

ABSTRACT
Intratumoural administration of unmethylated cytosine-phosphate-guanine motifs (CpG) to stimulate toll-like receptor (TLR)-9 has been shown to induce tumour regression in preclinical studies and some efficacy in the clinic. Because activated natural killer T (NKT) cells can cooperate with pattern-recognition via TLRs to improve adaptive immune responses, we assessed the impact of combining a repeated dosing regimen of intratumoural CpG with a single intratumoural dose of the NKT cell agonist α-galactosylceramide (α-GalCer). The combination was superior to CpG alone at inducing regression of established tumours in several murine tumour models, primarily mediated by CD8+ T cells. An antitumour effect on distant untreated tumours (abscopal effect) was reliant on sustained activity of NKT cells and was associated with infiltration of KLRG1+ NKT cells in tumours and draining lymph nodes at both injected and untreated distant sites. Cytometric analysis pointed to increased exposure to type I interferon (IFN) affecting many immune cell types in the tumour and lymphoid organs. Accordingly, antitumour activity was lost in animals in which dendritic cells (DCs) were incapable of signaling through the type I IFN receptor. Studies in conditional ablation models showed that conventional type 1 DCs and plasmacytoid DCs were required for the response. In tumour models where the combined treatment was less effective, the addition of tumour-antigen derived peptide, preferably conjugated to α-GalCer, significantly enhanced the antitumour response. The combination of TLR ligation, NKT cell agonism, and peptide delivery could therefore be adapted to induce responses to both known and unknown antigens.
KEYWORDS: Intratumoural therapy, CpG, NKT cells, abscopal effect
Introduction
Cancer immunotherapy has now become a treatment option for many cancer patients, with remarkable clinical responses observed for some treatment modalities, notably the use of immune checkpoint inhibitors, and the transfer of ex vivo-manipulated T cells.1–3 Despite these advances, only a minority of cancers respond to the most promising immunotherapeutic options. Indeed, durable complete responses indicative of long-term acquired immunity to tumour-associated antigens remain the exception in most settings, pointing to underlying mechanisms of resistance. Solid cancers pose a particular problem, as they typically develop a highly immunosuppressive microenvironment.4
One approach to relieve immunosuppression in accessible lesions is to deliver immunostimulatory compounds directly into the tumour, thereby stimulating cells of the infiltrate and encouraging appropriate cross-talk with draining lymphoid tissues.5 Studies have shown that intratumoural targeting of pattern recognition receptors, most notably TLRs, can enhance endogenous adaptive antitumour responses that have been constrained locally, and can unleash responses that become active on tumours at distant locations – an abscopal effect.6–8 The most advanced intratumoural treatment involves stimulation via TLR9 with synthetic oligodeoxynucleotides containing unmethylated cytosine-guanine motifs (CpG), which results in inhibition of tumour growth in multiple preclinical tumour models,9–13 at doses that fail when administered intravenously.14 Clinical benefits have been observed in trials of intratumoural CpG,15–17 whereas intravenous administration failed to provide any survival benefit.18 Following promising combination studies in animals,8,19 intratumoural TLR9 agonist administration is currently being explored clinically in combination with other therapies, including those with the tyrosine kinase inhibitor, ibrutinib20 and checkpoint inhibitors pembrolizumab, nivolumab, and ipilimumab (NCT03007732, NCT03445533, NCT03618641, NCT02680184).
Assuming that increased adaptive immunity induced by intratumoural CpG is largely mediated through stimulation of APCs engaged in restimulating activated T cells, or possibly even priming de novo responses, it is worth noting that pattern recognition alone may not maximize the stimulatory potential of APCs. For example, resting APCs triggered via pattern recognition receptors are known to upregulate IL-12p40 and augment CD40 expression, but it is only with CD40 ligation that significant increases in IL-12p35 are induced, leading to production of the bioactive IL-12p70 heterodimer.21 Thus, T cell-derived signals are required in addition to pattern recognition. Although APCs in the tumour microenvironment or in the draining lymphoid tissues are unlikely to be in a completely resting state, the concept that contemporaneous T cell-derived signals can feed back on APCs to augment pattern recognition may still hold. These signals can include T cell-derived cytokines, and other direct molecular interactions, in addition to triggering of CD40.22–24 One way to encourage this T cell function in all individuals, irrespective of MHC haplotype, is to exploit innate-like T cells with invariant TCR structures that are restricted by conserved antigen-presentation molecules, such as type I CD1d-restricted natural killer T cells (NKT cells). These cells respond to glycolipid antigens, with α-galactosylceramide (α-GalCer) known to be a potent agonist that is active in mouse models as well as in humans.25,26 Administration of α-GalCer rapidly activates NKT cells, which in turn provide molecular signals that activate APCs.27–29 Downstream, this leads to transactivation of natural killer (NK) cells and enhanced adaptive immune responses. Importantly, signals from activated NKT cells can cooperate with pattern recognition via TLRs to further enhance this process.30–32 A downside to this response, depending on route and timing of administration, is enhanced release of pro-inflammatory cytokines associated with toxicity.31
To take advantage of the combined stimulatory activity of NKT cell agonism and TLR ligation, while at the same time reducing the potentially toxic side effects of a systemic cytokine storm, we assessed the impact of combining intratumoural administration of α-GalCer with CpG on adaptive antitumour responses. Overall, we found that the combination of α-GalCer and CpG induced significantly stronger antitumour responses than either compound alone, including an abscopal effect, that was CD8+ T cell-mediated and associated with increased infiltration of NKT cells. For less immunogenic tumours, the injection of antigenic peptide provided an additional advantage, particularly if conjugated directly to α-GalCer to form a self-adjuvanted vaccine.
Methods
Mice
Female and male mice were bred and housed by the Biomedical Research Unit at the Malaghan Institute of Medical Research. All animal experiments were performed in accordance with relevant guidelines and regulations and were approved by Victoria University of Wellington animal ethics committee. Animals used included: C57BL/6J (originally from The Jackson Laboratory, Bar Harbor, ME, USA) and the CD45.1 congenic strain B6.SJL-Ptprca Pepcb/BoyJ (from Ozgene Pty, Bentley, WA, Australia), BALB/cJ mice (The Jackson Laboratory), Cd1d−/− mice33 and Traj18−/− mice (B6(Cg)-Traj18tm1.1Kro/J; The Jackson Laboratory).34 Ifnar1flox/flox mice35 were crossed with CD11c-cre mice (Tg(Itgax-cre,-EGFP)4097Ach), and then an F2 cross performed to give cre-positive Ifnar1ΔCD11c mice and cre-negative littermates used as controls (Ifnar1+/+). Experiments in Clec9a-DTR mice, which express the human diphtheria toxin (DT) receptor under the control of the Clec9a promoter,36 and Siglec-H-DTR mice which express the human DT receptor under the control of the Siglec-H promoter,36 were conducted in F1 crosses with C57BL/6J mice. Both were supplied by Nanyang Technological Unit, Singapore.
Cell lines, media, and reagents
Murine tumour cell lines included: the T cell lymphoma EL4 transfected to express chicken ovalbumin (OVA),37 E.G7-OVA; EL4-LA, originally derived from EL4, which was generated in-house; the melanoma line B16.F10; and the colon carcinoma CT26 (all originally from ATCC, Manassas, Virginia, USA). Cell lines were authenticated by genetic analysis of microsatellite markers (IDEXX BioResearch, Columbia, MO). Cells were cultured in complete Iscove’s modified Dulbecco’s medium (cIMDM) comprising IMDM supplemented with 5% fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, 50 mM 2-ME (all Gibco, Grand Island, NY, USA). The lung carcinoma line TC-1, which expresses human papilloma virus (HPV) E6 and E7 oncoproteins38 was maintained in RPMI supplemented with 10% FBS, 1% penicillin/streptomycin, 1% glutamax and 1% sodium pyruvate (all Gibco). For the transfected lines, 500 µg/ml geneticin (G-418; Gibco) was added for selection. Cells were harvested and resuspended in incomplete IMDM for subcutaneous (s.c.) engraftment. The class B CpG 1826 (5’ TCC ATG ACG TTC CTG ACG TT 3’; Trilink Biotechnologies, San Diego, California) was resuspended in sterile injection water to 1.5 mM and diluted in PBS to 98 µM for injection, with 40 µl (25 µg) used per dose. The NKT cell agonist α-GalCer was manufactured in-house as previously described.39 Solubilization was achieved by freeze-drying in the presence of sucrose, L-histidine, and Tween 20,40 and resuspending to 582 µM in sterile injection water; this was diluted in PBS to 58 µM for injection, with 40 µl (2 µg) used per dose. The α-GalCer-HPV E7 peptide conjugate was prepared as described41 using peptide FFRKGQAEPDRAHYNIVTFCCKCDS that was N-terminally modified with aminooxyacetic acid (AoAA) for oxime ligation (prepared in-house). The conjugate was dissolved in DMSO (5 mg/ml), freeze-dried in solubilization matrix as above, resuspended to 113 µM in water as frozen stock, and diluted in PBS to 58 µM for injection, with 40 µl used per dose. Peptide GQAEPDRAHYNIVTFCCKCDS was dissolved to 327 µM in DMSO, diluted in PBS to 58 µM for injection, with 40 µl used per dose. For in vivo cell ablation experiments, DT (Sigma-Aldrich, Auckland, New Zealand) was maintained as 1 mg/ml frozen stock and diluted in PBS for an i.p. injection of 15 ng/g of body weight administered on days 4, 5 and 8 post-tumour challenge. Information regarding CD8+ cell depletion and organ excision are provided in the Supplementary Materials and Methods.
