

Abstract
Glioblastoma (GBM) is a rapidly fatal malignancy typically treated with radiation and temozolomide (TMZ), an alkylating chemotherapeutic. These cytotoxic therapies cause oxidative stress and DNA damage, yielding a senescent-like state of replicative arrest in surviving tumor cells. Unfortunately, recurrence is inevitable and may be driven by surviving tumor cells eventually escaping senescence. A growing number of so-called “senolytic” drugs have been recently identified that are defined by their ability to selectively eliminate senescent cells. A growing inventory of senolytic drugs is under consideration for several diseases associated with aging, inflammation, DNA damage, as well as cancer. Ablation of senescent tumor cells after radiation and chemotherapy could help mitigate recurrence by decreasing the burden of residual tumor cells at risk of recurrence. This strategy has not been previously explored for GBM. We evaluated a panel of 10 previously described senolytic drugs to determine whether any could exhibit selective activity against human GBM persisting after exposure to radiation or TMZ. Three of the 10 drugs have known activity against BCL-XL and preferentially induced apoptosis in radiated or TMZ-treated glioma. This senolytic activity was observed in 12 of 12 human GBM cell lines. Efficacy could not be replicated with BCL-2 inhibition or senolytic agents acting against other putative senolytic targets. Knockdown of BCL-XL decreased survival of radiated GBM cells, whereas knockdown of BCL-2 or BCL-W yielded no senolytic effect.
Implications:
These findings imply that molecularly heterogeneous GBM lines share selective senescence-induced BCL-XL dependency increase the significance and translational relevance of the senolytic therapy for latent glioma.
Introduction
Glioblastoma (GBM) is the most common and lethal adult brain tumor. Standard treatment includes maximal surgical resection followed by radiation and temozolomide (TMZ; ref. 1). Treatment-resistant cells may persist in a latent state for months or years prior to inevitable recurrence. Because surgery is rarely performed until radiographic or symptomatic recurrence, relatively little is known about the molecular characteristics and potential drug sensitivity of the latent human glioma cells that ultimately give rise to recurrence (2–4). Prior work has demonstrated that radiation and TMZ induce a reversible senescent-like phenotype in malignant glioma (2, 3, 5–7). “Irreversible” senescence in nonmalignant cells can be induced by genotoxic or other cellular stress and is regulated by master regulators including CDKN2A (P16), P53, and P21 (CDKN1A). Senescent cells are characterized by mitotic arrest as well as altered transcriptional, metabolic, and secretory phenotypes (8). Malignant cells including GBM frequently accumulate genetic alterations that hamper induction and maintenance of senescence, including homozygous loss of CDKN2A (9), or mutations in P53 and associated pathways (10). Moreover, although P21 is generally upregulated in GBM, it has been shown to serve a potentially protumorigenic role in malignant cells (11). Despite these alterations, chemotherapy and radiation can induce a senescent-like phenotype in GBM, characterized by mitotic arrest, altered cellular morphology, increased β-galactosidase (β-gal) activity, upregulated p21, pRn and/or cyclin B1, histoneH3 S10 phosphorylation, and downregulated cyclin D1 (3, 12). Novel therapies are being explored to further promote such a senescent state of proliferative arrest to help maintain their nonproliferative state (13). Unfortunately, malignant cells are notorious for escaping any therapy-induced senescence-like state to reenter the cell cycle, leading to refractory tumor recurrence (14, 15). Recent advances in the understanding of senescence have been fueled by evidence that senescent cells underlie mechanisms of aging and aging-associated diseases (16). A growing array of so-called “senolytic” drugs has been identified that can ablate senescent cells by targeting antiapoptotic mechanisms, many of which are implicated in both senescence and cancer (Table 1; refs. 17, 18). Repurposing existing agents for senescence-associated diseases has facilitated rapid progression of senolytic strategies into clinical trials (19). A senolytic approach to GBM has not previously been specifically explored. We asked whether known senolytic drugs could be used to help eliminate human GBM cells surviving in a nonproliferative senescent-like state and identified increased BCL-XL dependency as a vulnerability in GBM cells surviving after radiation or TMZ.
Table 1.
(38).
| SCAPss | Inhibitors |
|---|---|
| 1. PI3Kδ, AKT, ROS-protective, metabolic | Quercetin, Fisetin, Piperlongumine |
| 2. MDM2, p53, p21, serpine | Quercetin, Fisetin, Dasatinib |
| 3. BCL-2 | Navitoclax, Venitoclax |
| 4. BCL-XL | Navitoclax, A1331852, A1155463 |
| 5. Ephrins, dependence receptors, tyrosine kinases | Dasatinib, Piperlongumine |
| 6. HIF1α | Quercetin, Fisetin |
| 7. HSP-90 | Onalespib |
Materials and Methods
Cell culture
Human patient-derived GBM lines used have been previously described and were cultured according to established protocols (20). Tumor lines maintained as patient-derived xenografts (PDX) are from the Mayo National PDX resource (20). Such lines are designated as “GBM6, GBM10, GBM12, GBM39, GBM76, GBM123, GBM164, GBM196” (see Table 2 for details regarding cell lines utilized). Protocols for implantation of patient-derived GBM cells, serial passage of flank tumor xenografts, and short-term explant culturing have been described previously (20). For short-term explant cultures, some lines are maintained in serum-containing media, and others in serum-free media as noted in Table 2. Some cell lines were maintained from time of harvest as in vitro cell lines rather than PDX lines, as reported previously (21). Such lines are designated as dBT114, 116, 120, 132 (differentiated brain tumor). Cell have been regularly passaged up 70%–80% confluency, and after radiation culture conditions were not changed after induction of senescence. GBM cell lines were maintained in passage 2–4 and dBT line were in passage 9–11 (Supplementary Table S1). Cell supernatants for Mycoplasma contamination have been assessed at regular intervals using MycoAlert Mycoplasma Detection Kit (catalog no.: LT07-118) using manufacturer protocols and yielded negative results. Cell authentication was confirmed by short tandem repeat analysis with each experiment relative to historic controls (20).
Table 2.
(27).
| Line | Sex | Age | Recurrence status | MGMT methylation | Subtype | IDH1 | CDKN2A | PTEN | EGFR | TP53 | Met | Tert Prom | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | M | 65 | Primary | No | Clas | WT | LL | L | A (v3) | M | A | M | |
| 10 | M | 41 | Recurrent | No | Mes | WT | LL | LM | A | A | M | ||
| 12 | M | 68 | Primary | Yes | Mes | WT | LL | AM | ML | M | |||
| 39 | M | 51 | Primary | Yes | Mes | WT | L | LM | A (v3) | A | M | MDM4 & PIKC32B Amp | |
| 43 | M | 69 | Primary | No | Mes | WT | LL | M | M | ||||
| 76 | M | 38 | Recurrent | Yes | Clas | WT | LL | LM | A (v3) | A | M | ||
| 114a | M | 68 | Primary | Yes | ND | WT | LL | L | A | M | |||
| 116a | F | 56 | Primary | Yes | Mes | WT | LL | LM | A | M | A | M | |
| 120a | M | 57 | Recurrent | No | PN | WT | A | LL | A | M | A | M | |
| 123 | F | 62 | Recurrent | No | PN | WT | L | A (v2) | M | M | CDK4&Myc Amp; ATRX mut | ||
| 132a | M | 75 | Primary | No | Clas | WT | L | L | A | M | M | ||
| 164 | F | 38 | Primary | Yes | PN | Mb | LL | L | A | — | PDGFR Amp; NF1 loss; ATRX trunc | ||
| 196 | F | 30 | Primary | Yes | ND | Mc | LL | L | A | MA | A | — | PDGFR & RB1 Amp |
Note: Subtypes: Classical (Clas), mesenchymal (Mes), proneural (P), not determined (ND); v2, v3 = EGFR variants.
