

Introduction
Blau syndrome (BS), described by Edward Blau in 1985, is a rare autosomal dominant form of a multi-system inflammatory condition which presents with arthritis, dermatitis, and uveitis.[1] The manifestations of BS are very generic, and therefore, identifying the disease can be very challenging, especially when the patient presents first to the dermatologist or ophthalmologist without clinically obvious systemic findings.[2] The inheritance pattern of BS can also be either familial or sporadic, thereby, leading to diagnostic delays in cases where there is no family history.[1,3] The diagnosis of BS requires a high degree of clinical suspicion and a tertiary level of care for appropriate management. BS is an important clinical entity where collaborative efforts of the pediatricians, pediatric rheumatologists, and ophthalmologists must converge for the diagnosis and optimum management of these children. This editorial highlights the varied clinical manifestations of the disease and the need for its widespread awareness.
Epidemiology and Demographics
Due to the rarity of the disease, its exact prevalence and epidemiological profile are not known. The literature published from the international registry combining data from 11 countries suggests that the median age of onset of the disease is 2 years (range: 3 months to 13 years).[4] The median age of onset of the ocular disease is 4.4 years (range: 6 months to 22 years).[3,4] Both genders can be equally susceptible. There are no data on the ethnicities which are at higher risk of the disease, or for more severe disease. Data from India are particularly lacking because of which the exact prevalence and distribution of the disease cannot be assessed.[5]
Ophthalmic Manifestations of BS
The most common ocular symptoms of BS include photophobia, pain, and redness in the eye. Children can present with symptoms of keratoconjunctivitis sicca such as a feeling of dryness, itching, and foreign body sensation. There could be a diminution of vision and headache and associated tearing from the eyes. Children with mild chronic anterior uveitis may have minimal symptoms and may present only when complications such as band-shaped keratopathy develop causing a decrease in visual acuity.[2,4,6,7,8,9,10]
Ocular findings are reported in over 60–80% of the patients with BS.[3] The manifestations are usually bilateral and detected using the slit-lamp examination with +90 diopter lens for fundus evaluation in addition to indirect ophthalmoscopy using +20 diopter lens. The most common manifestation is anterior uveitis [Figure 1]. Bilateral chronic panuveitis with multifocal choroiditis is common, but intermediate uveitis may also occur. The ocular manifestations of BS have been summarized in Table 1.[3,6,7,8,9,10,11,12]
Figure 1.
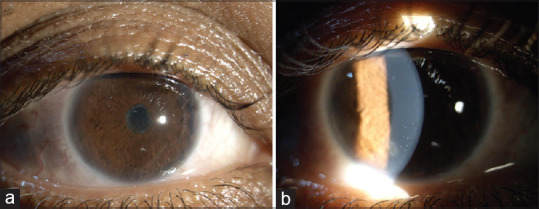
(a) An 11-year-old male child with Blau syndrome with anterior uveitis with 360-degree posterior synechiae and complicated cataract; (b) a 25-year-old female patient having significant anterior uveitis with cells and flare, and keratic precipitates due to underlying Blau syndrome
Table 1.
| Anterior segment findings | |
|---|---|
| Common | Bilateral granulomatous anterior uveitis |
| Band-shaped keratopathy | |
| Posterior synechiae | |
| Anterior chamber and anterior vitreous cells | |
| Complicated cataract | |
| Glaucoma (secondary) | |
| Uncommon | Hypopyon uveitis |
| Fibrin in the anterior chamber | |
| Iris nodules | |
|
| |
| Posterior segment findings | |
|
| |
| Common | Panuveitis |
| Multifocal choroidal scars | |
| Peripapillary nodules | |
| Uncommon | Retinal vascular leakage |
| Optic nerve head edema | |
| Cystoid macular edema | |
The usual clinical manifestations of BS include band-shaped keratopathy, anterior chamber cells and flare, posterior synechiae, complicated cataracts, and the advanced forms of the disease can have posterior segment involvement with vitritis, retinal vascular sheathing, and macular thickening. In these eyes, the fundus imaging reveals the presence of retinal vascular leakage due to vasculitis and macular edema.[1,3,6,8,10] On dilated fundus examination, peripapillary nodules have been observed in more than 75% of the eyes in one series by Carreño et al.[13] and should raise suspicion for BS. A subgroup of patients can develop severe panuveitis, which responds poorly to systemic corticosteroid and immunosuppressant therapies and progresses to permanent vision loss and phthisis bulbi.[14]
It is important to remember that none of the uveitic manifestations of BS are specific to the disease. Therefore, phenotype identification solely based on ocular examination can be misleading, since these features are also common in children with other conditions.
