

Abstract
Introduction:
Despite the impressive responses achieved with tyrosine kinase inhibitor (TKI) therapy, treatment resistance develops in 16–33% of patients of chronic myelogenous leukemia (CML). Of the BCR-ABL1 dependent mechanisms, mutations in the tyrosine kinase domain (TKD) are the commonest cause of resistance.
Material and Methods:
Allele specific oligonucleotide - polymerase chain reaction (ASO-PCR) was done for testing the six common TKD mutations, T315I, G250E, E255K, M244V, M351T, and Y253F.
Results and Conclusion:
TKD mutation study was done on 83 patients. Of these 44 (53%) were positive for one or more mutations. On analyzing specific mutations, E255K was the commonest mutation seen in 24 (29%) cases, followed by T315I in 23(28%) cases. Y253F mutation was not seen in the present study sample. In the present cohort of 83 patients, 29 (35%) cases were positive for single mutation, 12 (14%) had two mutations and 3 (4%) had three mutations.
KEY WORDS: ASO-PCR, imatinib resistance, tyrosine kinase domain mutations
Introduction
Chronic myelogenous leukemia (CML) is the first malignancy to be associated with a defined cytogenetic abnormality, that is, Philadelphia (Ph) chromosome, formed by reciprocal translocation between chromosome 9 and 22, that is, t (9;22)(q34;q11). This results in fusion between ABL1 gene at chromosome 9 and BCR gene at chromosome 22. The chimeric BCR-ABL1 oncogene encodes the BCR-ABL1 fusion protein with constitutional tyrosine kinase activity.[1]
CML is also the first malignancy for which targeted therapy, tyrosine kinase inhibitors (TKI) was introduced, which improved prognosis of CML considerably.[2] However, despite the impressive responses achieved with TKI, treatment resistance develops in 16–33% patients.[3] Resistance can be primary, when there is a failure to reach required hematological or cytogenetic or molecular responses within the recommended time point; or secondary, when there is loss of a previously obtained remission status.[4]
Resistance can be due to BCR-ABL1-dependent or BCR-ABL1-independent mechanisms. Amongst the BCR-ABL1-dependent mechanisms, poor patient compliance is most common cause, and of the BCR-ABL1-dependent mechanisms, point mutations in the tyrosine kinase domain (TKD), is most common cause.[5]
Incidence of TKD mutations in imatinib resistant CML patients range from 22% to 90%, depending on the methodology used to evaluate the mutations, for example, Sanger sequencing-based studies quoting lower incidence than those studies where mutational analysis was done using allele specific oligonucteotide PCR (ASO-PCR) or denaturing high pressure liquid chromatography (dHPLC).[6]
In this study, we evaluated the frequencies of six common TKD mutations (T315I, E255K, M244V, G250E, M315T, and Y253F) in TKI resistant patients using ASO-PCR.
Material and Methods
Study was approved by the Institutional Ethics Committee and was performed following the ethical standards of Helsinki.
Prospectively over a period of 18 months, 90 CML patients were enrolled for TKI resistance analysis, indication being either due to treatment failure or suboptimal response to TKI as per the European LeukemiaNet (ELN) recommendations [Table 1].[7]
Table 1.
European LeukemiaNet recommendations for BCR-ABL1 kinase domain mutation analysis[7]
| At diagnosis |
| Only in accelerated phase or blast crisis patients |
| During first-line imatinib therapy |
| In case of treatment failure |
| In case of an increase in BCR-ABL1 transcript levels leading to loss of major molecular response |
| In any other case of suboptimal response |
| During second-line dasatinib or nilotinib therapy |
| In case of hematologic or cytogenetic failure |
| Disease progression to accelerated or blast phase |
RNA extraction was done using QIAamp RNA blood mini kit (Qiagen, Germany). A 260/280 ratio of 1.9 to 2.1 was accepted as “pure” for RNA. The median RNA quantity was 112 ng (range, 34–468 ng) with a median 260/280 ratio of 2.0 (range, 1.9–2.1).
cDNA synthesis was done using High Purity cDNA Synthesis kit (Thermo Scientific, USA). Quality of synthesized cDNA was checked by cDNA-PCR amplification of wild type ABL housekeeping gene.
