

Key Points
Long-term gilteritinib therapy sustained remission, leading to better long-term survival than chemotherapy in relapsed/refractory FLT3+ AML.
The safety profile of gilteritinib was stable over a 2-year period, with no new or clinically significant safety signals.
Visual Abstract

Abstract
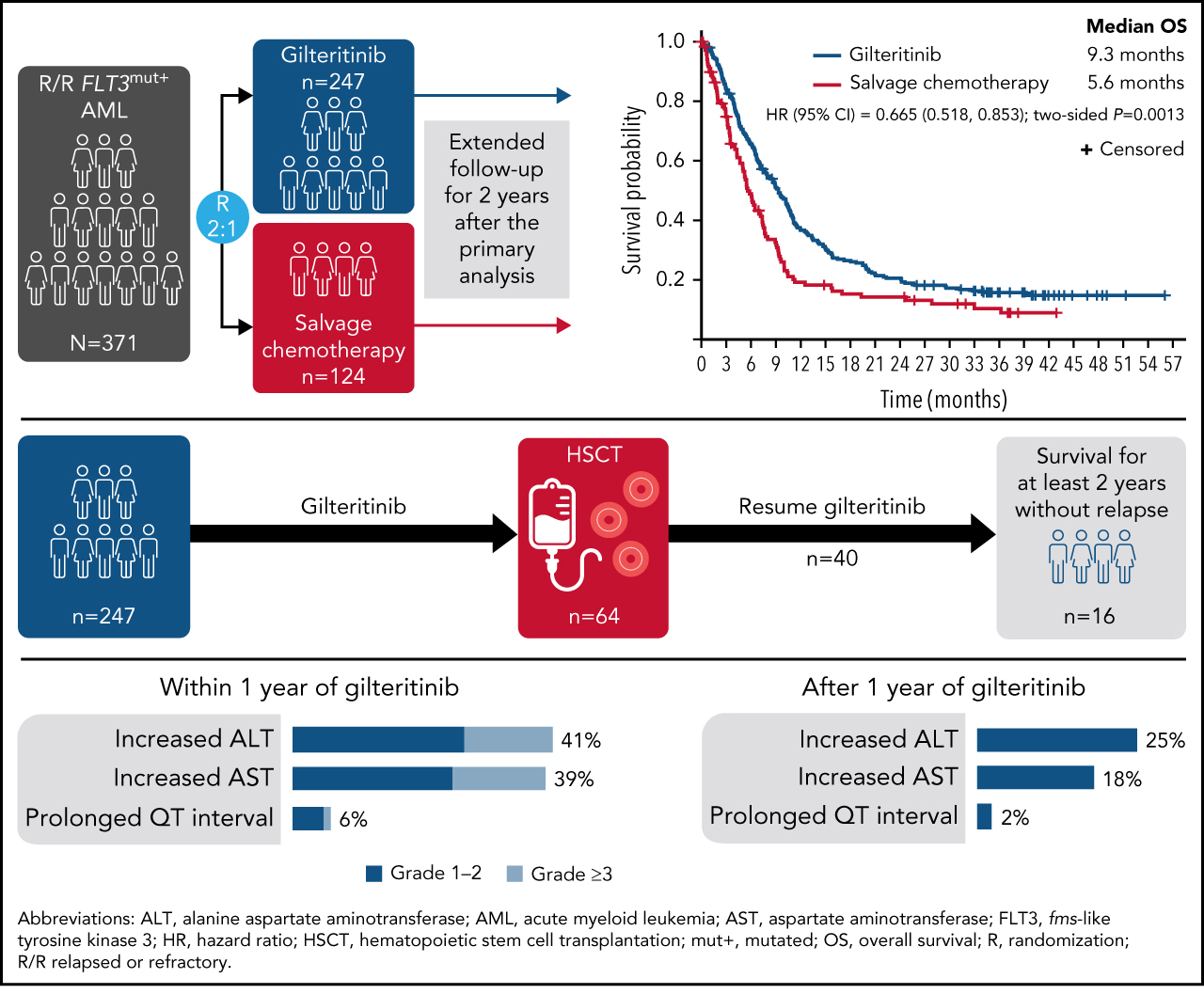
The phase 3 ADMIRAL (NCT02421939; Study ID: 2215-CL-0301) trial showed superior overall survival in patients with relapsed/refractory FLT3-mutation–positive acute myeloid leukemia (AML) randomized 2:1 to receive the oral FMS-like tyrosine kinase 3 inhibitor gilteritinib vs those randomized to receive salvage chemotherapy (SC). Here we provide a follow-up of the ADMIRAL trial 2 years after the primary analysis to clarify the long-term treatment effects and safety of gilteritinib in these patients with AML. At the time of this analysis, the median survival follow-up was 37.1 months, with deaths in 203 of 247 and 97 of 124 patients in the gilteritinib and SC arms, respectively; 16 gilteritinib-treated patients remained on treatment. The median overall survival for the gilteritinib and SC arms was 9.3 and 5.6 months, respectively (hazard ratio, 0.665; 95% confidence interval [CI], 0.518, 0.853; two-sided P = .0013); 2-year estimated survival rates were 20.6% (95% CI, 15.8, 26.0) and 14.2% (95% CI, 8.3, 21.6). The gilteritinib-arm 2-year cumulative incidence of relapse after composite complete remission was 75.7%, with few relapses occurring after 18 months. Overall, 49 of 247 patients in the gilteritinib arm and 14 of 124 patients in the SC arm were alive for ≥2 years. Twenty-six gilteritinib-treated patients remained alive for ≥2 years without relapse; 18 of these patients underwent transplantation (hematopoietic stem cell transplantation [HSCT]) and 16 restarted gilteritinib as post-HSCT maintenance therapy. The most common adverse events of interest during years 1 and 2 of gilteritinib therapy were increased liver transaminase levels; adverse event incidence decreased in year 2. Thus, continued and post-HSCT gilteritinib maintenance treatment sustained remission with a stable safety profile. These findings confirm that prolonged gilteritinib therapy is safe and is associated with superior survival vs SC. This trial was registered at www.clinicaltrials.gov as #NCT02421939.