Systemic manifestations of BS
The most important systemic manifestations of BS include granulomatous dermatitis which presents as a recurrent or persistent scaly/lumpy rash that can be felt under the skin on the torso, arms, and legs. Dermatitis may present with scaly ichthyosiform or papulo-erythematous rash. The rash is typically non-pruritic and it can persist for several years unless treated.[3,4,12,15] A leading international research group on BS[3] has observed that the mean age of developing dermatological manifestations, such as skin rash, is the first year.
Another prominent clinical sign of BS is boggy swelling and pain in the joints due to tenosynovitis involving the hands, feet, knees, wrists, ankles, and metacarpophalangeal joints [Figure 2]. Most children also develop early-onset boggy swelling of the joints (before 5 years of age).[3] Severe tenosynovitis can lead to joint contractures and permanent deformities such as camptodactyly (inability to straighten fingers).[3,4,7,10,12,14,15,16]
Figure 2.

(a) A 7-year-old boy with Blau syndrome with dry skin and maculopapular rash on the right forearm region; (b) an 11-year-old boy with Blau syndrome having tender boggy swelling of the wrist joints and small joints of the hands, and dermatitis on all four limbs; (c) a 45-year-old female with Blau syndrome having joint contractures and deformity involving the small joints of the hand
The kidneys and liver may be involved resulting in deranged enzyme profiles, hepatosplenomegaly, and renal insufficiency. Rare findings of BS include renal granulomas, portal fibrosis, and parotid gland swelling.[17,18] One of the hallmark features of BS is the development of non-caseating granulomas in various organ systems. Biopsies of the liver in patients with suspected BS can reveal non-caseating granulomas with an otherwise normal liver parenchyma. Special stains such as immunohistochemical staining for interleukin (IL)-17 can be used for aiding the diagnosis.[19]
Manifestations of BS: The Indian context
The systemic manifestations of the disease are like those reported from other countries and these include boggy joint swelling, symmetrical arthritis, and tenosynovitis. Ultrasonography of the joints revealing prominent tenosynovitis is a useful clinical indicator of BS as per Banday et al.[20] Jha et al.[21] have described a case of a 26-year-old male from north India who presented with progressive vision loss, joint deformities (polyarthritis), and stunted growth. This case was noteworthy due to peri-articular osteopenia and erosions noted on radiography.[21] In a report from north India, Jindal et al.[17] demonstrated disseminated granulomas in the liver and kidney detected in a patient at 21 years of age. The correct diagnosis of BS could only be established after two decades when the patient developed hepatic and renal lesions. Naik et al.[22] from south India have described a patient who had a history of diffuse rash involving the face, trunk, and extremities at 2 years of age and was diagnosed as having BS at 5 years of age when he presented with sudden onset of pain, redness, and diminution of vision in both eyes for 1 week; and was detected to have bilateral swelling of the proximal and distal interphalangeal joints.[22]
There is a paucity of literature on the ocular manifestations of BS from India. Recently, Babu and Rao.[9] have published a series of seven Indian patients (four females and three males) with BS. Two patients had sporadic mutations. All the patients had bilateral ocular disease with three having anterior uveitis, and another three having panuveitis. One case had a rare manifestation of subepithelial corneal opacities and a retinal granuloma in addition to uveitis. The mean age of diagnosis in this case series was 13.7 years (range: 3–25 years) and all had been misdiagnosed earlier as having either juvenile idiopathic arthritis (JIA) or sarcoidosis.[9]
Differential diagnosis of BS
The differential diagnosis of BS having systemic manifestations includes systemic sarcoidosis, JIA, and Behçet's disease. Its ocular manifestations can mimic ocular tuberculosis (OTB) and ocular sarcoidosis.[1,3,4,6,8,10,15,19,23] Children with BS can occasionally present with panuveitis, retinal vasculitis, and multifocal choroiditis - a feature common in OTB. In a previous study on OTB from India, it was observed that multifocal choroiditis with retinal vasculitis has a strong odd's ratio for predicting the diagnosis (40.99 [CI: 5.13–327.86]) of OTB.[24]
Since uveitis is a major clinical manifestation of BS, several leading clinician-scientists from around the world have formed collaborative consortia to highlight the key features to aid fellow colleagues in arriving at the diagnosis of BS in a timely manner.[4,10] Due to the rarity of the condition, the diagnosis of BS may be missed even by uveitis specialists; therefore, it is important to highlight the most common manifestations of the disease.