Allele Specific Oligonucleotide-Polymerase Chain Reaction (ASO-PCR) was done for testing the six common TKD mutations, T315I, G250E, E255K, M244V, M351T, and Y253F.
The primers and PCR reaction conditions were used as previously described,[8] with modifications, as outlined in Table 2. The PCR products run on agarose gel electrophoresis showed amplicon lengths for mutations- T315I = 158 bp, G250E = 255 bp, E255K = 194 bp, M244V = 227 bp, M351T = 236 bp, Y253F = 226 bp and wild kinase domain = 374 bp [Figure 1].
Table 2.
ASO-PCR for the tyrosine kinase domain mutations tested
| Primer sequence | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Type | Forward primer sequence 5’to 3’ | Reverse primer sequence 5’to 3’ | |||||
| T315I | GCC CCC GTT CTA TAT CAT CAT [mutant forward (MF)] | GGA TGA AGT TTT TCT TCT CCA G [mutant reverse (MR)] | |||||
| G250E | GAA GCA CAA GCT GGG CGA [mutant forward (MF)] | GCC AAT GAA GCC CTC GGA C [mutant reverse (MR)] | |||||
| E255K | GCG GGG GCC AGT ACG GGA [mutant forward (MF)] | GCC AAT GAA GCC CTC GGA C [mutant reverse (MR)] | |||||
| M244V | GAA CGC ACG GAC ATC ACC G [mutant forward (MF)] | GCC AAT GAA GCC CTC GGA C [mutant reverse (MR)] | |||||
| M351T | CCACTCAGATCTCGTCAGCCAC [mutant forward (MF)] | ATG CCC AAA GCT GGC TTT G [mutant reverse (MR)] | |||||
| Y253F | CTG GGC GGG GGC CAG TT [mutant forward (MF)] | GCC AAT GAA GCC CTC GGA C [mutant reverse (MR)] | |||||
| Wild Type | TGG TTC ATC ATC ATT CAA CGG TGG [Wild type forward (WF)] | GTT CCC GTA GGT CAT GAA CTC AG [Wild type reverse (WR)] | |||||
| The last nucleotide of the forward primers of each specific mutation is mutated | |||||||
|
| |||||||
| PCR reagents | |||||||
|
| |||||||
| Reagents | Mutation tested | ||||||
|
| |||||||
| E255K | Y253F | T315I | M244V | G250E | M351T | ||
|
| |||||||
| Distilled water (µL) | 19.0 | 19.2 | 15.4 | 14.5 | 19.2 | 14.5 | |
| Buffer (µL) | 2.5 | 2.5 | 2.0 | 2.0 | 2.5 | 2.0* | |
| dNTP (µL) | 0.4 | 0.5 | 0.4 | 0.5 | 0.5 | 0.5 | |
| MgCl2 (µL) | 0.1 | - | - | - | - | 0.2 | |
| Primers used | MF and MR | MF, WF WR | MF and MR | MF and WR | MF, WF and WR | MF and MR | |
| Primer volume (µL) | 0.3 | 0.3 | 0.5 | 0.5 | 0.3 | 0.5 | |
| Taq Polymerase (µL) | 0.4 | 0.4 | 0.3 | 0.5 | 0.4 | 0.5 | |
| Annealing temperature (°C) | 66 | 55 | 66 | 71.5 | 56 | 72 | |
| Product length (bp) | Wild KD | - | 374 | - | - | 374 | - |
| Mutant KD | 194 | 226 | 158 | 227 | 255 | 236 | |
|
| |||||||
| * MgCl2 depleted buffer was used. MF: Mutant forward; MR: Mutant reverse; WF: Wild type forward; WR: Wild type reverse. | |||||||
| PCR CONDITIONS | |||||||
|
| |||||||
| Pre-heating | 94°C for 5 minutes | ||||||
| Amplification x 30 cycles | Denaturation - 94°C for 25 seconds Annealing- At respective temperatures indicated above, for 25 seconds Extension- 72°C for 30 seconds. |
||||||
| Final extension | 72°C for 5 minutes | ||||||
Figure 1.
