

Abstract
Cancer cells shed naked DNA molecules into the circulation. This circulating tumor DNA (ctDNA) has become the predominant analyte for liquid biopsies to understand the mutational landscape of cancer. Coupled with next-generation sequencing, ctDNA can serve as an alternative substrate to tumor tissues for mutation detection and companion diagnostic purposes. In fact, recent advances in precision medicine have rapidly enabled the use of ctDNA to guide treatment decisions for predicting response and resistance to targeted therapies and immunotherapies. An advantage of using ctDNA over conventional tissue biopsies is the relatively noninvasive approach of obtaining peripheral blood, allowing for simple repeated and serial assessments. Most current clinical practice using ctDNA has endeavored to identify druggable and resistance mutations for guiding systemic therapy decisions, albeit mostly in metastatic disease. However, newer research is evaluating potential for ctDNA as a marker of minimal residual disease in the curative setting and as a useful screening tool to detect cancer in the general population. Here we review the history of ctDNA and liquid biopsies, technologies to detect ctDNA, and some of the current challenges and limitations in using ctDNA as a marker of minimal residual disease and as a general blood-based cancer screening tool. We also discuss the need to develop rigorous clinical studies to prove the clinical utility of ctDNA for future applications in oncology.
Introduction
Liquid biopsies have emerged as a new method for analyzing biomarkers from blood and other bodily fluids. Today, “liquid biopsy” usually refers to the identification and analysis of cell-free DNA (cfDNA) derived from patient plasma samples. cfDNAs are free-floating, naked DNA molecules that are shed into the circulation actively by cells, as well as by cells undergoing death by apoptosis and/or necrosis (1). Many studies have shown the potential benefit of using cfDNA for clinical management in such areas as identification of fetal genetic anomalies (2), solid organ transplant rejection (3), and identification of cancer mutations from blood (4). For cancer and oncology research, most studies evaluating cfDNA have examined DNA derived specifically from cancer cells, also known as circulating tumor DNA (ctDNA). In this relatively brief Review, it is not feasible to discuss all existing and emerging applications of cfDNA in helping guide clinical decision making. Numerous excellent recent reviews have highlighted the many clinical studies using ctDNA as integral and integrated biomarkers (5–7). Thus, we will focus on the technical aspects of ctDNA, including the challenges of using ctDNA in oncology across the spectrum from primary prevention to metastatic disease (Figure 1).
Figure 1. The potential utility of ctDNA across the spectrum of human cancers.

MRD, minimum residual disease. Adapted with permission from Cancers (108).
The field of oncology has evolved at a rapid pace over the past twenty years. Many cancers that were once uniformly fatal can now be treated effectively for years, and some appear curable even in the metastatic setting (8–10). Unquestionably the discoveries of genetic alterations that lead to human cancers have allowed targeting of these alterations for therapeutic gain. Historically, sequencing of a patient’s tumor to find mutations was laborious, time-consuming, and expensive and could only be performed on a limited scale. Newer technologies have largely overcome these barriers but have also led to new challenges. In this Review, we first describe the rationale of identifying genetic alterations in cancers that are amenable to targeted and other therapies, including a brief background on the principles of oncology and the goals and differences in treating solid-tumor patients with early-stage versus metastatic disease. We then briefly review the history of liquid biopsies, cfDNA, and ctDNA, and the various technologies and platforms for analyzing these substrates. We also include limited examples of past and current studies that have led to routine use of ctDNA in the management of patients with cancer. Finally, we present some of the challenges of achieving clinical utility of liquid biopsies/ctDNA to address pressing unmet needs in oncology.
Precision oncology in early-stage and metastatic cancer
Precision oncology arose from the idea that understanding the molecular underpinnings of a patient’s tumor creates an opportunity to use drugs to selectively target cancer cells harboring these genetic changes (11). These genetic alterations, which are generally gene mutations, amplifications, fusions, and loss of tumor suppressor genes, are termed “drivers,” since they are responsible for cancerous phenotypes including the ability to proliferate and metastasize (12, 13). However, drivers also represent the Achilles’ heel of cancers, as the majority of these genetic alterations are somatic, i.e., present only in the cancer cell. This affords the opportunity to develop therapies with exquisite specificity for cancer cells while leaving normal cells relatively unharmed. Although there are indeed heritable mutations/genetic alterations that predispose to cancer and are therefore found in every cell that contains DNA (e.g., germline mutations in genes such as BRCA1 and BRCA2), these represent a minority of all cancer cases (14). Notably, even in BRCA1 and BRCA2 gene mutation carriers, subsequent somatic genetic events must occur to transform a normal cell into a cancer cell (15) that can also result in therapeutic vulnerabilities. An example of this is PARP inhibitors, which have been approved for breast cancers in both the metastatic and the early-stage setting for patients with germline pathogenic BRCA1 or BRCA2 mutations (16–20), and for ovarian, pancreatic, and prostate cancers with BRCA1 or BRCA2 mutations (21–26).
The ability to assess molecular alterations in tumor tissues was limited in the past because of the cost and time of “first-generation sequencing.” With the advent of next-generation sequencing (NGS), rapid analysis of a cancer genome became feasible (27). Currently, however, most clinical cancer gene NGS assays consist of panels of 500 to 600 genes, though whole-exome and whole-genome NGS is possible. The reasons for this are pragmatic: currently there is little if any clinical actionability in obtaining more than 500 to 600 genes of a patient’s tumor via NGS, and performing whole-exome/whole-genome NGS increases costs and, importantly, turnaround time owing to the additional bioinformatics analyses required. That said, NGS of tumors along with the use of targeted therapies and immunotherapies has become standard of care for most if not all cancers in the metastatic setting and is also now indicated for select early-stage cancers such as non–small cell lung cancer and melanomas (28, 29). This topic is covered in more depth in other Reviews in this series (30), and therefore we will only touch upon this topic as needed for clarity and background.
To demonstrate how liquid biopsies are currently being used, and how their future use could address unmet needs, we will next review the goals and principles of systemic therapies as they relate to patients with solid tumors (31). For the majority of solid tumors, stage IV represents metastatic disease and is considered incurable in most cases. Therefore therapies are directed toward treating the cancer to extend and improve quality of life. Currently this is where the bulk of precision oncology efforts are focused: to find molecular alterations that have targeted therapies (11, 32, 33). Also, because metastatic disease is largely incurable, there tends to be more latitude in pursuing therapies that may not be standard of care but may realistically still afford a chance of benefit (11, 31, 32, 34). Such strategies, however, must be considered carefully and are nowadays often discussed at molecular tumor boards (33), and have led to guidelines to contextualize a framework for prioritizing actionable NGS results (35).
