | 0.69 | Yes | Yes | Yes | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFHQ01 | Spaethianum | tofieldiae | Spaethianum | sp. | No | Spaethianum | liriopes (97.5) | N/A | sp. | sp. | sp. | 0.69 | Yes | Yes | Yes | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFHR01 | Spaethianum | tofieldiae | Spaethianum | sp. | No | Spaethianum | liriopes (97.5) | N/A | sp. | sp. | sp. | 0.75 | Yes | Yes | Yes | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFHS01 | Spaethianum | tofieldiae | Spaethianum | sp. | No | Spaethianum | liriopes (97.5) | N/A | sp. | sp. | sp. | 0.69 | Yes | Yes | Yes | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFIV01 | Spaethianum | tofieldiae | Spaethianum | sp. | No | Spaethianum | liriopes (97.5) | N/A | sp. | sp. | sp. | 0.69 | Yes | Yes | Yes | 0 | 5 of 5 | 5 of 5 | 0 | N/A |
| LFIW01 | Spaethianum | incanum | Spaethianum | incanum | Yes | Spaethianum | incanum (99.86) | – | incanum | incanum | incanum | 7.07 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| LPVI01 | Graminicola | falcatum | Graminicola/caudatum | falcatum | Yes | Graminicola | falcatum (98.86) | 98.86 | sp. | sp. | sp. | 7.31 | No | No | No | 0 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| LTAN01 | Destructivum | higginsianum | Destructivum | higginsianum | Yes | Destructivum | higginsianum (99.75) | – | higginsianum | sp. | higginsianum | 0.61 | Yes | No | Yes | 4 | 5 of 5 | 5 of 5 | 0 | N/A |
| LUXP01 | Acutatum | acutatum | Acutatum | scovillei | No | Acutatum | scovillei (99.88) | 94.49 | scovillei | sp. | scovillei | 0.55 | Yes | No | Yes | 3 | 5 of 5 | 5 of 5 | 0 | N/A |
| LVCK01 | Acutatum | acutatum | Acutatum | acutatum | Yes | Acutatum | acutatum (99.88) | – | acutatum | acutatum | acutatum | 3.24 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| LZRM01 | Acutatum | godetiae | Acutatum | godetiae | Yes | Acutatum | godetiae (99.5) | – | godetiae | godetiae | godetiae | 1.27 | Yes | Yes | Yes | 1 | CHS, GAPDH, HIS, TUB2 | 3 of 4 | GAPDH = 0.40 | No |
| MASO02 | Orbiculare | lindemuthianum | Orbiculare | lindemuthianum | Yes | Orbiculare | lindemuthianum (99.55) | – | lindemuthianum | lindemuthianum | lindemuthianum | 4.66 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| MASP02 | Orbiculare | lindemuthianum | Orbiculare | lindemuthianum | Yes | Orbiculare | lindemuthianum (99.55) | – | lindemuthianum | lindemuthianum | lindemuthianum | 4.66 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| MJBS01 | Unassociated | orchidophilum | Unassociated | orchidophilum | Yes | Unassociated | orchidophilum (99.76) | – | orchidophilum | orchidophilum | orchidophilum | 9.69 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| MPGH01 | Dematium | chlorophyti | Unassociated | chlorophyti | Yes | Dematium | chlorophyti (99.19) | – | chlorophyti | chlorophyti | chlorophyti | 15.27 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| MQVQ01 | Graminicola | sublineola | Graminicola/caudatum | sublineola | Yes | Graminicola/caudatum | sublineola (99.79) | – | sublineola | sublineola | sublineola | 0.96 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| MRBI01 | Graminicola | graminicola | Graminicola/caudatum | graminicola | Yes | Graminicola/caudatum | graminicola (99.79) | – | graminicola | graminicola | graminicola | 5.18 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 4 of 4 | 0 | N/A |
| MVNS02 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.61) | – | fructicola | sp. | sp. | 0.25 | No | Yes | Yes | 4 | 5 of 5 | 3 of 4 | ACT = 0.08 | No |
| MWPZ01 | Destructivum | higginsianum | Destructivum | higginsianum | Yes | Destructivum | higginsianum (99.69) | – | higginsianum | sp. | higginsianum | 0.49 | Yes | No | Yes | 4 | 5 of 5 | 5 of 5 | 0 | N/A |
| MWUF01 | Gloeosporioides | gloeosporioides | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.53) | 96.05 | sp. | sp. | sp. | 0.16 | Yes | Yes | Yes | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.16 | No |
| NBAU02 | Truncatum | truncatum | Truncatum | sp. | No | Truncatum | truncatum (96.05) | – | sp. | sp. | sp. | 5.64 | Yes | Yes | Yes | 0 | 5 of 5 | 3 of 5 | ACT = 14.02, GAPDH = 0.40 | No |
| NJHP01 | Unassociated | sansevieriae | Agaves | sansevieriae | Yes | Unassociated | sansevieriae (99.87) | – | sansevieriae | sp. | sansevieriae | 0.66 | Yes | No | Yes | 2 | 5 of 5 | 5 of 5 | 0 | N/A |
| NOWE01 | Gloeosporioides | gloeosporioides | Truncatum | sp. | No | Truncatum | truncatum (95.61) | 79.81 | sp. | sp. | sp. | 6.08 | Yes | Yes | Yes | 0 | 5 of 5 | 3 of 5 | ACT = 14.04, TUB2 = 0.03 | No |
| NWBT01 | Destructivum | lentis | Destructivum | lentis | Yes | Destructivum | lentis (99.82) | – | lentis | lentis | lentis | 4.44 | Yes | Yes | Yes | 1 | 5 of 5 | 5/5 | 0 | N/A |
| NWMS01 | Gloeosporioides | musae | Gloeosporioides | musae | Yes | Gloeosporioides | musae (98.60) | – | sp. | sp. | sp. | 0.29 | No | No | No | 0 | CHS, GAPDH, TUB2 | 2 of 3 | TUB2 = 1.31 | No |
| PHOC01 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.62) | – | sp. | sp. | sp. | 0.18 | Yes | Yes | Yes | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.08 | No |
| PJEX01 | Destructivum | tanaceti | Destructivum | tanaceti | Yes | Destructivum | tanaceti (99.47) | – | tanaceti | tanaceti | tanaceti | 3.38 | Yes | Yes | Yes | 1 | ACT, CHS, TUB2 | 2 of 3 | CHS = 0.28 | No |
| PNFH01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| PNFI01 | Acutatum | fioriniae | Acutatum | fioriniae | Yes | Acutatum | fioriniae (99.82) | – | fioriniae | fioriniae | fioriniae | 3.56 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| PUHP01 | Destructivum | shisoi | Destructivum | shisoi | Yes | Destructivum | shisoi (100.00) | – | shisoi | shisoi | shisoi | 3.07 | Yes | Yes | Yes | 1 | ACT, CHS, TUB2 | 3 of 3 | 0 | N/A |
| QAPF01 | Orbiculare | sidae | Orbiculare | sp. | No | Orbiculare | Sidae (99.58) | – | sp. | sp. | sp. | 0.18 | Yes | Yes | Yes | 2 | 5 of 5 | 5 of 5 | 0 | N/A |
| QAPG01 | Orbiculare | spinosum | Orbiculare | spinosum | Yes | Orbiculare | spinosum (99.7) | – | spinosum | spinosum | spinosum | 1.20 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| QCWU01 | N/A | sp. | Gigasporum | gigasporum | No | Gigasporum | gigasporum (94.48) | N/A | sp. | sp. | sp. | 2.84 | No | No | No | 0 | CHS, GAPDH, HIS, TUB2 | 4 of 4 | 0 | No |
| QFRH01 | Gloeosporioides | gloeosporioides | Gloeosporioides | gloeosporioides | Yes | Gloeosporioides | gloeosporioides (99.05) | – | gloeosporioides | gloeosporioides | gloeosporioides | 1.86 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| QLYQ01 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.43) | – | sp. | sp. | sp. | 0.13 | Yes | Yes | Yes | 3 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.73 | No |
| QPMP01 | Gloeosporioides | tropicale | Gloeosporioides | tropicale | Yes | Gloeosporioides | fructicola (98.45) | 98.30 | sp. | sp. | sp. | 0.08 | No | No | No | 0 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | ACT = 1.46, GAPDH = 0.87 | No |
| QPMQ01 | Gloeosporioides | viniferum | Gloeosporioides | viniferum | Yes | Gloeosporioides | viniferum (98.99) | – | sp. | sp. | sp. | 0.68 | No | No | No | 0 | ACT, GAPDH, TUB2 | 1 of 3 | ACT = 1.04, GAPDH = 0.03 | No |
| QPMR01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (98.96) | – | sp. | sp. | sp. | 0.12 | Yes | Yes | Yes | 0 | ACT, CHS, GAPDH, TUB2 | 0 of 4 | ACT = 0.64, CHS = 1.0, GAPDH = 0.37, TUB2 = 0.44 | No |
| QPMS01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.02) | – | siamense | siamense | sp. | 0.36 | No | No | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | CHS = 0.41, TUB2 = 0.38 | No |
| QPMT01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.02) | – | siamense | siamense | sp. | 0.36 | No | No | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 2 of 4 | CHS = 0.41, TUB2 = 0.38 | No |
| QPMU01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.22) | – | sp. | sp. | sp. | 0.09 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.35, CHS = 0.50, TUB2 = 0.44 | No |
| QPMV01 | Gloeosporioides | fructicola | Gloeosporioides | fructicola | Yes | Gloeosporioides | fructicola (99.43) | – | sp. | sp. | sp. | 0.13 | No | No | No | 3 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.24 | No |
| QPMW01 | Gloeosporioides | fructicola | Gloeosporioides | fructicola | Yes | Gloeosporioides | fructicola (99.53) | – | sp. | sp. | sp. | 0.16 | No | No | No | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.16 | No |
| QPMX01 | Gloeosporioides | fructicola | Gloeosporioides | fructicola | Yes | Gloeosporioides | fructicola (99.53) | – | sp. | sp. | sp. | 0.16 | No | No | No | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.16 | No |
