

ABSTRACT
The NLRP3 (NLR family pyrin domain containing 3) inflammasome is involved in diverse inflammatory diseases, thus strict control of its activation is necessary to prevent excessive inflammation. Protein ubiquitination has been reported to regulate the assembly, protein expression and activation of the NLRP3 inflammasome. Until now, several deubiquitinases (DUBs) have been reported to affect the degradation of NLRP3 through the proteasome pathway. However, there is no research on DUBs regulating NLRP3 degradation through macroautophagy/autophagy. Here, we demonstrated the pivotal function of USP5 (ubiquitin specific peptidase 5) in restraining the activation of the NLRP3 inflammasome independent of its deubiquitinating enzyme activity. USP5 selectively promoted K48-linked polyubiquitination of NLRP3 and mediated its degradation through the autophagy-lysosomal pathway by recruiting the E3 ligase MARCHF7/MARCH7. Knockdown of USP5 facilitated the two-signal model of lipopolysaccharide and ATP-triggered IL1B/IL-1β production. Simultaneously, USP5 overexpression in vivo reduced IL1B and polymorphonuclear (PMN) infiltration in alum-induced peritonitis. Overall, the data revealed that USP5 is a key scaffold protein recruiting the E3 ligase MARCHF7 to NLRP3, and promoting autophagic degradation of NLRP3. The findings provide new insight into USP5 in the regulation of excessive activation of the NLRP3 inflammasome and inflammatory innate immune response.
Abbreviations
3-MA: 3-methyladenine; AIM2: absent in melanoma 2; ATG5: autophagy related 5; BafA1: bafilomycin A1; CASP1: caspase 1; CHX: cycloheximide; Co-IP: co-immunoprecipitation; CQ: chloroquine; DUBs: deubiquitinases; IL1B/IL-1β: interleukin 1 beta; LAMP1: lysosomal associated membrane protein 1; LPS: lipopolysaccharide; MARCHF7/MARCH7: membrane associated RING-CH-type finger 7; NFKB/NF-κB: nuclear factor kappa B; Nig.: nigericin; NLRC4: NLR family CARD domain containing 4; NLRP3: NLR family pyrin domain containing 3; PECs: peritoneal exudate cells; PMN: polymorphonuclear; PMs: peritoneal macrophages; PYCARD/ASC: PYD and CARD domain containing; TLRs: toll like receptors; TNF/TNF-α: tumor necrosis factor; Ub: ubiquitin; USP5: ubiquitin specific peptidase 5; WT: wild type.
KEYWORDS: Autophagy-lysosome pathway, deubiquitinase, MARCHF7, NLRP3 inflammasome, USP5
Introduction
NLRP3 (NLR family pyrin domain containing 3) inflammasome has a fundamental role in host defense against microbial pathogens and its deregulation may cause diverse inflammatory diseases. The NLRP3 inflammasome is a polyprotein complex composed of NLRP3, CASP1/caspase-1, and PYCARD/ASC (PYD and CARD domain containing) [1]. NLRP3 activation is a two-step process of priming and activation. After being recognized by TLRs (toll like receptors), extracellular and intracellular pathogen-associated molecular patterns (PAMPs) or danger/damage-associated molecular patterns (DAMPs) can activate NFKB/NF-κB (nuclear factor kappa B) signaling, causing the expression of precursors of IL1B/IL-1β (interleukin 1 beta) and IL18 (interleukin 18) and completing priming process [2]. The NLRP3 inflammasome can be further activated by extracellular ATP, potassium ion (K+) efflux, pore-forming toxins, mitochondrial reactive oxygen species, and destabilized lysosome [3]. These signals result in the assembly of the NLRP3 inflammasome complex and self-activation of the precursor CASP1, which then releases mature CASP1 (CASP1 p20). CASP1 p20 cleaves IL1B and IL18 to generate the mature forms [4,5], completing activation process.
The increased level of NLRP3 expression is a key step in inflammasome activation [6,7]. Moderate NLRP3 expression level can help the body resist exogenous microbial infection and endogenous cell damage. However, overaccumulation of NLRP3 protein can also trigger certain hereditary or acute inflammatory diseases, such as gout, atherosclerosis, and inflammatory bowel disease [5,8–10]. Regulation of NLRP3 expression can be achieved by modifying NLRP3 mRNA expression at the transcriptional level [11,12]. However, increasing evidence proved that post-translational modifications, especially protein ubiquitination, are important regulators of NLRP3 inflammasome assembly and activation [13]. Changes in protein ubiquitination caused by E3 ubiquitination ligases or deubiquitinases (DUBs) can promote protein degradation by the proteasome or autophagy-lysosomal system, thereby maintaining NLRP3 protein homeostasis. We previously reported that the E3 ubiquitin ligase TRIM31 attenuated NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3 [14]. NLRP3 is also found in autophagosomes and lysosomes upon inflammasome activation [15]. Autophagy-dependent degradation has been described in the regulation of NLRP3 expression [16]. However, there is no research on DUBs regulating NLRP3 degradation through autophagy.
Potential DUBs that could interact with NLRP3 have been screened [17]. The authors identified many NLRP3 interacting DUBs through co-immunoprecipitation (Co-IP) experiments in HEK293T cells [17]. After analyzing the protein localization of these DUBs, we found that USP5 may localize in lysosome, while other DUBs do not (Table S1). Given the fact that USP5 interacts with NLRP3 and NLRP3 can be degraded through autophagy-lysosome pathway, we hypothesized that USP5 may regulate NLRP3 degradation through the autophagy-lysosomal pathway. In this study, we showed that USP5 works as a scaffold to recruit MARCHF7 to promote K48-linked polyubiquitination and autophagic degradation of NLRP3. Knockdown of USP5 promoted the secretion of CASP1 p20 and IL1B p17, but had no effect on NFKB signaling. Overexpression of USP5 in vivo reduced IL1B and PMN infiltration in alum-induced peritonitis in which NLRP3 was reported to play protective role.
Results
USP5 specifically inhibits NLRP3 inflammasome activation
To investigate the effect of USP5 during NLRP3 inflammasome activation, the expression of Usp5 was first detected in peritoneal macrophages (PMs) stimulated by LPS and IL1B. Both the protein and mRNA levels of USP5 were increased in a time-dependent manner in PMs stimulated by LPS or IL1B (Figure 1A,B). The protein of USP5 was also increased in a concentration-dependent manner in PMs stimulated by LPS (Figure S1A). LAMP1 (lysosomal associated membrane protein 1) and LC3 are considered to be markers of lysosomes [18] or autophagosomes [19], respectively. Consistent with our prediction, confocal microscopy showed the co-localization of USP5 both with LAMP1 and LC3 (Figure S4A,B), confirming USP5 localizes in lysosomes and autophagosomes.
Figure 1.

USP5 specifically inhibits NLRP3 inflammasome activation. (A, B) RT-PCR analysis (A) or immunoblot analysis (B) of USP5 expression from mouse PMs stimulated with LPS or IL1B for various times. (C) ELISA of IL1B, TNF and IL6 in supernatants from mouse PMs silenced for Usp5, primed with LPS for various times, and followed by stimulated with ATP for 30 min. (D) ELISA of IL1B, TNF, and IL6 in supernatants from mouse PMs silenced for Usp5, primed with LPS for 8 h, and followed by stimulated with ATP, Nig., alum, poly(dA:dT) or flagellin for 30 min. (E–G) Immunoblot analysis of supernatants (SUP) or whole-cell lysates (WCL) from mouse PMs silenced for Usp5, primed with LPS, and followed by stimulated with ATP, alum, or Nig. for 30 min. (H) Densitometric analysis of protein expression levels in mouse PMs stimulated with LPS and followed by stimulated with ATP, alum, or Nig. for 30 min. Data are from three independent experiments (C,D, and H; mean ± SD of triplicate assays) or are representative of three independent experiments with similar results. *P < 0.05, **P< 0.01, ***P< 0.001, Student’s t-test.