For patients with early-stage cancer, the role of systemic therapies like chemotherapy is additional treatment after local therapy (surgery and/or radiation) in patients who would otherwise later relapse with incurable metastatic disease due to the presence of microscopic or minimal residual disease (MRD) (36). Although clinical trials clearly provide proof that these additional, also known as adjuvant, systemic therapies improve cure rates for early-stage cancers, it is also well known and accepted that significant populations of patients who are already cured after local therapies receive systemic therapies needlessly, thus being exposed to their potential toxicities. This, in fact, led to the development of a genomic test for breast cancer to help identify such patients (37). Moreover, considerable numbers of patients will still develop incurable metastatic disease at some future point despite having received adjuvant systemic therapies. Equally important, owing to the curative intent of therapy for early-stage disease (38), adding, removing, or replacing adjuvant therapies outside of a clinical trial is generally discouraged, as there is great concern that doing so could compromise the chance of cure. Thus, adding targeted therapies for cancers with certain mutations in early-stage disease will generally require a rigorous level of evidence from large prospective randomized controlled trials with multi-year follow-up, as was recently shown for PARP inhibitors and BRCA1- or BRCA2-derived breast cancers (18, 20). Hence a true unmet need for precision oncology and ctDNA use in early-stage cancers is determining who is cured, who is not, and whether additional therapies beyond standard of care could cure additional patients who still have MRD.
Liquid biopsies and ctDNA
The advent of liquid biopsies changed the manner in which oncologists approach cancer therapies. There are now many genomic and genetic alterations that influence treatment decisions, and liquid biopsies, specifically ctDNA, allow for a relatively easy, noninvasive way to identify these alterations. We acknowledge that “liquid biopsy” can also refer to other analytes in blood used to assess cancer burden and/or genomic and genetic features of a patient’s cancer, including circulating tumor cells (39), protein biomarkers (40), and cell-free RNA (cfRNA) (41). Development of new DNA detection technologies for ctDNA, as well as the ever-increasing number of targeted therapies and immunotherapies being approved for cancers, has allowed for a more refined approach to treating patients with various malignancies. Although this approach has been mostly used in patients with metastatic disease for the reasons outlined above, recent studies support the use of targeted therapies in the adjuvant setting (18, 29).
Though NGS of tumor tissue is generally preferred, often a tissue biopsy is unobtainable or of insufficient quantity or quality, and therefore ctDNA analysis using blood is the only available option. Additionally, there are clinical situations in which procurement of tumor tissue for NGS is delayed because of the time needed to set up a biopsy or surgery, as well as time for processing of tissues for pathologic assessment. These factors further add to the turnaround time for obtaining NGS results from tumors (42). In contrast, liquid biopsies generally require only phlebotomy using several blood tubes, and samples can be quickly sent off for NGS analysis. In addition, liquid biopsies allow for a fast and easy method of serial analysis over multiple time points that can help guide treatment decisions by identifying the emergence of mutations and other genetic alterations that predict for resistance or response to the next line of therapy (43, 44). Moreover, this approach can avoid targeted therapies and immunotherapies that would have little chance of benefit, thereby minimizing toxicities, financial burden, and other adverse events.
Although the ability to use cfDNA for clinical applications is relatively new, the existence of cfDNA was first described in 1948 (45). cfDNA refers to naked free-floating DNA molecules shed or released from cells, which occurs under normal circumstances in healthy tissues due to cell turnover and active shedding into the circulation (46). Presently, there is no clear known function of cfDNA, and many feel it may simply be a cellular waste product that eventually clears the circulation through the kidneys as urine cfDNA. In addition, the half-life of cfDNA is short, speculated to be 1 to 2 hours (47), maybe because of DNases in the blood that lead to quick degradation. cfDNA is typically found as short fragments approximately 160–180 base pairs in length and is now used for a number of clinical applications, such as assessing fetal genetic anomalies from maternal blood (48). Interestingly, cfDNA was initially proposed as a way to assess cancer patients’ overall tumor burden, as it was noted that, in general, higher overall cfDNA levels are detected in patients with cancer than in individuals without cancer (49). Ultimately, total cfDNA levels were not sensitive nor specific enough for following a patient’s overall tumor burden and response to therapies.
Regardless, ctDNA assessment has become relatively routine in patients with metastatic disease, and ctDNA generally can be present among cfDNA at variant allele fractions (VAFs) ranging from <0.1% to 10%, though it can be even higher (42, 50). VAFs represent the percentage of mutant DNA molecules relative to the total number of DNA molecules for a given gene. Numerous studies have demonstrated that higher VAFs are associated with increased tumor burden and worse prognosis (51–54). Importantly, therapeutic intervention can also lead to changes in the levels of ctDNA in the blood (47). For example, surgical resection of tumors can lead to a decrease in ctDNA, as can chemotherapy, targeted therapy, and immune therapies (42, 47, 55, 56). Collectively, these and now numerous clinical studies have demonstrated that changes in ctDNA levels correlate with therapies, both local and systemic. There have also been numerous correlative studies evaluating whether de novo or acquired mutations found in ctDNA can predict response or resistance to a given therapy (57, 58) and/or whether serial ctDNA monitoring can detect MRD (43, 59, 60). The majority of these studies retrospectively evaluated samples that had been collected prospectively. However, due to the rather rigorous requirements for optimal analyte preparation (61), described below, as well as the need for sufficient plasma DNA for analysis, there are caveats to these studies, as only a few were prospectively planned to incorporate ctDNA analyses. More recently, studies have demonstrated some limited clinical validation for MRD detection using newer approaches (59, 62) as well as using ctDNA as a primary blood-based cancer screening test (40, 63). These studies also highlighted some of the limitations of ctDNA, including that some cancer types may secrete or shed less DNA into the circulation for unknown reasons (64).