| QPMY01 | Gloeosporioides | fructicola | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.53) | – | sp. | sp. | sp. | 0.16 | Yes | Yes | Yes | 4 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.16 | No |
| QPNA01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.15) | – | siamense | siamense | sp. | 0.22 | No | No | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.36, CHS = 0.41, TUB2 = 0.53 | No |
| QPNB01 | Gloeosporioides | aenigma | Gloeosporioides | sp. | No | Gloeosporioides | Aenigma (99.87) | – | aenigma | sp. | sp. | 0.43 | No | Yes | Yes | 3 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | CHS = 0.25 | No |
| QRFY01 | Gloeosporioides | gloeosporioides | Gloeosporioides | sp. | No | Gloeosporioides | rhexiae (100), Jiangxiense (100) | 92.95 | sp. | sp. | sp. | 0.00 | Yes | Yes | Yes | 5 | TUB2 | 1 of 1 | 0 | N/A |
| QXIZ01 | Gloeosporioides | gloeosporioides | Gloeosporioides | sp. | No | Gloeosporioides | rhexiae (100), Jiangxiense (100) | 92.95 | sp. | sp. | sp. | 0.00 | Yes | Yes | Yes | 5 | TUB2 | 1 of 1 | 0 | N/A |
| RJJI01 | Gloeosporioides | siamense | Gloeosporioides | siamense | Yes | Gloeosporioides | siamense (99.87) | – | siamense | siamense | siamense | 0.77 | Yes | Yes | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 4 of 4 | 0 | N/A |
| RYZW01 | Orbiculare | trifolii | Orbiculare | trifolii | Yes | Orbiculare | trifolii (99.58) | – | trifolii | sp. | trifolii | 0.54 | Yes | No | Yes | 2 | 5 of 5 | 5 of 5 | 0 | N/A |
| SSNE01 | Gloeosporioides | fructicola | Gloeosporioides | fructicola | Yes | Gloeosporioides | fructicola (99.43) | – | sp. | sp. | sp. | 0.13 | No | No | No | 3 | ACT, CHS, GAPDH, TUB2 | 3 of 4 | ACT = 0.24 | No |
| VNWS01 | N/A | sp. | Gloeosporioides | gloeosporioides | No | Gloeosporioides | gloeosporioides (99.84) | N/A | gloeosporioides | gloeosporioides | gloeosporioides | 2.85 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| VNWT01 | N/A | sp. | Gloeosporioides | sp. | N/A | Gloeosporioides | fructicola (99.13) | N/A | sp. | sp. | sp. | 0.04 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.87, CHS = 0.50, GAPDH = 0.71 | No |
| VRTN01 | Truncatum | truncatum | Truncatum | sp. | No | Truncatum | truncatum (96.05) | – | sp. | sp. | sp. | 5.64 | Yes | Yes | Yes | 0 | 5 of 5 | 3 of 5 | ACT = 14.43, GAPDH = 0.40 | No |
| VUJX01 | Truncatum | truncatum | Truncatum | sp. | No | Truncatum | truncatum (95.92) | – | sp. | sp. | sp. | 5.76 | Yes | Yes | Yes | 0 | 5 of 5 | 3 of 5 | ACT = 14.43, GAPDH = 0.41 | No |
| WEZJ01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.09) | – | sp. | siamense | sp. | 0.16 | Yes | No | Yes | 1 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.36, CHS = 0.50, TUB2 = 0.59 | No |
| WEZK01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | fructicola (99.13) | 99.09 | sp. | sp. | sp. | 0.04 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.87, CHS = 0.50, GAPDH = 0.71 | No |
| WEZL01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.28) | – | sp. | sp. | sp. | 0.06 | Yes | Yes | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 1 of 4 | ACT = 0.35, CHS = 0.50, TUB2 = 0.59 | No |
| WEZM01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | chrysophilum (98.80) | 98.72 | sp. | sp. | sp. | 0.08 | Yes | Yes | Yes | 0 | ACT, CHS, GAPDH | 0 of 3 | ACT = 0.43, CHS = 0.28, GAPDH = 0.34 | No |
| WEZN01 | Gloeosporioides | siamense | Gloeosporioides | sp. | No | Gloeosporioides | siamense (99.04) | – | sp. | sp. | sp. | 0.04 | Yes | Yes | Yes | 2 | CHS, GAPDH, TUB2 | 2 of 3 | TUB2 = 0.59 | No |
| WEZO01 | Gloeosporioides | gloeosporioides | Gloeosporioides | gloeosporioides | Yes | Gloeosporioides | gloeosporioides (99.84) | – | gloeosporioides | gloeosporioides | gloeosporioides | 2.85 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| WIGM01 | Orchidearum | musicola | Orchidearum | musicola | Yes | Orchidearum | musicola (99.37) | – | musicola | musicola | musicola | 1.43 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| WIGN01 | Orchidearum | sojae | Orchidearum | sojae | Yes | Orchidearum | sojae (98.41) | – | sp. | sp. | sp. | 0.69 | No | No | No | 0 | 5 of 5 | 3 of 5 | CHS = 1.33, HIS = 0.24 | No |
| WIGO01 | Orchidearum | plurivorum | Orchidearum | plurivorum | Yes | Orchidearum | plurivorum (99.66) | – | plurivorum | plurivorum | plurivorum | 0.75 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| WOWK01 | Gloeosporioides | asianum | Gloeosporioides | asianum | Yes | Gloeosporioides | asianum (99.87) | – | asianum | sp. | asianum | 0.85 | Yes | No | Yes | 2 | ACT, CHS, GAPDH, TUB2 | 4 of 4 | 0 | N/A |
| WVTB01 | Gloeosporioides | gloeosporioides | Gloeosporioides | gloeosporioides | Yes | Gloeosporioides | gloeosporioides (99.10) | – | gloeosporioides | gloeosporioides | gloeosporioides | 1.91 | Yes | Yes | Yes | 1 | 5 of 5 | 5 of 5 | 0 | N/A |
| WWFS01 | Destructivum | destructivum | Destructivum | tabaci | No | Destructivum | tabaci (99.72) | 97.35 | tabaci | tabaci | tabaci | 1.32 | Yes | Yes | Yes | 1 | ACT, CHS, HIS, TUB2 | 3 of 4 | CHS = 0.29 | No |
Reports species complex membership for the genome accession when it’s member of a clade containing all species of that complex and supported by ≥70% bootstrap and ≥70% jackknife support in the maximum-likelihood (ML) tree and strict consensus maximum-parsimony (MP) trees resulting from analysis of the concatenated alignment. Note: MPGH01 species complex ID was marked as unknown because C. chlorophyti was not a well-supported member of the core C. dematium species complex.
Reports species name for the genome accession when member of a clade that includes members of one species and is supported by ≥70% bootstrap and ≥70% jackknife support in the ML tree and the strict consensus MP tree resulting from analysis of the concatenated alignment.
Reports the species complex name of the concatenated sequence that was most similar to the query’s (genome derived data) concatenated sequence.
Reports the taxon with the concatenated sequence that was most similar to the query’s concatenated sequence.
Percent similarity between taxon identified in the GenBank record and taxon identified based on sequence similarity; acc., accession.
Reports the database species that met the parameters for a similarity-based ID. If no taxon met the parameters, then the ID was reported at the genus level. A species level ID was determined when the most similar database concatenated sequence was at least 99% similar to the query (i.e., the GenBank accession’s concatenated sequence), and at least 0.2% more similar to the query than the next most similar database sequence.
If more than one with I ≥ 99%, it is listed as sp.
Cutoff was 0.42%, instead of 0.2% as in Similarity ID 1.
Reports the % I of first most similar taxon – % I of second most similar taxon.
Reports the number of database concatenated sequences that are at least 99% similar to the query.
Reports the loci that were available in common from the genome accession (i.e., the query) and for the taxon with the most similar concatenated sequence to the genome accession. This was reported as “5 of 5” when all five loci were available for the query and the taxon with the most similar concatenated sequence.
Reports the proportion of loci that indicate the same most similar taxon as the concatenated similarity result.
Reports the loci that are not most similar to the same database taxon as the concatenated sequence, and the % difference between the most similar taxon for the given locus and the sequence of the taxon whose concatenated sequence was most similar to the query’s concatenated sequence.
Reports whether similarity-based incongruence was corroborated with phylogeny. Reports whether the query (i.e., genome assembly) is in a well-supported sister relationship with different Colletotrichum species between any two single marker trees.
Among the 58 where submitter- and phylogeny-based identifications disagreed, 56 were identified to species level by the submitter, but only six by phylogeny; only two were identified to species level by phylogeny where the submitter identified the genome only to genus level (QCWU01 and VNWS01), and only four (LECP01, LECQ01, LUXP01, and WWFS01) were identified by phylogeny to species level in cases where the submitter also identified the genome to species level – all of which were identified to different names by method. Finally, identifications to species–complex level differed (not including comparisons where the accession’s identification was ambiguous) between submitter and phylogeny only once (NOWE01). The remaining three (JABGMY01, JABSTW01, and VNWTO10) were not identified to either the species complex or species level by the submitter.