To explore the physiological role of USP5, we designed a small interfering RNA (siRNA) targeting mouse Usp5 and transfected it into PMs. The knockdown efficiency of siRNA was validated at both the mRNA and protein levels (Figure S1B). We initially established a widely used two-signal model using LPS and ATP to detect the role of USP5 in the NLRP3 pathway. We found the secretion of IL1B was significantly increased in Usp5 knockdown PMs primed by LPS for different times and then treated with ATP (Figure 1C). However, the secretion of IL6 and TNF was not influenced by Usp5 siRNA transfection (Figure 1C). The mRNA expression of Il1b, Il6, and Tnf was also detected and no variances were observed between control siRNA transfected PMs and Usp5 siRNA transfected PMs (Figure S1C). Consistently, LPS-induced NFKB signaling and MAPK signaling including the phosphorylation of RELA/p65, MAPK/ERK, MAPK/JNK, and MAPK/p38 was comparable between control siRNA transfected PMs and Usp5 siRNA transfected PMs (Figure S1D,E).
To further corroborate the role of USP5 in the NLRP3 signaling pathway, two different activators of NLRP3, nigericin (Nig.) and alum were used. Similar to ATP stimulation, Nig. and alum induced IL1B secretion was also increased in Usp5 siRNA transfected PMs compared to that in control siRNA transfected PMs (Figure 1D). Again, LPS-induced secretion of IL6 and TNF was not affected by Nig. and alum (Figure 1D). To examine the specificity of the function of USP5 for the NLRP3 inflammasome, we utilized double-stranded DNA (poly(dA:dT)), a trigger of the AIM2 (absent in melanoma 2) inflammasome and flagellin, a trigger of the NLRC4 (NLR family CARD domain containing 4) inflammasome. We found the secretion of IL1B was comparable between control siRNA and Usp5 siRNA transfected PMs stimulated with poly(dA:dT) and flagellin (Figure 1D), indicating USP5 specifically regulates NLRP3 inflammasome. Further, immunoblotting analysis showed that Usp5 siRNA transfected PMs responded more efficiently than control siRNA transfected PMs to LPS plus ATP, LPS plus Nig. and LPS plus alum-induced content of IL1B p17 and CASP1 p20 in supernatants which indicated the activation of NLRP3 inflammasome (Figure 1E–H).
We also designed and synthesized a siRNA that target human USP5 and verified the interference efficiency in THP-1 cells (Figure S2A). Similar to the siRNA knockdown of Usp5 in PMs, transfection of USP5 siRNA in THP-1 cells resulted in elevated secretion of IL1B upon stimulation with LPS plus ATP (Figure S2B). Immunoblotting analysis also showed increased content of IL1B p17 and CASP1 p20 in supernatants (Figure S2C,D), indicating USP5 regulation of NLRP3 activation is not species specific. To summarize, these data suggested that USP5 specifically regulates the activation of the NLRP3 inflammasome, but not AIM2 or NLRC4 inflammasome.
USP5 interacts with NLRP3
To explore the mechanism by which USP5 inhibits NLRP3 activation, we examined whether USP5 interacts with the known NLRP3 inflammasome components. We expressed Flag-tagged USP5 with MYC-tagged full-length NLRP3, CASP1, and PYCARD in HEK293T cells and performed a co-immunoprecipitation (Co-IP) assay. Co-IP assays clearly showed the interaction between USP5 with NLRP3, but not with CASP1 or PYCARD (Figure 2A). Consistent with this, an interaction between endogenous USP5 and NLRP3 was evident in PMs (Figure 2B). Notably, the interaction between endogenous USP5 and NLRP3 increased upon stimulation with LPS. Confocal microscopy also showed the co-localization of USP5 with NLRP3, but not with AIM2 and NLRC4 (Figure 2C).
Figure 2.

USP5 interacts with NLRP3. (A) HEK293T cells expressing Flag-USP5 and MYC-NLRP3, MYC-CASP1 or MYC-PYCARD were lysed. Co-immunoprecipitation of Flag-USP5 with MYC-NLRP3, MYC-CASP1 or MYC-PYCARD from HEK293T cells. (B) Co-immunoprecipitation of endogenous USP5 with endogenous NLRP3, CASP1 or PYCARD from mouse PMs stimulated with LPS for indicated time periods. (C) HeLa cells transfected with GFP-USP5 and MYC-NLRP3, Flag-AIM2 or Flag-NLRC4 were fixed and incubated with a secondary antibody conjugated to Alexa Fluor 568. Colocalization between USP5 and NLRP3, AIM2 or NLRC4 were examined by Confocal microscopy. Scale bars: 10 µm. Intensity profiles of indicated proteins along the plotted lines, as analyzed by ImageJ line scan analysis. Nucleus were visualized with DAPI (blue). Confocal imaging results are representative of three independent experiments. (D) Schematic diagram of USP5 and its truncation mutants. (E) Flag-USP5 or its mutants and MYC-NLRP3 were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-MYC antibody and then immunoblotted with the indicated antibodies. (F) Schematic diagram of NLRP3 and its truncation mutants. (G) MYC-NLRP3 or its mutants and Flag-USP5 were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-MYC antibody and then immunoblotted with the indicated antibodies. Data are representative of three independent experiments with similar results.
USP5 consists of five individual domains designated cUBP, nUBP, USP, UBA1, and UBA2 [20]. To detect which domain(s) of USP5 were responsible for the interaction with NLRP3, five truncated mutants were constructed (Figure 2D). Co-IP results showed that USP5 mutants with only UBP domain or UBA domain lost the ability to interact with NLRP3, whereas USP5 mutant with only USP domain and USP5 mutants with UBP domain deletion or UBA domain deletion still interact with NLRP3 (Figure 2E). These findings indicated the USP domain on USP5 is required for the interaction between USP5 and NLRP3.
NLRP3 is composed of a C-terminal leucine-rich repeat (LRR), a central oligomerization domain termed NACHT, and an N-terminal pyrin domain (PYD) [21]. Co-IP experiments with three NLRP3 truncated mutants (Figure 2F) showed that NLRP3 mutant with PYD domain deletion lost the ability to interact with USP5 (Figure 2G). These collective findings indicated that USP5 interacts with NLRP3 through the USP motif on USP5 and the PYD domain on NLRP3.
USP5 promotes autophagic degradation of NLRP3
Considering the negative regulation of USP5 on the activation of NLRP3 inflammasome and interaction between USP5 and NLRP3, we speculated that USP5 may regulate NLRP3 protein expression. Indeed, we found overexpression of USP5 greatly attenuated NLRP3 protein expression in HEK293T cells and PMs, while the protein level of PYCARD and CASP1 was not changed upon USP5 overexpression (Figure 3A–C). Overexpression of USP5 also decreased NLRP3 protein expression, but not PYCARD and CASP1 protein expression in HeLa cells (Figure S3A). Knockdown of Usp5 significantly enhanced NLRP3 protein expression, and had no influence on CASP1 and PYCARD protein expression in LPS-primed mouse PMs (Figure 3D,E). Consistent with the fact that USP5 specifically regulates NLRP3 inflammasome, knockdown of Usp5 has no effect on the protein expression of AIM2 and NLRC4 (Figure 3D,E). Notably, the mRNA level of Nlrp3, Casp1, and Pycard did not change in Usp5 siRNA transfected PMs compared to that in control siRNA transfected PMs (Figure S3B), indicating USP5 regulates NLRP3 expression at the protein level, but not at the transcription level. To directly confirm that USP5 regulates NLRP3 degradation, PMs were first stimulated with LPS for 4 h prior to the treatment of protein synthesis inhibitor cycloheximide (CHX) for different times. We found the NLRP3 protein degradation was attenuated in Usp5 siRNA transfected PMs compared to that in control siRNA transfected PMs (Figure 3F,G). However, no differences in CASP1, PYCARD, AIM2, and NLRC4 protein degradation were observed between Usp5 siRNA and control siRNA transfected PMs (Figure 3F,G). These data indicated that USP5 decreases NLRP3 protein level by facilitating its degradation.
Figure 3.