Intratumoural treatment
For experiments with EL4-derivatives, mice were challenged with 1 × 106 cells s.c. in the flank. For B16.F10, 1 × 105 cells were used, for CT26, 2 × 105 cells, and for TC-1, 1 × 105 cells. Engraftment was on one or both flanks, as indicated in the text. When average tumour size reached 36 mm2 (calculated as the product of the two bisecting diameters), mice were subjected to intratumoural therapy; mice were anesthetized with isofluorane (Sigma-Aldrich) using a SomnoSuite anesthesia system (Kent Scientific), and compounds injected using 29 G needles. For some experiments, compounds were injected s.c. peritumourally, or s.c. on the contralateral limb from the tumour site. tumour growth was monitored every 1–2 d until ethical endpoint, when tumour size on either flank exceeded 200 mm2. Survival was recorded as time from tumour implantation to ethical endpoint.
Mass cytometry
For mass cytometry experiments, B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) congenic mice were engrafted with E.G7-OVA cells (CD45.2) subcutaneously on the flank and treated as indicated in the text. At the designated experimental time points (9 or 12 days after tumour injection), peripheral blood, spleen, tumour and draining lymph nodes (dLN) samples were collected for analysis. Methodology is supplied in Supplementary Materials and Methods. The optimized antibody panel used is in Supplementary Table 1 with metal-conjugated antibodies targeting CD45 to barcode samples according to treatment (Supplementary Table 2).
Multiparameter spectral flow cytometry
For multiparameter spectral flow cytometry, C57BL/6J mice were engrafted with 1 × 106 E.G7-OVA cells on both flanks, with only one tumour subjected to treatment. Tissues were collected and processed on day 12 after tumour challenge. The antibody staining panel was optimized before use, as described.42,43 Antibodies, fluorophores and clones are provided in Supplementary Table 3. Methodology is supplied in Supplementary Materials and Methods.
High-dimensional data analysis
For each tissue, mass cytometry data was cleaned by gating out cell doublets and dead cells. For the tumour samples, CD45.1-positive tumour cells were also gated out (Supplementary Figure 1), and de-barcoded to identify treatment groups, using FlowJo v10.5.3 software (FlowJo). Downsampling from live/singlet/CD45+ events was performed to give consistent event numbers across all samples. Data from all treatment groups was then combined for t-stochastic neighborhood embedding (t-SNE)44 to define clusters of phenotypic similarity using the Cytobank platform (https://www.cytobank.org/) (Cytobank, Mountain View, CA, USA).45 The gating strategy shown in Supplementary Figure 1 was overlaid to identify clusters representing major leucocyte populations. Heatmaps were then generated for different cell types, where changes in expression of selected markers in response to treatment were calculated as mean fold-change of log2-transformed median fluorescent intensity (MFI) values for treatment on a given day relative to mean values derived from the treatment associated with the lowest expression (across all treatments and days). For spectral flow cytometry, data for each tissue was cleaned as above using FlowJo v10.5.3 software, and T cells, NK cells and NKT cells analyzed separately, based on the gating strategy shown in Supplementary Figure 2. Files containing only the cells of interest were subjected to tSNE (Cytobank) and then the FlowSOM R package (https://github.com/SofieVG/FlowSOM)46 was used for cell subset clustering. This involved first determining the optimal number of clusters by identifying the smallest number of clusters that minimized intra-cluster signal variance, and then establishing the optimal cluster number for the full data set by taking the median of the optimal numbers for each individual marker. Marker enrichment modeling (MEM) analysis47 was then performed in R to quantify each of the markers expressed on each cluster (https://github.com/cytolab/mem). This analysis scaled the median magnitude of difference in marker expression between clusters, depending on the spread (interquartile variance) of the data, with these values then computed in comparison to the remaining cells in a given dataset. This meant MEM values could be positively enriched (positive values) or negatively enriched (negative values). The percentages of cells in the different clusters were calculated for each of the different treatments, and in each tissue, followed by permutational multivariate analysis of variance (PERMANOVA)48 using the vegan package in R, to assess the median and variances of the different treatment groups.49 The adonis function was first used to prepare the data to allow for visualization with the betadisper function; this generated dispersion plots, with the median as the center for each group. To determine the significance of differences between treatment groups the pairwiseAdonis package was implemented.50 To identify clusters of significance an indicator species analysis was performed using the indicspecies package in R, following separation of the data into intratumoural injected or contralateral data where applicable.
Multicolor immunohistochemistry
For multicolor immunohistochemistry experiments, B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) congenic mice were engrafted with EG7.OVA cells (CD45.2) on both flanks, with only one tumour subjected to treatment. tumour samples were collected and snap-frozen in OCT on day 12 after tumour cell challenge for analysis. Antibodies, fluorophores and clones are provided in Supplementary Table 4. Methodology is supplied in Supplementary Materials and Methods.
Conventional flow cytometry
For conventional flow cytometry experiments, blood samples were collected into 200 µl of 0.5 mM EDTA in PBS, centrifuged at 1500 x g for 4 min and the supernatant removed. Red blood cells (RBCs) were lysed with RBC lysis solution (QIAGEN, Valencia, CA, USA), the centrifugation was repeated, and the pellet resuspended in FACs buffer (PBS supplemented with 1% FCS, 0.01% NaN3, 2 mM EDTA) and plated in 96 well plate for staining. Methodology is supplied in Supplementary Materials and Methods. Antibodies, fluorophores and clones are provided in Supplementary Table 5.
Results
Intratumoural treatment with CpG and α-GalCer induces local and distal antitumour immune responses that are greater than observed with either agent alone
Antitumour effects of intratumoural CpG have been reported in mice using different multiple dosing regimens.10,13 We initiated our studies with engrafted E.G7-OVA tumours, a line derived from the murine T cell lymphoma EL4 that has been modified to express chicken ovalbumin (OVA) as a model of a tumour-associated neoantigen. Earlier studies had shown that E.G7-OVA is sensitive to a single intratumoural dose of 25 µg CpG 1826 (hereafter “CpG”) alone, but full regression was not achieved.9 In preliminary experiments, we established a multiple dosing regimen with three treatments given four days apart, where each treatment involved direct injection of 25 µg CpG on two consecutive days. We have previously reported that contemporaneous intravenous co-administration of α-GalCer with TLR agonists can enhance APC activation, cytokine release and adaptive immunity, and that cytokine release, in particular, can be enhanced further by introducing the NKT cell agonist several hours before the TLR agonist.30,31 Therefore, we evaluated variations on the intratumoural regimen, where a single dose of 2 µg of α-GalCer was either co-injected with the first dose of CpG, or where it was given 6 h earlier (by delaying the first CpG dose by 6 h) (Figure 1a). To examine the potential for an abscopal effect, tumours were engrafted on both flanks and allowed to become established at each site before intratumoural treatment was initiated at only one site. The impact of the combined treatments was compared to the use of each agent alone, with CpG given in the repeated schedule, and α-GalCer as a single dose. To control for the impact of injection alone, animals in the α-GalCer group, and untreated controls, received repeated sham injections with PBS vehicle with the same dosing schedule as used with CpG.
Figure 1.

Intratumoural treatment with α-GalCer significantly enhances local and distal antitumour immune responses to intratumoural CpG. (a) Treatment schedule. Treatment was either with repeated doses of 25 μg of CpG (green arrow), a single dose of 2 μg of α-GalCer (yellow arrow), or a combination of both where the first dose of CpG was either given at the time of α-GalCer, or 6 h after α-GalCer. PBS was injected as a vehicle control for all injections. (b) Growth of E.G7-OVA tumours plotted for each mouse per treatment group (n = 8–10), showing injected tumour and untreated tumour on contralateral flank. (c) Survival curves (to ethical endpoint) aggregated over several experiments (total n = 15–28), in animals with E.G7-OVA tumours on one flank. *p < .05, ***p < .001, log-rank Mantel-Cox test. (d) Growth of EL4-LA tumours plotted for each mouse per treatment group (n = 5–8 per group). (e) Survival curves aggregated over several experiments (total n = 10–13), in animals with EL4-LA tumours on one flank. ****p < .0001 for CpG vs each combined treatment.