ctDNA collection and processing
Blood, and specifically plasma, is the ideal analyte for ctDNA collection and is currently being used in the clinic predominantly for companion diagnostic purposes, meaning finding mutations that are linked to an FDA-approved therapy. Although plasma is now the preferred analyte for cfDNA analyses, cfDNA can be detected in serum, though serum cfDNA has decreased integrity compared with plasma cfDNA (65–67). However, other sources of cfDNA/ctDNA have been evaluated, including cerebrospinal fluid (68) and urine ctDNA (55). For most current commercial and research assays, cfDNA is generally isolated by separation of plasma from whole blood using double centrifugation protocols to maximally remove cells, followed by extraction of nucleic acids using various techniques (69). It should be noted that great care must be taken in processing cfDNA and that the use of dedicated equipment (centrifuges, pipettes, etc.), “clean rooms,” dedicated hoods, etc. is essential to avoid contamination of samples with aerosolized DNA, which is a major issue in dealing with NGS and other assays that measure mutations at 0.01% VAF or lower (70). Generally speaking, at least 20 mL of whole blood should be obtained to ensure enough plasma DNA for analysis, though this will vary depending on the nature of the assay (71). Ideally, cfDNA should be isolated within hours of blood collection to prevent lysis of white blood cells; otherwise cell lysis will liberate large amounts of cellular genomic DNA into the plasma component within the blood collection tube (61, 72, 73). Because of the wide variation in cfDNA concentration between individuals, the amount of cfDNA needed to truly reach the technical limit of detection (LOD) of any given platform can be problematic. For example, if the technical LOD for a given assay is one mutant DNA molecule in 10,000 wild-type DNA molecules, then to truly define a negative result, the sample must contain at least 10,000 DNA molecules and preferably severalfold above this amount whenever possible. In practice, obtaining 10,000 DNA molecules or “genome equivalents” is non-trivial. A genome equivalent (GE) is defined as one genome of an organism, which in the case of humans is the haploid genome. The haploid genome is approximately 3 billion base pairs, and has a molecular mass of approximately 3 picograms. Therefore, to obtain 10,000 GEs in a given sample, there must be approximately 30 nanograms of DNA, which is often challenging given the relatively low concentrations of plasma DNA found in human samples (74).
As mentioned above, analyte considerations must be taken into account in the processing of plasma DNA from blood, including contamination issues and lysis of white blood cells when blood samples are not processed within a few hours of collection. We and others have shown that Streck Cell-Free DNA BCT tubes can prevent cellular lysis, owing to cell membrane–stabilizing preservatives used in these tubes (61, 74). Indeed, standard EDTA tubes that are not processed within hours can lead to inaccurate quantification of mutational allele frequencies due to lysis of blood cells, thereby increasing the number of wild-type alleles. Therefore, proper handling and timely sample processing are key steps to ensure the integrity of cfDNA for downstream analysis.
Analysis of ctDNA
Methods to analyze ctDNA have historically used either PCR-based assays or NGS paired with specialized bioinformatics analysis. Although many PCR-based strategies and technologies have been created and are still available for cfDNA testing, a discussion is beyond the scope of this Review. Rather, we will briefly touch on the initial single-molecule PCR methods for analyzing cfDNA and how they evolved into the current use of NGS techniques.
Digital PCR was the initial method described by Kinzler and Vogelstein, which established the idea of single-molecule amplification that could allow assessment of rare mutant alleles in a background of many wild-type alleles (75). Although the term “digital” is often assumed to have an electronic connotation, it refers (to the binary nature of diluting samples such that each partition or compartment of the assay platform has either one or zero DNA molecules. The first high-throughput digital PCR method was called BEAMing (beads, emulsions, amplification, and magnetics; ref. 76). This somewhat cumbersome process was successfully employed to demonstrate that digital PCR could be used for ctDNA detection in metastatic disease and following response to therapies (4, 77). Subsequent commercial evolution of BEAMing came from droplet digital PCR (ddPCR), which enabled more uniform partitioning of DNA molecules in a technology platform that could be used by standard research laboratories (60). Many studies demonstrating the analytic and clinical validity of ddPCR followed (78–80). However, a major limitation of all digital PCR platforms is the finite number of mutations per assay that can be analyzed. But an advantage of digital PCR over current NGS methods is that the readout for digital PCR does not require any bioinformatics analyses and is akin to data collected and analyzed using a flow cytometer. This readout can significantly reduce turnaround time, which can be clinically useful. Moreover, the accuracy of digital PCR and the ability to easily run technical replicates allowed for an LOD of 0.01% to 0.001%, which at the time of digital PCR’s emergence was not possible with NGS. However, with the advent of “barcoding” of individual DNA molecules using molecular tags, along with new bioinformatics analyses, NGS of cfDNA evolved to overcome the limitations of errors introduced by PCR amplification and/or NGS, to allow for a technical LOD that equaled or even surpassed that of digital PCR (81).
NGS offers deeper and more comprehensive profiling methods to identify genetic alterations. Rather than querying for only known variants with molecular probes as with digital PCR, NGS has the capacity to find genetic perturbations in a relatively unbiased fashion, since sequencing, by its very nature, will identify all the base pairs of a given DNA molecule. The flexibility in selectively capturing and/or amplifying only regions of interest has led to approaches for sequencing only parts of a genome, including so-called “exome capture” and also gene panels (82). Unfortunately, NGS is prone to sequencing errors, due to the inherent nature of strand synthesis, as well as PCR amplification. In the past, this had hindered the use of NGS for ctDNA analysis, as allele fractions of ctDNA are often low, in the range of 0.01% to 1% ctDNA to cfDNA (42, 83).
A seminal breakthrough in NGS was the concept of “tagging” each individual DNA molecule with a molecular barcode. The advent of barcoding of each individual DNA fragment with unique identifiers (UIDs), coupled with bioinformatics analysis and higher-fidelity DNA polymerases, allowed for ultrasensitive detection of ctDNA (84). The first such technique was the Safe-Sequencing System (Safe-SeqS; ref. 84), followed by many other variations, including cancer personalized profiling by deep sequencing (CAPP-Seq; ref. 85), tagged-amplicon deep sequencing (TAm-Seq; ref. 86), and Duplex Sequencing (Duplex-Seq; ref. 87). All of these incorporate some form of barcoding of individual DNA molecules that enables ultrasensitive detection of mutations that otherwise would be either below the LOD and/or impossible to distinguish from NGS/PCR artifacts. The basic concept is shown in Figure 2A. The simplest method of generating a UID is to design primers containing degenerate, random sequences. As an example, one could design 30-mer primers with fixed 5′ and 3′ ends, each with 8 nucleotides, but the remaining 14 internal nucleotides would be designated “N” and randomly picked to be either A, C, G, or T during synthesis (Figure 2B). Thus each base pair position has a 1 in 4 chance of being a given nucleotide. When multiplied by the 14 internal nucleotides (1/4 to the 14th power), this yields a combination of 268,435,456 unique primer sequences, which is generally more than adequate to uniquely tag each individual DNA molecule if the starting material is 10,000 GEs. Thus, at a given molar amount of synthesized primers, one could generate an almost infinite number of UIDs based on the length of the variable nucleotides. Using this approach, after PCR amplification, true mutations at low allele fraction should theoretically be present at nearly 100% of NGS reads within a barcode “family”; in addition, presuming there is enough starting DNA template of more than one mutant molecule, multiple barcode families should also be present that harbor the same mutation. Statistical algorithms via bioinformatics analyses dictate the number of mutant molecules in a given family, as well as the need for multiple families with the mutation, in order for one to call a true mutation with confidence (84). On the other hand, PCR and NGS artifacts would be present only in a single barcode family and likely with only a few NGS reads, and would therefore be discarded as artifacts. The LOD using these approaches can be as low as one mutant molecule per millions of wild-type DNA molecules, and indeed, studies have demonstrated that barcoded NGS approaches can even measure error rates of high-fidelity DNA polymerases estimated to be 1 error in 4.4 × 107 nucleotides (84). The utilization and evolution of barcoding of DNA molecules have revolutionized the ability of NGS to detect rare mutations, and subsequently its use for clinical applications.