Pearls in detecting BS
Age: The mean age of presentation of BS is 4 years (range: 3 months to 58 years). However, in countries such as India, the mean age of presentation has been reported to be higher (13.7 years; range: 3 years to 25 years) because the disease may begin with largely asymptomatic mild ocular inflammation, which goes undetected. Reports from India suggest that BS gets detected late when the patient is already in the second decade of life.[17] The condition gets misdiagnosed as JIA or sarcoidosis for more than two decades before the diagnosis of BS is considered and confirmed by genetic testing.[17]
Inheritance: Patients with BS may be sporadic. Therefore, the absence of family history must not be taken as a feature against BS.[3,4,9,15]
Differentiating from JIA: More than 50% of the patients with BS can present with panuveitis and multifocal choroiditis. These findings are uncommon in JIA.[6,8] The involvement of the renal and liver tissue by non-caseating granuloma can differentiate BS from JIA.[19,25] Therefore, patients with hepatomegaly should be considered for surveillance biopsies.
Differentiating from Behçet's disease: Hypopyon and fibrin in the anterior chamber are rare in BS.[6,8] These features, in addition to retinal vasculitis with the characteristic fern-tree appearance on fluorescein angiography, are commonly encountered in Behçet's disease. The important diagnostic criteria for Behçet's disease include oral and genital ulcers, which have not been reported in BS.
Differentiating from sarcoidosis: BS must be differentiated from systemic sarcoidosis, which can be a very close mimicker. Both entities can have non-caseating granulomas in various organs. Children with sarcoidosis can have lymphadenopathy and pulmonary involvement which are not seen in BS (except a few case reports including one report of a patient with interstitial lung disease and another with an apical pulmonary nodule).[26,27] Children with BS can develop prominent synovial cysts which are not seen in sarcoidosis. On skin biopsy, the histology of the dermis and synovium reveal comma-shaped or worm-like bodies within the cytoplasm of the epithelioid cells in BS but not in sarcoidosis.[19] Optic nerve inflammation and choroidal granulomas are very common in sarcoidosis, but uncommon in BS.[6,8]
Differentiating from OTB: OTB must be differentiated from BS with the help of supportive laboratory evidence such as Mantoux test, interferon-gamma release assay (Quantiferon TB Gold®), and contrast-enhanced computerized chest tomography for Ghon's focus as outlined by the Collaborative Ocular Tuberculosis Study (COTS) group focusing specifically on children.[23] There is a significant overlap of ocular manifestations with BS, therefore, ocular examination alone is insufficient for differentiating between the two. Systemic inflammation resulting in tenosynovitis and dermatitis is rare in tuberculosis.
BS can present with ocular manifestations alone with delayed dermatological and joint involvement. The ophthalmologists should keep in mind the possibility of BS, especially in the presence of clinical features outlined above.
Ocular imaging (fluorescein angiography [FA], indocyanine green angiography [ICGA], and optical coherence tomography [OCT]) can be helpful in diagnosing BS and differentiating from other entities (see below).
Ocular imaging in BS
Ocular imaging can play a vital role in understanding the phenotypic presentation of ocular inflammation in BS, and picking clues that can differentiate BS from mimickers such as sarcoidosis and OTB. Children with JIA seldom have the chorioretinal disease (except macular edema), and therefore, have an unremarkable FA and ICGA.[28] Patients with OTB and sarcoidosis can have significant retinal vasculitis and optic nerve head inflammation with hyperfluorescence on FA (not reported in BS), active choroiditis (does not occur in BS), and choroidal granulomas which present with hypofluorescence on both FA and ICGA (not reported in BS).[28,29,30] Concilio et al.[31] have recently reported the relevance of anterior segment OCT (AS-OCT) in an 8-year-old girl with bilateral anterior granulomatous uveitis with underlying BS. AS-OCT in this patient revealed high-intensity reflective layers in the anterior cornea along with hyperreflective dots on the posterior corneal surface indicative of anterior chamber inflammation and deposition of inflammatory cells on the posterior corneal surface.