Introduction
The emergence of targeted therapies has expanded treatment options for patients with newly diagnosed, relapsed or refractory (R/R) acute myeloid leukemia (AML).1-5 Compared with chemotherapy alone, agents that inhibit FMS-like tyrosine kinase 3 (FLT3) kinase activity, administered alone or in combination with chemotherapy, have improved survival and response rates in patients with AML.1,2,4,6,7
Gilteritinib is a FLT3 inhibitor approved for patients with R/R FLT3-mutation–positive (FLT3+) AML based on the phase 3, randomized, controlled ADMIRAL trial (NCT02421939; Study ID: 2215-CL-0301), which reported the superiority of gilteritinib over salvage chemotherapy (SC) in this patient population.1 Gilteritinib resulted in significantly longer median overall survival (OS; 9.3 months) compared with SC (5.6 months; hazard ratio for death [HR], 0.64; 95% confidence interval [CI], 0.49, 0.83; two-sided P < .001). In addition, the combined rates of complete remission and complete remission with partial hematologic recovery (CR/CRh) were higher with gilteritinib than with SC, at 34.0% and 15.3%, respectively. Although the survival benefit of gilteritinib was maintained in this trial when data were censored for transplantation, long-term survival among patients with R/R AML is generally limited to those who undergo allogeneic hematopoietic stem cell transplantation (HSCT). Even when accounting for the 2:1 randomization design of the ADMIRAL trial, a greater proportion of patients randomized to receive gilteritinib than those assigned to SC underwent allogeneic HSCT during the trial (gilteritinib, n = 63; SC, n = 19). Forty-nine of the 63 transplanted gilteritinib-treated patients received post-HSCT maintenance with gilteritinib. After adjustment for exposure duration, gilteritinib was associated with fewer grade ≥3 and serious adverse events (AEs) compared with SC.
The primary analysis of the ADMIRAL trial was initiated after a prespecified number of survival events to determine an improvement in median OS (ADMIRAL trial primary end point) with gilteritinib; however, a substantial fraction of the study population had a short follow-up duration at the time of primary analysis, thus limiting the evaluation of long-term survival in these patients. Furthermore, the safety profile associated with the continued use of gilteritinib beyond 1 year is not well described. As the use of gilteritinib and other FLT3 inhibitors becomes more prevalent in the treatment of AML, an evaluation of outcomes associated with continued use of FLT3 inhibitors beyond 1 year and the impact of FLT3 inhibitors as post-HSCT maintenance therapy is warranted.
We therefore performed a post hoc analysis of data from the ADMIRAL trial, 2 years after the primary analysis, to build on results from the primary analysis with respect to the long-term efficacy of gilteritinib, to characterize patients in the gilteritinib arm who remained alive for ≥2 years without relapse, and to evaluate the incidence of late-occurring AEs of interest associated with continued treatment with gilteritinib beyond 1 year.
Subjects and methods
Patient population
The ADMIRAL trial enrolled adult patients (aged ≥ 18 years) with confirmed FLT3+ AML who were in their first relapse after achieving CR with initial induction therapy or were refractory to initial induction therapy. Patients were randomly assigned 2:1 to receive gilteritinib 120 mg/d or preselected high- or low-intensity SC. Further details regarding the patient population and treatment administration have been described in the primary publication.1
The study was conducted in accordance with the principles of the Declaration of Helsinki.