Intratumoural injection of CpG alone led to a modest antitumour response with reduced growth relative to the control group, while treatment with α-GalCer alone had no significant impact (Figure 1b). However, reduced tumour growth was observed with the combined treatment over either treatment alone, with complete rejections observed in the majority of animals. Notably, the untreated tumours on the contralateral flank showed the same pattern of response to treatment. Furthermore, in repeated experiments conducted in animals with tumours on one flank, an additional survival benefit was observed when the first dose of CpG was delayed (Figure 1c). This regimen was adopted for most of the later studies.
A similar experimental setup with tumours on both flanks was used to assess responses to EL4. Genetic analysis of microsatellite markers (IDEXX BioResearch, Columbia, MO) had shown that the line used had undergone some genetic drift from the original reference EL4 cell line, and is referred to here as EL4-LA. This cell line maintained features of classical EL4 cells, including morphology and phenotype by flow cytometry (data not shown). Treatment of mice engrafted with EL4-LA with the different agents was generally more effective than in mice engrafted with E.G7-OVA, suggesting it was more immunogenic. Again, the combined treatment was superior at reducing tumour growth compared to the single agents (Figure 1d), with contralateral untreated tumours responding similarly to the injected tumours. Over repeated experiments conducted in animals with tumours on one flank, a significant survival benefit was observed when introducing α-GalCer into the repeated CpG treatment schedule, with all but one of the 26 animals given combined regimen, either co-injected or 6 h apart, completely rejecting the tumour (Figure 1e).
To establish whether the efficacy of the α-GalCer and CpG combination was specifically dependent on intratumoural effects, antitumour activity was compared between groups treated via intratumoural, peritumoural, or subcutaneous (at a distal site) routes of administration. Of the three routes, all conducted with the delayed CpG regimen, intratumoural was the most effective, with the majority of the mice completely rejecting the tumours (Figure 2a). Both peritumoural and subcutaneous treatment induced a tumour growth delay, but ultimately the vast majority of animals succumbed to unchecked tumour growth. Therefore, the therapeutic agents have to be introduced to the tumour microenvironment to induce the immune changes associated with strong activity.
Figure 2.

Antitumour effect of CpG with α-GalCer requires direct injection, facilitating a CD8+ T cell-mediated response to undefined tumour antigens. (a) Survival curves showing CpG and α-GalCer treatment of E.G7-OVA tumours with the delayed CpG dosing schedule as in Figure 1, delivered intratumourally (i.t.), peritumourally (p.t.) or subcutaneously (s.c.), versus intratumoural PBS treatment (blue) (n = 5–8; **p < .01 ***p < .001). (b) Survivors from intratumoural CpG and α-GalCer treatment were either depleted of CD8+ cells with anti-CD8 (gray line) at 250 ng/mouse, or injected with isotype control mAb (pink), −2 and −1 days before rechallenge with E.G7-OVA. Survival relative to tumour growth in naive animals is shown (n = 5–9). Representative of two experiments. (c) Growth curves (left) and survival (right) for animals with E.G7-OVA tumours on both flanks subjected to combination treatment on one flank followed by administration with either anti-CD8 (gray) at 250 ng/mouse or isotype mAb (pink) on days 2,3 and 6 after treatment initiation (n = 8–9). (d) Survivors from intratumoural CpG and α-GalCer treatment of E.G7-OVA were rechallenged with EL4-LA. Growth curves (left) and survival (right) relative to EL4-LA in naïve animals are shown (n = 5–7). Representative of two experiments.
To test for the establishment of an adaptive immune response with memory, animals that had been challenged on one flank with EL4-LA, and were successfully treated with the combined treatment regimen, were re-challenged on the opposite flank. Half of these animals were administered an antibody to deplete CD8+ T cells before the tumour cells were injected, while the remainder were administered an isotype control antibody. All animals that received the control antibody rejected the second round of tumour challenge, whereas those that received anti-CD8 depleting antibody developed progressing tumours (Figure 2b), suggesting effective treatment was associated with a CD8+ T cell-mediated memory response.
To establish whether CD8+ T cells were also the main effectors in the immediate antitumour response to treatment, depleting anti-CD8 antibodies, or isotype control, were injected during the course of treatment of E.G7-OVA; tumours were engrafted on both flanks, with only one site treated. In mice that received the control antibody, regression of both treated and contralateral tumours was seen in the majority of mice, whereas there were no significant antitumour responses at either site in mice depleted of their CD8+ T cells (Figure 2c). Therefore, both the local antitumour response and abscopal effect involve CD8+ T cells.
The introduced OVA antigen was a potential immunological target for CD8+ T cell-mediated elimination. To test this, animals that had rejected E.G7-OVA were subsequently challenged with EL4.LA cells, which do not express OVA. While the EL4.LA tumours showed initial growth, they were eventually rejected in the majority of animals (Figure 2d). This suggests the antitumour activity against E.G7-OVA was broader than solely targeting the introduced neoantigen and included endogenous targets common to both tumour cell lines.
Intratumoural treatment with CpG and α-GalCer leads to broad changes in immune profile in the tumour and lymphoid tissues
Cytometry by time-of-flight (CyTOF) was used to investigate the immune profile in animals bearing E.G7-OVA tumours treated with either CpG, α-GalCer, or the combination with a delayed first dose of CpG. Analysis was performed on infiltrates from the treated tumour, the dLN of the treated tumour, blood and spleen, and was conducted at two timepoints; on day 9 after engraftment when all tumours were still progressing irrespective of treatment, and on day 12 when treatment-induced regression was first observed (Supplementary Figure 3). The cytometry data for each tissue were combined for tSNE to visualize clusters of phenotypically similar cells, with the expression of well-defined phenotypic markers used to define the major cell types represented (Figure 3a). Each cell type was then interrogated for treatment-related changes in expression of molecules related to function, including activation markers, and Ki67 as a marker of proliferation. Those cell types and markers that showed a significant treatment-related change are displayed (Figure 3b).
Figure 3.

Mass cytometry analysis of leukocytes after treatment. Mice bearing E.G7-OVA tumours were treated as described for Figure 1, with animals receiving vehicle, CpG, -GalCer, or the combination with the first CpG dose 6 h after -GalCer. Treatment was initiated on day 7 post tumour engraftment and the indicated tissues were collected at day 9 or day 12 after tumour engraftment (n = 3 per treatment group at each timepoint), and mass cytometry was performed on all samples with barcoding to discriminate treatments. (a) tSNE analysis was performed on each tissue to determine clusters of phenotypic similarity, with expert gating used to define the indicated cell types represented by the major clusters (using gating strategy in Supplementary Figure 1). (b) Heatmaps show expression of the indicated markers on the defined cell types, expressed as fold change of log2-transformed MFI over the treatment/day with the lowest expression values. Details are provided in Material and Methods.
One of the most notable treatment-induced changes in the tumour infiltrate was the enhanced expression of sca-1, which has been associated with exposure to type I IFN,51,52 and was increased on lymphocytes (T cells, B cells, NK cells and NKT cells; the latter defined as NK1.1+ T cells) and myeloid cells (DCs and monocyte/macrophages) early in the response (day 9) in the combined treatment group. Furthermore, the increased expression was largely retained on CD8+ T cells, NKT cells, NK cells and myeloid cells in the regression phase (day 11). Ly6C, another marker that has been associated with exposure to type I IFN,51,52 was increased on T cells with all treatments but remained high on CD4+ T cells with the combined treatment in the regression phase. On CD8+ T cells, the markers of activation, CD44, KLRG1 and PD-1, were all increased on day 9 with the combined treatment group, with this expression sustained to day 12. These cells also showed earlier and sustained expression of the proliferation marker Ki67. Another notable change in tumours with the combined treatment was early expression of Ki67 on B cells, and increased and sustained expression of CD138, a marker of antibody-producing plasmablasts or plasma cells. Overall, these data suggest combined treatment results in an increased frequency of activated adaptive immune cells, and a general pattern of higher exposure to type I IFN.
Many of the phenotypic changes that were prominent in the combined treatment group in the tumour at day 9 were also seen at this time point in the dLN, including increases in sca-1 and Ly6C expression. However, the only obviously upregulated activation marker on T cells was the early activation marker CD69, which was reduced back to background levels by the regression phase. Few phenotypic changes at this later timepoint were unique to the combination group, although, as was seen in the tumour, the B cells retained upregulated expression of CD138. Interestingly, DCs showed increased expression of CD86 and PD-L1 with combined treatment at day 9, indicating they were activated, and they also expressed increased levels of BST-2, which has been associated with exposure to type I IFN.53 These data point to an early role for the dLN in the effector response.
The blood and spleen exhibited many phenotypic changes at day 9, although few were unique to the combined treatment. Nonetheless, these changes highlight the systemic effect of the different treatments, causing modulation to immune cells in distant lymphoid tissues. The profiles observed for the combined treatment group were generally similar to treatment with CpG alone. Exceptions were the NK cells and NKT cells, which in the combination group retained some of the phenotypic changes to day 12, notably increased sca-1, BST-2 and Ki67 on NKT cells in the blood, and increased sca-1 on NK cells in blood and spleen.