Genetic testing and its implications
Genetic testing for nucleotide-binding oligomerization domain (NOD) 2 mutations is confirmatory for the diagnosis of BS. R334W mutation in the NOD2 gene (detected after amplifying the exon 4 of the NOD2 gene using polymerase chain reaction, and subjecting the sample to direct sequencing) is the most commonly encountered mutation described in the literature.[3,4,10,15] A few reports have identified other novel mutations in the NOD2 gene in BS. The other reported mutations include E600K, Y563S, and M513T among others.[3] Jain et al.[32] have described a previously unreported heterozygous mutation in exon 4 of the NOD2 gene in a 12-year-old girl child resulting in a glutamic acid to lysine substitution. Since the disease can be inherited in an autosomal dominant manner, the family should be screened for the condition. The penetrance of NOD2 gene mutations is very high. Genetic counseling must be offered to the family if one child has been diagnosed with BS.
Immunological perspectives in BS
International efforts have been made toward identifying the immunological profile in patients with BS. Various pro-inflammatory mediators are responsible for the systemic inflammation that occurs in patients with BS, with interferon (IFN)-γ being likely the chief molecule.[33] Studies suggest that nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) may play an important role in BS, along with complement factor B (Bf) that can activate the alternate complement pathway.[33]
Recent updates in the management of BS
Most children with BS require long-term immunosuppression and surgical interventions such as cataract/glaucoma surgeries, and occasionally, pulse intravenous corticosteroid therapy for unrelenting systemic/ocular inflammation.[3,4,9,10,15,17,22] The common immunosuppressive agents employed in the management of BS include methotrexate, azathioprine, mycophenolate mofetil, among others.[10,15,16] The use of anti-tumor necrosis factor (TNF)-α agents has been shown to ameliorate the pro-inflammatory cascade in patients with BS, and good results have been demonstrated in the Chinese and Japanese cohorts.[7,15] Agents such as adalimumab and infliximab can be used with beneficial results. A large series of 50 children from Japan has recently shown that early treatment with biological agents (consisting of monoclonal antibodies including anti-TNF-α agents infliximab and adalimumab) can avoid blindness in children with BS.[15]
A few cases of BS, however, may continue to worsen despite aggressive corticosteroid, immunosuppressive, and biological therapy. Therefore, close monitoring is necessary in all cases. Recently, successful liver transplantation has also been described in a patient with BS-related hepatic disease.[34]
Summary and Conclusions
More literature is now being published on BS from various international clinical centers, including India, and several cases that were previously labeled as early-onset sarcoidosis or JIA are now being correctly identified as BS. Availability of clinical data and genetic testing, and improved collaborative efforts between pediatricians, dermatologists, ophthalmologists, uveitis specialists, pediatric rheumatologists, clinical geneticists, and immunologists have helped achieve this. Increased awareness of BS among all treating doctors and continued collaborative efforts among these specialists are needed to improve outcomes in patients with BS.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors wish to acknowledge the contribution of Dr. Kalpana Babu Murthy, Head of the Department of Uveitis and Ocular Inflammation at Prabha Eye Clinic & Research Centre and Vittala International Institute of Ophthalmology, Bangalore for her help in providing clinical photographs.