Figure 2. Molecular barcoding for NGS libraries to improve detection of rare mutations.
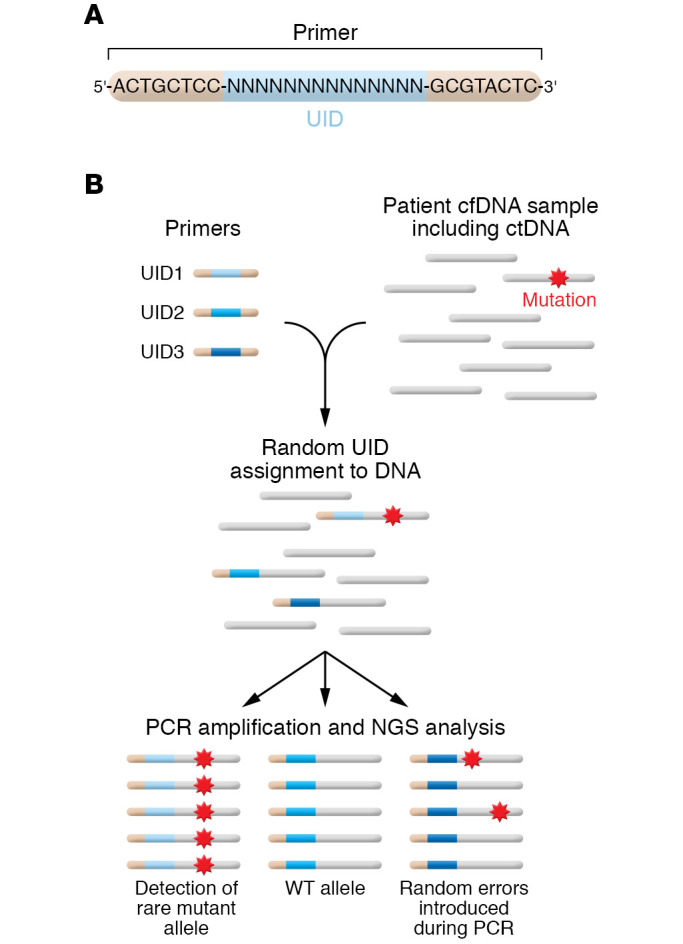
(A) Incorporation of random sequences for degenerate primers used to molecularly tag each DNA molecule. Each “N” can be either A, C, G, or T and is chosen randomly during synthesis. (B) Schema of Safe-SeqS (adapted with permission from Proceedings of the National Academy of Sciences of the USA; ref. 84). Clinically relevant mutations are present at very low frequency in patient samples. Barcoding of DNA can improve the signal-to-noise ratio in NGS analysis, because mutant tumor alleles containing the same UID will be amplified, whereas random errors resulting from PCR amplification will remain at low frequency.
Clinical utility
When discussing the current clinical utility of ctDNA for cancer care, one can envision two broad categories: qualitative and quantitative assessment. These are not mutually exclusive, but in general, only qualitative aspects of ctDNA have truly been utilized for clinical decision making thus far. If certain mutations are found in ctDNA of a patient with cancer, this opens the possibility of a targeted therapy — an on-label FDA-approved therapy, off-label use, and/or eligibility for a clinical trial. A recent example is mutant PIK3CA found in either tissue or ctDNA in patients with hormone receptor–positive, HER2-negative metastatic breast cancer, enabling the use of the PI3Kα inhibitor alpelisib (88), but there are many other examples beyond the scope of this Review. Qualitative aspects of ctDNA are also used to identify resistance mutations that serve as negative predictors of response to certain therapies. Examples include KRAS mutations, which predict for resistance to EGFR antibody–based therapies (89), as well as ESR1 mutations in hormone receptor–positive breast cancers that predict for resistance to certain endocrine therapies (78, 90, 91). Although current commercial ctDNA assays are less sensitive than their tissue counterparts, this is largely due to pragmatic issues, i.e., the time and expense that would be needed for the level of redundant NGS, and the amount of blood/plasma and the extensive bioinformatics analyses required, for ctDNA assessment to approach the level of sensitivity of tumor tissue NGS. However, with newer technologies and with the cost of NGS continually decreasing (92), it is tempting to speculate that ctDNA may soon achieve a sensitivity of mutation detection equal if not superior to that of its tumor tissue counterpart.
As more genes and regions of the genome are sequenced using NGS, other qualitative/semiquantitative information becomes available that may also inform current clinical decision making. Specifically, so-called tumor mutation burden (TMB) and high microsatellite instability (MSI-H) are two tissue-agnostic FDA-approved indications for immunotherapy with the anti–PD-1 antibody pembrolizumab (93, 94). The reason TMB leads to a higher likelihood of response is thought to be the higher chance that any given mutation will create a neoantigen that can then be recognized by the immune system once anti–PD-1 therapy is initiated. This concept is similar to buying more lottery tickets; the more tickets purchased, the higher the odds of winning. Statistically, one would need to buy a critical number of lottery tickets to significantly impact the likelihood of winning. Similarly, a high TMB (defined as 10 mutations per megabase of DNA) implies a high number of mutations found within a cancer, which has been shown to be predictive of response to pembrolizumab owing to the increased likelihood of neoantigens that can be recognized by the immune system with anti–PD-1 therapy (94). Similarly, MSI-H tumors are generally cancers that arise from loss of mismatch repair gene function, leading to instability of microsatellite regions (repetitive DNA sequences) in the human genome that generally results in high TMB (93). Although MSI-H usually leads to a high TMB, high TMB can arise through other mechanisms. MSI-H is measured by NGS via evaluation of designated microsatellite regions and computation of whether these have enough changes in individual DNA strands to be classified as microsatellite stable, low, or high (95). Since the number of microsatellite regions queried is limited, ctDNA can be and is used by most commercial vendors to assess MSI-H. Because accurate assessment of TMB requires a large number of DNA base pairs to be sequenced, assessing TMB through NGS of ctDNA was historically challenging owing to the limitations mentioned above. However, with the increasing numbers of genes being sequenced using ctDNA, many commercial tests have recently incorporated this information into their liquid biopsy assays and thus can now be used to assess TMB and the likelihood of response to pembrolizumab and perhaps other immunotherapies (96).