Comparisons of our phylogeny-based identifications with our similarity-based identifications for all 117 genome accessions indicated that they were nonidentical in 30 cases using SID1, 31 cases using SID2, and 20 using SID3. In all these cases, when one method identified a genome to the species level, the other method identified it only to genus level and vice versa. Among the 30 cases using SID1, 19 were identified to species level by similarity and 11 with phylogeny. Using SID2, 12 were identified to species level by similarity and 19 with phylogeny. Using SID3, nine were identified to species level by similarity and 11 with phylogeny.
Among the 58 genome accessions where submitter- and phylogeny-based identifications were nonidentical, 18 also differed at the species level between phylogeny and SID1 (Table 4). SID1 resulted in species-level identification for 17 of these 18 genomes and for only one by phylogeny (QCWU01). In all these 18 cases one method resulted in a species name and the other did not (e.g., QPNA01, C. siamense versus Colletotrichum sp.). The CA phylogeny shows zero support for seven of the 17 similarity-based species-level identifications (e.g., JABGLZ01 is member of a well-supported clade (BS = 92) that contains C. grevilleae and C. pseudotheobromicola, but not C. theobromicola). Among the remaining 10, phylogeny suggests that the similarity-based identification could be correct (e.g., JABSTW01 and JABGMY01 are sister taxa within a polytomy containing 17 species, including C. rhexiae). Examination of the single case where phylogeny resulted in a species name, when similarity did not, shows that although the sequences from QCWU01 are most similar to that of C. gigasporum, similarity is very low for each (concatenated comparison = 94.48%), suggests that QCWU01 is from an undescribed species whose closest known relative is C. gigasporum.
Among the 58 genome accessions where submitter- and phylogeny-based identifications differed, only 12 of the 18 that differed between SID1 and phylogeny also differed between SID2 and phylogeny and 10 for SID3 and phylogeny. This suggests that SID3 criteria result in higher rates of agreement with phylogeny than those of SID1 or SID2. SID2 identified 11 of these 12 to species level, whereas SID3 identified nine of 10 to species level. In each of these comparisons one method identified the accession to species level while the other method identified it to only genus level.
Topological- and similarity-based congruence were evaluated among individual marker loci. The main results are as follows with additional detail described in the Supplementary Text S1. In this context, we define incongruence as a situation where at least one independent marker locus from the database taxon that is most similar to the concatenated sequence of the query taxon is not most similar to the same database taxon. We independently evaluated incongruence in situations where this difference was >0.0, >0.50, and >1.0% (Table 4).
The number of genome accessions with at least one incongruent locus based on similarity with a % similarity difference > 0 was 53 (45.3%). There were 32 (27.4%) with a % similarity difference > 0.50, and 16 (13.7%) with a % similarity difference > 1.00 (Table 4). Of these 16, four had at least one locus with a % similarity difference > 2.00; these same four (NBAU02, NOWE01, VRTN010, and VUJX01) had a % similarity difference > 14.00, all of which were identified as members of clade Truncatum by phylogeny.
Topological incongruence was not corroborated for any of the 53 genome accessions that indicated similarity-based incongruence for at least one locus. However, during these investigations, significant topological incongruence was revealed for up to five of six species of clade Truncatum. High levels of similarity-based incongruence were observed in the ACT sequences of NBAU02, NOWE01, VRTN010, and VUJX01.
The fungal protein distance tree curation tool used by the RefSeq group at NCBI displayed a Colletotrichum-subtree (Supplementary Fig. S3) topology that compared well with the SNP tree topology of Colletotrichum genomes in Eaton et al. (2021). A diverse group of 35 genomes in the SNP tree overlapped with the genomes displayed in the protein distance tree and the phylogenetic placements were in the same clades in both trees. The recently published genome-scale phylogeny of the kingdom Fungi (Li et al. 2021) contained 24 Colletotrichum genomes that overlapped with genomes present in the NCBI Fungi protein distance tree. The NCBI Fungi protein distance tree displayed the same phylogenetic placements of the Colletotrichum species complexes as in the published multialignment concatenation-based approach. It was therefore (with good support) easy to find the misidentified NOWE01 assembly labeled as C. gloeosporioides in clade Truncatum and the correctly placed genomes of clade Gloeosporioides (Supplementary Fig. S3). Beyond the Colletotrichum taxa, the Fungi protein distance tree compared well at higher ranks (Robbertse et al. 2019) with the previously published yeast genome-scale phylogeny (Shen et al. 2018) and the recent Fungi phylogeny (Li et al. 2021), both of which also made use of the OrthoDB (https://www.orthodb.org/) orthologs (Fungi v.9) for their analysis.
Discussion
Greater clarity is needed in Colletotrichum species descriptions focused on updates to sequence submissions and voucher annotations to improve public data repositories. Curators for the NCBI Taxonomy often deal with the challenges of releasing species names accurately tied to sequence records in the NCBI and International Nucleotide Sequence Database Collaboration (https://www.insdc.org/) databases. Besides numerous misspellings and orthographic variants, a particularly challenging area is in releasing unpublished names after they are validly published. Numerous records are not updated by submitters after a new species name is published (Schoch et al. 2017) and this problem is quite noticeable in an actively researched genus like Colletotrichum. Figure 1 indicates the accumulation of new species names in the genus over time as well as their public release in NCBI Taxonomy upon sequence submission to the International Nucleotide Sequence Database Collaboration databases.
A recent paper made numerous additional recommendations on best practices to follow during the process of describing a new species (Aime et al. 2021). This included a set of recommendations to perform after an article is published to ensure that new names are documented accurately, and we recommend readers follow the suggestions therein. We also want to highlight recommendations to treat voucher information on public sequence records with special care. Submitting type material identifiers from public collections, as stated in species descriptions, with sequence data will ensure that these type of records are accessible to serve as references.
We took a conservative approach to incorporating Colletotrichum names in NCBI’s Taxonomy and into the analyses of this study by only accepting names after evaluating their original species descriptions in the primary literature and determining their validity. For example, several names were excluded because their descriptions lacked critical typification information; and holotypes were not designated in the descriptions of several species, including C. australisinense, C. bannaense, C. ledongense, and C. philodendricola. Associating strains with these names was guided exclusively by whether this information was properly documented in the original description or in a later epitypification.
We incorporated species complex names in NCBI Taxonomy based on validation by our phylogenetic analyses where each species-complex circumscription commonly used in the literature during the last decade was treated as a hypothesis. These analyses confirmed the monophyly of most these species complexes as previously circumscribed with the exceptions noted above, including the C. caudatum complex, whose species were all well supported members of clade Graminicola. In the spirit of recognizing the major clades of Colletotrichum, we recognize species of the C. caudatum species complex (sensu Crouch 2014) only as members of clade Graminicola. We recognize clade Spaethianum as synonymous with the C. spaethianum species complex in NCBI Taxonomy because each was recovered in our MP strict consensus tree and most optimal ML tree, and was well supported by at least BS analysis, unlike the C. destructivum and C. dematium species complexes. We elected to associate names included in clade Agaves, sensu Bhunjun et al. (2021), with clade Agaves in NCBI Taxonomy despite the issue that no sequence data exist from the type strain of C. agaves to verify its phylogenetic position within Colletotrichum. Nevertheless, we use Agaves as clade/species complex name, rather than selecting another, knowing that species-complex is utilized as an informal rank not governed by the Code.
We took a conservative approach in this study to associating sequences with names because a goal was to identify and populate RTL only with sequences that we could validate as derived from strains associated with verified names, and eliminate sequences with indicators that call into question the quality or traceability to its type strain source material. For example, we excluded several species, including C. alcornii, C. axonopodi, C. baltimorense, C. caudatum, C. citri, C. citricola, C. somersetense, and C. zoysiae because their ITS1 and/or ITS2 sequences were too long or too short based on RTL genus-length quality standards.
ITS sequences can be used to identify Colletotrichum unknowns to species-complex level in most but not all cases. Misidentified sequences in GenBank are misleading or uninformative when only identified to Kingdom level. To prevent Colletotrichum species from going undetected or misidentified it is best to use the curated RTL Colletotrichum ITS dataset in alignments to verify the identity or placement of sequences. Alternatively, BLAST users should search against this dataset in the dedicated RTL BLAST database on the recently updated BLAST web interface (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) instead of using the default GenBank Nucleotide (Nr/Nt) database (Sayers et al. 2020). The Taxonomy Lineage Report displays the lineage information of the top hits and may indicate the appropriate species complex if there is a clear high-to-low distribution in the top hits above 95% identity. However, if the query is equally different from all the complex members and top hits are ~96.5%, for example, then the query sequence may not necessarily belong to the same species complex as the top hits. In the case of uncertainty, it is then better to use a phylogenetic method (which takes position-specific information into account), because BLAST results provide only a summarized and limited analysis of the relationship between sequences. Except for ITS sequences from two species (C. riograndense and C. orchidis), all other RTL ITS sequences from 224 species clustered by the species complex to which they belong, in a strict consensus MP tree. It is possible that the observed variation in these two sequences may not be biological, considering the overall consistency with which ITS copies from type material and verified ITS copies originating from genome assemblies clustered with ITS copies of the same complex.
When all Colletotrichum ITS records in GenBank were compared against the RTL Colletotrichum ITS dataset, ~97% were confirmed as likely originating from a Colletotrichum fungus (≥440 alignment and ≥92% identity). The rest had additional sequence quality issues or better alignment and identity to other Fungi. The genus most frequently mistaken for Colletotrichum was Fusarium and, indeed, it has been mentioned by others (Shivaprakash et al. 2011) that (without close inspection) the conidia of Fusarium and Colletotrichum spp. can be confused.