References
- 1.Rose CD, Martin TM, Wouters CH. Blau syndrome revisited. Curr Opin Rheumatol. 2011;23:411–8. doi: 10.1097/BOR.0b013e328349c430. [DOI] [PubMed] [Google Scholar]
- 2.Rosenbaum JT, Planck SR, Davey MP, Iwanaga Y, Kurz DE, Martin TM. With a mere nod, uveitis enters a new era. Am J Ophthalmol. 2003;136:729–32. doi: 10.1016/s0002-9394(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 3.Rosé CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P, et al. Blau syndrome: Cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford) 2015;54:1008–16. doi: 10.1093/rheumatology/keu437. [DOI] [PubMed] [Google Scholar]
- 4.Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: An international registry. Arthritis Rheum. 2006;54:3337–44. doi: 10.1002/art.22122. [DOI] [PubMed] [Google Scholar]
- 5.Janarthanan M, Poddar C, Sudharshan S, Seabra L, Crow YJ. Familial Blau syndrome: First molecularly confirmed report from India. Indian J Ophthalmol. 2019;67:165–7. doi: 10.4103/ijo.IJO_671_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pillai P, Sobrin L. Blau syndrome-associated uveitis and the NOD2 gene. Semin Ophthalmol. 2013;28:327–32. doi: 10.3109/08820538.2013.825285. [DOI] [PubMed] [Google Scholar]
- 7.Wu S, Zhong L, Sun Z, Zhu T, Song H, Sui R. Ocular features in Chinese patients with Blau syndrome. Ocul Immunol Inflamm. 2020;28:79–85. doi: 10.1080/09273948.2019.1569239. [DOI] [PubMed] [Google Scholar]
- 8.Suresh S, Tsui E. Ocular manifestations of Blau syndrome. Curr Opin Ophthalmol. 2020;31:532–7. doi: 10.1097/ICU.0000000000000705. [DOI] [PubMed] [Google Scholar]
- 9.Babu K, Rao AP. Clinical profile in genetically proven Blau syndrome: A case series from South India. Ocul Immunol Inflamm. 2021;29:250–6. doi: 10.1080/09273948.2020.1746353. [DOI] [PubMed] [Google Scholar]
- 10.Sarens IL, Casteels I, Anton J, Bader-Meunier B, Brissaud P, Chédeville G, et al. Blau syndrome-associated uveitis: Preliminary results from an international prospective interventional case series. Am J Ophthalmol. 2018;187:158–66. doi: 10.1016/j.ajo.2017.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Nascimento H, Sousa JM, Fernández DG, Salomão GH, Sato EH, Muccioli C, et al. Blau-Jabs syndrome in a tertiary ophthalmologic center. Ophthalmic Surg Lasers Imaging Retina. 2018;49:70–5. doi: 10.3928/23258160-20171215-12. [DOI] [PubMed] [Google Scholar]
- 12.Poline J, Fogel O, Pajot C, Miceli-Richard C, Rybojad M, Galeotti C, et al. Early-onset granulomatous arthritis, uveitis and skin rash: Characterization of skin involvement in Blau syndrome. J Eur Acad Dermatol Venereol. 2020;34:340–8. doi: 10.1111/jdv.15963. [DOI] [PubMed] [Google Scholar]
- 13.Carreño E, Guly CM, Chilov M, Hinchcliffe A, Arostegui JI, Lee RW, et al. Optic nerve and retinal features in uveitis associated with juvenile systemic granulomatous disease (Blau syndrome) Acta Ophthalmol. 2015;93:253–7. doi: 10.1111/aos.12544. [DOI] [PubMed] [Google Scholar]
- 14.PaÇ Kisaarslan A, SÖzerİ B, Şahİn N, Özdemİr ÇİÇek S, GÜndÜz Z, Demİrkaya E, et al. Blau syndrome and early-onset sarcoidosis: A six case series and review of the literature. Arch Rheumatol. 2019;35:117–27. doi: 10.5606/ArchRheumatol.2020.7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical characteristics and treatment of 50 cases of Blau syndrome in Japan confirmed by genetic analysis of the NOD2 mutation. Ann Rheum Dis. 2020;79:1492–9. doi: 10.1136/annrheumdis-2020-217320. [DOI] [PubMed] [Google Scholar]
- 16.Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012;12:44–51. doi: 10.1016/j.autrev.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 17.Jindal AK, Pilania RK, Suri D, Gupta A, Gattorno M, Ceccherini I, et al. A young female with early-onset arthritis, uveitis, hepatic, and renal granulomas: A clinical tryst with Blau syndrome over 20 years and case-based review. Rheumatol Int. 2021;41:173–81. doi: 10.1007/s00296-019-04316-6. [DOI] [PubMed] [Google Scholar]