USP5 promotes autophagic degradation of NLRP3. (A) MYC-NLRP3, MYC-CASP1 and MYC-PYCARD were transfected in HEK293T cells together with gradient amount of Flag-USP5. NLRP3, CASP1 and PYCARD protein level were detected by western blot. (B) Immunoblot analysis of extracts from mouse PMs reconstituted with vectors for MmUSP5, then stimulated with LPS (200 ng/mL) for various times. (C) Densitometric analysis of protein expression levels in mouse PMs stimulated with LPS. (D) Immunoblot analysis of extracts from mouse PMs silenced for Usp5, then stimulated for various times with LPS. (E) Densitometric analysis of protein expression levels in mouse PMs stimulated with LPS. (F, G) Immunoblot analysis of extracts from mouse PMs silenced for Usp5 stimulated with LPS for 4 h, and then treated for various times with cycloheximide (CHX, 100 ug/mL). NLRP3, PYCARD, CASP1, AIM2 and NLRC4 expression levels were quantitated by measuring band intensities using “ImageJ” software. The values were normalized to ACTB. (H) Immunoblot analysis of extracts from HEK293T cells transfected with MYC-NLRP3 and Flag-USP5 expression plasmid then treated with MG132 (10 mM), BafA1(1 µM), 3-MA (10 mM) or chloroquine (CQ, 10 µM) for 6 h.(I) Immunoblot analysis of extracts from mouse PMs reconstituted with vectors for MmUSP5 stimulated with LPS (200 ng/mL) for 4 h and then treated with MG132 (10 mM), BafA1(1 µM), 3-MA (10 mM) or chloroquine (10 µM) for 6 h before western blot.(J) Western blot was performed in lysates of HEK293T cells or HEK293T ATG5 KO cells expressing Flag-USP5 and MYC-NLRP3. (K) Immunoblot analysis of extracts from mouse PMs silenced for ATG5, then with LPS (200 ng/mL) for 4 h. Data are from three independent experiments (C, E, and G; mean ± SD of triplicate assays) or are representative of three independent experiments with similar results. *P < 0.05, **P< 0.01, ***P< 0.001, Student’s t-test.
The ubiquitin-proteasome system (UPS) and the autophagy-lysosomal system are the major pathways that regulate protein degradation [22]. To explore which pathway mediates USP5-induced degradation of NLRP3 protein, various inhibitors were used. We found the autophagy inhibitors 3-methyladenine (3-MA), bafilomycin A1 (BafA1), and chloroquine (CQ) suppressed USP5-induced NLRP3 degradation in HEK293T cells and PMs, while the proteasome inhibitor MG132 did not (Figure 3H,I). Similar results were obtained in HeLa cells (Figure S3C). Simultaneously, USP5 had no significant effect on LC3 conversion, which means that it had no detectable impact on overall autophagic flux (Figures 3H,I and S3C). Previous studies have demonstrated that NLRP3 can co-locate with autophagosomes and lysosomes after inflammasome activation [15]. Interestingly, we found partial co-localization between USP5, NLRP3, and LC3 or USP5, NLRP3, and LAMP1 after inflammasome activation (Figure S4A,B). Overall, these results indicated that USP5 induces NLRP3 degradation through autophagy-lysosomal system.
Autophagy-related proteins (ATGs) are key linker proteins in the autophagy pathway, among which ATG5 is integral in both canonical and non-canonical autophagy [23]. To directly confirm USP5 mediates NLRP3 protein degradation through autophagy, ATG5 knockout (KO) HEK293T cells were used. We found overexpression of USP5 failed to degrade NLRP3 in ATG5 KO cells (Figure 3J). Similar result was obtained in PMs transfected with Atg5 siRNA (Figure 3K). Altogether, these results demonstrated that USP5 could promote the degradation of NLRP3 through the autophagy-lysosome pathway.
USP5 increases K48-linked ubiquitination of NLRP3
The ubiquitin system plays an integral role in the control of NLRP3 inflammasome activation. NLRP3 can be ubiquitinated with both K48 and K63 linkage in macrophages [14,16]. Given that USP5 is a deubiquitinating enzyme, we first examined whether USP5-mediated degradation of NLRP3 depends on its enzymatic activity. It has been reported that USP5 stabilizes TUFM depends on its deubiquitinase activity [24]. As shown in Figure 4B, WT USP5 plasmid but not the enzymatic activity mutant USP5C335A plasmid was able to cleave TUFM ubiquitination (Figure 4A,B). We transfected plasmids of WT USP5 or mutant USP5C335A into HEK293T cells together with MYC-tagged NLRP3 and examined the expression of NLRP3 protein. We found both the WT USP5 and the mutant USP5C335A led to a significant reduction in NLRP3 protein expression (Figure 4C). Similar results were obtained in PMs (Figure 4D) and HeLa cells (Figure S3D). These data indicated that the degradative function of USP5 on NLRP3 does not require USP5 enzymatic activity.
Figure 4.

USP5 increases K48-linked ubiquitination of NLRP3. (A) Schematic diagram of WT USP5, USP5 enzymatically inactive mutant (USP5C335A). (B) Immunoblot analysis of lysates from HEK293T cells transfected with HA-tagged ubiquitin (HA-Ub), Flag-TUFM and WT GFP-USP5 or GFP-USP5C335A, followed by IP with anti-HA, probed with anti-Flag. (C) Immunoblot analysis of extracts from HEK293T cells transfected MYC-NLRP3 with Flag-USP5 or Flag-USP5C335A expression plasmids. (D) Immunoblot analysis of extracts from mouse PMs reconstituted with vectors for MmUSP5 or MmUSP5C335A stimulated with LPS (200 ng/mL) for 4 h before western blot. (E) Immunoblot analysis of lysates from HEK293T cells transfected with HA-tagged ubiquitin (HA-Ub), MYC-NLRP3 and WT GFP-USP5 or GFP-USP5C335A, followed by IP with anti-HA, probed with anti-MYC. (F) Pulldown (with GST beads and TUBE) and immunoblot analysis of extracts from mouse PMs silenced for Usp5 and then stimulated with LPS. (G) Immunoblot analysis of lysates from HEK293T cells transfected with HA-tagged K48-linked ubiquitin (K48-Ub) or HA-tagged K63-linked ubiquitin (K63-Ub), MYC-NLRP3 and Flag-USP5, followed by IP with anti-HA, probed with anti-MYC. (H) Immunoblot analysis of lysates from mouse PMs silenced for Usp5, followed by IP with anti-NLRP3, probed with anti-Ub, K48-Ub or K63-Ub. Data are representative of three independent experiments with similar results.
Next, we examined whether USP5 could modulates NLRP3 ubiqutination. GFP-tagged WT USP5 or USP5C335A plasmids were transfected into HEK293T cells together with MYC-tagged NLRP3 and HA-tagged ubiquitin plasmids, and then NLRP3 ubiquitination was examined. We found overexpression of USP5 could increase NLRP3 polyubiquitination (Figure 4E). Consistent with the data that USP5C335A promoted the degradation of NLRP3, overexpression of USP5C335A also led to a significant increase in NLRP3 ubiquitination (Figure 4E). Meanwhile, endogenous ubiquitin chains of NLRP3 were analyzed by enriching the whole ubiquitin chains by GST‐Ub‐TUBE. Consistent with the above results, knockdown of Usp5 greatly decreased LPS-induced polyubiqutination of NLRP3 (Figure 4F). We then defined the type of linkages in the polyubiquitin chains attached to NLRP3 mediated by USP5. HEK293T cells were transfected with the ubiquitin mutant K48 and K63, in which all of the lysine residues were replaced by arginine substitutions, except for the one at position 48 and 63, respectively. Overexpression of USP5 significantly increased NLRP3 ubiquitination in WT and K48 ubiquitin transfected cells, but not in K63 ubiquitin transfected cells (Figure 4G), indicating USP5 mainly promotes K48-linked ubiquitination of NLRP3. To concurrently exclude any artificial effects driven by overexpression and to explore whether USP5-mediated NLRP3 K48-linked polyubiquitination could occur under physiological conditions, polyubiquitination of endogenous NLRP3 protein was examined. LPS stimulation could increase both of K48- and K63-linked polyubiquitination of endogenous NLRP3 (Figure 4H). While siRNA knockdown of Usp5 expression greatly decreased LPS-induced K48-lined polyubiqutination of NLRP3, and K63-linked ubiquitination of NLRP3 was unchanged (Figure 4H). Together, these data revealed that USP5 mediated K48-linked ubiquitination independent on its enzymatic activity.
USP5 recruits MARCHF7 to promotes K48-linked ubiquitination of NLRP3
Since USP5 promoted NLRP3 polyubiquitination independent of its enzymatic activity, we hypothesized that USP5 likely works as a scaffold to recruit an E3 ligase for K48-linked polyubiquitination and degradation of NLRP3. Several E3 ligases involved in the polyubiquitination and degradation of NLRP3, such as TRIM31 [14], FBXL2 [25], ARIH2 [26], CUL1 [27] and MARCHF7 [16] have been reported. Among them, MARCHF7 was reported to mediate K48-linked ubiquitination and autophagic degradation of NLRP3 in dopamine (DA)-treated bone marrow-derived macrophages [16]. Thus, we investigated whether USP5-mediated ubiquitination of NLRP3 and degradation was dependent on MARCHF7 in LPS-treated macrophages.