UNITE (https://unite.ut.ee/repository.php) is another resource that curates ITS sequences but their approach is different. It is focused on the clustering of all GenBank and UNITE ITS sequences and generating species hypothesis clusters at different distances. These clusters and associated names are then consumed by metabarcoding software pipelines to identify environmental sequences generated by next-generation sequencing. UNITE’s main focus is not detailed type material–name–sequence verification and curation of associated public collection identifiers. Instead, they state that the NCBI RTL is used as one source to inform their taxonomic annotation of similar sequences (Nilsson et al. 2019). However, in the case of species complexes, a species hypothesis cluster may contain more than one sequence from type material of different species. Thus, sequences identical to one of the type material sequences may be assigned the species name of the cluster, which is not necessarily the same name associated with the identical type material sequence (Robbertse et al. 2017). In addition, the classification that UNITE uses does not include informal species complex name designations.
RIP likely plays an important role in the variable quality of ITS sequences seen in Colletotrichum genome assemblies. As seen before in the review of Trichoderma sequences (Robbertse et al. 2017), ITS copies included in genome assemblies should be carefully evaluated for quality and origin to exclude using assembly sequences from contaminants or chimeras. The biological process of RIP in Colletotrichum (Bengtsson-Palme et al. 2013; Lelwala et al. 2019; Rao et al. 2018) and other ascomycetes (van Wyk et al. 2021) should also be taken into consideration when doing an ITS BLAST search against a genome assembly. Sometimes the RIP mutation altered the sequence so much that, in our review, the full 5.8S gene was not recognized in, for example, WVTB01000107.1 (Supplementary Fig. S4). However, in this case an alignment using a BLAST search with the complete ITS region was produced, but with low % identity (85%) for NR_160754.1 against WVTB01000107.1. During the usual RefSeq curation, the low identity of the ITS type material sequence in the WVTB01 (C. gloeosporioides) assembly was (initially) a concern. However, during this intensive review it became evident that the low identity can be attributed to the process of RIP. The submitted identity of WVTB01 was also confirmed by the phylogenetic and similarity-based analyses using the protein coding type marker loci (Fig. 6) and can be considered for reference assembly in the RefSeq database. The quality of ITS copies included in genome assemblies should be carefully evaluated and confirm that all copies indeed originate from the targeted organism and not from contaminants or chimeras.
The Fungi protein distance tree curation tool used by the RefSeq group at NCBI is searchable by any rank below the kingdom. Genome assemblies are highlighted if the searched rank is associated with the genome’s species in the NCBI Taxonomy classification. For example, searching the Fungi tree for the rank “Colletotrichum gloeosporioides species complex” and zooming into the clades containing the highlighted leaves display the misidentified GCA_002901105.1 assembly labeled as C. gloeosporioides in clade Truncatum and the correctly placed genomes from clade Gloeosporioides in clade Gloeosporioides (Supplementary Fig. S3). It has also been previously reported that this genome assembly was incorrectly deposited as C. gloeosporioides (Rogério et al. 2020). Another genome misidentification was reported by Shin et al. (2019), where the genome from strain KC05 was reidentified as C. scovillei using combined sequence analyses of multiple genes. Other genomes that need attention regarding their submitted identification have been indicated in the interactive protein distance tree (Supplementary Fig. S3). The RefSeq assembly GCF_011075155.1 (GenBank: GCA_011075155.1 [JAAJBS01]) from C. scovillei is an example of a genome serving as a reference for evaluation of closely related genomes. By comparison, the LUXP01 assembly is incorrectly labeled as C. acutatum (Supplementary Fig. S3). This conclusion is also supported by the comparison with the marker gene loci from type material (Table 4) and the phylogenetic analysis, which verified the identity of another genome assembly LVCK01 from C. acutatum that is more distantly placed from the RefSeq C. scovillei genome (Supplementary Fig. S3). Identification of two RefSeq assemblies (QPNA01 and VUJX01) using the phylogeny- and similarity-based identifications discussed in this article were inconclusive to the species level, but this result can be attributed to likely sequence-name bookkeeping or sequence errors in type marker data and the conservative approach being taken here. It is also interesting to note that the RJJI01 assembly of C. siamense from type material was the only assembly confirmed as C. siamense by the marker loci phylogeny- and similarity-based identifications, while other assemblies were not. However, a recent in-depth examination of the species boundary of C. siamense s.l. was conducted including molecular and wet-lab analyses (Liu et al. 2016), concluding that C. siamense s.l. is a single species (with no obstruction to gene flow) instead of a species complex. Gan et al. (2017) also supported the wider species concept of C. siamense, and in their analysis included sequence data from the type strain ICMP 18578 (source of assembly RJJI01) and the strain Cg363 (source of assembly QPNA01). The average genome-wide nucleotide identity between the type strain assembly and the QPNA01 assembly is 96% and with an average of 96.1% to all C. siamense assemblies in GenBank.
Marker selection for final phylogenetic analyses was informed by several rounds of ML analyses with various marker sets. Although we present verified sequence data here for ACT, ApMat, Apn2, CAL, CHS-1, GAPDH, GS, HIS, Mat1Apn2, SOD, and TUB2, sequences for some are unavailable for most Colletotrichum species (Table 2). For example, verified ApMat and GS are available primarily only for species of the clade Gloeosporioides, whereas verified Apn2 and Mat1Apn2 are only available for species of clades Graminicola and Caudatum. We settled on including only markers with high coverage across the genus, because we did not find convincing evidence that adding the other markers to the CA would be of greater value than excluding them due to potential issues related to gaps in character sampling among major clades. One example may be that ML trees resulting from analysis of the CA versus analysis of all loci indicated different intrageneric positions of clade Acutatum; however, its position was poorly supported in both trees. We also did not find significant improvements to resolution or node support when analyzing species complexes alone with custom marker sets. For example, ML analysis of a concatenated alignment of ACT, ApMat, CAL, CHS-1-1, GAPDH, GS, HIS, SOD, and TUB2 for all members of clade Gloeosporioides similarly did not result in noteworthy improvements to resolution or node confidence relative to analysis of the CA.
We also took a conservative approach when presenting and interpreting well-supported phylogenetic relationships relative to many previous studies of evolutionary relationships within Colletotrichum by presenting the MP strict consensus tree topology and overlaying only support values from MP JK and ML BS analyses >70%. There were no well-supported incongruities between the most optimal ML tree and the MP strict consensus tree that were consequential to the questions addressed in this study; nevertheless, each is indicated in Figure 6. We found a general pattern of low BS support at nodes basal to those corresponding with recognized species complexes and higher values at nodes nearest to the tips. This pattern was expected, given that marker selection for Colletotrichum has been optimized for species discovery and recognition (Sharma et al. 2013, 2014; Silva et al. 2012); however, it has resulted in a high degree of uncertainty in the pattern of early divergences and the relationships among its major clades. Although lack of resolution among basal divergences is less impactful to this study than resolution at the tips, knowledge of these details would likely provide important insights into various tangential topics, such as the evolution of morphological characters (Sánchez-García et al. 2020), pathogenicity (Laraba et al. 2021), or host relationships (Wang et al. 2019).
All 15 recognized species complexes were well supported with the exceptions of C. dematium and C. dracaenophilum, which were poorly supported by JK and BS, and C. caudatum, the species of which are nested within clade Graminicola. These results contrast with the findings of Bhunjun et al. (2021), who found more robust MP and ML BS support for the C. dematium and C. dracaenophilum species complexes. Also, we identified a clade Gigasporum containing C. arxii, C. gigasporum, C. jishouense, C. magnisporum, C. pseudomajus, C. radicis, C. serranegrense, and C. vietnamense; however, Bhunjun et al. (2021) found C. chiangraiense as another member of clade Gigasporum, whereas this species occupies an unsupported sister relationship with clade Boninense in our MP tree and with a grade containing C. citrus-medicae + clade Boninense + clade Agaves in our ML tree. These discrepancies may be because of differences in strain/reference sequence selection, marker selection, or alignment strategy. For example, we included only verified strains in analyses, concatenated and analyzed two additional marker loci HIS and CAL, and eliminated much of the GAPDH alignment from analysis based on Guidance2 output, which indicated those positions were unreliable for phylogenetic analysis.
Although a higher degree of confidence exists for species relationships within the major clades than among major clades, plenty of uncertainty remains within. This is most well illustrated in clade Gloeosporioides, where most species lack well-supported sister-species relationships. Given the addition of the informative markers ApMat and GS to analysis of this complex does not result in much additional resolution for species relationships, which may suggest that some recognized species boundaries may be overly restrictive or unjustified without additional data. Species names in Colletotrichum have rapidly accumulated over the last 10 years after a temporary plateau, beginning in the mid-1960s, was reached (Fig. 1). This includes an increase within clade Gloeosporioides, where some names have since been synonymized (Liu et al. 2016). Other studies investigating species boundaries have not noted evidence for such species conflation in clade Gloeosporioides (Bhunjun et al. 2021; Weir et al. 2012), but analyses with larger intraspecific sampling may be required to further address this hypothesis. Alternatively, lack of resolution within clade Gloeosporioides, and others, may be a function of recent and rapid radiation events and lack of coalescence and accumulation of synapomorphies at the marker loci considered (Maddison and Knowles 2006; Matute and Sepulveda 2019). A molecular dating study by Bhunjun et al. (2021) estimated the stem ages of the 14 major clades of Colletotrichum range from 55.8 mya for clade Orbiculare to 14.8 mya for clades Magnum and Orchidearum. The stem age of clade Gloeosporioides was estimated at 31.7 mya, notably older than several other species complexes with higher levels of resolution and support for their described species.
We found that character sampling was uneven among species and species complexes across Colletotrichum. This resulted in some challenges when selecting markers for estimating Colletotrichum phylogeny and for species identification. Additional data collection from markers with missing data from the type strains identified in this study may help to improve the results of all future studies of Colletotrichum systematics, specifically resolution among closely related species and improved determinations of species boundaries.