Abbreviations: M, mutant; A, amplified or net gain (>2n); L, loss; LL, homozygous deletion.
a,dBT (“differentiated brain tumor—aka, maintained in culture).
bHeterozygous IDH R132H mutation.
cHomozygous IDH R132H mutation.
Senescence induction
After plating, cells were maintained in 10 cm2 culture dishes for 2–4 days until >50% confluent. They were then treated with TMZ for 7 days or radiation (cesium-137 irradiator) at various doses as indicated (5, 22). Most experiments were performed with 15 Gy to induce senescence. Radiation and TMZ each caused death of a variable percentage of cells in the ensuing days. Senescent tumor cells used for experiments are considered to be those that survive following TMZ or radiation treatments. Except for radiation doses of 4 Gy or below, no visible proliferation occurred within 1 month after treatment with TMZ or radiation. For initial screening of senolytic drugs, cells were maintained for at least 20 days after radiation or TMZ prior to replating cells into black-walled optical 96-well plates (5,000 cells/well) prior to drug treatments.
Analysis of cell proliferation
GBM39 was treated with 0, 4, 8, 15, and 20 Gy radiation as described above, and 3 days after that, cells then plated in 96-well plates. After allowing cells to adhere overnight, they were placed into the IncuCyte (Thermo Fisher Scientific Series 8000 WJ Incubator). Images were captured every 4 hours over 40 days for automated quantification of cellular confluence.
Senolytic drug screen
Human GBM cells were exposed to 10, 15, or 20 Gy radiation, as indicated above. All GBM lines tested (Table 2) yielded a subpopulation of surviving nonproliferative cells used for subsequent experiments, with the exception of GM43, which yielded insufficient surviving cells for senescence experiments. All senolytic drugs used in this study were dissolved in DMSO (Table 1). Control (0 nmol/L) cells were treated with the same dose of DMSO as cells with the highest drug concentration. Unless specified otherwise, cells were maintained in drug-containing media for 4 days prior to evaluation of cell viability using the CellTiter-Glo assay (Promega, catalog no. G7570). This assay utilizes an ATP-dependent luciferase reaction to generate luminescence proportional to viable cell numbers. For all experiments, luminescence values are normalized to the 0 nmol/L control for that cell line and radiation dose. All dose–response graphs are depicted as means and SD of three technical replicates at each concentration. Representative data are shown for experiments performed in independent replicates; complete normalized data for all assays performed (>10,000) are provided in online Supplementary Data.
Variables impacting BCL-XL inhibitor sensitivity