With the CyTOF data pointing to an involvement of T cells, NKT and NK cells, we examined these populations further by spectral flow cytometry, using fluorescent OVA257-265 peptide-loaded H-2Kb pentamers (OVA pent) to assess whether OVA-specific CD8+ T cells played any role in responses to E.G7-OVA tumours, and antibodies to NK1.1 and fluorescent CD1d tetramers loaded with the α-GalCer analog PBS-57 to specifically examine the involvement of NK cells and NKT cells. Assessment was on day 12 after tumour implantation, which was 5 days after initiation of treatment, allowing T cell expansion to occur, and was conducted on both treated and untreated tumours in an attempt to separate local from systemic effects. The analysis included a panel of 20 antibodies (Supplementary Table 3), with high-dimensional analysis performed using tSNE and FlowSOM software to assign clusters of phenotypic similarity for each cell-type, and MEM used to quantify markers expressed (Figure 4 and Supplementary Figures 4–6). This analysis did not show any clear expansion of OVA pent+ clusters – indeed very few of these cells were detected in any tissue (data not shown). Manual expert gating focusing specifically on OVA pent+ cells also did not reveal any trends in the proportion of OVA-specific CD8+ T cells associated with treatment (Supplementary Figure 7).
Figure 4.
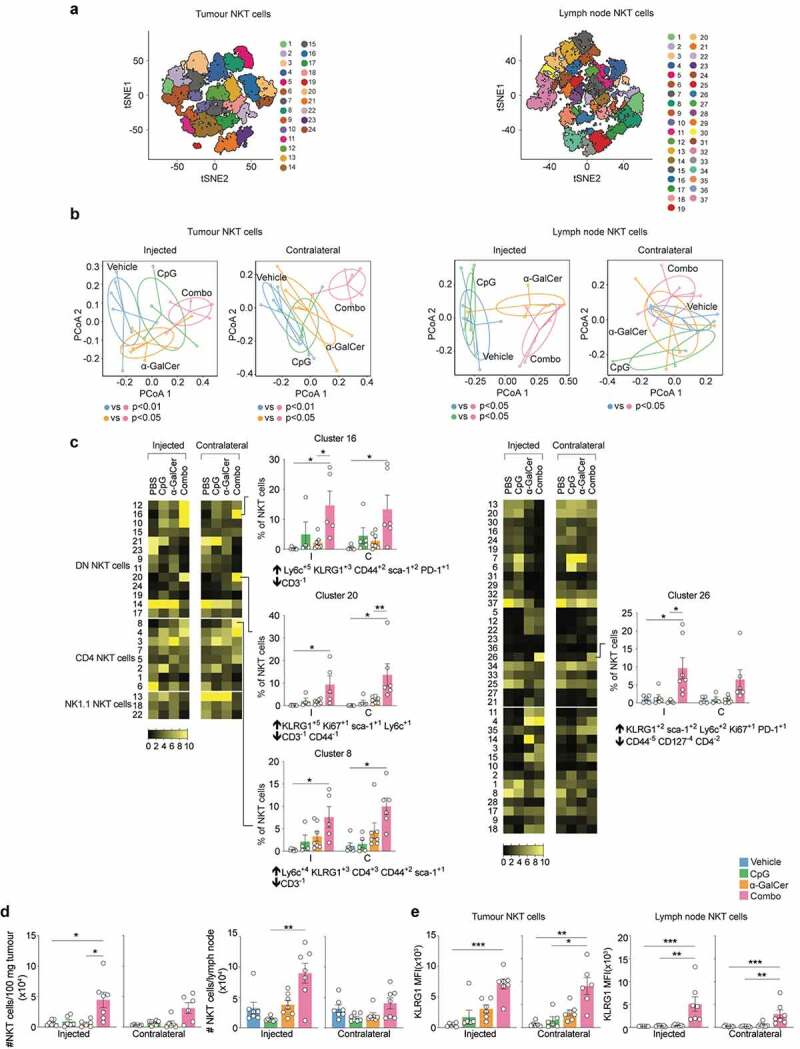
Spectral flow cytometry analysis of leukocytes after treatment. Treatment was as in Figure 3, except E.G7-OVA tumours were implanted on both flanks, with only one subjected to intratumoural injection (n = 7 per treatment group). Tissues were collected for analysis 12 days after tumour injection. The gating strategy in Supplementary Figure 2 was used to define T cells, NK cells and NKT cells (a) tSNE analysis was performed for each cell type, with FlowSOM then conducted on tSNE axes to assign clusters (numbered). (b) PERMANOVA was conducted to assess dissimilarity in the composition of clusters with treatment (further results in Supplementary Figures 4–6). Derived dissimilarity plots from the analysis of NKT cell clusters in each tissue are shown, along with significance of pairwise comparisons between the treatments. (c) Heatmaps showing proportion of NKT cells in each cluster compared by treatment conditions. Graphs and MEM labels are shown for clusters where there were significant differences between the combination treatment group and the other groups. Up arrows indicate positively enriched markers, and down arrows indicate negatively enriched markers *p < .05 **p < .01 ***p < .001; Indicator species analysis. (d) Assessment of NKT cell number in the tumour and tumour dLN by manual gating. NKT cells defined as CD3ε+ CD1d tetramer+ cells. (e) Assessment of KLRG1 expression on NKT cells by manual gating. Kruskal-Wallis with Dunn’s multiple comparisons test; *p < .05, **p < .01, ***p < .001.
To investigate whether the overall profile of T cell, NK cell and NKT cell cluster frequencies differed with treatment, PERMANOVA analysis was conducted on each cell type (Figure 4a). Dispersion plots were generated to show median and variance of the proportions of clusters in the treatment groups; subsequent pairwise comparisons were conducted between each of the treatments to assess whether any dissimilarity observed between treatments was significant (Figure 4b and Supplementary Figures 4–6). In the spleen, for all three cell types examined, there were treatment-related dissimilarities in cluster profiles relative to untreated vehicle controls, suggesting treatment-induced changes in phenotype (Supplementary Figure 4). However, there was no dissimilarity between the combination treatment and individual treatments, suggesting further investigation to define clusters associated with efficacious combination treatment was unlikely to be enlightening. In tumour and draining lymph nodes, the T cell- and NK cell-cluster profiles associated with CpG alone and the combination treatment showed dissimilarity from untreated controls; however, they were not significantly different from each other, suggesting that induced changes in phenotype in both cases were dominated by the CpG stimulus. In contrast, analysis of NKT cell clusters in tumour and lymph nodes suggested features unique to the combination treatment could be detected, as only the combination treatment resulted in a profile of clusters with dissimilarity to vehicle (Figure 4b). We therefore assessed the treatment-associated frequencies of each NKT cell cluster in these tissues, with adjustment for multiple testing (Figure 4c). Combined treatment was associated with significant increases in individual NKT cell clusters expressing KLRG1 in the tumour, at both injected and contralateral sites, suggesting a systemic effect. These KLRG1hi clusters were either NK1.1− CD4− (DN) NKT cells, or NK1.1− CD4+ NKT cells. A KLRG1hi cluster was also increased in the draining lymph nodes. Along similar lines, manual gating on NKT cells showed an increased cell number of NKT cells in the tumour and dLN of combination treated mice (Figure 4d), and confirmed an increased expression of KLRG1 on NKT cells (Figure 4e), but no significant changes in other cell types could be identified (Supplementary Figures 7–10). Thus, overall, the most profound changes induced by the combination treatment related to modulation of NKT cells, with changes seen over and above those induced with the NKT cell agonist α-GalCer alone, notably in the tumours themselves.
To further investigate the immune cell infiltration of tumours after treatment, immunohistochemistry was performed on tissue sections from E.G7-OVA tumours harvested 12 days after tumour engraftment (Figure 5). Vehicle-injected tumours and their contralateral tumours displayed minimal evidence of host-derived CD45.1+ cell infiltration and little evidence of CD8+ T cells (defined as CD45.1+ CD3ε+ CD8+) (Figure 5a,b). In comparison, the α-GalCer and CpG injected tumours displayed a significantly greater number of both CD45.1+ cells and CD8+ T cells (Figure 5c, i & j). There was a slight increase in these cell types in the contralateral treated tumours as well (Figure 5d, i & j); however, at the point of harvest these tumours had not displayed a similar decrease in size as the treated ones, likely reflecting a lag in the immune response, with no statistically significant changes in cellular composition observed. Assessment of the number of macrophages (defined as F4/80+ CD45.1+ cells) demonstrated a significant increase in the α-GalCer and CpG injected tumours compared to vehicle injected tumours (Figure E-H & K). In some tumour zones, voids were present (Figure 5c) and quantification of the total number of voids per section suggested a greater frequency in the treated tumours; however, no significant increase was observed (Figure 5l).
Figure 5.