- 18.Shen M, Moran R, Tomecki KJ, Yao Q. Granulomatous disease associated with NOD2 sequence variants and familial camptodactyly: An intermediate form of NOD2-associated diseases? Semin Arthritis Rheum. 2015;45:357–60. doi: 10.1016/j.semarthrit.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 19.James G. Blau's syndrome and sarcoidosis. Lancet. 1999;354:1035. doi: 10.1016/S0140-6736(05)76645-9. [DOI] [PubMed] [Google Scholar]
- 20.Banday AZ, Mukherjee S, Kumrah R, Sun D, Jindal AK. Prominent tenosynovitis on ultrasonography: A useful sign for the diagnosis of Blau syndrome. Eur J Rheumatol. 2022;9:58–9. doi: 10.5152/eurjrheum.2020.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jha S, Mittal S, Kumar RR, Dhooria A, Rawat A, Dhir V. An unusual cause of deforming erosive arthritis in an adult. Rheumatology (Oxford) 2020;59:602. doi: 10.1093/rheumatology/kez312. [DOI] [PubMed] [Google Scholar]
- 22.Naik AU, Annamalai R, Biswas J. Uveitis in sporadic Blau syndrome: Long-term follow-up of a refractory case treated successfully with adalimumab. Indian J Ophthalmol. 2018;66:1483–5. doi: 10.4103/ijo.IJO_629_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Testi I, Agrawal R, Mahajan S, Agarwal A, Gunasekeran DV, Raje D, et al. The Collaborative Ocular Tuberculosis Study (COTS)-1: A multinational descriptive review of tubercular uveitis in pediatric population. Ocul Immunol Inflamm. 2020 doi: 10.1080/09273948.2020.1781197. In Press. [DOI] [PubMed] [Google Scholar]
- 24.Gupta A, Bansal R, Gupta V, Sharma A, Bambery P. Ocular signs predictive of tubercular uveitis. Am J Ophthalmol. 2010;149:562–70. doi: 10.1016/j.ajo.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal K, Barua S, Adhicari P, Das S, Marak R. Early-onset sarcoidosis and juvenile idiopathic arthritis: A diagnostic dilemma. Indian J Dermatol Venereol Leprol. 2016;82:542–5. doi: 10.4103/0378-6323.183626. [DOI] [PubMed] [Google Scholar]
- 26.Becker ML, Martin TM, Doyle TM, Rosé CD. Interstitial pneumonitis in Blau syndrome with documented mutation in CARD15. Arthritis Rheum. 2007;56:1292–4. doi: 10.1002/art.22509. [DOI] [PubMed] [Google Scholar]
- 27.Su J, Liu D. Blau syndrome with pulmonary nodule in a child. Australas J Dermatol. 2021;62:217–20. doi: 10.1111/ajd.13551. [DOI] [PubMed] [Google Scholar]
- 28.Carlsson E, Beresford MW, Ramanan AV, Dick AD, Hedrich CM. Juvenile idiopathic arthritis associated uveitis. Children (Basel) 2021;8:646. doi: 10.3390/children8080646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agarwal A, Afridi R, Agrawal R, Do DV, Gupta V, Nguyen QD. Multimodal imaging in retinal vasculitis. Ocul Immunol Inflamm. 2017;25:424–33. doi: 10.1080/09273948.2017.1319494. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal A, Mahajan S, Khairallah M, Mahendradas P, Gupta A, Gupta V. Multimodal imaging in ocular tuberculosis. Ocul Immunol Inflamm. 2017;25:134–45. doi: 10.1080/09273948.2016.1231332. [DOI] [PubMed] [Google Scholar]
- 31.Concilio M, Cennamo G, Giordano M, Fossataro F, D’Andrea L, Ciampa N, et al. Anterior segment-optical coherence tomography features in Blau syndrome. Photodiagnosis Photodyn Ther. 2021;34:102278. doi: 10.1016/j.pdpdt.2021.102278. [DOI] [PubMed] [Google Scholar]
- 32.Jain L, Gupta N, Reddy MM, Mittal R, Barik MR, Panigrahi B, et al. A novel mutation in helical domain 2 of NOD2 in sporadic Blau syndrome. Ocul Immunol Inflamm. 2018;26:292–4. doi: 10.1080/09273948.2016.1207789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takada S, Kambe N, Kawasaki Y, Niwa A, Honda-Ozaki F, Kobayashi K, et al. Pluripotent stem cell models of Blau syndrome reveal an IFN-?-dependent inflammatory response in macrophages. J Allergy Clin Immunol. 2018;141:339–49.e11. doi: 10.1016/j.jaci.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 34.Sinharay R, McKeown L, Phillips C, Li A, Duckworth A, Hall F, et al. First report of liver transplantation in Blau syndrome: The challenges faced in this rare granulomatous liver disease. Transpl Immunol. 2021;65:101378. doi: 10.1016/j.trim.2021.101378. [DOI] [PubMed] [Google Scholar]