To evaluate the radiation dose effect on BCL-XL inhibitor sensitivity, we radiated GBM39 with 0, 1, 2, 4, 8, and 15 Gy. Four days after radiation, cells were plated in black walled 96-well plates overnight and drugs were added the next day. Seven days after drug treatment, cell viability was measured by Cell Titer-Glo. To determine the impact of varying duration of drug exposure, we plated 15 Gy radiated cells 7 days after radiation, with the minimum drug exposure time being 1 hour and the maximum 96 hours. Cells were plated in black-walled 96-well plates, and drugs were added the next day as described above. At the designated timepoint, drug-containing media were removed, rinsed once, and replaced with drug-free media for the remainder of the experiment. The cell viability assay was performed after 96 hours.
qRT-PCR
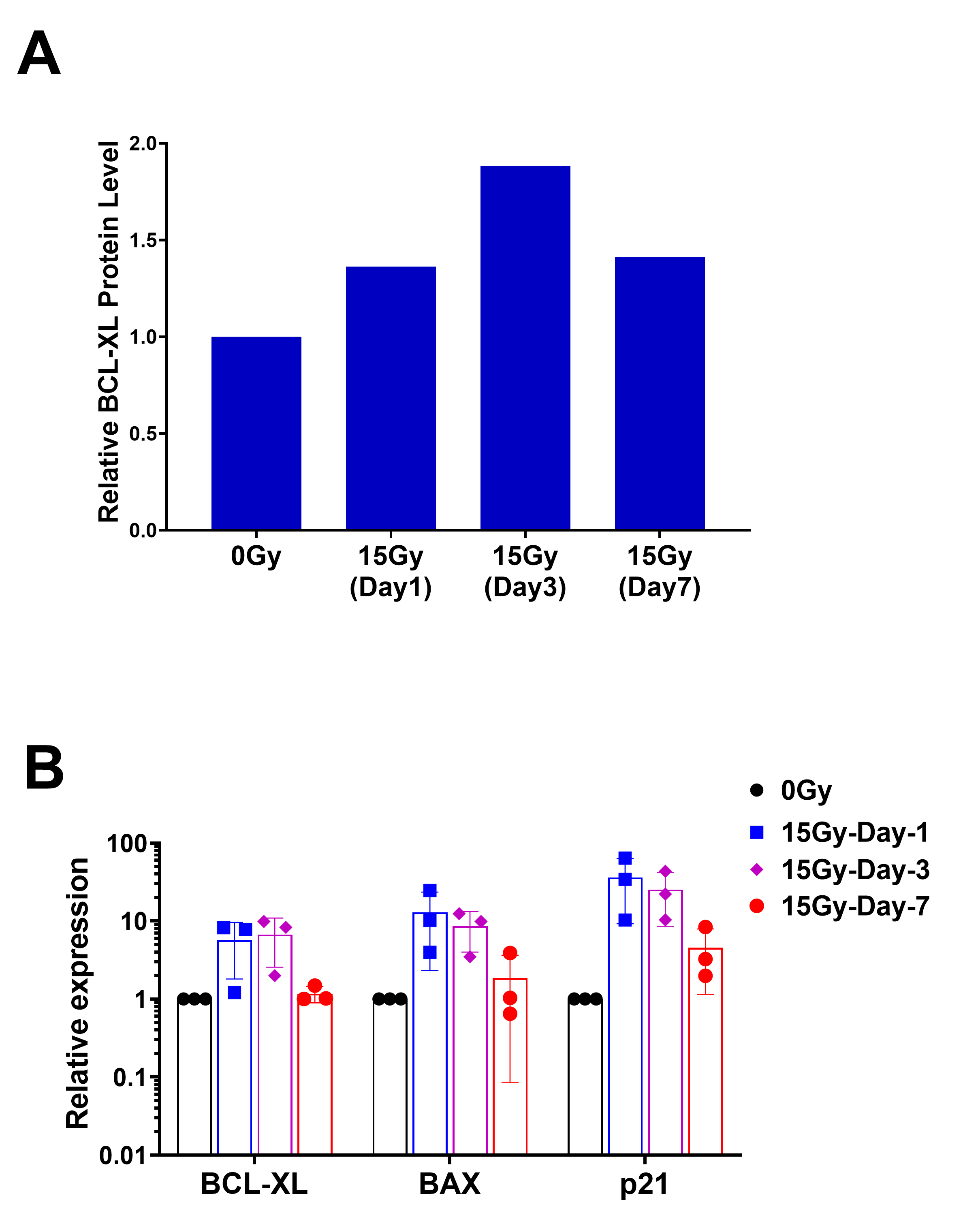
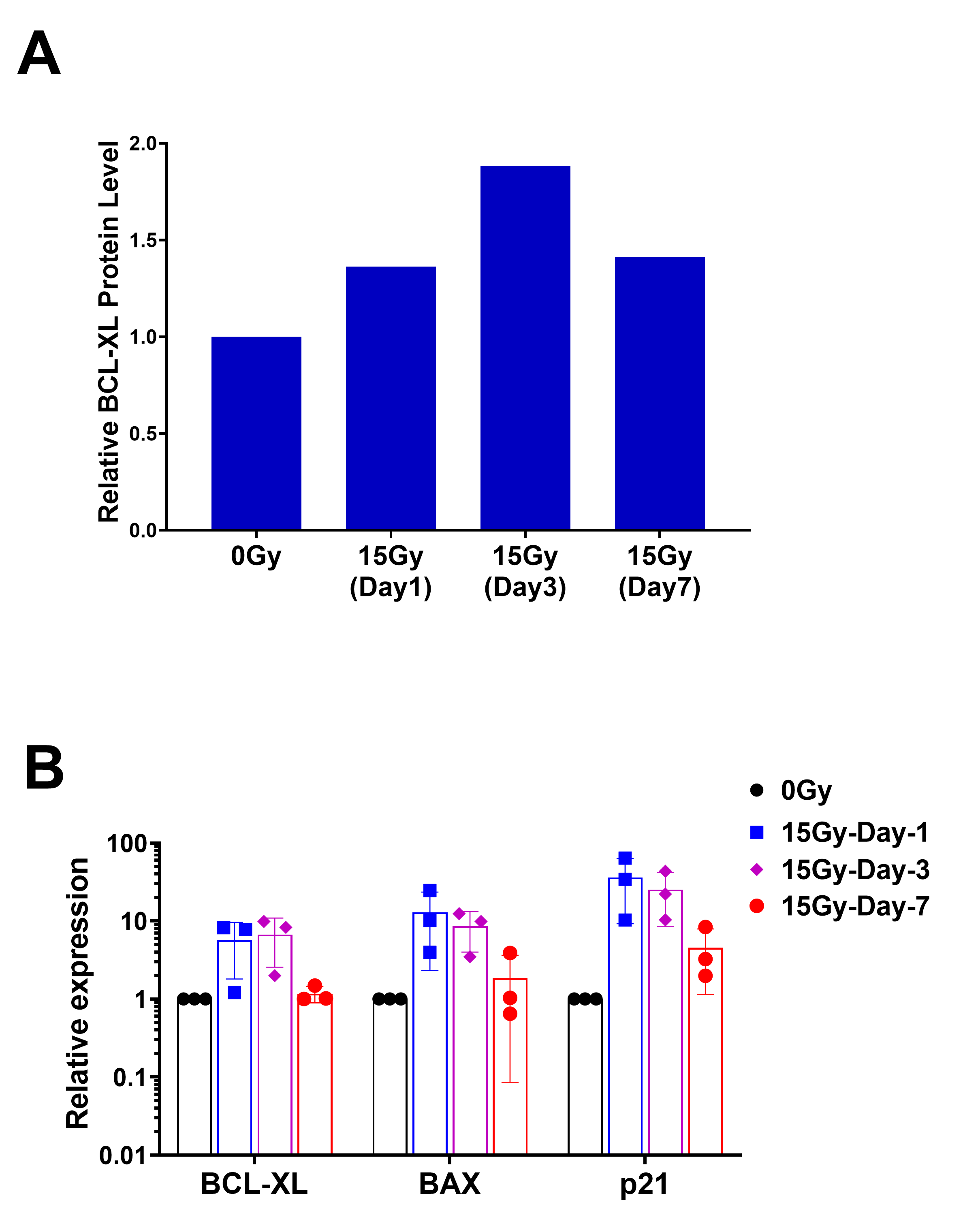
RNA was extracted from irradiated or non-irradiated cells at variable time points. Briefly, cells were washed with PBS before being homogenized with TRIzol reagent (Invitrogen). RNA precipitation was performed at −20°C overnight. Resulting RNA pellets were dissolved in RNase-free water and concentration was measured by absorbance at 260 nm (A260) using a Nanodrop2000. cDNA synthesis was performed with 1 µg of total RNA using a M-MLV reverse transcriptase kit (Thermo Fisher Scientific) as per the manufacturer's protocol. A total of 25 ng of cDNA was used for real-time PCR by the Taqman gene expression assay targeting IL6 (Hs00174131_m1), BCL-2 (Hs00608023_m1), and BCL-XL (Hs001691412_m1) on an ABI 7500/7500-Fast Real-Time PCR System (Applied Bioscience). The relative expression of each gene was determined by the ΔΔCT method.
Senescence-associated β-gal activity
Senescence-associated (SA-β-gal) staining Kit (Cell Signaling Technology, #9860) was used as an indicator of relative senescence after radiation as per the manufacturer's directions. Briefly, cells were fixed for 10 minutes in β-gal fixative solution (10% 100× Fixative Solution; 90% H2O) and washed with PBS. The cells were then stained with β-gal Staining Solution (93% 1× Staining Solution; 1% 100× Solution A, 1% 100× Solution B, 5% X-gal. Wells without samples were filled with PBS, and the plate was wrapped in parafilm to prevent evaporation. The plate was left overnight in a dry incubator at 37°C. The next day, cells were examined under a microscope for β-gal–positive cells (blue staining). PBS was added to the wells, and the plate was placed on the rocker (speed = 30/minute) for 5 minutes. The plate was washed three times. Staining was performed in proliferating, and chemo-radiation treated GBMcells. To follow development of senescence over time, β-gal staining was performed at days 0, 7, and 14 following 15 Gy radiation. For each timepoint and condition, staining was done in multiple wells.