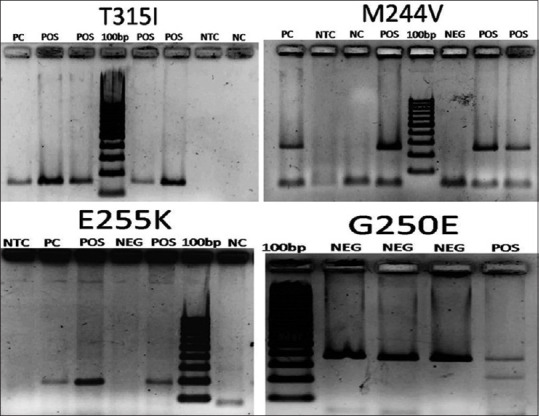
Agarose gel electrophoresis photographs of cases tested for TKD mutations with ASO-PCR. Amplicon lengths- T315I = 158 bp, M244V = 227 bp, E255 K = 194 bp, G250E = 255 bp, Wild kinase domain = 374 bp. (G250E ASO-PCR was biplexed with wild type kinase domain). NC: Negative control (Non –CML sample), NTC: Negative for template control (PCR mastermix without any template added), PC: Positive control, NEG: Negative for mutation, POS: Positive for mutation tested
Results
Ninety CML patients were prospectively tested for TKD mutation. Median age of patients was 38 (range, 14–70) years. The cohort included 57 males and 33 females with a male: female ratio of 1.7:1.
Excluding three patients who were analyzed at baseline, the median duration of TKI exposure in the rest was 42 (range, 3–171) months. The most common indication for TKD mutation analysis was molecular response failure (57%), followed by loss of complete cytogenetic response (16%). Table 3 outlines the indications for TKD mutation analysis in patients tested.
Table 3.
Indications for TKI resistance testing in patient cohort for TKD mutation
| Patients (n=90) | Percentage (%) | |
|---|---|---|
| Baseline accelerated phase | 2 | 2 |
| Baseline blast crisis | 1 | 1 |
| Progression to accelerated phase | 5 | 6 |
| Progression to blast crisis | 6 | 7 |
| Loss of complete hematologic response (CHR) | 14 | 16 |
| Cytogenetic response failure | 5 | 6 |
| Cytogenetic response failure with loss of CHR | 1 | 1 |
| Molecular response failure | 51 | 57 |
| Molecular and cytogenetic response failure | 4 | 4 |
| Molecular response failure with loss of CHR | 1 | 1 |
Seven patient's cDNA did not yield optimal results for housekeeping gene, therefore, TKD mutation was done on 83 patients’ samples. Of these 53% (n = 44) were positive for one or more mutations. On analyzing specific mutations, E255K was the most common mutation seen in 29% (n = 24) cases, followed by T315I seen in 28% (n = 23) cases. Y253F mutation was not seen in any of the patients tested. The relative frequencies of the individual mutations tested is outlined in Table 4.
Table 4.
Frequency of individual kinase domain mutations tested in 83 patients
| Kinase domain mutation | Frequency of mutation. |
|---|---|
| E255K | 24 (29%) |
| T315I | 23 (28%) |
| M244V | 11 (13%) |
| G250E | 3 (4%) |
| M351T | 1 (1%) |
| Y253F | 0 |
In our cohort of 83 patients, 35% (n = 29) cases were positive for single mutation, 14% (n = 12) cases had two mutations, and 4% (n = 3) cases had three mutations. The overall mutation profile of our cohort is depicted in Figure 2.