Where ctDNA is still lacking is in the ability to use its quantitative aspects to guide clinical decisions. As mentioned previously, there have been and continue to be large numbers of studies using post hoc or planned analysis to evaluate the prognostic and predictive capability of ctDNA, in which plasma samples are analyzed at various time points retrospectively. However, despite these studies and their clinical validation, clinic utility is still lacking. Here, we are using Henry and Hayes’s definition of clinical utility (97), i.e., high-level evidence that quantitative detection of ctDNA can actually lead to changes and interventions that will positively affect outcomes for patients with cancer. Although such studies are ongoing and are the basis of many current clinical trials (98), these studies require long-term follow-up of outcomes akin to other screening/biomarker studies to definitively prove that disease-free, progression-free, and/or overall survival is meaningfully affected by the detection of ctDNA in undiagnosed people and/or patients with cancer. As an example, although quantitative assessment of circulating tumor cells (CTCs) has been clinically validated as a prognostic marker for metastatic breast cancer, its clinical utility has yet to be proven, and indeed, one study evaluating changing chemotherapy treatments based on a high number of CTCs did not show any benefit, i.e., showed lack of clinical utility (99). Further, any test that has been clinically validated as truly detecting cancer using ctDNA is ultimately not helpful for managing the disease until clinical utility has been proven — i.e., does acting on the results of the test improve outcomes in a clinically meaningful way?
Demonstrating clinical utility for ctDNA is especially challenging in studies evaluating ctDNA use as a primary cancer screening test, as well as for MRD in the adjuvant setting. As mentioned, a number of validation studies have been published or are ongoing, but to demonstrate that acting on these ctDNA results leads to improved outcomes such as the curing of more disease and/or improvement of overall survival will require intervention studies with long-term follow-up. Key to developing ctDNA assays for primary cancer screening and, in some instances, follow-up detection in the curative setting for MRD is knowledge that the ctDNA detected is coming from a given cancer type. For example, if one were to detect a TP53 mutation in an asymptomatic person after cfDNA analysis, it would be unclear from which cell type this mutation arose, given that TP53 mutations are found in a variety of human cancers. More perplexing, some such mutations, including TP53, may arise from clonal hematopoiesis (CH) (100). CH is relatively easy to detect using cfDNA but is often an incidental finding, as it has been shown that many asymptomatic individuals develop CH as a result of aging (101). Although CH may be the precursor to preleukemic syndromes, many individuals with CH will not experience any hematologic disease, and in fact, CH seems to have more correlation with cardiovascular risk than with hematologic cancer (102). In addition to the above concerns, the current sensitivity of ctDNA NGS assays can detect cancer cells prior to detection by radiographic scans; therefore, it is unclear what if anything should be done upon discovery of a ctDNA mutation in an asymptomatic person. To begin to address this vexing issue, recently investigators have explored the use of epigenetic DNA modifications and physical DNA fragmentation patterns to identify not only ctDNA, but which tissue of origin the ctDNA was derived from (103, 104). Bisulfite treatment followed by NGS is typically how one can distinguish methylated from unmethylated DNA (105), but newer approaches using immunoprecipitation have also been published and show high sensitivity and specificity for a given tumor type (106). These promising approaches set the stage for future clinical utility studies that may realize the full potential of ctDNA and liquid biopsies.
Challenges and future directions
Despite the bright future and strong momentum of ctDNA as a tool to guide cancer prevention and therapy, many challenges remain. Beyond the need to prove clinical utility, decreasing turnaround time, decreasing costs, and improving sensitivity and specificity for MRD and primary cancer screening remain as technical, but not insurmountable, obstacles. As with most technologies, increased speed of NGS along with decreased costs continues to be the norm, such that one can easily envision that these issues will soon be resolved (92). However, the use of ctDNA for MRD, and as a primary cancer screening test, is more difficult to address. As mentioned, there are limited clinical validation studies attesting to the positive predictive value of ctDNA for these use indications. Detection of ctDNA generally is a harbinger of MRD as a primary screen and/or after curative intent therapies for patients with cancer. Again, further long-term studies are needed to prove that such detection is actionable and can result in clinically meaningful interventions that provide robust evidence of utility. On the other hand, there has been little or no validation regarding the negative predictive value of these tests, which implies that absence of ctDNA does not necessarily equate with lack of cancer. Thus, MRD may still exist in this scenario, and in the case of patients with cancer treated with curative intent, absence of ctDNA cannot currently be equated with cure. There are many technical reasons for this, and much of it depends on the nature of current assays designed to detect MRD. As an example, many ctDNA assays used to “track” MRD use a bespoke approach, meaning that tumor tissue from the patient is subjected to NGS and individual mutations are used as markers to follow response to therapies and/or MRD (62). In contrast, a generalized ctDNA assay using common mutated genes, and/or methylated versus unmethylated DNA that has been validated across various cancer types, may serve as a tissue-agnostic and independent approach for using blood to track and monitor ctDNA without the need for NGS of tumor tissue (59).
Both bespoke and generalized approaches have advantages and disadvantages and are not necessarily mutually exclusive. Although the bespoke assay lends further confidence and presumably sensitivity and specificity of individual mutations to be used as markers of MRD, this approach can be hampered by inadequacy (in both quantity and quality) of tissue samples as well as by the time needed to obtain tumor tissue, perform NGS to identify markers, and then develop individual markers for each patient. In certain clinical situations, this may not be practical or possible. On the other hand, although non-bespoke assays may forego the use of tissue NGS, a negative result may be indeterminant, since the mutated/methylated genes being queried may not be present in the genetic makeup of a given patient’s cancer.
Further obfuscation comes from the fact that a given plasma sample may not have enough GEs to decisively show that a negative result is truly free of ctDNA. There has been increasing interest in overcoming the limited number of GEs in a plasma sample by increasing the number of mutations or amount of methylated DNA for tracking of MRD (62). The concept here is that “more shots on goal” may allow for any rare ctDNA molecule to be identified because, for a given sample of plasma DNA, there may be only a few ctDNA molecules that may or may not contain a specific marker that is being tracked (Figure 3). Thus, rather than following one or two mutations, which would require tens of thousands of GEs, if enough mutations/methylated DNA markers are queried, the odds of detecting ctDNA may greatly increase even with limited amounts of plasma DNA. Although this approach was shown, in a limited manner, to have a very high positive predictive value (107), negative predictive value data are still lacking and may require a dramatic increase in the number of tracked markers to truly determine whether a negative result on a liquid biopsy test can allow reasonable confidence in identifying patients who are cured of their disease.