The DNA sequence reference set presented in Table 1 indicated its value as an important identification resource by revealing errors and other shortcomings related to the submitter-based identifications of the Colletotrichum genome accessions in the Assembly database at NCBI. GenBank is populated most densely with sequences from clade Gloeosporioides (51) and clade Acutatum (26). However, the number of unique species names identified within these groups (with “sp.” counted once per clade) is much lower at eight and seven, respectively, indicating that both are represented by multiple individuals of a single species. In total, 41 discrete Colletotrichum species are distributed among 11 of its 14 major clades plus the unassociated species C. orchidophilum, C. chlorophyti, and C. nigrum (Fig. 6). These data indicate that only 17.6% of the 233 verified species in this study are represented with genome data in GenBank, and these data are unevenly distributed among the major lineages of Colletotrichum (Fig. 7). We encourage submissions of genome sequence data that were generated from well-identified specimens that have been accessioned into a public biorepository (culture collections and herbaria), particularly from clades that are most underrepresented, such as Magnum (0) and Boninense (1), and members of the C. dracaenophilum and C. dematium species complexes (0).
Fig. 7.
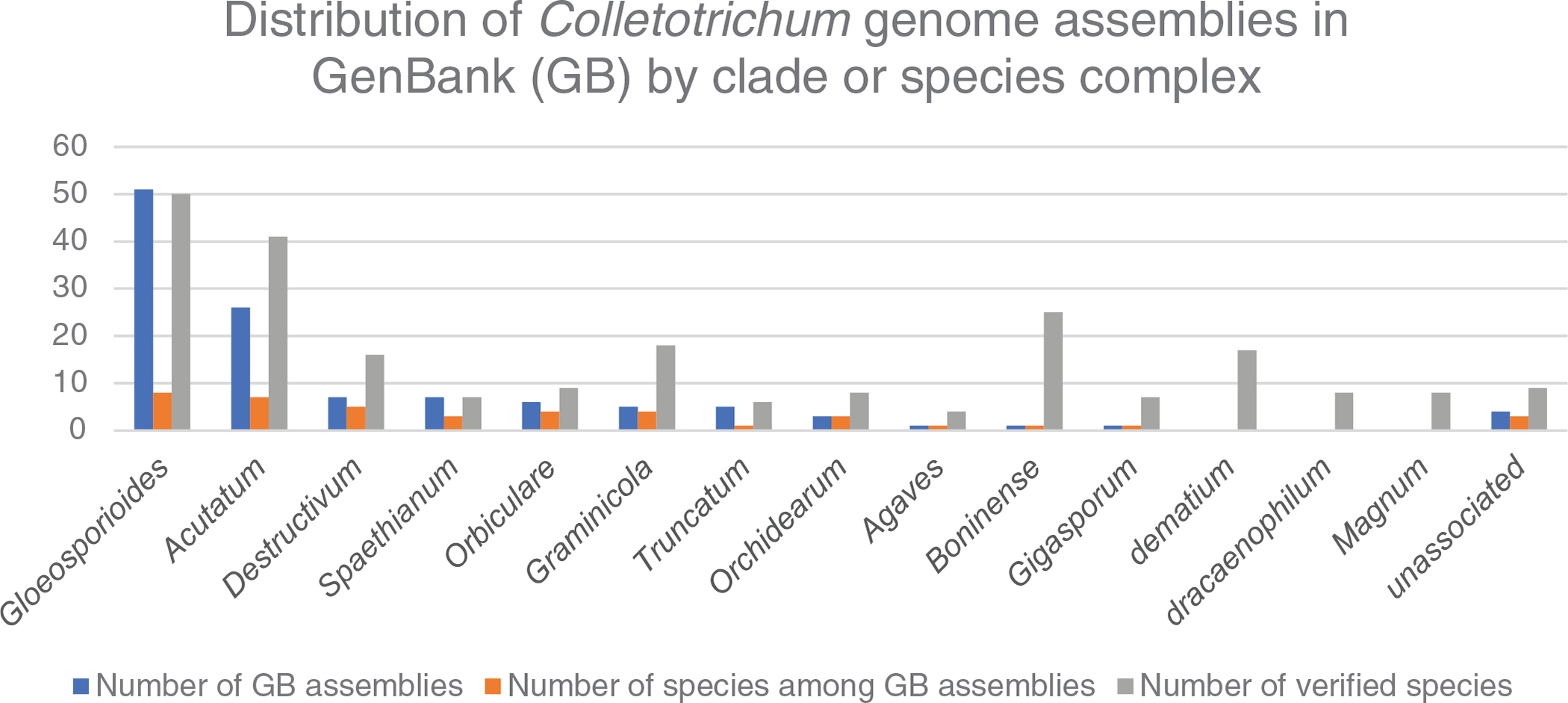
The number of Colletotrichum genome assemblies in GenBank (GB) and the number of species these assemblies represent relative to the number of species names verified during this study. Species per complex were counted based on our phylogenetic identifications; genus level IDs (“Colletotrichum spp.”) were counted as one species per complex.
We advocate for using multilocus phylogenetic analysis to estimate evolutionary relationships among lineages of Colletotrichum, the presentation of consensus trees (e.g., instead of hand-picked single most parsimonious trees), and resampling analyses such as JK and BS to determine levels of character support, where low levels should be interpreted as indication that the relationship is unlikely and should not garner recognition that could be misconstrued as support. We also advocate for phylogenetic species recognition (PSR; Harrington and Rizzo 1999), as a tool for species identification in Colletotrichum, but with some caveats.
Although the PSR may fail to detect recently diverged species because of incomplete lineage sorting (ILS) and lack of accumulated synapomorphies, which may lead to an underestimate of species diversity (Kizirian and Donnelly 2004), it relies on the presence of synapomorphies, a residual signature of descent with modification, to associate an “unknown” with a verified taxon while not requiring reciprocal monophyly (Taylor et al. 2000). This permits identification of an unknown that falls within a well-supported monophyletic unit containing a single type specimen irrespective of the monophyly of its sister species, which in many cases may be a polytomy containing multiple type specimens or a paraphyletic grade. The Genealogical Concordance Phylogenetic Species Recognition method (Avise and Ball 1990; Taylor et al. 2000), a widely utilized species recognition method for Fungi (Hilario et al. 2021; Laurence et al. 2014; Liu et al. 2016; Samarakoon et al. 2018; Xu et al. 2018), may help to address species recognition in Colletotrichum, but we find it operationally impractical to apply in this use-case because several large individual trees would need to be painstakingly compared and different phylogenetic optimality criteria do not yield 100% congruent topologies. Furthermore, our study does not contain sufficient intraspecific sampling to robustly evaluate species boundaries, which we agree would be ideal.
We found that comparisons of pairwise sequence similarity using concatenated sequences provided a rapid proxy for identifying most Colletotrichum genomes using the Classify Sequences plugin (Geneious; https://www.geneious.com/tutorials/sequence-classifier/). The best performing criteria were those of SID3 with only 21 disagreements with phylogeny-based ID, indicating that similarity thresholds determined by empirical data can improve the accuracy of species identification in Colletotrichum. Nevertheless, SID3 provided over-confidence in 11 of these 21 disagreements by determining a species-level ID for each, despite lack of phylogenetic support for the same conclusion.
Complex patterns of similarity were identified among single markers for nearly half (53) of the 117 genomes. These were distributed in cases where identifications by similarity (SID3) agreed and disagreed with those by phylogeny. The largest proportion of cases with these patterns resulted in genus-level identification by both methods (33). This may suggest that although no significant topological incongruence was detected among single marker trees, these patterns may still obscure species recognition regardless of the identification method used.
Analyses of the 64 genomes for which no complex patterns of similarity were detected among loci resulted in species-level identification agreement between phylogeny and SID3 in 93.8% (60) of these cases versus in only 71.7% (38) of cases with such complex patterns. Furthermore, 78.1% (50) of the genomes without this complex pattern were identified to species level by phylogeny whereas only 22.6% (12) of genomes with this complex pattern were identified to species level by phylogeny. This indicates further that similarity-based incongruence among loci of Colletotrichum species may cause the difficulty associated with identifying these genomes to the species level. However, at least one other factor is at play here, and it obscures our ability to determine the scope of the impact made on identifications by incongruent patterns of similarity among loci. The total number of genus-level IDs based on phylogeny among all 117 genomes was nearly half (56); however, we do not have evidence to suggest that genomes without these complex patterns would remain difficult to identify if additional sequence data were available, as we hypothesize is true for those having these patterns. For example, three or fewer of the five loci used for analyses were unavailable for 10 of the 53 with complex patterns and only 20 (35.7%) were represented by the full complement of loci included in the CA. On the other hand, there was twice the proportion (70.3%) of genomes represented by a full complement of loci for those without these complex patterns. Therefore, we suggest that only after having a dataset to analyze without missing data might one be able to determine the impact of these complex patterns as they relate to our ability to identify them to the species level. An additional layer of complexity is that 33 of the 56 (58.9%) genomes that could not be identified to species level by phylogeny were also members of clade Gloeosporioides (51 genomes from this clade in our dataset). This suggests that recognized species in this clade may be too narrowly circumscribed and contain fewer phylogenetic species than is assumed. Furthermore, it’s possible that if these were more broadly circumscribed, there would be no such complex patterns of similarity among their loci.