We first transfected siRNAs specific to mouse Marchf7 into PMs, and the Marchf7 knockdown was efficient (Figure S5A). We found the secretion of IL1B was increased in Marchf7 knockdown PMs primed by LPS and then treated with ATP (Figure 5A). Marchf7 knockdown also enhanced NLRP3 protein expression in LPS-primed mouse PMs, and have no effect on CASP1 and PYCARD protein expression (Figure 5B,C). Contrarily, MARCHF7 overexpression could reduce NLRP3 protein expression (Figure 5D,E). These results indicated that MARCHF7 also regulates LPS- and ATP-activated NLRP3 inflammasome. To directly confirm MARCHF7 regulates USP5-mediated NLRP3 ubiquitination and degradation, we transfected siRNA targeting human MARCHF7 into HEK293T cells, and the efficiency of MARCHF7 knockdown was verified (Figure S5B). We found that USP5-mediated K48-linked ubiquitination of NLRP3 and its degradation was ablated by MARCHF7 knockdown (Figure 5F,G), confirming MARCHF7 is required for USP5-mediated NLPR3 ubiquitination. At the same time, when the siRNA of Usp5 and the siRNA of Marchf7 were co-transfected into PMs, the secretion of IL1B not changed (Figure 5H).
Figure 5.
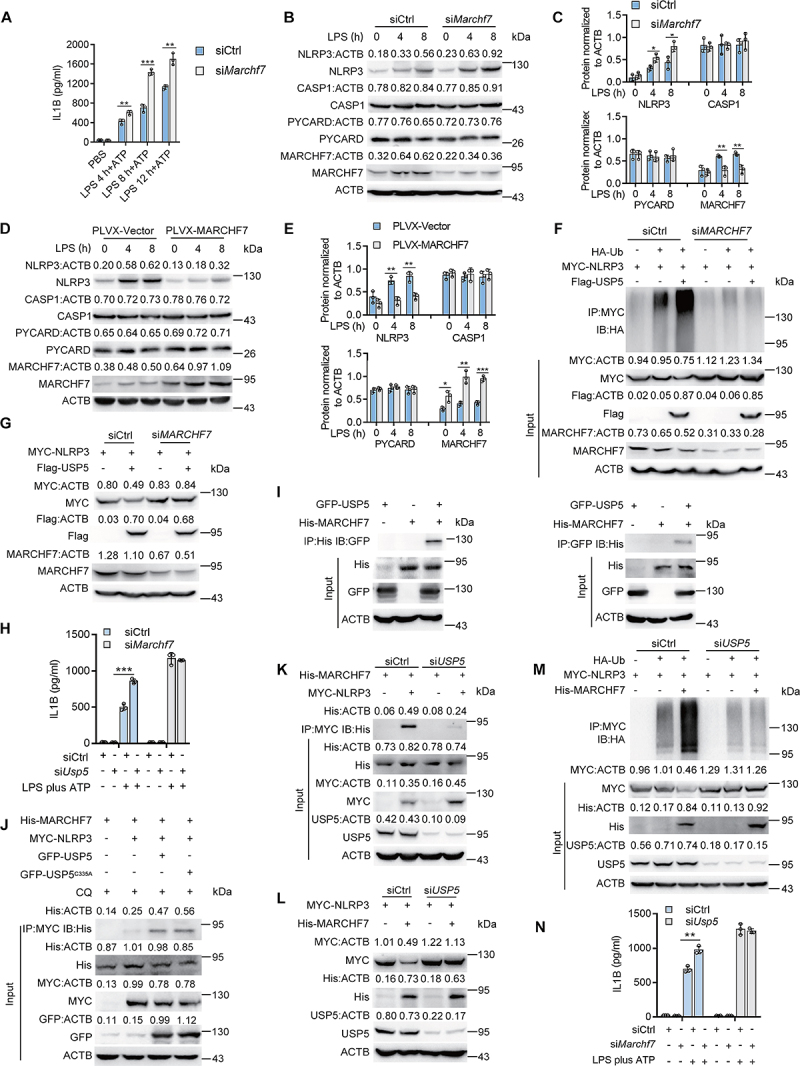
USP5 recruits MARCHF7 to promotes K48-linked ubiquitination of NLRP3. (A) ELISA of IL1B in supernatants from mouse PMs silenced for Marchf7, primed with LPS for various times, and followed by stimulated with ATP for 30 min. (B) Immunoblot analysis of extracts from mouse PMs silenced for Marchf7, then stimulated for various times with LPS. (C) Densitometric analysis of protein expression levels in mouse PMs stimulated with LPS. (D) Immunoblot analysis of extracts from mouse PMs reconstituted with vectors for MmMARCHF7, then stimulated with LPS (200 ng/mL) for various times. (E) Densitometric analysis of protein expression levels in mouse PMs stimulated with LPS. (F) Immunoblot analysis of lysates from HEK293T cells or HEK293T cells silenced for MARCHF7 transfected with HA-tagged K48-linked ubiquitin (K48-Ub), MYC-NLRP3 and Flag-USP5, followed by IP with anti-MYC, probed with anti-HA. (G) Co-IP was performed in lysates of HEK293T cells or HEK293T cells silenced for MARCHF7 expressing Flag-USP5 and MYC-NLRP3. (H) ELISA of IL1B in supernatants from mouse PMs silenced for Usp5 or both silenced for Usp5 and Marchf7, primed with LPS for various times, and followed by stimulated with ATP for 30 min.(I) HEK293T cells expressing His-MARCHF7 and GFP-USP5 were lysed. Co-immunoprecipitation of His-MARCHF7 with GFP-USP5 from HEK293T cells. (J) Co-immunoprecipitation and immunoassay of extracts of HEK293T cells transfected with MYC-NLRP3, His-MARCHF7 and GFP-USP5 or GFP-USP5C335A treated with chloroquine (10 µM) for 6 h. Co-immunoprecipitation of His-MARCHF7 with MYC-NLRP3 from HEK293T cells. (K) HEK293T cells or HEK293T cells silenced for USP5 expressing His-MARCHF7 and MYC-NLRP3 were lysed. Co-immunoprecipitation of MYC-NLRP3 with His-MARCHF7 from HEK293T cells. (L) Western blot was performed in lysates of HEK293T cells or HEK293T cells silenced for USP5 expressing His-MARCHF7 and MYC-NLRP3. (M) Immunoblot analysis of lysates from HEK293T cells or HEK293T cells silenced for USP5 transfected with HA-tagged K48-linked ubiquitin (K48-Ub), MYC-NLRP3 and His-MARCHF7, followed by IP with anti-MYC, probed with anti-HA. (N) ELISA of IL1B in supernatants from mouse PMs silenced for Usp5 or both silenced for Marchf7 and Usp5, primed with LPS for various times, and followed by stimulated with ATP for 30 min. Data are from three independent experiments (A, C, E, H, and N; mean ± SD of triplicate assays) or are representative of three independent experiments with similar results. *P < 0.05, **P< 0.01, ***P< 0.001, Student’s t-test.
We next investigated how USP5 regulates NLRP3 polyubiquitination through MARCHF7. Co-transfection and Co-IP assays showed that USP5 could interact with MARCHF7 (Figure 5I). Reciprocal pull-down achieved the same results (Figure 5I). As an E3 ligase for NLRP3, we confirmed the interaction between MARCHF7 and NLRP3 (Figure S5C). Interestingly, we showed that overexpression of USP5 increased the interaction between MARCHF7 and NLRP3 (Figure 5J). Notably, the USP5 enzymatic activity mutant USP5C335A could also promote the interaction between MARCHF7 and NLRP3 (Figure 5J), confirming the above results that the deubiquitinating enzyme activity is not required for the USP5-mediated polyubiquitination and degradation of NLRP3. Reciprocal pull-down achieved the same results (Figure S6D). In contrast, siRNA knockdown of USP5 expression in HEK293T cells impaired the binding of MARCHF7 and NLRP3 (Figure 5K). These results demonstrated that USP5 is required for MARCHF7 binding to NLRP3.