Protein analysis by Western blotting
Cells grown in 6-well plates, 10 cm2 dishes or T-25 flasks were washed with PBS, trypsinized and collected in 1.5 microcentrifuge tubes as a cell pellet. The cell pellet was then lysed using lysis buffer composed of 10% RIPA lysis buffer, 4% Protease Inhibitor cocktail, 1% Phosphatase Inhibitor cocktail-2, 1% Phosphatase Inhibitor cocktail-3, and 84% molecular grade water. The cell palate with the lysis buffer then sonicated for 30 minutes in a water bath sonicator (1-minute sonication every other minute for a total of 30 minutes). The whole lysate was centrifuged for 10 minutes at 17,000 × g to collect the supernatant as the final protein lysate. The concentration of the final protein lysate was then measured using a BCA kit (Thermo Fisher Scientific). Proteins extracted from cells using lysis buffer were separated in an SDS-PAGE along with the protein ladder (Life Technologies) using 4%–12% Bis-Tris gel (Thermo Fisher Scientific). Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad). The membrane was blocked in 5% fat-free milk (Cell Signaling Technology) for 30 minutes, washed three times (5 minutes each) using Tris-buffered saline with tween20, and probed with different antibodies (Cell Signaling Technology).
Gene knockdown using siRNA
siRNA sequences were either designed manually using commercially available software or purchased as already designed sequences (Horizon-Thermo Fisher Scientific). The siRNA was resuspended in 1× siRNA buffer (Dharmacon) or in other nuclease-free solutions. Cells were plated overnight at the optimal density in 6-well plates (4 × 105 per well) or 96-well plates (2 × 104 per well) in antibiotic-free medium. The next day, the transfection complex was prepared by mixing the siRNA (either for the gene of interest or the negative siRNA control) and Lipofectamine RNAiMAX (Invitrogen) in serum-free medium. A total of 250 µL from the transfection complex were added to each well of the 6-well plates and 10 µL for each well of 96-well plates. One day posttransfection, RNA was collected to be analyzed by qRT-PCR to confirm gene silencing. Two to three days posttransfection, protein was collected to be analyzed by SDS-PAGE and Western blotting to confirm gene silencing. Three days after transfection, cell viability in response to gene silencing was measured using a Cell Titer-Glo assay (Promega) and the cell survival ratio was calculated compared with negatively silenced control cells.
Caspase activity assay
Sham or 15 Gy radiated GBM39 cells were treated with 0 or 1,000 nmol/L A1331852 7 days after radiation. Caspase 3/7 activity assay was performed using the Caspase-Glo 3/7 assay (Promega, catalog no. G8090) according to the manufacturer's protocol. Analysis was performed 24 hours after A1331852 treatment. Simultaneously plated cells were incubated for 4 days prior to cell viability assay to demonstrate the correlation between caspase activity and resultant cell viability. Luminescence values were normalized to the 0 nmol/L control of each radiation dose group.
Statistical analysis
Multiple linear regression was used to assess the relationship of drug dose and treatment (e.g., control, radiation, TMZ) with cell inhibition within each cell line. For these models, cell inhibition was the outcome and drug dose, treatment, and the drug dose-by-treatment interaction were the independent predictors. If the drug dose-by-treatment interaction was significant (P < 0.05), pairwise comparisons were made at each drug dose between treatment groups using a two-tailed t test. IC50 was calculated via nonlinear regression (curve fit) of dose–response inhibition curves. Statistical analyses were performed using GraphPad prism 8.4.2.
Data availability statement
Data were generated by the authors and included in the article. Raw data for all graphs are provided as Supplementary Data.
Results
Chemoradiation induces a senescence-like state of sustained proliferative arrest
We sought to test the hypothesis that GBM could be relatively more sensitive to senolytic ablative therapies when induced into a senescent-like state of proliferative arrest following radiation (7, 8, 23). No phenotypic marker is perfectly sensitive or specific for senescence. Indeed, significant overlap exists between phenotypic characteristics of malignant and senescent cells (3, 6, 24, 25). However, while malignant cells are defined in part by proliferative behavior, senescent cells, by definition, do not divide. We evaluated single-fraction radiation doses on human GBM cells and determined that radiation doses of 8 Gy or higher lead to sustained loss of proliferative activity. Representative findings are illustrated in Fig. 1A, demonstrating confluency as a function of radiation dose in GBM39, quantified over time using the Incucyte live cell imaging system. Nonproliferative senescent-like cultures demonstrated evidence of increased SA-β-gal staining, consistent with a senescent phenotype induced by radiation (Fig. 1B; ref. 8). Subsequent experiments utilized 12 additional human GBM lines (Table 2). Among these, only one line (GBM43) failed to yield sufficient numbers of surviving cells after radiation (10–20 Gy) for use in downstream senolytic experiments.
Figure 1.

Radiation of GBM cells in vitro induces a senescent-like phenotype susceptible to senolytic. A, Change in GBM39 confluency in vitro over 40 days following radiation at doses ranging from 0 to 20 Gy, error bars are ± SD from technical replicates. B, Increased β-gal in GBM39 over 14 days following 15 Gy radiation. C, Senolytic drug screening was performed 21 days after 15 Gy radiation in GBM39. Cells were exposed to drugs for 4 days prior to analysis via CellTiter-Glo. Purple and black lines denote the dose–response curve for 15 and 0 Gy radiated cells, respectively. Luminescence values are normalized to 0 nmol/L control for each radiation dose. Navitoclax and A1331852 had a lower IC50 in radiated cells; *, P < 0.05. Data shown are the mean ± SD of three technical replicates; similar results were obtained GBM76, GBM6, and GBM123 (see Supplementary Figs. S1 and S2 and Supplementary Data).
Radiated GBM cell lines are selectively vulnerable to anti-BCL-2 family agents
A growing variety of drugs have been reported to demonstrate senolytic activity. Consistent with a degree of overlapping biology between senescence and malignancy, some senolytic drugs have known antitumor activity (25, 26). We investigated whether prior radiation increases sensitivity to senolytic agents. The senolytic drugs tested and the senescent cell–associated antiapoptotic pathways (SCAP) they disable are summarized in Table 1 (26). Of the initial candidate senolytic drugs tested in GBM39 (Fig. 1C), only navitoclax and A1331852 demonstrated markedly consistently improved ability to kill senescent GBM cell. Navitoclax targets BCL-2 and BCL-XL, whereas A1331852 targets BCL-XL only (27, 28). Representative data for GBM39 (newly diagnosed P53-WT GBM) are depicted in Fig. 1. Very similar results were observed in other GBM cell lines including recurrent GBM (GBM76; Supplementary Fig. S1) and IDH-mutant GBM (GBM196; Supplementary Fig. S2). Of note, the P53-mutant status did not interfere with radiation-induced induction of proliferative arrest, SA-β-gal activity nor sensitivity to BCL-XL inhibition (Supplementary Fig. S2).