Treatment with intratumoural α-GalCer and CpG induces extensive infiltration of immune cells. Mice (CD45.1+) were engrafted with E.G7-OVA (CD45.2+) tumours on both flanks, and either treated intratumourally with α-GalCer and CpG (with delayed first dose) on one flank, as described for Figure 1, or with PBS according to the same schedule. tumours were collected on day 12 post tumour engraftment, 5 days after the start of treatment for analysis by immunohistochemistry. Antibody staining was against the molecular markers indicated. Average values from 10 images per section are plotted with n = 4 mice per group. Staining for CD45.1 and CD8α are shown in the top row (a-d) and staining with CD45.1 and F4/80 shown in the bottom row (e-h). Double positive cells (i.e. CD45.1+CD8α+ or CD45.1+F4/80+ cells) appear as white cells in the images. Scale bar = 50 µm. (i) Quantification of CD45+ cells per total nuclei per slide is shown. One-way ANOVA with Tukey’s multiple comparison test; *p < .05. (j) Number of CD8+ T cells (defined as CD45+CD3+CD8+) and (k) F4/80 cells (defined as CD45.1+F4/80+) per nuclei per section. (l) Frequency of voids (defined as intact 0.003–0.044 mm2 empty circular structures) per section. One-way ANOVA with Tukey’s multiple comparison test was performed.
Response to intratumoural treatment with CpG and α-GalCer requires NKT cells
To assess the role of NKT cells further, three different approaches to blocking NKT cell function were used. In the first approach, a CD1d-blocking antibody was administered to mice undergoing treatment. This was initiated 3 days after intratumoural α-GalCer administration to avoid blocking any early NKT cell-mediated APC activation events, so that the analysis was focussed on the role of NKT cells in the later effector phase of the response. In the second approach, analysis was conducted in CD1d deficient (CD1d−/−) animals that are devoid of all CD1d-restricted T cells, including the type I NKT cells known to be α-GalCer-reactive. Finally, analysis was conducted in mice that lack the ability to form the TCR-α chain expressed by type I NKT cells, yet otherwise retain an intact TCR repertoire (Traj18−/−),34 and so were devoid of α-GalCer-reactive cells. All analyses were conducted in mice with E.G7-OVA tumours on each flank, with only one subjected to treatment.
Interestingly, late administration of CD1d blocking antibody during treatment significantly reduced antitumour activity and survival (Figure 6a), suggesting an ongoing requirement for NKT cell/CD1d interactions for sustained efficacy. The impact was more apparent on distal tumours, with these tumours significantly larger than the directly treated tumours when measured on day 12. Indeed, it was primarily the unchecked growth of the distal tumours that caused a significant reduction in overall survival relative to animals that received the isotype control antibody. This trend for the outcome to be worse in distal tumours was also seen in the host strains devoid of NKT cells throughout treatment; in both CD1d−/− and Traj18−/− mice the treated tumours showed reduction in growth relative to controls, presumably mediated by the CpG, but growth of the untreated distal tumours was largely unaffected, meaning there was no significant survival benefit to treatment (Figure 6b) and c. As earlier experiments had shown CpG alone was capable of inducing some abscopal activity in wild-type mice (Figure 1b), these data suggest NKT cells are required for the abscopal effect even in the absence of an exogenous NKT cell agonist like α-GalCer.
Figure 6.

Effective treatment with abscopal effect requires NKT cells. (a) Growth curves of E.G7-OVA on both flanks in PBS-treated mice (blue) relative to those subjected to treatment with CpG and α-GalCer with either anti-CD1d (gray) or isotype control (pink) injections initiated three days after the α-GalCer dose (n = 8–9). Representative of two similar experiments. Middle panels show paired analysis of tumour size at day 12 after tumour engraftment for injected versus untreated contralateral tumours in each mouse. **p < .01, ***p < .001; Wilcoxon test. Far right panels show survival curves. ***p < .001 log-rank Mantel-Cox test. (b) Similar analyses of combination treatment (pink) versus PBS-treated controls (blue) in CD1d-deficient mice (n = 17), or (c) in Traj18-deficient mice (n = 6–13).
Response to combined intratumoural treatment requires type I IFN signaling by DCs and involves distal lymphoid tissues
Upregulation of type I IFN-inducible markers on immune cells after treatment pointed to the involvement of members of this cytokine family in the antitumour response. To investigate this, we examined whether IFN-α could be detected in tumour-bearing mice with or without treatment. Mice were treated once with the agonists alone or in combination, and tumours removed 6 h later to assess IFN-α levels in tissue homogenates. Even in the vehicle-treated animals, measurable quantities of IFN-α were detected in the tumour, whereas no measurable levels could be detected at the same time in the serum with or without treatment (Figure 7a). Of note, significantly more IFN-α was detected in tumours after treatment with the combination of CpG and α-GalCer compared to controls.
Figure 7.

Combined intratumoural treatment requires type I IFN signaling by DCs and involves distal lymphoid tissues. (a) Assessment of IFN-α levels in E.G7-OVA tumour tissue homogenates or serum 6 h after indicated intratumoural treatments. Aggregate of two experiments. Individual values per mouse are plotted with mean ± SEM. **p < .01 one-way ANOVA with Tukey’s multiple comparisons test. (b) Survival in PBS-treated mice (blue) relative to those subjected to treatment with CpG and α-GalCer with blocking anti-IFNAR1 (gray; n = 8) or isotype control (pink; n = 8) initiated two days before start of treatment. ***p < .001. Representative of two experiments. (c) Survival in PBS-treated mice versus combined treatment in animals without expression of IFNAR1 on DCs (Ifnar1ΔCD11c), or IFNAR1-intact controls (Ifnar1+/+). Statistical test between combination treatment groups shown (n = 6–7). (d) Survival in PBS-treated mice versus combined treatment in Clec9a-DTR animals where Clec9a+ cells were depleted with DT (n = 7) or not (n = 7). DT was administered i.p. at 15 ng/g of body weight on days 1 and 2 prior to intratumoural treatment. Statistical test between combination treatment groups shown; ***p < .001. Representative of two experiments. (e) Survival in PBS-treated mice versus combined treatment in Siglec-H-DTR animals where Siglec-H+ cells were depleted with DT (n = 7) or not (n = 8). DT was administered as noted in (D). Statistical test between combination treatment groups shown; **p < .01. Representative of two experiments. (f) Survival in PBS-treated mice versus combined treatment in animals subjected to surgery to remove inguinal lymph nodes, or sham surgery, 7 days before engraftment E.G7-OVA engraftment on the same flank (n = 7–8). Statistical test between combination treatment groups shown; ***p < .001. Representative of two experiments. (g) Survival in animals with spleens removed, or sham surgery, 7 days before engraftment E.G7-OVA (n = 9–10). Statistical test between combination treatment groups shown; ***p < .001. (h) Assessment of serum ALT levels 18 h after indicated intratumoural treatments versus intravenous α-GalCer at the same dose. Individual values per mouse are plotted with mean ± SEM. ***p < .001. Representative of two experiments.
To determine whether type I IFN played a role in antitumour activity, a blocking antibody was administered to prevent signaling via the IFN-α/β receptor during treatment. Mice administered the anti-IFNAR1 antibody completely lost their ability to prevent tumour growth, whereas significant antitumour responses were seen in those given an isotype control antibody (Figure 7b). Furthermore, the antitumour efficacy of the combination treatment was abrogated in mice lacking expression of IFN-α/β receptor on DCs due to Cre-mediated excision of IFNAR1 on CD11c+ cells (Figure 7c), suggesting type I IFN-mediated stimulation of DCs is critical for treatment efficacy.
To establish which subsets of DCs were involved in effective treatment, two different conditional ablation models were used; one where conventional DCs of the “cDC1” sub-type could be ablated with DT due to transgenic expression of the human DT receptor from the Clec9a promoter, and another where pDCs could be ablated in animals expressing DTR from the Siglec H promoter. When Clec9a+ cells were depleted, the therapeutic efficacy of the combination of CpG and α-GalCer was significantly reduced, with only a slight delay in tumour growth observed (Figure 7d). The residual activity may reflect incomplete ablation of cDC1s, as DT treatment was unable to induce full deletion, with ~13% retained in the experiment shown. Administration of DT itself was not responsible for the reduced antitumour activity, as efficacy was maintained in the presence of DT in non-transgenic animals (data not shown). Taken together with the observation that CD8+ T cell are key effectors in the response, it is likely the significantly reduced antitumour efficacy in animals depleted of cDC1s reflected a loss in capacity for cross-presentation, which is heightened in these cells. Depletion of pDCs also resulted in reduced antitumour efficacy of the treatment (Figure 7e). However, in repeated experiments, escape from therapeutic control occurred later than in mice depleted of cDC1s, perhaps suggesting lower effector burst size or persistence. As pDCs are major producers of type I IFN, this is consistent with known roles for type I IFN in both APC activation and T cell memory formation.54,55 Again, ablation was not complete, with ~20% pDCs retained; these remaining cells may be responsible for the residual activity observed.