Finally, we checked whether USP5 is required for MARCHF7-mediated NLRP3 polyubiqutination and degradation. Overexpression of MARCHF7 could decrease the NLRP3 protein level, while knockdown USP5 expression in HEK293T cells restored NLRP3 expression (Figure 5L). Importantly, MARCHF7-induced NLRP3 polyubiquitination was greatly attenuated upon USP5 siRNA transfection (Figure 5M). We also found that when the siRNA of Usp5 and the siRNA of Marchf7 were co-transfected into PMs, the secretion of IL1B not changed (Figure 5N). Overall, the results indicated that USP5 recruited MARCHF7 to promote K48-linked polyubiquitination of NLRP3 and subsequent degradation.
USP5 overexpression reduces IL1B and PMN infiltration in alum-induced peritonitis in vivo
To investigate the physiological function of USP5, we established a USP5 overexpression model through injection of an expression plasmid encoding Flag-tagged mouse USP5 (Flag-MmUSP5) with in vivo transfection reagent intravenously. The expression of USP5 was confirmed in various organs collected from the treated mice (Figure S6). We first investigated the anti-inflammatory function of USP5 in vivo using an IL1B-dependent mouse peritonitis model [11]. Intraperitoneal injection of alum can induce the production of PMs in mice [28,29]. As shown in Figure 6A, the overexpression of USP5 could specifically inhibit the secretion of IL1B in vivo, while the production of other pro-inflammatory cytokines, such as TNF and IL6, was not affected (Figure 6A). In addition, less CASP1 p20 and IL1B p17 were also observed in the lavage fluid from USP5 overexpressing mice (Figure 6B). Next, the recruitment of inflammatory cells in the lavage fluid was analyzed. Alum-induced recruitment of PECs (peritoneal exudate cells) were reduced in the mice overexpressing USP5 (Figure 6C). Recruitment of neutrophils and LY6C+ monocytes were also inhibited by overexpression of USP5 (Figure 6D–G). Collectively, these findings further proved that USP5 could inhibit NLRP3 inflammasome and subsequent immune cell accumulation in mouse peritonitis in vivo.
Figure 6.
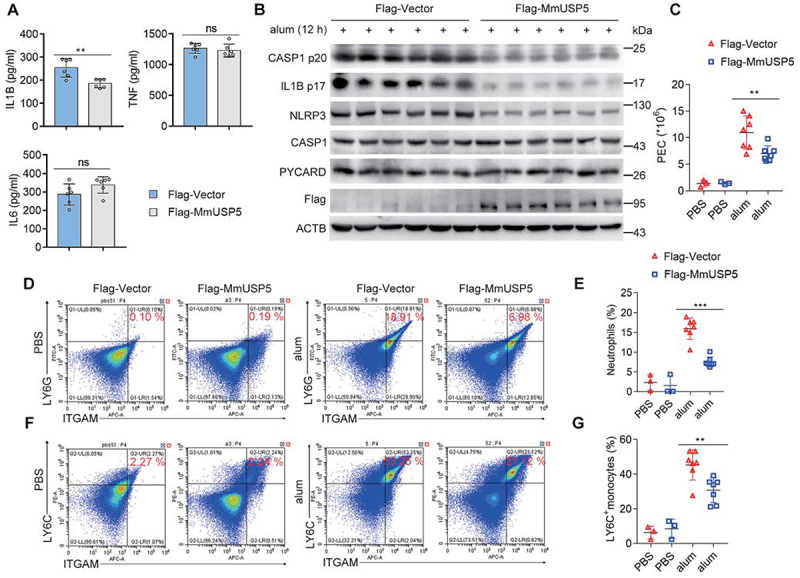
USP5 overexpression suppresses alum-induced peritonitis in vivo. (A) ELISA analysis (six mice per group) of serum levels of IL1B, TNF and IL6 from WT or USP5 overexpression mice after i.p. alum injection for 12 h. (B) WT or USP5 overexpression mice were i.p. injected with alum for 12 h. PECs were lysed and analyzed by immunoblot. (C) Absolute numbers of PECs. (D) Expression of ITGAM/CD11b and LY6G in PECs harvested from alum-injected WT or USP5 overexpression mice. Quadrants and numbers among an indicated percentage of cells in each gate (ITGAM+ LY6G+, ITGAM+ LY6G−, ITGAM− LY6G+, and ITGAM− LY6G−). (E) Percentage of ITGAM+ LY6G+ (neutrophils) among PECs harvested from alum-treated WT or USP5 overexpression mice (mice: WT-PBS, n = 3; USP5 overexpression-PBS, n = 3; WT-alum, n = 7; USP5 overexpression-alum, n = 7). (F) Expression of ITGAM and LY6C in PECs from WT or USP5 overexpression mice injected with alum. Quadrants and numbers among an indicated percentage of cells in each gate (ITGAM+ LY6C+, ITGAM+ LY6C−, ITGAM− LY6C+, and ITGAM− LY6C−). (G) Percentage of ITGAM+ LY6C+ (LY6C+monocytes) among PECs harvested from alum-treated WT or USP5 overexpression mice (mice: WT-PBS, n = 3; USP5 overexpression-PBS, n = 3; WT-alum, n = 7; USP5 overexpression-alum, n = 7). Data are shown as mean ± SD in (A, C, E, and G). *P < 0.05, **P< 0.01, ***P< 0.001; (A) Student’s t-test; (C, E, and G) Two-way analysis of variance (ANOVA). Similar results were obtained in three independent experiments.
Discussion
NLRP3 inflammasome can be activated by various danger signals, including bacterial products, such as muramyl dipeptide (MDP) [30] or LPS, as well as particulate materials, such as silicium dioxide or titanium dioxide [31,32]. The overactivated NLRP3 inflammasome is associated with many diseases, so the activation of NLRP3 inflammasomes needs to be strictly controlled [33]. Here we show a pivotal function of USP5 in negatively regulating the NLRP3 inflammasome by acting on the second activation step (Figures 1, S1 and S2). USP5 could selectively promote K48-linked polyubiquitination of NLRP3 (Figure 4) and mediate NLRP3 degradation in autophagy (Figures S1, S3, S4, Figures 2 and 3) by recruiting the E3 ligase MARCHF7 (Figure 5). The roles of USP5 were also confirmed in the alum-induced peritonitis (Figure 6) in vivo. In fact, USP5 overexpression could reduce IL1B and PMN infiltration in alum-induced peritonitis, which was consistent with our previous results that USP5 could negatively regulate NLRP3 inflammasome.
Over the past decade, a large number of studies have shown that NLRP3 inflammasome activity is extensively regulated by deubiquitination. As positive regulators, USP7 and USP47 facilitates PYCARD speck formation and NLRP3 inflammasome activation by deubiquitinating NLRP3 in an unknown mechanism [34]. Also, BRCC3, a member of the JAMM deubiquitination enzyme family, promotes IL1B secretion by cleaving its K63-linked polyubiquitin chain on LRR domain of the NLRP3 [17]. With the assistance of UAF1, USP1 removes K48-linked polyubiquitin chain on NLRP3 and inhibits its degradation in proteasome pathway [35]. In addition, USP19 inhibits the proteasomal degradation of inflammasome-independent NLRP3 by cleaving its polyubiquitin chains and stabilizes NLRP3 promoted M2-like macrophage polarization [36]. By contrast, A20 can inhibit the activation of NLRP3 inflammasome by suppressing NFKB pathway, mainly playing a role in the transcription step of NLRP3 inflammasome [37]. However, whether deubiquitinating enzymes (DUBs) could promote the degradation of NLRP3 especially in lysosomal pathway and restrain the activation of NLRP3 is still not reported. Here, we found that USP5 is involved in the autophagy degradation of NLRP3 as an important DUB.
USP5 has been found to play important roles in multiple cellular signal pathways, such as in DNA repair [38], stress reactions [39] and cancer progression [24,40–42]. However, the function of USP5 in inflammasome pathways as an innate immunity regulator has not been studied. In this paper, we investigate the function of USP5 in NLRP3 inflammasome signaling for the first time. Most of the reported DUBs can remove the ubiquitination of the substrates. But at the same time, DUBs can enhance the ubiquitination of certain target proteins. The functional patterns used by DUBs to change the different types of ubiquitination of its substrates can be summarized into six working patterns, which are dependent or independent of their enzyme activities [43]. USP5 has been reported to regulate function of SMURF1 [44], HDAC2 [45], OSBPL8/ORP8 [46], and TUFM [24] depending on its deubiquitinase activity. Interestingly, in our report, USP5 could promote, but not cleave, the K48-linked ubiquitination level of NLRP3 and degrade NLRP3 (Figure 4). In previous study, USP5 inhibits IFNB expression and supports VSV replication by recruiting STUB1 to degrade DDX58/RIG-I [43]. These inspire us that USP5 may act as a critical scaffold protein and recruit a specific E3 ligase to regulate the activation of NLRP3 inflammasome.