Figure 3. Rationale for increasing genome equivalents versus increasing the number of tracking mutations to identify ctDNA.
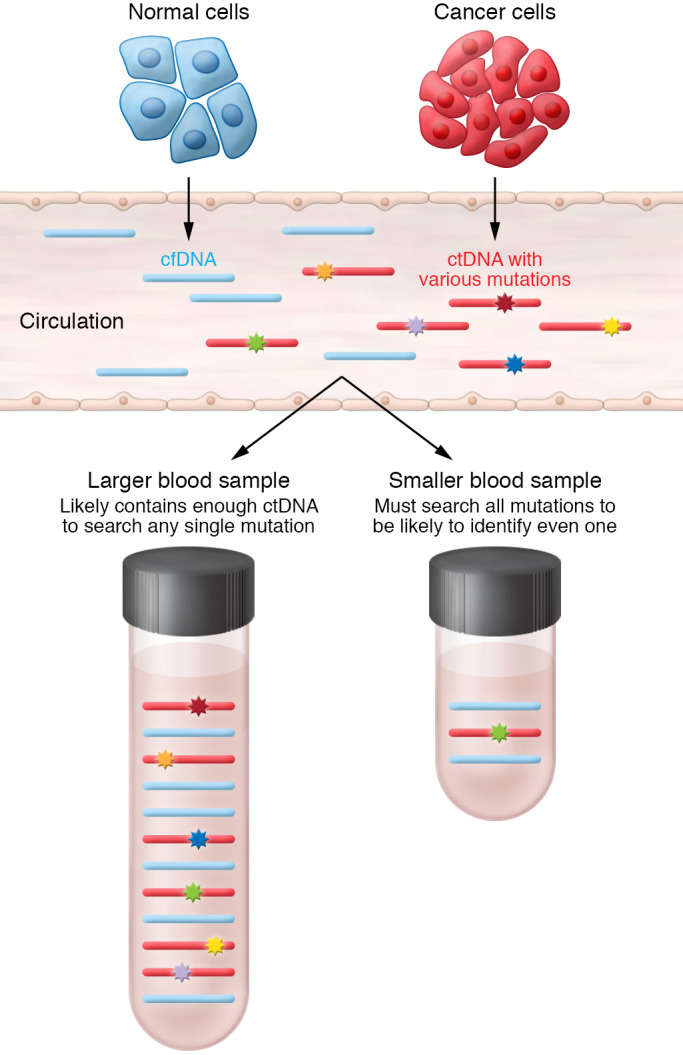
In this example, the tumor/cancer cells have six distinct mutations (colored stars) that are being shed into the circulation along with normal DNA from normal cells (in blue). If an assay only queries for a single mutation, then a large amount of DNA is required to ensure a high likelihood that the mutation will be in the sample (bottom left). On the other hand, if plasma DNA is limited, then there may be only a single mutation in the sample (green star), and therefore querying for all DNA mutations is needed such that there is high likelihood that any mutation will be identified (bottom right).
Conclusions
In summary, liquid biopsies with ctDNA are a relatively new approach to help guide clinical decision making for the prevention, diagnosis, and treatment of human cancers. Applications of ctDNA as an indicator of disease status and mutational landscape are expanding, enabling oncologists to make more informed decisions with precision. Although liquid biopsies have become standard of care in select circumstances, there are still challenges and areas in which to grow its clinical utility for optimizing cancer care. Excitingly, one can envision ctDNA’s future role as a cancer screening test; as a companion diagnostic for obtaining targeted therapies and immunotherapies; for following response to therapies; and as a marker of MRD to determine which patients are potentially cured and which may benefit from additional therapies. Yet all of these applications and more can be easily obtained from a patient’s blood sample. Truly the future is bright for precision oncology with liquid biopsies at the forefront of this revolutionary way to make health care personal.
Acknowledgments
It is impossible to cite and give credit to all the many deserving studies and investigators who have pioneered the use of cfDNA, both in the laboratory and in the clinic. We apologize to our colleagues whose important work we could not include. This work was supported by the NIH (T32CA119925 to DKD). BHP is supported by the Breast Cancer Research Foundation, the Komen Foundation, and the NIH (CA214494/CA194024). We also appreciate the support of the Canney Foundation, the Marcie and Ellen Foundation, Donna and John Hall, the Cornelius A. Craig Chair, Steve Kandell, Amy and Barry Baker, the Vanderbilt-Ingram Cancer Center support grants NIH CA068485 and U54CA163072, and the Breast Cancer Specialized Program of Research Excellence (SPORE) grant NIH CA098131.
Version 1. 06/15/2022
Electronic publication
Footnotes
Conflict of interest: BHP is a paid consultant for The Jackson Laboratory, EQRx, Sermonix, Hologics, and Guardant Health and is a paid scientific advisory board member with ownership interest for Celcuity Inc. BHP also has research contracts with GE Healthcare, Lilly, and Pfizer. Under separate licensing agreements between Horizon Discovery Ltd. and Johns Hopkins University, BHP is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by Johns Hopkins University in accordance with its conflict-of-interest policies.
Copyright: © 2022, Dang et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2022;132(12):e154941. https://doi.org/10.1172/JCI154941.
Contributor Information
Donna K. Dang, Email: donna.dang@vumc.org.
Ben H. Park, Email: ben.h.park@vumc.org.