While the immune phenotype analysis showed intratumoural treatment served to change the local tumour microenvironment it remained possible the efficacy relied on key cellular interactions in distal lymphoid organs. To examine this, antitumour activity was assessed in mice where key dLNs, or the spleen, were removed by surgery before tumour challenge and treatment. The adjacent inguinal lymph nodes were identified as the main dLNs by tracing drainage of intratumourally injected ink. When these lymph nodes were removed, there was a negative impact on treatment efficacy in the E.G7-OVA model (data not shown) and complete loss of activity when the more immunogenic EL4-LA tumours were used (figure 7f). This was not an effect of the surgery itself, as controls were subjected to sham surgery alone without significant impediment to treatment efficacy. tumours in mice with surgically removed spleens also did not respond to treatment (Figure 7g). Therefore, the efficacy of the combined intratumoural treatment may require access of antigens, or the agonists themselves, to the lymph and blood. To assess whether there was significant access to the blood, we investigated whether liver enzymes were increased in serum, as it is known that systemic administration of α-GalCer into mice leads to a transient phase of hepatotoxicity caused by activation of liver-resident NKT cells.56 Serum was collected 18 h after mice were treated intratumourally with the CpG and α-GalCer, alone or in combination, and levels of ALT determined. Animals injected intravenously with α-GalCer served as a positive control, with significantly raised ALT levels observed as expected (Figure 7h). In contrast, mean levels of ALT were not significantly increased after intratumoural treatment compared to vehicle-treated animals. However, some individual scores exceeded a threefold increase from the normal range (range:14–56 IU/L representing 2.5th and 97.5th percentiles57); it therefore remains possible that some low-level leakage of α-GalCer is how NKT cell activity is recruited into the antitumour response. Of note, these results also highlight a safety benefit to intratumoural treatment, with the strong antitumour activity associated with a lowered risk of potentially deleterious systemic effects.
Intratumoural treatment is improved by introducing peptide antigens
To test the utility of the combined intratumoural treatment regimen, experiments were conducted in animals challenged with cells from different cancer types, including CT26 colon carcinoma (Figure 8a), B16.F10 melanoma (Figure 8b), and TC-1 lung carcinoma (Figure 8c). In each case, the combined treatment had superior antitumour activity to single agents, with significant reductions in tumour growth, although few full regressions were achieved in these models. We therefore explored whether efficacy could be improved by introducing known tumour-associated antigens into the stimulatory microenvironment created by the treatment. This was achieved in two ways; by simply co-injecting antigenic peptides, or by chemically linking antigenic peptides to α-GalCer. The latter was achieved by conjugation of the peptides to a prodrug derivative of α-GalCer via a cleavable linker. Once acquired by antigen-presenting cells, intracellular enzymatic cleavage of the linker by cathepsins releases both the peptide and the active form of α-GalCer.58 The two different approaches were assessed in mice bearing TC-1 tumours on each flank (Figure 8d). This cell-line expresses HPV E6 and E7 oncoproteins and is a commonly used model of HPV-associated malignancies, with an H-2 Db-binding peptide from HPV E7 defined as a useful target epitope for immunotherapy. Simply injecting this peptide alone intratumourally was insufficient to elicit any responses; nor was co-administration of peptide with α-GalCer (Figure 8d). As was shown earlier, the combination of CpG and α-GalCer significantly reduced growth but did not induce full regressions. However, when peptide was co-administered with the combination treatment, tumour regression was induced in some mice. The most efficacious treatment was when the α-GalCer-E7 peptide-conjugate was injected with CpG, inducing full regression in the majority of treated tumours, including all of the contralateral tumours. Injection of the conjugate alone could not account for the improved antitumour activity (Figure 8e), as combination with CpG was required to see a significant survival advantage.
Figure 8.

Intratumoural treatment is improved by introducing peptide antigens. Survival for indicated intratumoural treatments in mice engrafted with (a) CT26 colon carcinoma (n = 7; BALB/cJ hosts), (b) B16.F10 melanoma (n = 5–8, C57BL/6 J hosts), or (c) TC-1 lung cancer cell lines (n = 5–8; C57BL/6 J hosts). *p < .05, **p < .01, ***p < .001, ****p < .0001 relative to CpG alone. Minimum of two similar experiments performed per tumour line. (d) Growth curves (left) and survival (right) in the TC-1 tumour model with additional groups where HPV E7 peptide was injected intratumourally either alone, in combination with α-GalCer and CpG (given at time of α-GalCer dose), or as a covalent conjugate with α-GalCer (n = 5–9). Statistical tests relative to α-GalCer and CpG (6 h) treatment group shown. (e) Assessment of intratumoural HPV E7 peptide-conjugate with CpG compared to treatment with the single agents (n = 5–8).
Discussion
We have demonstrated that the efficacy of repeated intratumoural CpG treatment on established solid tumours can be significantly enhanced by intratumoural administration of a single dose of the NKT cell agonist α-GalCer. This resulted not only in the regression of treated tumours, but also regression of untreated tumours at distal sites. The most important effectors in the response to the combined treatment were CD8+ T cells, presumably responding to as-yet undefined antigens expressed in the different tumour models tested. In situations where at least one of the antigens targeted by the CD8+ T cells was known, it was possible to increase responses further by also introducing targeted peptide epitopes directly into the tumour, preferably attached to α-GalCer.
Initial mass cytometry analyses showed that the combined treatment of CpG and α-GalCer increased expression of sca-1 and Ly6C on many different immune cell types, including T cells, suggesting broad exposure to type I IFNs.51,52 Indeed, efficacy was lost when type I IFN signaling was blocked, most specifically when IFNAR1 was absent on CD11c+ cells, indicating that a critical role for type I IFN was modulating DC function. Indeed, the antitumour effect was strongly dependent on cDC1 cells, which have a heightened propensity for cross-presentation to CD8+ T cells.59 Given that CD8+ T cells were involved in both the immediate antitumour response and a memory response at re-challenge, it is likely that type I IFN has a role in promoting this function.60,61 The immediate source of the type I IFN was not identified, although it was notable that significant levels of IFN-α were observed in uninjected tumours (but absent in the serum), suggesting that engraftment of tumours alone was sufficient to initiate a type I IFN response. This may reflect underlying immunity in response to tumour challenge, possibly even involving adaptive cells, although this was not explored further. Importantly, the combined intratumoural treatment served to increase IFN-α levels, which may have been one of the key triggers of effective immunity. It remains unclear whether this served to reset an existing antitumour response or help initiate a new one. A potential source of the IFN-α are pDCs, which are powerful inducers of type I IFN in response to infection, and to TLR ligation with nucleic acid agonists such as CpG.54,55 However, when hosts were depleted of pDCs before treatment, the initial antitumour response proceeded as normal, with many tumours regressing to some extent. Nonetheless, the tumours all eventually progressed, suggesting that the absence of pDCs (and by implication, pDC-derived type I IFN) during the initial priming phase did not prevent initiation of the T cell response, but did compromise the development of lasting immunity. This effect may be via direct stimulation of CD8+ T cells by type I IFN provoking clonal expansion and memory formation.55,62,63
The antitumour response against E.G7-OVA was improved when the administration α-GalCer preceded the initial dose of CpG by 6 h. Our earlier studies had shown that when an NKT cell agonist was administered intravenously before a TLR ligand, a “conditioned” environment was created that enabled a significantly enhanced cytokine response to the TLR ligand, characterized by spikes in serum TNF, IL-12p70 and IFN-γ.31 Although these earlier studies had not been conducted with CpG, a similar effect on cytokine induction may have been seen in the intratumoural setting. The improved antitumour activity with delayed TLR stimulation may therefore be a response to the immediate antitumour effects of exposure to cytokine, rather than improved T cell function. However, another possible interpretation is that pDCs were conditioned by NKT cells to produce more type I IFN with TLR9 ligation, which in turn improved the function of DCs engaged in T cell priming.
Antibody-mediated CD8 depletion studies highlighted a key role for CD8+ T cells in the antitumour activity of the combined treatment, which was supported by mass cytometry analysis showing enhanced activation and evidence of proliferation of CD8+ T cells and CD4+ T cells in tumours and dLNs at day 9, when tumours were yet to start regressing. However, by day 12, when the tumours had started to regress, expression of activation and proliferation markers were not as evident. In fact, when analysis was conducted by flow cytometry on day 12, no clear pattern of activation marker upregulation was detected. Nonetheless, analysis by immunohistochemistry showed increased CD8+ T cells in tumours administered the combined treatment, consistent with T cell-mediated antitumour activity.