MARCHF7 is the only known E3 ligase, which could lead to the lysosomal degradation of NLRP3 in the existence of dopamine [16]. However, our location prediction analysis of MARCHF7 did not reveal a direct lysosomal location (data not shown). Thus, the detailed mechanism by which MARCHF7 mediated autophagic degradation of NLRP3 needs further study. Since USP5 is located in the lysosome (Figure S4), we hypothesized that USP5 may recruit MARCHF7 and degrade NLRP3. To validate the role of MARCHF7 in USP5-mediated NLRP3 polyubiquitination and degradation, we demonstrated that MARCHF7 also plays a role in the classic two-step activation (LPS plus ATP stimulation) of inflammasome firstly, and an obvious combination between USP5 and MARCHF7 was easily detected. Secondly, USP5 failed to enhance the K48-linked ubiquitination of NLRP3 and subsequently abolished its function in promoting the degradation of NLRP3 when MARCHF7 was knockdown, which indicated that USP5 mediated-NLRP3 degradation relied on MARCHF7. Thirdly, knockdown of USP5 could greatly inhibit the interaction between MARCHF7 and NLRP3, and decrease the degradation and ubiquitination of NLRP3 by MARCHF7, which indicated that MARCHF7-mediated polyubiquitination and autophagic degradation of NLRP3 dependent on USP5 (Figure 5). Taken all together, our findings indicated USP5 is a key scaffold protein recruiting E3 ligase MARCHF7 to NLRP3, and promoting K48-linked polyubiquitination followed by autophagy-mediated degradation of NLRP3. At the same time, this scaffolding function of USP5 is not specific, because it can also recruit STUB1 to increase the ubiquitination of DDX58. The results extend previous studies by providing more details of the role for MARCHF7 in the regulation of NLRP3 inflammasome activation especially in the absence of dopamine.
In summary, we identified the first DUB which promoting NLRP3 degradation in autophagy-lysosomal system, and propose a working model to illustrate how USP5 negatively regulates NLRP3 inflammasome activation (Figure 7). Our results link DUB, E3 ubiquitin ligase, and autophagy system to a complex regulatory network that sensitively regulates the activation of the NLRP3 inflammasome. This complex control may play a critical role in preventing excessive NLRP3 activation and uncontrolled inflammatory diseases. The NLRP3 inflammasome is an important focus of biomedical research and a therapeutic target for a variety of diseases. Our findings provide a new insight into USP5 in the regulation of excessive activation of NLRP3 inflammasome and inflammatory innate immune response. The deepening of this study will also contribute to a better understanding of related diseases and provide important evidence that will inform the development of new therapies.
Figure 7.

Model of USP5 participating in NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. USP5 could promote K48-linked polyubiquitination of NLRP3 and mediate NLRP3 degradation in autophagy by recruiting E3 ligase MARCHF7, inhibiting the inflammasome signaling pathway.
Materials and methods
Reagents and antibodies
LPS (Escherichia coli, 055:B5; L6529), 3-MA (M9281), ATP (A1852), MG132 (C2211), CHX (66-81-9), and chloroquine (CQ; C6628) were purchased from Sigma-Aldrich and were used at a final concentration of 200 ng/mL, 10 mM, 5 mM, 10 µM, 100 ug/mL, and 10 µM, respectively. IL1B (211-11B) was from PeproTech and was used at a final concentration of 10 ng/ mL. Imject™ Alum Adjuvant (77161) was obtained from Thermo Fisher Scientific and was used at a final concentration of 200 µg/mL. Nigericin (tlrl-nig), flagellin (tlrl-epstfla), poly(dA:dT) (tlrl-patn-1), and bafilomycin A1 (BafA1; tlrl-baf1) were from Invivogen and were used at a final concentration of 10 μM, 200 ng/mL, 200 ng/mL, and 1 µM, respectively. Poly(dA:dT) and flagellin were transfected into macrophages. Anti-CASP1 p45 and p20 (AG-20B-0042-C100), anti-NLRP3 (for western; AG-20B-0014-C100) were from AdipoGen. Anti-USP5 (sc-390943), anti-ATG5 (sc-133158), anti-MARCHF7 (sc-166945), anti-Ub (sc-8017), anti-ACTB (sc-8432), protein G agarose (sc2003) used for IP, and horseradish peroxidase-conjugated goat anti mouse(sc-2005) or rabbit (sc-2004) secondary antibodies were from Santa Cruz Biotechnology. Anti-IL1B p31 (12242S) and p17(85658S), anti-LC3 (4108); anti-p-MAPK/JNK (9252S), anti-p-RELA/p65 (3033S), anti-p-MAPK/p38 (4511S), anti-p-MAPK/ERK (4695S), anti-MAPK/JNK (9252S), anti-RELA/p65 (8242S), anti-MAPK/p38 (8690S), anti-MAPK1/ERK2-MAPK3/ERK1 (4370S), anti-K48-ub (8081S), anti-GFP (2956S), and anti-K63-ub (12930S) were from Cell Signaling Technology. Anti-LAMP1(ab25630), anti-NLRP3 (for immunofluorescence staining; ab4207); Alexa Fluor 405 donkey anti goat (ab175665), anti-AIM2 (ab204995), and anti-NLRC4 (ab201792) were from Abcam. Rabbit anti-MYC (A190-105A) was from Bethl Laboratories. Rabbit anti-Flag (PA1-984B) was from Invivogen. Rabbit anti-HA (600-401-384) was from Rockland. Rabbit anti-His (66005-1-Ig) and rabbit anti-V5 (14440-1-AP) were from Proteintech. Mouse anti-MYC (TA150121) and mouse anti-HA (TA100012) were from Origene. Mouse anti-Flag (F9291) was from Sigma-Aldrich; Alexa Fluor 488 (A11001) and Alexa Fluor 568 (A11004) goat anti mouse secondary Ab, and Alexa Fluor 488 (A11034) and Alexa Fluor 568 (A11011) goat anti rabbit secondary Ab were from Thermo Fisher Scientific.
Mice and cell culture
C57BL/6 mice used to prepare PMs were obtained from Joint Ventures Sipper BK Experimental Animal (Shanghai, China). Mice were housed in individually ventilated cages. Mice were culled using cervical dislocation isolated from the other mice. To obtain mouse PMs, the mice were injected with starch by intraperitoneal injection 3 days in advance, and peritoneal exudate cells were obtained 3 days later. After 2 h, unless adherent cells were removed, adherent monolayer cells were used as PMs. Human HeLa (CCL-2), HEK293T(CRL-1573), and THP-1 cells (TIB-202) were obtained from the American Type Culture Collection/ATCC. HeLa and HEK293T were cultured at 37°C in an atmosphere of 5% CO2 in DMEM (HyClone GE Healthcare, SH30022.01) supplemented with 10% fetal bovine serum (FBS; Thermo Scientific, 10082147), 100 U/mL penicillin and100 µg/mL streptomycin (Thermo Scientific, 15140122). THP-1 cells were cultured in RPMI-1640 medium (HyClone GE Healthcare, SH30027.01) supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, THP-1 cells were differentiated to a macrophage-like state by incubating with 20 nM PMA (Sigma-Aldrich, P1585) overnight.
Construction and transfection of plasmids
NLRP3, CASP1, and PYCARD cDNA were amplified from THP-1 cells or PMs by standard PCR and cloned in pCMV-N-MYC plasmids (Beyotime Biotechnology, D2756). USP5 was cloned in pCMV-2-Flag (Sigma-Aldrich, E7398), pCDNA3.1 (invitrogen, V79020), and pEGFP-C1 (Clontech, 6084-1) plasmids. Expression vector for AIM2-Flag and NLRC4-Flag were provided by Dr. Xiaopeng Qi (Shandong University). Deletion and truncation cDNA were amplified from THP-1 cells and subcloned into pCMV2-Flag plasmids. Point mutations were generated using the KOD-Plus-Mutagenesis kit (Toyobo, 956900). MARCHF7 (CH889469) and TUFM (CH854204) were purchased from Vigene Biosciences, and HA-ubiquitin and other plasmids were described previously [14]. All constructs were confirmed by DNA sequencing. Plasmids were transiently transfected into HEK293T cells using Lipofectamine 2000 reagent (Invitrogen, 11668019) according to the manufacturer’s instructions.