Data analyses
The primary analysis of the ADMIRAL trial was triggered by a prespecified number of survival events that were sufficient to power a survival analysis with a power of 90% to detect a 35% improvement in median OS at a one-sided α value of 0.0245. At the time of primary analysis, median follow-up was 17.8 months. For this follow-up analysis, we performed a data cut on September 20, 2020, 2 years after the primary analysis of the ADMIRAL trial. Statistical modeling was not used to determine the timing of this analysis.
Assessments
Response and survival outcomes were assessed in patients who underwent HSCT during the ADMIRAL trial. Treatment response was assessed by using modified International Working Group criteria, as described in the primary publication.1,8 Complete definitions of treatment response parameters are presented in supplemental Table 1 (available on the Blood Web site). The complete composite remission (CRc) rate was defined as the sum of patients who achieved CR, CR with incomplete hematologic recovery, and CR with incomplete platelet recovery. FLT3 mutation status was assessed at enrollment (baseline) by a central laboratory using a polymerase chain reaction–based assay (LeukoStrat CDx; Invivoscribe Technologies, Inc.) according to published methods.9 The median FLT3–internal tandem duplication (ITD) allelic ratio (defined as the ratio of FLT3-ITD to wild-type FLT3 DNA) was established at 0.77 based on analyses in 335 evaluable patients; high and low FLT3-ITD allelic ratios were defined as ≥0.77 and <0.77, respectively. Next-generation sequencing was used to detect AML-associated mutations at enrollment, as previously described by using the ArcherDX myeloid panel.1
AEs were assessed and graded by using the Common Terminology Criteria for Adverse Events (version 4.03). Specific AEs of interest included cardiac AEs (eg, arrhythmia, cardiac failure, pericarditis/pericardial effusion); musculoskeletal AEs (eg, muscle damage or weakness); increased liver enzyme levels; gastrointestinal (GI) perforation, hemorrhage, or obstruction; pericarditis/pericardial effusion; and posterior reversible encephalopathy syndrome (PRES). The incidence of cutaneous squamous cell carcinoma and differentiation syndrome was also evaluated.
Statistical analyses
Descriptive statistics were used to assess continuous variables. Categorical data are reported as frequencies and percentages. Survival and response outcomes were assessed in the intention-to-treat population, which included all patients randomly assigned to treatment. Safety outcomes, including drug exposure, were assessed in the safety analysis set, defined as all patients who received ≥1 dose of the study drug. The Kaplan-Meier method and the Greenwood formula were used to estimate OS. The HR and CIs were used to determine differences in OS between groups. Because the statistical analysis plan did not include provisions for multiplicity correction with respect to evaluation of secondary outcomes or subgroup analyses, these results are reported as point estimates with 95% CIs. Statistical analyses were performed with SAS version 9.3 or higher software (SAS Institute, Inc.).
Results
Patient disposition and treatment exposure
As of the September 20, 2020, data cutoff date, there were 203 deaths (82%) in the gilteritinib arm and 97 deaths (78%) in the SC arm; 16 patients in the gilteritinib arm remained on treatment (Figure 1A). A total of 49 patients in the gilteritinib arm had been alive for ≥2 years. Overall, 26 of the 49 patients in the gilteritinib arm were living, without relapse, for ≥2 years; 16 of these patients remained on gilteritinib therapy (Figure 1B). The most common reasons for discontinuation of gilteritinib were lack of efficacy/AML progression/relapse (54%; n = 134 of 246), death (13%; n = 38 of 246, primarily due to AEs [11%; n = 27 of 246] or disease progression [3%; n = 7 of 246]), and AEs (13%; n = 31 of 246). Subsequent treatment with tyrosine kinase inhibitors, either alone or in combination with chemotherapy, was administered in 11 patients who were still in remission after achieving CRc (gilteritinib, n = 4; SC, n = 7). Patients in the gilteritinib arm subsequently received commercially available gilteritinib (n = 3) or quizartinib (n = 1). Patients in the SC arm subsequently received sorafenib only (n = 2), sorafenib and gilteritinib (n = 1), sorafenib with decitabine (n = 1), quizartinib (n = 1), midostaurin with azacitidine (n = 1), or another unknown/investigational FLT3 inhibitor (n = 1). At the time of data cutoff, 64 patients in the gilteritinib arm and 19 patients in the SC arm had undergone HSCT; in both arms, most patients who underwent transplantation had been preselected for high-intensity chemotherapy (Figure 1C). A total of 40 patients had resumed gilteritinib treatment post-HSCT.
Figure 1.


Patient disposition. (A) Disposition according to treatment received. (B) Disposition according to transplantation status: patients remaining alive for ≥2 years without relapse in the gilteritinib arm. (C) Disposition by preselected chemotherapy: patients remaining alive for ≥2 years without relapse in the gilteritinib arm. Due to limited follow-up of patients in the SC arm, data related to posttransplant relapse and survival were not available. ITT, intention-to-treat.