In terms of the events that may have led to effective antitumour activity, we speculate that intratumoural CpG is more likely to have had direct impact on local APCs, which then traffic with antigen to the dLNs to stimulate T cells. In contrast, α-GalCer may need to leach from the tumour to lymphoid tissues, where NKT cells reside in large numbers, in order to facilitate APC-licensing events. The glycolipid may have accessed the lymph nodes directly via the lymphatics or have been transported by APCs. Changes to profiles of NKT cell clusters were also seen in the spleen, likely reflecting systemic exposure to α-GalCer, although perhaps surprisingly, some changes were also seen with CpG alone. As CpG is unlikely to have directly activated NKT cells, these CpG-induced changes may reflect exposure to local cytokines and may suggest some CpG also gained access to the spleen. Although either compound may have accessed the spleen directly from the blood, splenic access could also be facilitated by dead or dying cells carrying the compounds, meaning the antigenic material and stimulatory compounds could be transferred together to resident splenic APCs to induce T cell stimulation. Indeed, transfer of α-GalCer incorporated into membranes to resident APCs has been shown to occur when α-GalCer-loaded tumours cells have been injected intravenously.64 Analysis by mass cytometry conducted early after treatment initiation, showed increased activation of DCs in both the lymph nodes and spleen, together with evidence of exposure to type I IFN, again implying that α-GalCer and/or CpG leach from the tumour to lymphoid organs to stimulate the antitumour response. The fact that surgical removal of the spleen abrogated the antitumour effect of the combined treatment indicates that the spleen plays an important role in initiating and/or sustaining the antitumour response. However, simply administering CpG and α-GalCer at a different nonmalignant subcutaneous site, where lymphoid tissues could also be accessed, was insufficient to induce the powerful antitumour responses observed with intratumoural administration. This indicates repeated modulation of the local tumour microenvironment (perhaps with the associated coordinated drainage of antigen and adjuvant compounds) plays a key role.
Perhaps the starkest change in immune profile observed with combination treatment was increased frequency of NKT cells in tumours undergoing rejection. While repeated local CpG treatment may create an environment that supports NKT cell infiltration or local proliferation, these cells also accumulated at the distal untreated tumours that underwent regression with the combined treatment. Furthermore, blocking CD1d-NKT cell interactions after the initial α-GalCer-mediated stimulus compromised the abscopal effect more than the local effect, and in NKT cell deficient animals, where only CpG should be active, the weak abscopal effect induced by CpG alone was also compromised. Thus, our studies suggest that NKT cells may contribute to the establishment of effective immunity at the distal sites, even in the absence of an exogenously administered agonist. Expression of KLRG1 was a key feature of the NKT cell clusters in the tumour and dLNs following administration of the combined treatment. It has previously been reported that KLRG1+ NKT cells accumulate as long-lived effectors in the lung following vaccination with α-GalCer-loaded DCs.65 These cells exhibited a limited CDR3β distribution, most likely as the result of clonal expansion, and responded rapidly to re-challenge several weeks to months after first exposure. As the KLRG1+ NKT cells contributed to long-term resistance to tumour challenge, the authors of this earlier work postulated that selective expansion of these cells should be explored as an antitumour therapy. The intratumoural treatment outlined here may have achieved this goal.
Finally, it was shown here that the antitumour activity of intratumoural α-GalCer and CpG could be improved in less responsive tumour models by introducing tumour-associated MHC class I-binding peptide antigens at the time α-GalCer was administered, and this was particularly effective when the peptides were chemically conjugated via an enzymatically cleavable linker to the NKT cell agonist. Conjugation may simply serve to retain the peptide for longer periods, potentially as a slow-release mechanism. However, with the linker requiring intracellular cathepsin cleavage to release the peptide, this strategy potentially leads to the preferential presentation of peptide and α-GalCer by the same APCs, thereby encouraging the licensing activities of NKT cell/APC interactions that lead to stimulation of stronger antigen-specific T cell responses. Furthermore, following i.v. administration, conjugates of this design have been shown to increase the establishment of tissue-resident CD8+ T cells, notably in the liver.41 This was correlated with low-level NKT cell-mediated hepatic inflammation, which was postulated to have encouraged residency of T cells that had been primed elsewhere. Whether the accumulation of NKT cells in the tumour seen here similarly contributes to some form of T cell residency that aids tumour rejection requires further investigation.
Supplementary Material
Acknowledgments
We thank the personnel of the Biomedical Research Unit of the Malaghan Institute of Medical Research for animal husbandry, the Hugh Green Cytometry Centre for support with flow cytometry, and the support staff at Sydney Cytometry and the Ramaciotti Facility for Human Systems Biology for their assistance with the mass cytometry studies.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This research was supported by the New Zealand Ministry of Business, Innovation and Employment (RTVU1603), the Maurice Wilkins Centre, the New Zealand Health Research Council (Project 14-500) and Avalia Immunotherapies. IF Hermans was supported by the Thompson Family Foundation and Hugh Dudley Morgans Fellowship. DIG is supported by an NHMRC Senior Principal Research Fellowship (1117766).
Disclosure statement
KK Prasit, IF Hermans and GF Painter are named inventors on a provisional patent application related to data and synthetic conjugate compounds described here. IF Hermans and GF Painter are founding scientists and shareholders of the biotech start-up Avalia Immunotherapies Limited. DI Godfrey is a member of the scientific advisory board, and a shareholder of Avalia Immunotherapies Limited.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplementary information. The data presented in this report are available from the corresponding author on reasonable request.
Synopsis
Intratumoural administration of the toll-like receptor (TLR)-9 agonist has been shown to induce tumour regression in preclinical studies and some efficacy in the clinic. By combining a repeated dosing regimen of intratumoural CpG with a single intratumoural dose of the NKT cell agonist α-galactosylceramide (α-GalCer), significantly improved antitumour activity was induced in several murine tumour models, including regression of untreated tumours at distant sites. In tumour models where the combined treatment was less effective, the inclusion of tumour antigen-derived peptide, preferably conjugated to α-GalCer, significantly enhanced the antitumour response.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2081009
References
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–18. doi: 10.1056/nejmoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/nejmoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/nejmoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin MZ, Jin WL, Hong W, Huang M, Wu M, Zhao X.. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):1–16. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marabelle A, Tselikas L, de Baere T, Houot R.. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28:xii33–xii43. doi: 10.1093/annonc/mdx683. [DOI] [PubMed] [Google Scholar]
- 6.Jiang X, Muthusamy V, Fedorova O, Kong Y, Kim DJ, Bosenberg M, Pyle AM, Iwasaki A. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J Exp Med. 2019;216(12):2854–2868. doi: 10.1084/jem.20190801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aznar MA, Planelles L, Perez-Olivares M, Molina C, Garasa S, Etxeberría I, Perez G, Rodriguez I, Bolaños E, Lopez-Casas P, et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J Immunother Cancer. 2019;7(1):116. doi: 10.1186/s40425-019-0568-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, Coffman RL, Guiducci C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A. 2016;113(46):E7240–E7249. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humbert M, Guery L, Brighouse D, Lemeille S, Hugues S. Intratumoral CpG-B promotes antitumoral neutrophil, CDC, and T-cell cooperation without reprograming tolerogenic pDC. Cancer Res. 2018;78(12):3280–3292. doi: 10.1158/0008-5472.CAN-17-2549. [DOI] [PubMed] [Google Scholar]
- 10.Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. 2012;188(4):1592–1599. doi: 10.4049/jimmunol.1101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auf G, Carpentier AF, Chen L, Le Clanche C, Delattre JY. Implication of macrophages in tumor rejection induced by CpG-oligodeoxynucleotides without antigen. Clin Cancer Res. 2001;7:3540–3543. [PubMed] [Google Scholar]
- 12.Sharma S, Karakousis CP, Takita H, Shin K, Brooks SP. Intra-tumoral injection of CpG results in the inhibition of tumor growth in murine Colon-26 and B-16 tumors. Biotechnol Lett. 2003;25(2):149–153. doi: 10.1023/A:1021927621813. [DOI] [PubMed] [Google Scholar]
- 13.Lonsdorf AS, Kuekrek H, Stern BV, Boehm BO, Lehmann PV, Tary-Lehmann M. Intratumor CpG-oligodeoxynucleotide injection induces protective antitumor T cell immunity. J Immunol. 2003;171(8):3941–3946. doi: 10.4049/jimmunol.171.8.3941. [DOI] [PubMed] [Google Scholar]
- 14.Lou Y, Liu C, Lizée G, Peng W, Xu C, Ye Y, Rabinovich BA, Hailemichael Y, Gelbard A, Zhou D, et al. Antitumor activity mediated by CpG: the route of administration is critical. J Immunother. 2011;34(3):279–288. doi: 10.1097/CJI.0b013e31820d2a05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai WYZ, Kim Y, Hoppe RT, Shah S, Horning SJ, Tibshirani R, Levy R. Preliminary report on a phase I/II study of intratumoral injection of PF-3512676 (CpG 7909), a TLR9 agonist, combined with radiation in recurrent low-grade lymphomas. Blood. 2006;108(11):2716. doi: 10.1182/blood.v108.11.2716.2716. [DOI] [Google Scholar]