RNA interference assay and RNA quantitation
The siRNAs were synthesized as follows: murine Usp5: 5ʹ-GGAAUUCUUCCUACAUCUUTTAAGAUGUAGGAAGAAUUCC-3ʹ, human USP5: 5ʹ-GCAGAUGGGUGAUCUACAATTUUGUAGAUCACCCAUCUGC-3ʹ for siUSP5-1;5ʹ-GGAGCUGACGUGUACUCAUTTAUGAGUACACGUCAGCUCC-3ʹ for siUSP5-2;5ʹ-GCCUGACAUUGUCAGAGAUTTAUCUCUGACAAUGUCAGGC-3ʹ for siUSP5-3. Unless specified, siUSP5-2 was used for the following experiment. Murine Marchf7 was 5′- GGACUUAUGUAGAAUUUGU-3. Human MARCHF7 was 5ʹ-GCACUUGGGAGUAAUUUGA-3ʹ for siMARCHF7-1, and 5ʹ-GCACACGUGUCCGAUUUAU-3ʹ for siMARCHF7-2. If not specified, a mixture of the two siRNAs were used. Murine Atg5 was 5ʹ-CCTTTGGCCTAAGAA GAAA-3ʹ for siAtg5-1, and 5ʹ- CATCTGAGCTACCCGGATA-3ʹ for siAtg5-2. If not specified, a mixture of the two siRNAs were used. The negative control was 5ʹ-UUCUCCGAACGUGUCACGU-3ʹ. These siRNA duplexes were transfected into mouse PMs or THP-1 cells using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, 13778150) according to the manufacturer’s instructions.
Total RNA from cells were extracted using the RNA fast 200 kit (Fastagen Biotech, 220011) and was reverse transcribed using reverse transcriptase (TaKaRa Bio, RR047A). A LightCycler and SYBR RT-PCR kit (Roche, 06924204001) were used for real-time quantitative RT-PCR (qRT-PCR) analysis. Specific primers used for RT-PCR assays were the sequences of primers used for RT-PCR was 5ʹ-CATTTGGAAGATTACCCGCCC-3ʹ and 5ʹ-CTCTGGCTGGTCCTTCTTAGC-3ʹ for mouse Nlrp3; 5ʹ-ACTGACTGGGACCCTCAAGT-3ʹ and 5ʹ-GCAAGACGTGTACGAGTGGT-3ʹ for mouse Casp1; 5ʹ-GCAACTGCGAGAAGGCTATG-3ʹ and 5ʹ-GCTCCTGTAAGCCCATGTCT-3ʹ for mouse Pycard; 5ʹ-GGGAGACTGGCTACCCCTTA-3ʹ and 5ʹ-AGTGAGACAAGTGCTCTGCC-3ʹ for mouse Usp5; 5ʹ-ACCTTCCAGGATGAGGACATGA-3ʹ and 5ʹ-AACGTCA
CACACCAGCAGGTTA-3 0 for Il1b; 5‘-GCCACCACGCTCTTCTG
TCT-3ʹ and 5ʹ-TGAGGGTCTGGGCCATAGAAC-3ʹ for mouse Tnf; 5ʹ-ACAACCACGGCCTTCCCTAC-3ʹ and 5ʹ-
CATTTCCACGATTTCCCAGA-3ʹ for mouse Il6; and 5ʹ-CCACACCCGCCACCAGTTCG-3ʹ and 5ʹ-TACAGCCCGGGGAGCATCG-3ʹ for mouse Actb. Data were normalized to ACTB expression in each sample.
Co-IP and immunoblot analysis
Whole-cell extracts were lysed in IP buffer (1.0% [vol:vol] Nonidet P40 [Solarbio, N8030], 50 mM Tris HCl [Solarbio, Z9912], pH 7.4, 50 mM EDTA [Solarbio, E1170], 150 mM NaCl), and a protease inhibitor mixture (Roche diagnostics GmbH, 11836170001). After centrifugation for 10 min at 12,000 g, supernatants were collected and incubated with protein G Plus-Agarose Immunoprecipitation reagent (Santa Cruz Biotechnology, sc-2003) together with IP antibodies. After of incubation overnight, beads were washed four times with immunoprecipitation buffer. Immunoprecipitates were eluted by boiling with 1% (wt:vol) SDS sample buffer and boiled at 100°C for 5 min then followed by western blot. Equal amounts of extracts were separated by SDS-PAGE, and then they were transferred onto nitrocellulose membranes and then blotted with specific Abs.
ELISA
The concentrations of IL1B (Dakewe Biotech Company Ltd., 1210122), TNF (Dakewe Biotech Company Ltd., 1217202), and IL6 (Dakewe Biotech Company Ltd., 1210602) were measured using ELISA kits according to the manufacturer’s instructions.
Ubiquitination assays
To evaluate ubiquitination of NLRP3 in HEK293T cells, the cells were transfected with MYC-NLRP3, HA-Ub (WT), HA-K48(K48), HA-K63(K63), and GFP-tagged WT or mutant USP5. Whole-cell extracts were Immunoprecipitated with anti-HA and analyzed by immunoblotting with anti-MYC antibody. To evaluate ubiquitination of endogenous NLRP3, macrophages were stimulated with LPS (200 ng/mL) then whole-cell lysates were IPed with anti-NLRP3 and analyzed by immunoblotting with anti-Ub antibody or incubated with GST-Ub-TUBE (LifeSensors, UM101) and pulled down with GST beads (Qiagen, 30950). The GST beads were washed with lysis buffer containing 500 mM NaCl for three times and subject to PAGE electrophoresis and immunoblot analysis.
Immunofluorescence staining and confocal analysis
HeLa cells were co-transfected with GFP-USP5 and MYC-NLRP3 for 24 h. The culture medium on the upper layer of the 12-well plate was removed and washed twice in 1× PBS (Santa Cruz Biotechnology, sc-281692) for 5 min. PBS was added slowly along the wall of the plate to avoid washing away the cells. One milliliter of 4% paraformaldehyde (Beyotime Biotechnology, P0098) was added to each well with slow shaking at room temperature for 20 min. After washing three times, the 0.1% Triton X-100 (Beyotime Biotechnology, P0096) was added to each well for 15 min, followed by three washes with 1× PBS. Cells were blocked for 1 h in 1% BSA (Beyotime Biotechnology, P0102) and incubated with primary antibody overnight in 1% BSA, followed by incubation with Alexa Fluor 568 (Thermo Fisher Scientific, A11004) goat anti mouse secondary Ab, Alexa Fluor 568 (Thermo Fisher Scientific, A11011) goat anti rabbit secondary Ab, Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific, A11001) goat anti mouse secondary Ab, Alexa Fluor 488 (Thermo Fisher Scientific, A11034) goat anti rabbit secondary Ab or Alexa Fluor 405(abcam, 175665) donkey anti goat secondary Ab. Nuclei were counterstained with anti-fade fluorescence mounting medium with DAPI (abcam, ab104139) or anti-fade fluorescence mounting medium (abcam, ab104135). Cells were examined by confocal laser microscopy using a model LSM780 microscope (Carl Zeiss, Oberkochen, Germany).
MmUSP5 and MmMARCHF7 overexpression lentivirus
pLVX-IRES-Puro-MmUSP5 and pLVX-IRES-Puro-MmMARCHF7 (mouse MARCGF7) were generated by subcloning the MmUSP5-coding sequence and MmMARCHF7-coding sequence into the pLVX-IRES-Puro vector, and the empty pLVX-IRES-Puro was used as a control. Plasmids encoding MmUSP5C335A was generated using the KOD-Plus-Mutagenesis kit. The lentivirus was produced by transient transfection of the pLVX-IRES-Puro-MmUSP5 and -MmMARCHF7 construct or control vector into HEK293T cells using Lipofectamine 2000 with pLVX-IRES-Puro, psPAX2, and pMD2. G.
In vivo peritonitis
WT or USP5 overexpressing mice (6–7 weeks old) were injected intraperitoneally with 700 µg of alum for 12 h. Peritoneal cavities were washed with 6 mL of PBS. The peritoneal fluids were harvested and concentrated for ELISA analysis and PECs were analyzed by fluorescence-activated cell sorting as described previously [14].
In vivo transfection
Plasmids were administered via tail vein injection. Flag-Vector or Flag-MmUSP5 plasmids were injected into mice along with the DNA transfection reagent (Entranster-in vivo; Engreen, 18668-11) by tail vein injection (40 µg/mouse) on the day before the alum model was started to ensure stable expression of plasmids in mice.