References
- 1.Bronkhorst AJ, et al. Characterization of the cell-free DNA released by cultured cancer cells. Biochim Biophys Acta. 2016;1863(1):157–165. doi: 10.1016/j.bbamcr.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 2.Fan HC, et al. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487(7407):320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schütz E, et al. Graft-derived cell-free DNA, a noninvasive early rejection and graft damage marker in liver transplantation: a prospective, observational, multicenter cohort study. PLoS Med. 2017;14(4):e1002286. doi: 10.1371/journal.pmed.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diehl F, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102(45):16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moding EJ, et al. Detecting liquid remnants of solid tumors: circulating tumor DNA minimal residual disease. Cancer Discov. 2021;11(12):2968–2986. doi: 10.1158/2159-8290.CD-21-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siravegna G, et al. How liquid biopsies can change clinical practice in oncology. Ann Oncol. 2019;30(10):1580–1590. doi: 10.1093/annonc/mdz227. [DOI] [PubMed] [Google Scholar]
- 7.Donaldson J, Park BH. Circulating tumor DNA: measurement and clinical utility. Annu Rev Med. 2018;69:223–234. doi: 10.1146/annurev-med-041316-085721. [DOI] [PubMed] [Google Scholar]
- 8.Hunter N, et al. Undetectable tumor cell-free DNA in a patient with metastatic breast cancer with complete response and long-term remission. J Natl Compr Canc Netw. 2020;18(4):375–379. doi: 10.6004/jnccn.2019.7381. [DOI] [PubMed] [Google Scholar]
- 9.Robert C, et al. 1082MO 5-year characterization of complete responses in patients with advanced melanoma who received nivolumab plus ipilimumab (NIVO+IPI) or NIVO alone. Ann Oncol. 2020;31(suppl 4):734–735. [Google Scholar]
- 10.Melenhorst JJ, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. 2022;602(7897):503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalton WB, et al. Personalized medicine in the oncology clinic: implementation and outcomes of the Johns Hopkins Molecular Tumor Board. JCO Precis Oncol. 2017;2017(1):1–19. doi: 10.1200/PO.16.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 13.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 14.McClain MR, et al. Adjusting the estimated proportion of breast cancer cases associated with BRCA1 and BRCA2 mutations: public health implications. Genet Med. 2005;7(1):28–33. doi: 10.1097/01.GIM.0000151155.36470.FF. [DOI] [PubMed] [Google Scholar]
- 15.Konishi H, et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A. 2011;108(43):17773–17778. doi: 10.1073/pnas.1110969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 17.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 18.Tutt ANJ, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N Engl J Med. 2021;384(25):2394–2405. doi: 10.1056/NEJMoa2105215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robson M, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 20.Litton JK, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–763. doi: 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González-Martín A, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–2402. doi: 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 22.Hussain M, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383(24):2345–2357. doi: 10.1056/NEJMoa2022485. [DOI] [PubMed] [Google Scholar]
- 23.Ray-Coquard I, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–2428. doi: 10.1056/NEJMoa1911361. [DOI] [PubMed] [Google Scholar]
- 24.Coleman RL, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–2415. doi: 10.1056/NEJMoa1909707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golan T, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore K, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–2505. doi: 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 27.Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013;98(6):236–238. doi: 10.1136/archdischild-2013-304340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dummer R, et al. Five-year analysis of adjuvant dabrafenib plus trametinib in stage III melanoma. N Engl J Med. 2020;383(12):1139–1148. doi: 10.1056/NEJMoa2005493. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y-L, et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2020;383(18):1711–1723. doi: 10.1056/NEJMoa2027071. [DOI] [PubMed] [Google Scholar]
- 30.Waarts MR, et al. Targeting mutations in cancer. J Clin Invest. 2022;132(8):e154943. doi: 10.1172/JCI154943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markman M. Realistic goals of cancer therapy: effective and humane care. Cleve Clin J Med. 1994;61(6):468–472. doi: 10.3949/ccjm.61.6.468. [DOI] [PubMed] [Google Scholar]
- 32.Jain NM, et al. Framework for implementing and tracking a molecular tumor board at a National Cancer Institute-Designated Comprehensive Cancer Center. Oncologist. 2021;26(11):e1962–e1970. doi: 10.1002/onco.13936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamborero D, et al. Support systems to guide clinical decision-making in precision oncology: The Cancer Core Europe Molecular Tumor Board Portal. Nat Med. 2020;26(7):992–994. doi: 10.1038/s41591-020-0969-2. [DOI] [PubMed] [Google Scholar]
- 34.Johnson DB, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist. 2014;19(6):616–622. doi: 10.1634/theoncologist.2014-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mateo J, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT) Ann Oncol. 2018;29(9):1895–1902. doi: 10.1093/annonc/mdy263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Citron ML. Dose-dense chemotherapy: principles, clinical results and future perspectives. Breast Care (Basel) 2008;3(4):251–255. doi: 10.1159/000148914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paik S, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 38.Frei E., 3rd Curative cancer chemotherapy. Cancer Res. 1985;45(12 pt 1):6523–6537. [PubMed] [Google Scholar]
- 39. Paoletti C, Hayes DF. Circulating tumor cells. In: Stearns V, ed. Novel Biomarkers in the Continuum of Breast Cancer. Cham: Springer International Publishing; 2016:235–258. [Google Scholar]
- 40.Cohen JD, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson MH, et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat Commun. 2021;12(1):2357. doi: 10.1038/s41467-021-22444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parsons HA, et al. Individualized molecular analyses guide efforts (IMAGE): a prospective study of molecular profiling of tissue and blood in metastatic triple-negative breast cancer. Clin Cancer Res. 2016;23(2):379–386. doi: 10.1158/1078-0432.CCR-16-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Murillas I, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka N, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11(8):1913–1922. doi: 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mandel P, Metais P. Nuclear acids in human blood plasma. C R Seances Soc Biol Fil. 1948;142(3-4):241–243. [PubMed] [Google Scholar]
- 46.Stroun M, et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001;313(1–2):139–142. doi: 10.1016/s0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- 47.Diehl F, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med. 2019;25(3):439–447. doi: 10.1038/s41591-018-0334-x. [DOI] [PubMed] [Google Scholar]
- 49.Leon SA, et al. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37(3):646–650. [PubMed] [Google Scholar]
- 50.Christenson ES, et al. Single-nucleotide polymorphism leading to false allelic fraction by droplet digital PCR. Clin Chem. 2017;63(8):1370–1376. doi: 10.1373/clinchem.2017.273177. [DOI] [PubMed] [Google Scholar]