We evaluated all 53 genomes with complex patterns of similarity among their loci for corroborating phylogenetic evidence of incongruity (i.e., well-supported incongruence among individual gene trees) and were unable to find a single instance. However, well-supported incongruence was detected among single-marker ML phylogenies representing several species within clade Truncatum, including C. aciculare, C. conoides, C. pandanicola, C. jasminigenum, and C. truncatum. Additionally, congruent positions of C. jasminigenum and C. curcumae could not be corroborated within the GAPDH tree relative to other trees that indicated their positions within Truncatum. Given that these incongruities are restricted to publicly available sequences and were not detected in any of the clade Truncatum genome accessions, we cannot rule these patterns out as artifacts of errors made during the sequence submission process.
In summary, we conducted an exhaustive review of Colletotrichum nomenclature and taxonomy and provide a comprehensive set of verified name – type strain – DNA sequence associations from up to 11 nuclear protein coding loci and ITS for 238 species. We evaluated ITS sequence quality for all verified names for which a sequence was available (236), and verified that 226 met the quality standards of the RTL project at NCBI. This effort ensures that the Colletotrichum user community has public and common access to verified name–strain associations for each Colletotrichum species and access to all associated and validated DNA reference sequences.
Our analyses of Colletotrichum genomes confirm in fine detail the utility of a core set of traditional protein-coding marker-loci and ITS as suitable for phylogeny estimation and species identification in Colletotrichum. For example, we confirmed that these protein-coding loci are single-copy, phylogenetically congruent, and sufficiently variable, and that their concatenation improves phylogenetic resolution while helping to avoid errors associated with single marker analyses. However, parsing ITS regions from genome assemblies to represent the sequenced organism should be done with caution because we found 42% of Colletotrichum assemblies that included ITS regions contained issues including contamination, RIP, low sequence quality, or misassembly.
We also verified the phylogenetic positions of each Colletotrichum species and species complex to high degrees of confidence, where possible, using multiple methods and stringent criteria. These data were not sufficient to resolve all nodes with confidence, particularly higher-level relationships, such as those among many species complexes. We used this information to update circumscriptions of each major clade of Colletotrichum and updated NCBI Taxonomy to reflect this understanding.
Complex patterns of sequence similarity among loci throughout Colletotrichum were identified and described, which are likely an artifact of ILS that can prevent species identification regardless of analysis method. We showed that multilocus sequence similarity appears to be a good predictor of phylogenetic identification; however, the complex patterns of similarity found among loci in many genome assemblies indicated clearly that species-level identification should not be based on single-locus similarity analyses (e.g., global pairwise alignments, BLAST, etc.). In cases where multiple single-locus similarity results are not all in agreement, phylogenetic analysis of multiple markers in context with an appropriate taxon sampling should be relied on exclusively for identification purposes. Although this may yield occasional genus-level/species complex-level IDs, they should be accurate – a result that is best suited to inform the plant health community, including PPOs who need to utilize this information to make well-informed quarantine decisions in the interest of protecting agriculture and native systems from alien-pest invasions.
Finally, we evaluated submitter identifications of all 117 Colletotrichum genome assemblies accessioned in GenBank and show that half disagreed with our phylogeny-based identifications, nine of which were clearly misidentified. We foresee a future where genome-level sequence data will be leveraged for species identification purposes and encourage the Colletotrichum user community to submit genome sequences from verified type strains and well-identified specimens that span the diversity of this large genus. This will help us to enter a next and more informative phase of biodiversity exploration of this important genus.
Supplementary Material
Acknowledgments
The work of Stacy Ciufo in generating the species curves from NCBI Taxonomy and Species Fungorum is appreciated. We thank Michael Waring for assistance with the databasing sequences and to the U.S. Department of Agriculture’s Pathways Programs, which provided financial support for his internship; Pete Touhey of the U.S. Department of Agriculture’s APHIS/PPQ National Identification Services for providing historical interception and identification data of fungal pathogens at U.S. Ports-of-Entry; and Biomatters/Geneious, particularly Matt Kearse, for collaborating with the U.S. Department of Agriculture to create the Sequence Classifier Plugin and for assistance with troubleshooting and bug fixes.
Literature Cited
- Aime MC, Miller AN, Aoki T, Bensch K, Cai L, Crous PW, Hawksworth DL, Hyde KD, Kirk PM, Lücking R, May TW, Malosso E, Redhead SA, Rossman AY, Stadler M, Thines M, Yurkov AM, Zhang N, and Schoch CL 2021. How to publish a new fungal species, or name, version 3.0. IMA Fungus 12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung SLL, Liu HF, Pei DF, Lu BB, Oo MM, and Deng JX 2020. Morphology and molecular characterization of a fungus from the Alternaria alternata species complex causing black spots on Pyrus sinkiangensis (Koerle pear). Mycobiology 48:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC, and Ball RM 1990. Principles of genealogical concordance in species concepts and biological taxonomy. Oxf. Surv. Evol. Biol. 7:45–67. [Google Scholar]
- Barbera P, Kozlov AM, Czech L, Morel B, Darriba D, Flouri T, and Stamatakis A 2019. EPA-ng: Massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sanchez-Garcia M, Ebersberger I, de Sousa F, Amend AS, Jumpponen A, Unterseher M, Kristiansson E, Abarenkov K, Bertrand YJK, Sanli K, Eriksson KM, Vik U, Veldre V, and Nilsson RH 2013. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4:914–919. [Google Scholar]
- Bhunjun CS, Phukhamsakda C, Jayawardena RS, Jeewon R, Promputtha I, and Hyde KD 2021. Investigating species boundaries in Colletotrichum. Fungal Divers. 107:107–127. [Google Scholar]
- Blackwell M 2011. The Fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 98:426–438. [DOI] [PubMed] [Google Scholar]
- Cai F, and Druzhinina IS 2021. In honor of John Bissett: Authoritative guidelines on molecular identification of Trichoderma. Fungal Divers. 107:1–69. [Google Scholar]
- Callahan BJ, McMurdie PJ, and Holmes SP 2017. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11:2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon PF, Damm U, Johnston PR, and Weir BS 2012. Colletotrichum – Current status and future directions. Stud. Mycol. 73:181–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantino PD, and de Queiroz K 2020. International Code of Phylogenetic Nomenclature (PhyloCode), 1st ed. CRC Press, Boca Raton, FL. [Google Scholar]
- Chapman D, Purse BV, Roy HE, and Bullock JM 2017. Global trade networks determine the distribution of invasive non-native species. Glob. Ecol. Biogeogr. 26:907–917. [Google Scholar]
- Crouch JA 2014. Colletotrichum caudatum s.l. is a species complex. IMA Fungus 5:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crous PW, Groenewald JZ, Slippers B, and Wingfield MJ 2016. Global food and fibre security threatened by current inefficiencies in fungal identification. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20160024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm U, Cannon PF, Liu F, Barreto RW, Guatimosim E, and Crous PW 2013. The Colletotrichum orbiculare species complex: Important pathogens of field crops and weeds. Fungal Divers. 61:29–59. [Google Scholar]
- Damm U, Cannon PF, Woudenberg JH, and Crous PW 2012a. The Colletotrichum acutatum species complex. Stud. Mycol. 73:37–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm U, Cannon PF, Woudenberg JH, Johnston PR, Weir BS, Tan YP, Shivas RG, and Crous PW 2012b. The Colletotrichum boninense species complex. Stud. Mycol. 73:1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm U, O’Connell RJ, Groenewald JZ, and Crous PW 2014. The Colletotrichum destructivum species complex – Hemibiotrophic pathogens of forage and field crops. Stud. Mycol. 79:49–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm U, Sato T, Alizadeh A, Groenewald JZ, and Crous PW 2019. The Colletotrichum dracaenophilum, C. magnum and C. orchidearum species complexes. Stud. Mycol. 92:1–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm U, Woudenberg JHC, Cannon PF, and Crous PW 2009. Colletotrichum species with curved conidia from herbaceous hosts. Fungal Divers. 39:45–87. [Google Scholar]
- Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, and Foster GD 2012. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton MJ, Edwards S, Inocencio HA, Machado FJ, Nuckles EM, Farman M, Gauthier NA, and Vaillancourt LJ 2021. Diversity and cross-infection potential of Colletotrichum causing fruit rots in mixed-fruit orchards in Kentucky. Plant Dis. 105:1115–1128. [DOI] [PubMed] [Google Scholar]