To discriminate further between the dependence of radiated human GBM upon BCL-XL and BCL-2, we evaluated navitoclax and A1331852, as well as A1155463, a BCL-XL–selective inhibitor, and venetoclax, a BCL-2–selective inhibitor (29). For reference, the previously published Ki values for each of these four drugs for antiapoptotic BCL-2 family members (BCL-XL, BCL-2, and BCL-W) are shown in Supplementary Table S2. To date, no drug is available that selectively inhibits BCL-W. Ablation of radiated cells was seen across all lines (GBM10, GBM39, GBM76, GBM123) and for all drugs, except venetoclax (Fig. 2). Three of the four cell lines showed no selective ablation with venetoclax, but lower viability was seen with high-dose venetoclax in radiated GBM10 cells. These data suggested BCL-XL to be the most relevant therapeutic target for ablation of previously radiated senescent-like GBM. To evaluate further the reproducibility of BCL-XL dependence in radiated human GBM, we tested A1331852 and/or navitoclax in several additional molecularly diverse cell lines with and without prior 10, 15, or 20 Gy radiation (Supplementary Fig. S3). The molecular characteristics of the cell lines utilized are summarized in Table 2. Among the 13 human GBM cell lines evaluated, meaningful analysis was possible in all, with the exception of GBM43, for which insufficient cells survived radiation to permit testing. Results of the pharmacologic BCL-XL inhibition studies performed are summarized in Supplementary Table S1 and Fig. 5. While the IC50 of BCL-XL inhibition with and without radiation varied markedly among lines, radiated GBM was reproducibly more susceptible to BCL-XL inhibition than nonradiated GBM.
Figure 2.

Radiated GBM cell lines are selectively vulnerable to BCL-XL blockade. GBM39, GBM76, GBM10, and GBM123 were used to evaluate the senolytic activity of BCL-2-family inhibitors, including the BCL-XL–specific inhibitors (A1331852, A1155463), the selective BCL-2 inhibitor (venetoclax), and dual inhibitor of both BCL-XL and BCL-2 (navitoclax). Dose–response curves shown for control nonradiated (black) and 15 Gy radiated (purple) cells. Cells were exposed to drugs for 4 days, starting 21 days after radiation. 15 Gy radiated cells demonstrated higher sensitivity than nonradiated cells to the BCL-XL–selective inhibitors (A1331852, A1155463), and navitoclax (inhibits BCL-XL and BCL-2) but, not venetoclax (BCL-2–selective inhibitor). For all groups, luminescence values are normalized individually to 0 nmol/L control. Data shown are means ± SD of three technical replicates at each concentration. Data shown are representative of multiple confirmatory experiments. Complete data for each cell line and condition are available in Supplementary Data.
Figure 5.

Senescent GBM is selectively vulnerable to BCL-XL inhibitors. A, Relative survival of senescent and proliferating all GBM cell lines tested with 1 µmol/L A1331852 treatment. B, Comparison of selective vulnerability of p53 WT and mutant senescent glioma to BCL-XL inhibition. C, Demonstrating senolytic effect of A1331852 across the primary and recurrent gliomas with or without chemoradiation. D, Data shown summarize the results obtained utilizing six p53-WT (blue labels) and six p53-mutant GBM lines (red labels) subjected to varying senescence induction paradigms followed by exposure to BCL-XL inhibitors (A1331852 or A1155463) or the BCL-XL+BCL-2 inhibitor, navitoclax. Each data point depicts the mean fold change in IC50. Bold underline = GBM lines from patients with recurrent disease.
Timepoint of BCL-XL inhibitor sensitivity
In the previous experiments, radiated cells were maintained for 2–4 weeks after radiation prior to senolytic drug testing. While the senescence process often takes weeks, the apoptotic pathways regulated by BCL-2 family members are dynamically regulated within days following DNA damage. As such, we used GBM39 to ask if a minimum period of time after radiation must elapse following radiation prior to onset of BCL-XL sensitivity. Using a timed assay following radiation, GBM39 was treated with the BCL-XL inhibitors, A1331852 or A1155463, for 4 days, starting 1, 5, or 9 days after radiation. Analysis was performed at the end of the 4-day drug exposure. While efficacy was seen upon treatment starting 1 day after radiation, more complete cell ablation was observed with a leftward shift of the dose–response curve after 4 days had elapsed since radiation and prior to starting treatment 5 or 9 days following radiation (Fig. 3A; Supplementary Fig. S4B).
Figure 3.

GBM vulnerability to BCL-XL inhibition depends on postirradiation timepoint, radiation dose, and duration of inhibitor exposure. A, Sensitivity to BCL-XL inhibition at 4 (blue), 8 (purple), and 12 (red) days after 15 Gy radiation. All cohorts were exposed to drugs for the same amount of time (4 days) and were analyzed on the fifth day after plating. Very similar results were obtained with the alternative BCL-XL inhibitor A1155463, and after induction of senescence with TMZ—see Supplementary Fig. S4. B, Impact of prior radiation dose on BCL-XL inhibitor sensitivity: A1331852 treatment was initiated 4 days following various doses of radiation. C, Duration of drug exposure impacts GBM39 vulnerability to A1331852, applied 7 days after radiation for 1 to 96 hours with equal total culture duration prior to analysis. Luminescence values are normalized individually by 0 nmol/L control. Graphs show means ± SD of technical replicates at each concentration. D–I, Radiated GBM cells are selectively vulnerable to BCL-XL knockdown. BCL-XL, BCL-W, and BCL-2 were knocked down via siRNA in GBM39, 7 days after 0 Gy, or 15 Gy radiation (D–F). Quantified data from western blots are normalized to GAPDH. Control cells were transfected with a scrambled construct. Images for the Western blots used for quantification in A are provided in Supplementary Fig. S6. CellTiter-Glo analysis was performed to evaluate relative survival after knockdown with siRNA constructs against BCL-XL, BCL-W, and BCL-2 (G–I). Relative luminescence for knockdowns is shown in the 0 and 15 Gy groups, normalized to control (scrambled) knockdowns. Error bars show SD. J, Representative data demonstrating elevated caspase 3/7 activity 24 hours A1331852 exposure, quantified using Caspase-Glo assay. Experiment performed 7 days after 0 or 15 Gy radiation. Cells from the same experiment were analyzed for viability after 4 days of A1331852 exposure using CellTiter-Glo. Error bars = SD.
Dependency on drug exposure time and radiation dose
We next asked whether a minimal radiation dose was required to induce susceptibility to cell death with BCL-XL inhibition. Radiation doses of 4 Gy or higher in GBM39 promoted sensitivity to A1331852, with increasing efficacy up to 15 Gy (Fig. 3B). For most cell lines, radiation doses of 10–20 Gy yielded similar results (Supplementary Fig. S3; Supplementary Table S1). In anticipation of future therapeutic dosing, we next evaluated what duration of continuous BCL-XL inhibitor exposure is required to observe senolytic effects. To accomplish this, GBM39 was exposed to drugs for varying durations from 0–96 hours, with the drug then being washed off and cells maintained in normal growth media thereafter until time of analysis. Although an impact was observed even within 1 hour of treatment, maximal impact was seen in cells that had received sustained exposure to BCL-XL inhibition for 96 hours (Fig. 3B).