Figure 2.
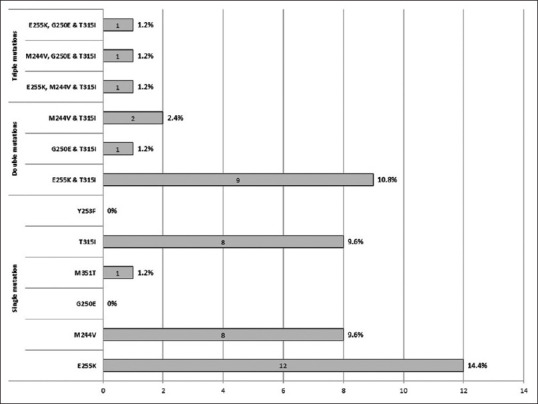
Mutation profile of TKD mutation positive patients
Discussion
Although with TKI therapy, responses have improved dramatically in CML patients, still not all CML patients on TKI therapy show optimal responses. Excluding compliance and individual differences in drug metabolizing mechanisms, molecular-genetic alterations in the BCR-ABL1 fusion transcript results in TKI resistance. These BCR-ABL1-dependent mechanisms of TKI resistance occur most commonly due to mutations occurring in the TKD of these molecules.
TKD mutations cause TKI resistance due to ineffective binding of TKI to the kinase domain of BCR-ABL1. The main categories of TKD mutations that correlate with clinical resistance to TKI are at the mutations occurring at: Imatinib binding site, P-loop (ATP binding site), Catalytic (C) domain, and Activation (A) loop. Among these, P-loop mutations are the most common (48%) and they destabilize the conformation required for imatinib binding and have been associated with an increased transforming potential and a worse prognosis regardless of their sensitivity to imatinib. In addition, P-loop mutations have been reported to be associated with a worse prognosis in comparison with other categories of mutations. Till date more than 100 TKD mutations have been documented. However, some type of mutations are more frequent then others. Since each of the TKD mutations have variable levels of response to TKI, it is important to detect them, so as to take appropriate therapeutic decision.[6,9]
In this study, tyrosine kinase domain mutational analysis was done in 83 patients, among which 44 (53%) were positive for the mutations tested. With frequencies of 29% and 28% respectively, E255K and T315I were the most common mutations seen. In addition, coexistence of two/three mutations was seen in 14% and 4% of mutation positive cases, respectively. T315I mutation was invariably seen in all these cases with multiple mutations.
Incidence of TKD mutation reported has been from 19 to 60%. Although more than 100 different mutations involving different amino acids have been reported in the BCR-ABL1 kinase domain, only subset of these (G250E, Y253H, E255K/V, V299L, T315I, F317L/I, F359V/I/C, H396R, E450G/V, E459K) are associated with TKI failure.[10,11]
Multiple mutations were seen in 34% of our patients, however, if they are polyclonal or compound mutations could not be studied. “A compound mutant arises when two mutations are acquired by the same BCR-ABL1 molecule, thus by the same clone, as opposed to polyclonality where two clones acquire a single mutation each.”[12]
Khorashad et al.[13] using a cloning and sequencing method, found that 70% (33/47) of double mutations detected by direct sequencing were compound mutations, and concluded that compound mutations are common in patients with sequencing evidence for 2 BCR-ABL1 mutations and frequently reflect a highly complex clonal network, the evolution of which may be limited by the negative impact of missense mutations on kinase function.