Flow cytometry
Single-cell suspensions of PECs were prepared and the cells were stained with antibodies against surface markers. The PECs were assessed with the markers 7AAD (BioLegend, 559925), LY6G (BioLegend, 127605) and ITGAM/CD11b (BioLegend, 17-0012-81) for analysis of the recruitment of neutrophils and with 7AAD, LY6C (BioLegend, 128007) and ITGAM for analysis of monocytes. Data were acquired on a LSRFortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software.
Statistical analysis
Statistical significance between groups was determined by the two-tailed Student’s t-test and two-way ANOVA test. Differences were considered significant at P < 0.05. Kaplan–Meier survival curves were generated and analyzed for statistical significance using GraphPad Prism 5.0 (GraphPad, La Jolla, CA, USA). Pilot studies were used to estimate the sample size to ensure adequate power. There was no exclusion of data points or mice. No randomization or blinding was used.
Ethics statement
All animal experiments were undertaken in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Ethics Committee of Scientific Research of Shandong University Qilu Hospital, Jinan, Shandong Province, China (Permit number: KYLL-2017(KS)-361).
Supplementary Material
Acknowledgments
We thank Dr. Xiaopeng Qi (Shandong University) for the expression plasmids for Flag-NLRC4 and Flag-AIM2.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China [31730026, 81930039, 81525012 to C.G., 31900680 to B.L. and 82001678 to T.C.]. This work was also supported by the National Postdoctoral Program for Innovative Talents [BX201700146 to B.L.] and Shandong Provincial Natural Science Foundation [ZR2018BC021 to B.L.].
Disclosure statement
The authors declare no competing interests.
Data availability
Data in support of the findings of this study are available from the corresponding author upon request.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Skeldon AM, Faraj M, Saleh M.. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92(4):304–313. [DOI] [PubMed] [Google Scholar]
- [3].Kang MJ, Jo SG, Kim DJ, et al. NLRP3 inflammasome mediates interleukin-1beta production in immune cells in response to Acinetobacter baumannii and contributes to pulmonary inflammation in mice. Immunology. 2017;150(4):495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Deuteraiou K, Kitas G, Garyfallos A, et al. Novel insights into the role of inflammasomes in autoimmune and metabolic rheumatic diseases. Rheumatol Int. 2018;38(8):1345–1354. [DOI] [PubMed] [Google Scholar]
- [5].Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. [DOI] [PubMed] [Google Scholar]
- [6].Van De Veerdonk FL, Netea MG, Dinarello CA, et al. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 2011;32(3):110–116. [DOI] [PubMed] [Google Scholar]
- [7].Dowling JK, O’Neill LA. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol. 2012;47(5):424–443. [DOI] [PubMed] [Google Scholar]
- [8].Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Youm YH, Grant RW, McCabe LR, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–477. [DOI] [PubMed] [Google Scholar]
- [11].Huai W, Zhao R, Song H, et al. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat Commun. 2014;5:4738. [DOI] [PubMed] [Google Scholar]
- [12].Fernandes-Alnemri T, Kang S, Anderson C, et al. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. 2013;191(8):3995–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. FEBS J. 2019;287(281):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Song H, Liu B, Huai W, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Spalinger MR, Lang S, Gottier C, et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy. 2017;13(9):1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yan Y, Jiang W, Liu L, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160(1–2):62–73. [DOI] [PubMed] [Google Scholar]
- [17].Py BDF, Kim M-S, Vakifahmetoglu-Norberg H, et al. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49(2):331–338. [DOI] [PubMed] [Google Scholar]
- [18].Hasegawa H, Hsu A, Tinberg CE, et al. Single amino acid substitution in LC-CDR1 induces Russell body phenotype that attenuates cellular protein synthesis through eIF2alpha phosphorylation and thereby downregulates IgG secretion despite operational secretory pathway traffic. MAbs. 2017;9(5):854–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Flores-Bellver M, Bonet-Ponce L, Barcia JM, et al. Autophagy and mitochondrial alterations in human retinal pigment epithelial cells induced by ethanol: implications of 4-hydroxy-nonenal. Cell Death Dis. 2014;5:e1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Avvakumov GV, Walker JR, Xue S, et al. Two ZnF-UBP domains in isopeptidase T (USP5). Biochemistry. 2012;51(6):1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108(4):615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pehote G, Vij N. Autophagy augmentation to alleviate immune response dysfunction, and resolve respiratory and COVID-19 exacerbations. Cells. 2020;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ye X, Zhou XJ, Zhang H. Exploring the role of Autophagy-Related Gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front Immunol. 2018;9:2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu X, Huang A, Cui X, et al. Ubiquitin specific peptidase 5 regulates colorectal cancer cell growth by stabilizing Tu translation elongation factor. Theranostics. 2019;9(14):4208–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Han S, Lear TB, Jerome JA, et al. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem. 2015;290(29):18124–18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kawashima A, Karasawa T, Tago K, et al. ARIH2 ubiquitinates NLRP3 and negatively regulates NLRP3 inflammasome activation in macrophages. J Immunol. 2017;199(10):3614–3622. [DOI] [PubMed] [Google Scholar]
- [27].Wan P, Zhang Q, Liu W, et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. 2019;33(4):5793–5807. [DOI] [PubMed] [Google Scholar]
- [28].Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223. [DOI] [PubMed] [Google Scholar]
- [29].Jin J, Yu Q, Han C, et al. LRRFIP2 negatively regulates NLRP3 inflammasome activation in macrophages by promoting Flightless-I-mediated caspase-1 inhibition. Nat Commun. 2013;4:2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Martinon F, Agostini L, Meylan E, et al. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14(21):1929–1934. [DOI] [PubMed] [Google Scholar]
- [31].Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178(8):5271–5276. [DOI] [PubMed] [Google Scholar]
- [32].Ruiz PA, Moron B, Becker HM, et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut. 2017;66(7):1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stutz A, Kolbe CC, Stahl R, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med. 2017;214(6):1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Palazon-Riquelme P, Worboys JD, Green J, et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018;19:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song H, Zhao C, Yu Z, et al. UAF1 deubiquitinase complexes facilitate NLRP3 inflammasome activation by promoting NLRP3 expression. Nat Commun. 2020;11(1):6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu T, Wang L, Liang P, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. 2020. DOI: 10.1038/s41423-020-00567-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vande Walle L, Van Opdenbosch N, Jacques P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512(7512):69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nakajima S, Lan L, Wei L, et al. Ubiquitin-specific protease 5 is required for the efficient repair of DNA double-strand breaks. PLoS One. 2014;9(1):e84899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nostramo R, Varia SN, Zhang B, et al. The catalytic activity of the Ubp3 deubiquitinating protease is required for efficient stress granule assembly in Saccharomyces cerevisiae. Mol Cell Biol. 2016;36(1):173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dayal S, Sparks A, Jacob J, et al. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem. 2009;284(8):5030–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu Y, Wang WM, Zou LY, et al. Ubiquitin specific peptidase 5 mediates histidine-rich protein Hpn induced cell apoptosis in hepatocellular carcinoma through P14-P53 signaling. Proteomics. 2017;17:12. [DOI] [PubMed] [Google Scholar]
- [42].Kim SY, Kwon SK, Lee SY, et al. Ubiquitin-specific peptidase 5 and ovarian tumor deubiquitinase 6A are differentially expressed in p53+/+ and p53−/- HCT116 cells. Int J Oncol. 2018;52(5):1705–1714. [DOI] [PubMed] [Google Scholar]
- [43].Liu Q, Wu Y, Qin Y, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv. 2018;4(5):eaar2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Qian G, Zhu L, Huang C, et al. Ubiquitin specific protease 5 negatively regulates the IFNs-mediated antiviral activity via targeting SMURF1. Int Immunopharmacol. 2020;87:106763. [DOI] [PubMed] [Google Scholar]
- [45].Du Y, Lin J, Zhang R, et al. Ubiquitin specific peptidase 5 promotes ovarian cancer cell proliferation through deubiquitinating HDAC2. Aging (Albany NY). 2019;11(21):9778–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Z, Gao W, Zhou L, et al. Repurposing brigatinib for the treatment of colorectal cancer based on inhibition of ER-phagy. Theranostics. 2019;9(17):4878–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data in support of the findings of this study are available from the corresponding author upon request.