Elimination of TMZ-treated senescent glioma
Radiation and TMZ are both routinely administered to patients with GBM. Both may induce senescence and modulate apoptotic machinery. To determine whether TMZ (100 µmol/L) exposure similarly induces senescence and selective susceptibility to BCL-XL inhibition, we pretreated the GBM cell lines with TMZ for 20 days, leading to induction of SA-β-gal activity, upregulation of P21 expression and downregulation of Ki67, consistent with observed proliferative arrest in vitro. Using GBM39, we found that prior TMZ exposure for 14 days induced TMZ and BCL-XL inhibitor dose-dependent sensitivity to the BCL-XL–specific inhibitors A1331852 and A1155463 (Supplementary Fig 4D); GBM76 and GBM39 both showed comparatively minimal sensitivity to BCL-2–specific inhibition with venetoclax (Fig. 4). Because patients are generally treated with both TMZ and radiation, these were tested in combination. We failed to demonstrate a substantively additive effect between radiation and TMZ, perhaps suggesting a ceiling effect above which further cytotoxic insult will not further increase sensitivity to BCL-XL inhibition.
Figure 4.

TMZ exposure induces selective vulnerability to BCL-XL inhibitors. GBM76 and GBM39 were treated with TMZ (100 µmol/L) for 7days followed by 14 days of TMZ-free media prior to treatment with BCL-2 family inhibitors as shown. TMZ-treated cells demonstrated selective vulnerability to BCL-XL inhibitors (A1331852, A1155463, navitoclax), but not to the BCL-2–specific inhibitor (venetoclax). For all experiments, luminescence values are normalized individually to 0 nmol/L control. All data are means ± SD of three technical replicates at each concentration.
On the basis of the previously published selectivity of A1331852 and A1155463 to BCL-XL and venetoclax to BCL-2 (Supplementary Table S2), we predicted that knockdown of BCL-XL, but not BCL-2, would impede survival of radiated cells. We utilized siRNA constructs and scrambled controls and evaluated their impact on survival in radiated and nonradiated GBM. Compared with scrambled controls, BCL-2 and BCL-W knockdown elicited no impact on cell survival, whereas BCL-XL knockdown significantly decreased survival of radiated cells (Fig. 3D–I).
Finally, in one of our unutilized GBM39 cultures at 6 weeks following 8 Gy radiation, we observed spontaneous regrowth consistent with escape from senescence, with cell doubling time mirroring that of the original parent culture. This raised the question of whether prior cytotoxic therapy is sufficient to permanently induce sensitivity to BCL-XL blockade, or whether cells must remain in a senescent-like nonproliferative state to maintain sensitivity. Cells in the “escaped” culture reverted to quite poor sensitivity to BCL-XL inhibition (Supplementary Fig. S5). This is consistent with the observation that gliomas derived from patients with both primary and recurrent lesions (Fig. 5) proved relatively insensitive to BCL-XL inhibition until placed into a state of proliferative arrest with either TMZ or radiation.
Discussion
We here demonstrate that human GBM cells surviving in a senescent-like state after chemotherapy or TMZ are relatively more susceptible to BCL-XL blockade than naïve proliferative GBM. Conventional cancer therapies, such as radiation and alkylating chemotherapies, act through induction of oxidative stress and DNA damage (30, 31). These can exert therapeutic impact through multiple mechanisms: (i) lethal cell damage, (ii) inducing a senescent-like state of proliferative arrest that impedes further growth (32), (iii) promoting a state of tumor quiescence to attenuate proliferation, and/or (iv) altering the tumor microenvironment or tumor-immune response to achieve indirectly some combination of the first three goals (33).
The invariable recurrence of gliomas highlights the inadequate levels of lethal cell damage achieved by standard chemoradiation. Recent work showed that cancer cells with an initially senescent phenotype following chemotherapy escaped senescence upon p53 inhibition, giving rise to tumor stem cells that were more aggressive and resilient than those present in the original tumor (34–38). If true for glioma, senolytic therapies could help eliminate senescent glioma cells before they have the chance to reemerge as proliferative and predictably highly treatment-resistant recurrent gliomas.
Interestingly four of the GBM lines used (10, 76, 120, 123) were derived from a patient with recurrent tumors who had previously undergone chemotherapy and radiation (39). Although GBM123 was among the weaker responders, all others showed similarly augmented response to BCL-XL inhibition after reinduction of senescence with radiation or TMZ. Also of note, certain lines tested (including GBM6 and GBM10) have been reported as being radioresistant, with nominal prolongation of survival in xenografts upon radiation. Nevertheless, these were amenable to sufficient proliferative arrest in vitro to permit augmented ablation by BCL-XL inhibition.
p53 is a key regulator of cell-cycle arrest in the context of radiation-induced DNA damage and senescence, and is the most commonly mutated gene across human cancers (40, 41). Although several lines with some of the most robust relative sensitivity to BCL-XL (in radiated vs. nonradiated cells) were wild-type (WT) for p53 (e.g., GBM10, GBM39, GBM76, GBM114), highly significant induction was also observed in p53-mutant cell lines (GBM6, GBM12, GBM132, GBM196). Similarly, no clear p53 pattern was found in cells with modest radiation-induced BCL-XL inhibitor sensitivity: p53-mutant GBM123 showed only 2.5× induction of A1331852 sensitivity (results were more robust for A1155 and navitoclax; Fig. 3), yet p53-WT GBM164 showed a minimally significant difference between radiated and nonradiated cells after either A1331852 or navitoclax. That 11 of 12 tested lines showed significant induction of sensitivity to one or more BCL-XL inhibitors with radiation supports this broadly reproducible phenomenon across molecularly diverse GBM.
It is likely that additional stimuli besides radiation and alkylating chemotherapy may augment sensitivity to BCL-XL inhibition. Massler and colleagues, previously reported induction of synthetic lethality with BCL-XL inhibition in IDH-mutant gliomas, or upon induction of the on cometabolite D2-HG produced by IDH-mutant gliomas (42). Their study specifically utilized GBM164, which they observed to have robust sensitivity to BCL-XL. Indeed, among the 12 lines tested, GBM164 was one of the two most sensitive in the absence of radiation. However, the most sensitive line was GBM12, which is IDH WT. We additionally evaluated another line that, unlike GBM196, is homozygous for IDH1-R132H, in contrast to GBM164, which is heterozygous for IDH1-R132H. Accordingly, GBM196 generates higher levels of D2-HG, yet demonstrated approximately100× lower baseline sensitivity to A1331852 than GBM164. While we did not independently evaluate the impact of D2-HG on BCL-XL inhibition in this study, data obtained in this study would have not provided a rationale for doing so. Rather, our data indicate that baseline sensitivity to BCL-XL inhibition may vary widely among both IDH-mutant and IDH-WT cell lines. However, in 90% of cases, radiation exposure was observed to enhance radiation sensitivity. Notably, baseline sensitivity to BCL-XL was not predictive of the degree of final BCL-XL sensitivity achievable after exposure to radiation or TMZ. Indeed, certain lines with negligible baseline sensitivity (e.g., GBM39) were induced by radiation to be among the most sensitive after radiation.