- 16.Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8(10):1250–1257. doi: 10.1158/2159-8290.CD-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank MJ, Reagan PM, Bartlett NL, Gordon LI, Friedberg JW, Czerwinski DK, Long SR, Hoppe RT, Janssen R, Candia AF, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 2018;8(10):1258–1269. doi: 10.1158/2159-8290.CD-18-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jahrsdörfer B, Weiner GJ. CpG oligodeoxynucleotides as immunotherapy in cancer. Update Cancer Ther. 2008;3(1):27–32. doi: 10.1016/j.uct.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houot R, Levy R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood. 2009;113(15):3546–3552. doi: 10.1182/blood-2008-07-170274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sagiv-Barfi I, Kohrt HE, Burckhardt L, Czerwinski DK, Levy R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood. 2015;125(13):2079–2086. doi: 10.1182/blood-2014-08-593137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reis E Sousa C. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13(4):453–462. doi: 10.1016/S1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 22.Schoenberger SP, Toes REM, Van Dervoort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393(6684):480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 23.Bennett SRM, Carbone FR, Karamalis F, Miller JFAP, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186(1):65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393(6684):474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 25.Burdin N, Brossay L, Koezuka Y, Smiley ST, Grusby MJ, Gui M, Taniguchi M, Hayakawa K, Kronenberg M. Selective ability of mouse CD1 to present glycolipids: alpha-galactosylceramide specifically stimulates Vα14+ NKT lymphocytes. J Immunol. 1998;161:3271–3281. [PubMed] [Google Scholar]
- 26.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, et al. CD1d-Restricted and TCR-Mediated Activation of Vα14 NKT Cells by Glycosylceramides. Science. 1997;278(5343):1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M, Nakayama T, Taniguchi M, Koezuka Y, Tsuji M. Natural killer T cell ligand α-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med. 2002;195(5):617–624. doi: 10.1084/jem.20011889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii SI, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198(2):267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171(10):5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 30.Hermans IF, Silk JD, Gileadi U, Masri SH, Shepherd D, Farrand KJ, Salio M, Cerundolo V. Dendritic cell function can be modulated through cooperative Actions of TLR ligands and invariant NKTacells. J Immunol. 2007;178(5):2721–2729. doi: 10.4049/jimmunol.178.5.2721. [DOI] [PubMed] [Google Scholar]
- 31.Osmond TL, Farrand KJ, Painter GF, Ruedl C, Petersen TR, Hermans IF. Activated NKT cells can condition different splenic dendritic cell subsets to respond more effectively to TLR engagement and enhance cross-priming. J Immunol. 2015;195(3):821–831. doi: 10.4049/jimmunol.1401751. [DOI] [PubMed] [Google Scholar]
- 32.Corgnac S, Perret R, Zhang L, Mach J-P, Romero P, Donda A. iNKT/CD1d-antitumor immunotherapy significantly increases the efficacy of therapeutic CpG/peptide-based cancer vaccine. J Immunother Cancer. 2014;2(1):39. doi: 10.1186/S40425-014-0039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YH, Chiu NM, Mandal M, Wang N, Wang CR. Impaired NK1+ T cell development and early IL-4 production in CD1-deficient mice. Immunity. 1997;6(4):459–467. doi: 10.1016/S1074-7613(00)80289-7. [DOI] [PubMed] [Google Scholar]
- 34.Chandra S, Zhao M, Budelsky A, De Mingo Pulido A, Day J, Fu Z, Siegel L, Smith D, Kronenberg M. A new mouse strain for the analysis of invariant NKT cell function. Nat Immunol. 2015;16(8):799–800. doi: 10.1038/ni.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prigge JR, Hoyt TR, Dobrinen E, Capecchi MR, Schmidt EE, Meer N. Type I IFNs act upon hematopoietic progenitors to protect and maintain hematopoiesis during pneumocystis lung infection in mice. J Immunol. 2015;195(11):5347–5357. doi: 10.4049/jimmunol.1501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piva L, Tetlak P, Claser C, Karjalainen K, Renia L, Ruedl C. Cutting Edge: Clec9A+ Dendritic cells mediate the development of experimental cerebral malaria. J Immunol. 2012;189(3):1128–1132. doi: 10.4049/jimmunol.1201171. [DOI] [PubMed] [Google Scholar]
- 37.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54(6):777–785. doi: 10.1016/S0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 38.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 39.Lee A, Farrand KJ, Dickgreber N, Hayman CM, Jürs S, Hermans IF, Painter GF. Novel synthesis of α-galactosyl-ceramides and confirmation of their powerful NKT cell agonist activity. Carbohydr Res. 2006;341(17):2785–2798. doi: 10.1016/j.carres.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Giaccone G, Punt CJA, Ando Y, Ruijter R, Nishi N, Peters M, Von Blomberg BME, Scheper RJ, Van der Vliet HJJ, Van den Eertwegh AJM, et al. A phase I study of the natural killer T-cell ligand α-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8(12):3702–3709. [PubMed] [Google Scholar]
- 41.Holz LE, Chua YC, de Menezes MN, Anderson RJ, Draper SL, Compton BJ, Chan STS, Mathew J, Li J, Kedzierski L, et al. Glycolipid-peptide vaccination induces liver-resident memory CD8+ T cells that protect against rodent malaria. Sci Immunol. 2020;5(48):1–13. doi: 10.1126/sciimmunol.aaz8035. [DOI] [PubMed] [Google Scholar]
- 42.Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, Price KM. Panel design and optimization for high-dimensional immunophenotyping assays using spectral flow cytometry. Curr Protoc Cytom. 2020;92(1). doi: 10.1002/cpcy.70. [DOI] [PubMed] [Google Scholar]
- 43.Ferrer‐Font L, Small SJ, Lewer B, Pilkington KR, Johnston LK, Park LM, Lannigan J, Jaimes MC, Price KM. Panel optimization for high‐dimensional immunophenotyping assays using full‐spectrum flow cytometry. Curr Protoc. 2021;1(9). doi: 10.1002/CPZ1.222. [DOI] [PubMed] [Google Scholar]
- 44.Amir EAD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. ViSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31(6):545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010;53(1). doi: 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytom Part A. 2015;87(7):636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 47.Diggins KE, Greenplate AR, Leelatian N, Wogsland CE, Irish JM. Characterizing cell subsets in heterogeneous tissues using marker enrichment modeling. Nat Methods. 2017;14(3):275–278. doi: 10.1038/nmeth.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 49.Oksanen J, Blanchet FG, Kindt R, Legendrem P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, et al. Community Ecology. 2020. Package. [Google Scholar]
- 50.Martinez Arbizu P PairwiseAdonis: pairwise multilevel comparison using adonis. 2020. [Google Scholar]
- 51.DeLong JH, Hall AO, Konradt C, Coppock GM, Park J, Harms Pritchard G, Hunter CA. Cytokine- and TCR-mediated regulation of T cell expression of Ly6C and Sca-1. J Immunol. 2018;200:ji1701154. doi: 10.4049/jimmunol.1701154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dumont FJ, Coker LZ. Interferon‐α/β enhances the expression of Ly‐6 antigens on T cells in vivo and in vitro. Eur J Immunol. 1986;16(7):735–740. doi: 10.1002/eji.1830160704. [DOI] [PubMed] [Google Scholar]
- 53.Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006;177(5):3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- 54.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23(1):275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 55.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202(5):637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osman Y, Kawamura T, Naito T, Takeda K, Van Kaer L, Okumura K, Abo T. Activation of hepatic NKT cells and subsequent liver injury following administration of α-galactosylceramide. Eur J Immunol. 2000;30(7):1919–1928. doi:. [DOI] [PubMed] [Google Scholar]
- 57.Otto GP, Rathkolb B, Oestereicher MA, Lengger CJ, Moerth C, Micklich K, Fuchs H, Gailus-Durner V, Wolf E, De Angelis MH. Clinical chemistry reference intervals for C57BL/6J, C57BL/6N, and C3HeB/FeJ mice (Mus musculus). J Am Assoc Lab Anim Sci. 2016;55:375–386. [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson RJ, Compton BJ, Tang CW, Authier-Hall A, Hayman CM, Swinerd GW, Kowalczyk R, Harris P, Brimble MA, Larsen DS, et al. NKT cell-dependent glycolipid-peptide vaccines with potent anti-tumour activity. Chem Sci. 2015;6(9):5120–5127. doi: 10.1039/c4sc03599b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Den Haan JMM, Lehar SM, Bevan MJ. CD8+ but not CD8- dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685–1695. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. Cutting edge: CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol. 2006;176(8):4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 63.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176(8):4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 64.Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii SI. Cross-presentation of glycolipid from tumor cells loaded with α-galactosylceramide leads to potent and long-lived T cell-mediated immunity via dendritic cells. J Exp Med. 2007;204(11):2641–2653. doi: 10.1084/jem.20070458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shimizu K, Sato Y, Shinga J, Watanabe T, Endo T, Asakura M, Yamasaki S, Kawahara K, Kinjo Y, Kitamura H, et al. KLRG+ invariant natural killer T cells are long-lived effectors. Proc Natl Acad Sci U S A. 2014;111(34):12474–12479. doi: 10.1073/pnas.1406240111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as online supplementary information. The data presented in this report are available from the corresponding author on reasonable request.