- Farr DF, Aime MC, Rossman AY, and Palm ME 2006. Species of Colletotrichum on Agavaceae. Mycol. Res. 110:1395–1408. [DOI] [PubMed] [Google Scholar]
- Farris JS, Kallersjo M, Kluge AG, and Bult C 1994. Testing significance of incongruence. Cladistics 10:315–319. [Google Scholar]
- Gan P, Nakata N, Suzuki T, and Shirasu K 2017. Markers to differentiate species of anthracnose fungi identify Colletotrichum fructicola as the predominant virulent species in strawberry plants in Chiba Prefecture of Japan . J. Gen. Plant Pathol. 83:14–22. [Google Scholar]
- Harrington TC, and Rizzo DM 1999. Defining species in the fungi. Pages 43–70 in: Structure and Dynamics of Fungal Populations. Worrall JJ, ed. Kluwer Academic, Dordrecht, the Netherlands. [Google Scholar]
- Hawksworth DL, and Lücking R 2017. Fungal diversity revisited: 2.2 to 3.8 million species. Pages 79–95 in: The Fungal Kingdom. Heitman J, Howlett BJ, Crous PW, Stukenbrock EH, James TY, and Gow NAR, eds. ASM Press, Washington, DC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbett D, Abarenkov K, Koljalg U, Opik M, Chai B, Cole J, Wang Q, Crous P, Robert V, Helgason T, Herr JR, Kirk P, Lueschow S, O’Donnell K, Nilsson RH, Oono R, Schoch C, Smyth C, Walker DM, Porras-Alfaro A, Taylor JW, and Geiser DM 2016. Sequence-based classification and identification of Fungi. Mycologia 108:1049–1068. [DOI] [PubMed] [Google Scholar]
- Hilário S, Goncalves MFM, and Alves A 2021. Using genealogical concordance and coalescent-based species delimitation to assess species boundaries in the Diaporthe eres complex. J. Fungi (Basel) 7:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inderbitzin P, Robbertse B, and Schoch CL 2020. Species identification in plant-associated prokaryotes and fungi using DNA. Phytobiomes J. 4:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TY, Stajich JE, Hittinger CT, and Rokas A 2020. Toward a fully resolved fungal tree of life. Annu. Rev. Microbiol. 74:291–313. [DOI] [PubMed] [Google Scholar]
- Kamfwa K, Gepts P, Hamabwe S, Nalupya ZK, Mukuma C, and Lungu D 2021. Characterization of Colletotrichum lindemuthianum races in Zambia and evaluation of the CIAT Phaseolus core collection for resistance to anthracnose. Plant Dis. 105:3939–3945. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodadadi F, Gonzalez JB, Martin PL, Giroux E, Bilodeau GJ, Peter KA, Doyle VP, and Acimovic SG 2020. Identification and characterization of Colletotrichum species causing apple bitter rot in New York and description of C. noveboracense sp. nov. Sci. Rep. 10:11043. https://www.nature.com/articles/s41598-020-66761-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizirian D, and Donnelly MA 2004. The criterion of reciprocal monophyly and classification of nested diversity at the species level. Mol. Phylogenet. Evol. 32:1072–1076. [DOI] [PubMed] [Google Scholar]
- Laraba I, McCormick SP, Vaughan MM, Geiser DM, and O’Donnell K 2021. Phylogenetic diversity, trichothecene potential, and pathogenicity within Fusarium sambucinum species complex. PLoS One 16:e0245037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence MH, Summerell BA, Burgess LW, and Liew ECY 2014. Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol. 118:374–384. [DOI] [PubMed] [Google Scholar]
- Lelwala R, Korhonen P, Young N, Scott J, Ades P, Gasser R, and Taylor P 2019. Comparative genome analysis indicates high evolutionary potential of pathogenicity genes in Colletotrichum tanaceti. PLoS One 14:e0212248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leray M, Knowlton N, Ho SL, Nguyen BN, and Machida RJ 2019. GenBank is a reliable resource for 21st century biodiversity research. Proc. Natl. Acad. Sci. USA 116:22651–22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Steenwyk JL, Chang Y, Wang Y, James TY, Stajich JE, Spatafora JW, Groenewald M, Dunn CW, Hittinger CT, Shen X, and Rokas A 2021. A genome-scale phylogeny of the kingdom Fungi. Curr. Biol. 31:1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Cai L, Crous PW, and Damm U 2014. The Colletotrichum gigasporum species complex. Persoonia 33:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wang M, Damm U, Crous PW, and Cai L 2016. Species boundaries in plant pathogenic fungi: A Colletotrichum case study. BMC Evol. Biol. 16:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng X, Khaskheli MI, Sun X, Chang X, and Gong G 2020. Identification of Colletotrichum species associated with blueberry anthracnose in Sichuan, China. Pathogens 9:718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SW, Kroken S, Lee BN, Robbertse B, Churchill ACL, Yoder OC, and Turgeon BG 2003. A novel class of gene controlling virulence in plant pathogenic ascomycete fungi. Proc. Natl. Acad. Sci. USA 100:5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbe CM, Denman S, Cannon PF, Groenewald JZE, Lamprecht SC, and Crous PW 2004. Characterization of Colletotrichum species associated with diseases of Proteaceae. Mycologia 96:1268–1279. [PubMed] [Google Scholar]
- Lücking R, Aime MC, Robbertse B, Miller AN, Aoki T, Ariyawansa HA, Cardinali G, Crous PW, Druzhinina IS, Geiser DM, Hawksworth DL, Hyde KD, Irinyi L, Jeewon R, Johnston PR, Kirk PM, Malosso E, May TW, Meyer W, Nilsson HR, Opik M, Robert V, Stadler M, Thines M, Vu D, Yurkov AM, Zhang N, and Schoch CL 2021. Fungal taxonomy and sequence-based nomenclature. Nat. Microbiol. 6:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking R, Aime MC, Robbertse B, Miller AN, Ariyawansa HA, Aoki T, Cardinali G, Crous PW, Druzhinina IS, Geiser DM, Hawksworth DL, Hyde KD, Irinyi L, Jeewon R, Johnston PR, Kirk PM, Malosso E, May TW, Meyer W, Opik M, Robert V, Stadler M, Thines M, Vu D, Yurkov AM, Zhang N, and Schoch CL 2020. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, and Knowles LL 2006. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 55:21–30. [DOI] [PubMed] [Google Scholar]
- Matute DR, and Sepulveda VE 2019. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 131:103249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson LA, and Mooney HA 2007. Invasive alien species in an era of globalization. Front. Ecol. Environ. 5:199–208. [Google Scholar]
- Nilsson R, Larsson K, Taylor A, Bengtsson-Palme J, Jeppesen T, Dmitry S, Kennedy P, Picard K, Glöckner F, Tedersoo L, Saar I, and Kõljalg U 2019. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47: D529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson KH, and Koljalg U 2006. Taxonomic reliability of DNA sequences in public sequence databases: A fungal perspective. PLoS One 1:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norphanphoun C, Hongsanan S, Gentekaki E, Chen YJ, Kuo CH, and Hyde KD 2020. Differentiation of species complexes in Phyllosticta enables better species resolution. Mycosphere 11:2542–2628. [Google Scholar]
- Raja HA, Miller AN, Pearce CJ, and Oberlies NH 2017. Fungal identification using molecular tools: A primer for the natural products research community. J. Nat. Prod. 80:756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Sharda S, Oddi V, and Nandineni MR 2018. The landscape of repetitive elements in the refined genome of chilli anthracnose fungus Colletotrichum truncatum. Front. Microbiol. 9:2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbertse B, Schoch CL, and Brover V 2019. Using a full genome protein distance tree to review fungal genome classifications at NCBI RefSeq, Curation of Fungi Genomes Project. Presented at the 30th Fungal Genetics Conference, 12–17 March 2019, Pacific Grove, CA. [Google Scholar]
- Robbertse B, Strope PK, Chaverri P, Gazis R, Ciufo S, Domrachev M, and Schoch CL 2017. Improving taxonomic accuracy for fungi in public sequence databases: Applying ‘one name one species’ in well-defined genera with Trichoderma/Hypocrea as a test case. Database (Oxford) 2017:bax072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogério F, Boufleur TR, Ciampi-Guillardi M, Sukno SA, Thon MR, Massola NS Jr., and Baroncelli R 2020. Genome sequence resources of Colletotrichum truncatum, C. plurivorum, C. musicola, and C. sojae: Four species pathogenic to soybean (Glycine max). Phytopathology 110:1497–1499. [DOI] [PubMed] [Google Scholar]
- Samarakoon MC, Gafforov Y, Liu NG, Maharachchikumbura SSN, Bhat JD, Liu JK, Promputtha I, and Hyde KD 2018. Combined multi-gene backbone tree for the genus Coniochaeta with two new species from Uzbekistan. Phytotaxa 336:43–58. [Google Scholar]
- Sánchez-García M, Rybergc M, Khanc FK, Vargad T, Nagy LG, and Hibbett DS 2020. Fruiting body form, not nutritional mode, is the major driver of diversification in mushroom-forming fungi. Proc. Natl. Acad. Sci. USA 117:32528–32534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangdee A, Sachan S, and Khankhum S 2011. Morphological, pathological and molecular variability of Colletotrichum capsici causing anthracnose of chilli in the North-east of Thailand. Afr. J. Microbiol. Res. 5:4368–4372. [Google Scholar]
- Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, Canese K, Comeau DC, Funk K, Kim S, Klimke W, Marchler-Bauer A, Landrum M, Lathrop S, Lu Z, Madden TL, O’Leary N, Phan L, Rangwala SH, Schneider VA, Skripchenko Y, Wang J, Ye J, Trawick BW, Pruitt KD, and Sherry ST 2021. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 49:D10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Beck J, Brister JR, Bolton EE, Canese K, Comeau DC, Funk K, Ketter A, Kim S, Kimchi A, Kitts PA, Kuznetsov A, Lathrop S, Lu ZY, McGarvey K, Madden TL, Murphy TD, O’Leary N, Phan L, Schneider VA, Thibaud-Nissen F, Trawick BW, Pruitt KD, and Ostell J 2020. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 48:D9–D16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäffer AA, McVeigh R, Robbertse B, Schoch CL, Johnston A, Underwood BA, Karsch-Mizrachi I, and Nawrocki EP 2021. Ribovore: Ribosomal RNA sequence analysis for GenBank submissions and database curation. BMC Bioinformatics 22:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch CL, Aime MC, de Beer W, Crous PW, Hyde KD, Penev L, Seifert KA, Stadler M, Zhang N, and Miller AN 2017. Using standard keywords in publications to facilitate updates of new fungal taxonomic names. IMA Fungus 8:A70–A73. [Google Scholar]
- Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, Leipe D, McVeigh R, O’Neill K, Robbertse B, Sharma S, Soussov V, Sullivan JP, Sun L, Turner S, and Karsch-Mizrachi I 2020. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database (Oxford) 2020:baaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch CL, Robbertse B, Robert V, Vu D, Cardinali G, Irinyi L, Meyer W, Nilsson RH, Hughes K, Miller AN, Kirk PM, Abarenkov K, Aime MC, Ariyawansa HA, Bidartondo M, Boekhout T, Buyck B, Cai Q, Chen J, Crespo A, Crous PW, Damm U, De Beer ZW, Dentinger BTM, Divakar PK, Duenas M, Feau N, Fliegerova K, Garcia MA, Ge ZW, Griffith G, Groenewald JZ, Groenewald M, Grube M, Gryzenhout M, Gueidan C, Guo LD, Hambleton S, Hamelin R, Hansen K, Hofstetter V, Hong SB, Houbraken J, Hyde KD, Inderbitzin P, Johnston PR, Karunarathna SC, Koljalg U, Kovacs GM, Kraichak E, Krizsan K, Kurtzman CP, Larsson KH, Leavitt S, Letcher PM, Liimatainen K, Liu JK, Lodge DJ, Luangsa-ard JJ, Lumbsch HT, Maharachchikumbura SSN, Manamgoda D, Martin MP, Minnis AM, Moncalvo JM, Mule G, Nakasone KK, Niskanen T, Olariaga I, Papp T, Petkovits T, Pino-Bodas R, Powell MJ, Raja HA, Redecker D, Sarmiento-Ramirez JM, Seifert KA, Shrestha B, Stenroos S, Stielow B, Suh SO, Tanaka K, Tedersoo L, Telleria MT, Udayanga D, Untereiner WA, Uribeondo JD, Subbarao KV, Vagvolgyi C, Visagie C, Voigt K, Walker DM, Weir BS, Weiss M, Wijayawardene NN, Wingfield MJ, Xu JP, Yang ZL, Zhang N, Zhuang WY, and Federhen S 2014. Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for Fungi. Database (Oxford) 2014:bau061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Dentinger B, Dieguez-Uribeondo J, Divakar PK, Douglas B, Duenas M, Duong TA, Eberhardt U, Edwards JE, Elshahed MS, Fliegerova K, Furtado M, Garcia MA, Ge ZW, Griffith GW, Griffiths K, Groenewald JZ, Groenewald M, Grube M, Gryzenhout M, Guo LD, Hagen F, Hambleton S, Hamelin RC, Hansen K, Harrold P, Heller G, Herrera G, Hirayama K, Hirooka Y, Ho HM, Hoffmann K, Hofstetter V, Hognabba F, Hollingsworth PM, Hong SB, Hosaka K, Houbraken J, Hughes K, Huhtinen S, Hyde KD, James T, Johnson EM, Johnson JE, Johnston PR, Jones EB, Kelly LJ, Kirk PM, Knapp DG, Koljalg U, Kovacs GM, Kurtzman CP, Landvik S, Leavitt SD, Liggenstoffer AS, Liimatainen K, Lombard L, Luangsa-Ard JJ, Lumbsch HT, Maganti H, Maharachchikumbura SS, Martin MP, May TW, McTaggart AR, Methven AS, Meyer W, Moncalvo JM, Mongkolsamrit S, Nagy LG, Nilsson RH, Niskanen T, Nyilasi I, Okada G, Okane I, Olariaga I, Otte J, Papp T, Park D, Petkovits T, Pino-Bodas R, Quaedvlieg W, Raja HA, Redecker D, Rintoul TL, Ruibal C, Sarmiento-Ramirez JM, Schmitt I, Schussler A, Shearer C, Somtome K, Stefani FOP, Stenroos S, Stielow B, Stockinger H, Suetrong S, Suh SO, Sung GH, Suzuki M, Tanaka K, Tedersoo L, Telleria MT, Tretter E, Untereiner WA, Urbina H, Vagvolgyi C, Vialle A, Vu TD, Walther G, Wang QM, Wang Y, Weir BS, Weiss M, White MM, Xu J, Yahr R, Yang ZL, Yurkov A, Zamora JC, Zhasng N, Zhuang WY, and Schindel D 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 109:6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Kumar N, Weir BS, Hyde KD, and Shenoy BD 2013. The ApMat marker can resolve Colletotrichum species: A case study with Mangifera indica. Fungal Divers. 61:117–138. [Google Scholar]
- Sharma G, Pinnaka AK, and Shenoy BD 2014. Resolving the Colletotrichum siamense species complex using ApMat marker. Fungal Divers. 71:247–264. [Google Scholar]
- Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, Boudouris JT, Schneider RM, Langdon QK, Ohkuma M, Endoh R, Takashima M, Manabe R, Cadez N, Libkind D, Rosa CA, DeVirgilio J, Hulfachor AB, Groenewald M, Kurtzman CP, Hittinger CT, and Rokas A 2018. Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175:1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Han JH, Park HH, Fu T, and Kim KS 2019. Optimization of polyethylene glycol-mediated transformation of the pepper anthracnose pathogen Colletotrichum scovillei to develop an applied genomics approach. Plant Pathol. J. 35:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaprakash M, Appannanavar S, Dhaliwal M, Gupta A, Gupta S, Gupta A, and Chakrabartil A 2011. Colletotrichum truncatum: An unusual pathogen causing mycotic keratitis and endophthalmitis. J. Clin. Microbiol. 49:2894–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DN, Talhinhas P, Varzea V, Cai L, Paulo OS, and Batista D 2012. Application of the Apn2/MAT locus to improve the systematics of the Colletotrichum gloeosporioides complex: An example from coffee (Coffea spp.) hosts. Mycologia 104:396–409. [DOI] [PubMed] [Google Scholar]
- Soares VF, Velho AC, Carachenski A, Astolfi P, and Stadnik MJ 2021. First report of Colletotrichum karstii causing anthracnose on Strawberry in Brazil. Plant Dis. 105:3295. [Google Scholar]
- Steenkamp ET, Wingfield MJ, McTaggart AR, and Wingfield BD 2018. Fungal species and their boundaries matter – Definitions, mechanisms and practical implications. Fungal Biol. Rev. 32:104–116. [Google Scholar]
- Taylor JW, and Hibbett DS 2013. Toward sequence-based classification of fungal species. IMA Fungus 4:A33–A34. [Google Scholar]
- Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, and Fisher MC 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 31:21–32. [DOI] [PubMed] [Google Scholar]
- Thiéry O, Vasar M, Jairus T, Davison J, Roux C, Kivistik PA, Metspalu A, Milani L, Saks U, Moora M, Zobel M, and Opik M 2016. Sequence variation in nuclear ribosomal small subunit, internal transcribed spacer and large subunit regions of Rhizophagus irregularis and Gigaspora margarita is high and isolate-dependent. Mol. Ecol. 25:2816–2832. [DOI] [PubMed] [Google Scholar]
- Thines M, Aoki T, Crous PW, Hyde KD, Lücking R, Malosso E, May TW, Miller AN, Redhead SA, Yurkov AM, and Hawksworth DL 2020. Setting scientific names at all taxonomic ranks in italics facilitates their quick recognition in scientific papers. IMA Fungus 11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turland NJ, Wiersema JH, Barrie FR, Greuter W, Hawksworth DL, Herendeen PS, Knapp S, Kusber W-H, Li D-Z, Marhold K, May TW, McNeill J, Monro AM, Prado J, Price MJ, and Smith GF 2018. International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted at the Nineteenth International Botanical Congress, Shenzhen, China, July 2017. In: Regnum Vegatabile, Vol. 159. Koeltz Botanical Books, Oberreifenberg, Germany. [Google Scholar]
- Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, and Hyde KD 2015. The Diaporthe sojae species complex: Phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol. 119:383–407. [DOI] [PubMed] [Google Scholar]
- Vaghefi N, Shivas RG, Sharma S, Nelson SC, and Pethybridge SJ 2021. Phylogeny of cercosporoid fungi (Mycosphaerellaceae, Mycosphaerellales) from Hawaii and New York reveals novel species within the Cercospora beticola complex. Mycol. Prog. 20:261–287. [Google Scholar]
- van Wyk S, Harrison C, Wingfield B, De Vos L, van der Merwe N, and Steenkamp E 2019. The RIPper, a web-based tool for genome-wide quantification of repeat-induced point (RIP) mutations. PeerJ 7:e7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wyk S, Wingfield B, De Vos L, van der Merwe N, and Steenkamp E 2021. Genome-wide analyses of repeat-induced point mutations in the ascomycota. Front. Microbiol. 11:622368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Hao XY, Wang L, Xiao B, Wang XC, and Yang YJ 2016. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 6:35287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Jiang Y, Deane DC, He FL, Shu WS, and Liu Y 2019. Effects of host phylogeny, habitat and spatial proximity on host specificity and diversity of pathogenic and mycorrhizal fungi in a subtropical forest. New Phytol. 223:462–474. [DOI] [PubMed] [Google Scholar]
- Weir BS, Johnston PR, and Damm U 2012. The Colletotrichum gloeosporioides species complex. Stud. Mycol. 73:115–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Ge ZW, LoBuglio KF, and Pfister DH 2018. Otidea species from China, three new species with comments on some previously described species. Mycol. Prog. 17:77–88. [Google Scholar]
- Yakoby N, Kobiler I, Dinoor A, and Prusky D 2000. pH regulation of pectate lyase secretion modulates the attack of Colletotrichum gloeosporioides on avocado fruits. Appl. Environ. Microbiol. 66:1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan JY, Jayawardena MMRS, Goonasekara ID, Wang Y, Zhang W, Liu M, Huang JB, Wang ZY, Shang JJ, Peng YL, Bahkali A, Hyde KD, and Li XH 2015. Diverse species of Colletotrichum associated with grapevine anthracnose in China. Fungal Divers. 71:233–246. [Google Scholar]
- You BJ, Choquer M, and Chung KR 2007. The Colletotrichum acutatum gene encoding a putative pH-responsive transcription regulator is a key virulence determinant during fungal pathogenesis on citrus. Mol. Plant-Microbe Interact. 20:1149–1160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.