As such, while some molecular therapies can be selectively targeted to specific tumor subtypes, our data with only 12 cell lines that are divergent across phenotypes for EGFR, PTEN, CDKN2A, and p53, as well as gender, age, MGMT methylation, molecular subtype, and recurrence status, revealed no specific molecular phenotype that would obviously portend poor response to BCL-XL inhibition. Rather, we speculate that the therapy-induced sensitivity to BCL-XL dependency may be mechanistically linked to intracellular processes subserving and maintaining a molecular mediator of proliferative arrest and could thus be synergistic with therapies that activate appropriate mitotic checkpoint machinery (43). Among master regulators of cellular senescence, a mechanistic role of p16INK4A is largely excluded by virtue of most tested lines being homozygous null for CDKN2A, which encodes p16INK4A, an inhibitor of CDK4/6 (22, 44, 45). This could, by implication, reduce the probability of synergy between BCL-XL inhibitors with CDK4/6 inhibitors; yet also induce senescence, at least in regards to BCL-XL inhibition. Conversely, p21CIP1 is almost ubiquitously conserved in glioma, is frequently overexpressed in glioma, and is upregulated by radiation-induced DNA damage, providing an attractive avenue for future mechanistic investigations. While p21CIP1 is upregulated by p53, which is mutant in many GBMs, p21CIP1 is also regulated by other pathways and the interactions between mutant p53 and p21CIP1 can be paradoxical in cancer.
Recent work demonstrated that systemic navitoclax was effective for prolonging survival of GBM164 in a xenograft model as well as getting rid of senescent cells do (42, 46, 47). If BBB penetration were equivalent across cell lines, this would suggest that GBM114, GBM116, GBM120, GBM76, GBM39, and GBM12 should exhibit comparable or better sensitivity to BCL-XL inhibition after chemotherapy and/or radiation, whereas even radiated GBM132, GBM10, GBM123, and GBM196 each maintained an in vitro IC50 >10× that of nonradiated GBM164 and may thus be more resistant to BCL-XL inhibition. Efficacy of navitoclax as a senolytic was recently demonstrated in a tauopathy model of Alzheimer's disease, suggesting that BBB disruption may not be required for efficacy (48). Moreover, to the extent that radiation-induced senescence may contribute to radiation-induced cognitive impairment, concurrent ablation of senescent host cells could provide a cognitively beneficial adjunct to senescent tumor ablation (49).
While senescence serves to attenuate cell-intrinsic proliferation, SASP factors produced by senescent cells have been shown to promote tumor infiltration and recruitment of protumorigenic tumor-associated macrophages. We and others have observed increased aggressiveness of human glioma implanted into the previously radiated brain. Whether this phenomenon can be attenuated with senolytics is under investigation. However, the phenomenon of “senescence spreading” has been proposed as a means via which senescent cells could actually attenuate proliferation of adjacent tumor cells. The diversity of responses to BCL-XL inhibition in absence of clear phenotypic correlates suggests empiric studies will be required to discriminate among these possibilities.
The current study provides our first efforts to harness the senescent-like biology of treated human GBM to facilitate the clearance of dormant tumor cells. An obvious limitation of this work is that in vitro cultures offer notoriously poor surrogates of the in vivo human glioma microenvironment. Although we provide pilot data from a single in vivo experiment utilizing CED for BCL-XL inhibitor delivery and observed decreased bioluminescence, further work will be needed to test whether BCL-XL sensitivity is indeed higher in senescent than nonsenescent cells in vivo, and whether treatment can improve survival. Future work to critically characterize the molecular phenotype of senescent glioma will be important to optimally leverage vulnerabilities. Access to senescent human glioma tissue directly from human patients may be hampered by the fact that surgery is rarely performed except as treatment for actively proliferating disease. Moreover, senescent tumor cells may be present with relatively low abundance in latent disease and cannot be expanded given proliferative arrest. It is quite possible that the complex tumor microenvironment could endow malignant cells with diverse sources of trophic support that are lacking in vitro, potentially requiring adjuvant strategies not anticipated from in vitro analyses to achieve optimal senescent GBM ablation in vivo (50).
In conclusion, we demonstrate that radiation and TMZ endow glioma with increased dependence upon BCL-XL, blockade of which may facilitate ablation of latent glioma cells that survive prior cytotoxic therapy. Because radiation and TMZ are standard of care for GBM, further work is needed to determine whether BCL-XL inhibition could be leveraged to help forestall glioma recurrence.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
T.C. Burns was supported by NIH K12 NRCDP, R21 NS19770, the Minnesota Partnership for Biotechnology and Genomics, Lucius & Terrie McKelvey, Regenerative Medicine Minnesota, Humor to Fight the Tumor, and the Mayo Clinic Cancer Center. S. Burma was supported by grants from the NIH (R01CA258381 and R01CA246807) and by a National Aeronautics and Space Administration Award (NNX16AD78G). J.L.Kirkland and T. Tchkonia were supported by NIH R37 AG013925, P01 AG062413, and R33 AG061456 (Translational Geroscience Network). J.N. Sarkaria was supported by NIH U01CA227954 and U54 CA210180.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 835
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Disclosures
A.E. Warrington reports grants from NIH during the conduct of the study. T. Tchkonia reports grants from NIH; other support from Connor Fund, Robert & Arlene Kogod, and Robert & Theresa Ryan during the conduct of the study; in addition, T. Tchkonia has a patent for US 10,251,376 issued, licensed, and with royalties paid from Unity Biotechnologies and a patent for US 10,213,426 issued, licensed, and with royalties paid from Unity Biotechnologies. S. Burma reports grants from NIH and grants from NASA during the conduct of the study; and grants from NIH outside the submitted work. J.N. Sarkaria reports grants from GlaxoSmithKline, Bristol Myers Squibb, Curtana, Forma, AbbVie, Actuate, Boehringer Ingelheim, Bayer, Celgene, Cible, Wayshine, Boston Scientific, AstraZeneca, Black Diamond, Karyopharm, Fusion; other support from ADC Therapeutics, Travecta, and Aztek outside the submitted work. J.L. Kirkland reports grants from NIH; other support from Connor Fund, Robert & Arlene Kogod, Robert & Theresa Ryan, and Noaber Foundation during the conduct of the study; in addition, J.L. Kirkland has a patent for US 10,251,376 issued, licensed, and with royalties paid from Unity Biotechnologies and a patent for US 10,213,426 issued, licensed, and with royalties paid from Unity Biotechnologies. T.C. Burns reports a patent for 63/187,726 pending. No disclosures were reported by the other authors.