- 51.O’Leary B, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8(11):1390–1403. doi: 10.1158/2159-8290.CD-18-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boons G, et al. Longitudinal copy-number alteration analysis in plasma cell-free DNA of neuroendocrine neoplasms is a novel specific biomarker for diagnosis, prognosis, and follow-up. Clin Cancer Res. 2022;28(2):338–349. doi: 10.1158/1078-0432.CCR-21-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benhaim L, et al. Circulating tumor DNA is a prognostic marker of tumor recurrence in stage II and III colorectal cancer: multicentric, prospective cohort study (ALGECOLS) Eur J Cancer. 2021;159:24–33. doi: 10.1016/j.ejca.2021.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Torquato S, et al. Genetic alterations detected in cell-free DNA are associated with enzalutamide and abiraterone resistance in castration-resistant prostate cancer. JCO Precis Oncol. 2019;3:PO.18.00227. doi: 10.1200/PO.18.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Husain H, et al. Monitoring daily dynamics of early tumor response to targeted therapy by detecting circulating tumor DNA in urine. Clin Cancer Res. 2017;23(16):4716–4723. doi: 10.1158/1078-0432.CCR-17-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nabet BY, et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell. 2020;183(2):363–376. doi: 10.1016/j.cell.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Razavi P, et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat Cancer. 2020;1(4):382–393. doi: 10.1038/s43018-020-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quigley D, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7(9):999–1005. doi: 10.1158/2159-8290.CD-17-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikh AR, et al. Minimal residual disease detection using a plasma-only circulating tumor DNA assay in patients with colorectal cancer. Clin Cancer Res. 2021;27(20):5586–5594. doi: 10.1158/1078-0432.CCR-21-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beaver JA, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20(10):2643–2650. doi: 10.1158/1078-0432.CCR-13-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toro PV, et al. Comparison of cell stabilizing blood collection tubes for circulating plasma tumor DNA. Clin Biochem. 2015;48(15):993–998. doi: 10.1016/j.clinbiochem.2015.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kasi PM, et al. BESPOKE study protocol: a multicentre, prospective observational study to evaluate the impact of circulating tumour DNA guided therapy on patients with colorectal cancer. BMJ Open. 2021;11(9):e047831. doi: 10.1136/bmjopen-2020-047831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nadauld LD, et al. The PATHFINDER study: assessment of the implementation of an investigational multi-cancer early detection test into clinical practice. Cancers (Basel) 2021;13(14):3501. doi: 10.3390/cancers13143501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan KA, et al. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51(4):781–784. doi: 10.1373/clinchem.2004.046219. [DOI] [PubMed] [Google Scholar]
- 66.Jung M, et al. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin Chem. 2003;49(6):1028–1029. doi: 10.1373/49.6.1028. [DOI] [PubMed] [Google Scholar]
- 67.Lui YY, et al. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48(3):421–427. doi: 10.1093/clinchem/48.3.421. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A. 2015;112(31):9704–9709. doi: 10.1073/pnas.1511694112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Higgins MJ, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18(12):3462–3469. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong L, et al. Accurate quantification of supercoiled DNA by digital PCR. Sci Rep. 2016;6:24230. doi: 10.1038/srep24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johansson G, et al. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol Detect Quantif. 2019;17:100078. doi: 10.1016/j.bdq.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Warton K, et al. Evaluation of Streck BCT and PAXgene stabilised blood collection tubes for cell-free circulating DNA studies in plasma. Mol Diagn Ther. 2017;21(5):563–570. doi: 10.1007/s40291-017-0284-x. [DOI] [PubMed] [Google Scholar]
- 73.Pallisgaard N, et al. Controls to validate plasma samples for cell free DNA quantification. Clin Chim Acta. 2015;446:141–146. doi: 10.1016/j.cca.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 74.Meddeb R, et al. Quantifying circulating cell-free DNA in humans. Sci Rep. 2019;9(1):5220. doi: 10.1038/s41598-019-41593-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dressman D, et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100(15):8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Diehl F, et al. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3(7):551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 78.Chu D, et al. ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin Cancer Res. 2015;22(4):993–999. doi: 10.1158/1078-0432.CCR-15-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang P, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res. 2016;22(5):1130–1137. doi: 10.1158/1078-0432.CCR-15-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pender A, et al. Efficient genotyping of KRAS mutant non-small cell lung cancer using a multiplexed droplet digital PCR approach. PLoS One. 2015;10(9):e0139074. doi: 10.1371/journal.pone.0139074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hunter N, et al. TBCRC 040: Pathologic response evaluation and detection in circulating tumor DNA (PREDICT DNA): initial results piloting a tissue-biopsy independent method of identifying and monitoring tumor-specific mutations in early stage breast cancer. Cancer Res. 2020;80(4 suppl):P6-10-05 (abstract) doi: 10.1158/1538-7445.SABCS19-P6-10-05. [DOI] [Google Scholar]
- 82.Zhang JX, et al. A deep learning model for predicting next-generation sequencing depth from DNA sequence. Nat Commun. 2021;12(1):4387. doi: 10.1038/s41467-021-24497-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beaver JA, Park BH. Detecting plasma tumor DNA in early-stage breast cancer—reply. Clin Cancer Res. 2015;21(15):3570. doi: 10.1158/1078-0432.CCR-15-0994. [DOI] [PubMed] [Google Scholar]
- 84.Kinde I, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(23):9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chaudhuri AA, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7(12):1394–1403. doi: 10.1158/2159-8290.CD-17-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forshew T, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 87.Kennedy SR, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat Protoc. 2014;9(11):2586–2606. doi: 10.1038/nprot.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andre F, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–1940. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 89.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toy W, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robinson DR, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mardis ER. The impact of next-generation sequencing on cancer genomics: from discovery to clinic. Cold Spring Harb Perspect Med. 2019;9(9):a036269. doi: 10.1101/cshperspect.a036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marabelle A, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 95.Bonneville R, et al. Detection of microsatellite instability biomarkers via next-generation sequencing. Methods Mol Biol. 2020;2055:119–132. doi: 10.1007/978-1-4939-9773-2_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Georgiadis A, et al. Noninvasive detection of microsatellite instability and high tumor mutation burden in cancer patients treated with PD-1 blockade. Clin Cancer Res. 2019;25(23):7024–7034. doi: 10.1158/1078-0432.CCR-19-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Henry NL, Hayes DF. Uses and abuses of tumor markers in the diagnosis, monitoring, and treatment of primary and metastatic breast cancer. Oncologist. 2006;11(6):541–552. doi: 10.1634/theoncologist.11-6-541. [DOI] [PubMed] [Google Scholar]
- 98.Carr D, et al. All-cause mortality as the primary endpoint for the GRAIL/National Health Service England multi-cancer screening trial. J Med Screen. 2021;29(1):3–6. doi: 10.1177/09691413211059638. [DOI] [PubMed] [Google Scholar]
- 99.Smerage JB, et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol. 2014;32(31):3483–3489. doi: 10.1200/JCO.2014.56.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nadauld LD, et al. The PATHFINDER study: assessment of the implementation of an investigational multi-cancer early detection test into clinical practice. Cancers (Basel) 2021;13(14):3501. doi: 10.3390/cancers13143501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cristiano S, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mattox AK, et al. Bisulfite-converted duplexes for the strand-specific detection and quantification of rare mutations. Proc Natl Acad Sci U S A. 2017;114(18):4733–4738. doi: 10.1073/pnas.1701382114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nuzzo PV, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med. 2020;26(7):1041–1043. doi: 10.1038/s41591-020-0933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coombes RC, et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res. 2019;25(14):4255–4263. doi: 10.1158/1078-0432.CCR-18-3663. [DOI] [PubMed] [Google Scholar]
- 108.Mari R, et al. Liquid biopsies for ovarian carcinoma: how blood tests may improve the clinical management of a deadly disease. Cancers (Basel) 2019;11(6):774. doi: 10.3390/cancers11060774. [DOI] [PMC free article] [PubMed] [Google Scholar]