We used a highly sensitive ASO-PCR to detect clinically significant 6 most frequent TKD mutations in this study. However, there are many methods for TKD mutation analysis, including Next-Generation sequencing (NGS), Sanger sequencing, dHPLC, pyrosequencing, high resolution melting curve analysis, ASO-PCR. Of these, Sanger sequencing is most commonly used for TKD analysis, because of its advantage of detection of all mutations, their characterization and bidirectional conformation of mutations, however, it has low sensitivity (15–25%). ASO-PCR has high sensitivity (0.01–0.001%), but has limitation of characterizing only known mutations and is labor intensive if screening for multiple mutations.[10]
NGS based testing for detection of TKD mutations has advantage over Sanger sequencing and ASO-PCR in terms of quantitation of mutation allele frequency, determination of sub-clones, differentiation between polyclonal and compound mutations and being a high throughput sensitive technique. However, it has limitations for use in routine diagnostic monitoring due to the high cost of equipment and reagents, requirement of infrastructure, bioinformatics tools and technical expertise for analysis.[14]
Studies, however, show the greater utility of NGS in future clinical practice, as detection of low-level mutation can modify therapeutic decisions. It has been shown that NGS can detect low-level mutations/emerging resistant mutants earlier than Sanger sequencing and thereby aid in timely change in type of TKI therapy in CML-CP patients. Also, NGS may aid in therapeutic decision by detecting low-level mutations in AP/BC patients at diagnosis and in patients with treatment discontinuation, who relapse and fail to achieve major molecular response.[14,15] However, currently use of NGS for detection of TKD mutations is limited to specialized centers.
There is scarcity of data on TKD mutation from our sub-continent, current study provides data on the incidence of TKD mutation in CML patients from India. Also, we tested for 6 most frequent TKD mutations using ASO-PCR and found that 53% patients were positive for any one/more of the mutations tested. Therefore, if facility for sequencing is not available, then using simple ASO-PCR for these 6 common mutations can be done and cause of TKI resistance can be identified in about 50% of the cases. ASO-PCR is a simple and cost-effective test with high sensitivity; therefore, it can be used as a useful alternative in situations where more sophisticated tests are not available.
Declaration of patient consent
The authors certify that appropriate patients’ consents were obtained.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–56. [PubMed] [Google Scholar]
- 2.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 3.Ai J, Tiu RV. Practical management of patients with chronic myeloid leukemia who develop tyrosine kinase inhibitor-resistant BCR-ABL1 mutations. Ther Adv Hematol. 2014;5:107–20. doi: 10.1177/2040620714537865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milojkovic D, Apperley J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. 2009;15:7519–27. doi: 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- 5.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 6.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: By the GIMEMA working party on chronic myeloid leukemia. Clin Cancer Res. 2006;12:7374–9. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 7.Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872–84. doi: 10.1182/blood-2013-05-501569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang HY, Hwang JY, Kim SH, Goh HG, Kim M, Kim DW. Comparison of allele specific oligonucleotide-polymerase chain reaction and direct sequencing for high throughput screening of ABL kinase domain mutations in chronic myeloid leukemia resistant to imatinib. Haematologica. 2006;91:659–62. [PubMed] [Google Scholar]
- 9.Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: Recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118:1208–15. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 10.Alikian M, Gerrard G, Subramanian PG, Mudge K, Foskett P, Khorashad JS, et al. BCR-ABL1 kinase domain mutations: Methodology and clinical evaluation. Am J Hematol. 2012;87:298–304. doi: 10.1002/ajh.22272. [DOI] [PubMed] [Google Scholar]
- 11.Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O’Brien S, et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110:4005–11. doi: 10.1182/blood-2007-03-080838. [DOI] [PubMed] [Google Scholar]
- 12.Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:49. doi: 10.1186/s12943-018-0780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor–resistant CML: Frequency and clonal relationships. Blood. 2013;121:489–98. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soverini S, Abruzzese E, Bocchia M, Bonifacio M, Galimberti S, Gozzinni A, et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: A position paper. J Hematol Oncol. 2019;12:131. doi: 10.1186/s13045-019-0815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker WT, Lawrence RM, Ho M, Irwin DL, Scott HS, Hughes TP, et al. Sensitive detection of BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. J Clin Oncol. 2011;29:4250–9. doi: 10.1200/JCO.2011.35.0934. [DOI] [PubMed] [Google Scholar]