Authors' Contributions
M. Rahman: Conceptualization, data curation, formal analysis, validation, methodology, writing–original draft, project administration, writing–review and editing. I. Olson: Conceptualization, investigation, writing–original draft. M. Mansour: Data curation, formal analysis, investigation, methodology. L.P. Carlstrom: Conceptualization, validation, writing–original draft. R. Sutiwisesak: Methodology. R. Saber: Data curation, methodology. K. Rajani: Conceptualization, methodology. A.E. Warrington: Supervision, investigation, methodology, writing–review and editing. A. Howard: Methodology. M. Schroeder: Resources, supervision. S. Chen: Data curation, formal analysis, methodology. P.A. Decker: Formal analysis. E.F. Sananikone: Writing–review and editing. Y. Zhu: Resources, methodology. T. Tchkonia: Conceptualization. I.F. Parney: Resources. S. Burma: Writing–review and editing. D. Brown: Supervision. M. Rodriguez: Supervision, project administration, writing–review and editing. J.N. Sarkaria: Resources, supervision. J.L. Kirkland: Conceptualization, supervision, writing–review and editing. T.C. Burns: Conceptualization, resources, data curation, supervision, funding acquisition, validation, writing–review and editing.
References
- 1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 2. Adamski V, Hempelmann A, Flüh C, Lucius R, Synowitz M, Hattermann K, et al. Dormant glioblastoma cells acquire stem cell characteristics and are differentially affected by temozolomide and AT101 treatment. Oncotarget 2017;8:108064–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Filippi-Chiela EC, Thomé MP, Bueno e Silva MM, Pelegrini AL, Ledur PF, Garicochea B, et al. Resveratrol abrogates the temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the temozolomide-induced senescence in glioma cells. BMC Cancer 2013;13:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sughrue ME, Sheean T, Bonney PA, Maurer AJ, Teo C. Aggressive repeat surgery for focally recurrent primary glioblastoma: outcomes and theoretical framework. Neurosurg Focus 2015;38:E11. [DOI] [PubMed] [Google Scholar]
- 5. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One 2013;8:e55665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pawlowska E, Szczepanska J, Szatkowska M, Blasiak J. An interplay between senescence, apoptosis and autophagy in glioblastoma multiforme-role in pathogenesis and therapeutic perspective. Int J Mol Sci 2018;19:889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ye L, Wang C, Yu G, Jiang Y, Sun D, Zhang Z, et al. Bmi-1 induces radioresistance by suppressing senescence in human U87 glioma cells. Oncol Lett 2014;8:2601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeon HY, Kim JK, Ham SW, Oh SY, Kim J, Park JB, et al. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol 2016;37:5857–67. [DOI] [PubMed] [Google Scholar]
- 9. Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996;13:1065–72. [PubMed] [Google Scholar]
- 10. Van Meir EG, Kikuchi T, Tada M, Li H, Diserens AC, Wojcik BE, et al. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res 1994;54:649–52. [PubMed] [Google Scholar]
- 11. Georgakilas AG, Martin OA, Bonner WM. p21: a two-faced genome guardian. Trends Mol Med 2017;23:310–9. [DOI] [PubMed] [Google Scholar]
- 12. Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010;18:244–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hashizume R, Zhang A, Mueller S, Prados MD, Lulla RR, Goldman S, et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol 2016;18:1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553:96–100. [DOI] [PubMed] [Google Scholar]
- 15. Yeh AC, Ramaswamy S. Mechanisms of cancer cell dormancy–another hallmark of cancer? Cancer Res 2015;75:5014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, Fielder E, et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 2019;29:1061–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015;14:644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 2019;47:446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Himes BT, Peterson TE, de Mooij T, Garcia LMC, Jung MY, Uhm S, et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro Oncol 2020;22:967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiao Y, Feng Y, Wang X. Regulation of tumor suppressor gene CDKN2A and encoded p16-INK4a protein by covalent modifications. Biochemistry 2018;83:1289–98. [DOI] [PubMed] [Google Scholar]
- 23. Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg 2006;105:111–8. [DOI] [PubMed] [Google Scholar]
- 24. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest 2018;128:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Short S, Fielder E, Miwa S, von Zglinicki T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019;41:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc 2017;65:2297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017;9:955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016;15:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gentile M, Petrungaro A, Uccello G, Vigna E, Recchia AG, Caruso N, et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs 2017;26:1307–16. [DOI] [PubMed] [Google Scholar]
- 30. Davalli P, Marverti G, Lauriola A, D'Arca D. Targeting oxidatively induced DNA damage response in cancer: opportunities for novel cancer therapies. Oxid Med Cell Longev 2018;2018:2389523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther 2013;14:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mikula-Pietrasik J, Niklas A, Uruski P, Tykarski A, Ksiazek K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol Life Sci 2020;77:213–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rajani KR, Carlstrom LP, Parney IF, Johnson AJ, Warrington AE, Burns TC. Harnessing radiation biology to augment immunotherapy for glioblastoma. Front Oncol 2018;8:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ahmed AU, Auffinger B, Lesniak MS. Understanding glioma stem cells: rationale, clinical relevance and therapeutic strategies. Expert Rev Neurother 2013;13:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Almog N. Molecular mechanisms underlying tumor dormancy. Cancer Lett 2010;294:139–46. [DOI] [PubMed] [Google Scholar]
- 36. Gulaia V, Kumeiko V, Shved N, Cicinskas E, Rybtsov S, Ruzov A, et al. Molecular mechanisms governing the stem cell's fate in brain cancer: factors of stemness and quiescence. Front Cell Neurosci 2018;12:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rossari F, Zucchinetti C, Buda G, Orciuolo E. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis - clinical implications. Cell Oncol 2020;43:155–76. [DOI] [PubMed] [Google Scholar]
- 38. Triana-Martinez F, Loza MI, Dominguez E. Beyond tumor suppression: senescence in cancer stemness and tumor dormancy. Cells 2020;9:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vaubel RA, Tian S, Remonde D, Schroeder MA, Mladek AC, Kitange GJ, et al. Genomic and phenotypic characterization of a broad panel of patient-derived xenografts reflects the diversity of glioblastoma. Clin Cancer Res 2020;26:1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsuchida N, Gao C, Tsuchida Y, Nakajima T, Nishigaki R. [ DNA damage-induced signal pathway of p53 as a tumor suppressor and the gene mutation in human cancer]. Tanpakushitsu Kakusan Koso 2000;45:1742–51. [PubMed] [Google Scholar]
- 41. Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med 2016;6:a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 2017;8:1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dalton WB, Yang VW. Role of prolonged mitotic checkpoint activation in the formation and treatment of cancer. Future Oncol 2009;5:1363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McKenzie HA, Fung C, Becker TM, Irvine M, Mann GJ, Kefford RF, et al. Predicting functional significance of cancer-associated p16(INK4a) mutations in CDKN2A. Hum Mutat 2010;31:692–701. [DOI] [PubMed] [Google Scholar]
- 45. Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 2011;10:2497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020;583:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019;574:268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018;562:578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schindler MK, Forbes ME, Robbins ME, Riddle DR. Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys 2008;70:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gonzalez-Meljem JM, Apps JR, Fraser HC, Martinez-Barbera JP. Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer 2018;118:1283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine 2017;21:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 2015;7:279ra240. [DOI] [PubMed] [Google Scholar]
- 53. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19:202–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors and included in the article. Raw data for all graphs are provided as Supplementary Data.