Among patients in the safety analysis set who had received at least one dose of gilteritinib, 19.5% (n = 48 of 246) had ≥12 months of gilteritinib exposure (supplemental Table 2), and 10.2% (n = 25 of 246) had ≥24 months of gilteritinib exposure. Median relative dose intensity ranged from 98% to 100% at exposure durations of <6 months to ≥12 months. The proportions of patients who required dose decreases or dose interruptions increased with exposure duration.
OS, duration of remission, and cumulative relapse rates
At the time of data cutoff, the median time to follow-up for OS was 37.1 months. The median OS in the gilteritinib and SC arms remained unchanged at 9.3 and 5.6 months, respectively (HR, 0.665; 95% CI, 0.518, 0.853; two-sided P = .0013) (Figure 2), which was expected based on the number of survival events recorded at the time of the primary analysis. Estimated survival at 1, 2, and 3 years was longer for each time point in the gilteritinib arm. With longer follow-up, the survival benefit of gilteritinib was maintained in the FLT3-ITD mutation subgroup (HR, 0.662; 95% CI, 0.511, 0.858) (supplemental Figure 1A) and in patients with a high FLT3-ITD allelic ratio (HR, 0.519; 95% CI, 0.363, 0.742) (supplemental Figure 1C). A survival benefit was not observed in the FLT3-TKD subgroup (HR, 0.684; 95% CI, 0.301, 1.552) (supplemental Figure 1B) or in patients with a low FLT3-ITD allelic ratio (HR, 0.821; 95% CI, 0.563, 1.197) (supplemental Figure 1D). The survival benefit of gilteritinib was also maintained in patients preselected for high-intensity chemotherapy (HR, 0.697; 95% CI, 0.507, 0.958) (supplemental Figure 1E) or low-intensity chemotherapy (HR, 0.584; 95% CI, 0.399, 0.857) (supplemental Figure 1F). However, given the small numbers of patients in these subgroups, these findings do not imply definitive treatment differences. In the gilteritinib arm, the median duration of CR was 23.0 months (interquartile range [IQR], 4.9 months-not evaluable); median durations of CRc and CR/CRh in the gilteritinib arm were 4.6 months (IQR, 1.9-24.0 months) and 10.0 months (IQR, 2.8 months-not evaluable), respectively.
Figure 2.

OS in patients with R/R FLT3+ AML (intention-to-treat population).
The 2-year cumulative relapse rates in gilteritinib-treated patients who achieved a best response of CR or CRc were 52.6% and 75.7%, respectively. For all postbaseline assessments, the cumulative incidence of relapse after a best response of CR and after a best response of CRc in patients treated with gilteritinib is shown (Figure 3A-B). The majority of relapses after CRc in the gilteritinib arm occurred within 12 months of enrollment, and relapses rarely occurred after 18 months from enrollment (Figure 3B). We also evaluated the cumulative incidence of relapse in the gilteritinib arm from the time of randomization and saw no marked change in relapse incidence (supplemental Figure 2). A meaningful assessment of cumulative relapse rates in the SC arm could not be performed because bone marrow samples were only collected up to the end of treatment, and nearly all patients in the SC arm had discontinued treatment after ≤2 treatment cycles.
Figure 3.

Cumulative incidence of relapse in patients with R/R FLT3+ AML in the gilteritinib arm. (A) Cumulative incidence of relapse after CR. (B) Cumulative incidence of relapse after CRc.
Outcomes in gilteritinib arm patients who were alive for ≥2 years without relapse
The median duration of follow-up was 39.5 months for the 26 gilteritinib-treated patients who were alive for ≥2 years without relapse. Baseline characteristics of these 26 patients are presented in supplemental Table 3. Eighteen of these patients underwent HSCT during the trial, and 16 resumed gilteritinib post-HSCT. Regardless of gilteritinib treatment duration, most patients who remained alive without relapse for ≥2 years were aged <65 years (84.6%; n = 22 of 26), preselected for high-intensity chemotherapy before randomization (76.9%; n = 20 of 26), and had not received prior treatment with midostaurin or sorafenib (96.1%; n = 25 of 26). An equal number of patients had high or low baseline FLT3-ITD allelic ratios (46.2%; both n = 12); 12 patients (46.2%) had baseline NPM1 mutations. All 8 gilteritinib-treated patients who did not undergo HSCT had relapsed disease at baseline, and almost all of these 8 patients (87.5%; n = 7 of 8) were aged <65 years and had undergone HSCT before study entry (supplemental Table 4). Most of these patients had also been preselected for high-intensity chemotherapy (62.5%; n = 5 of 8), had a low FLT3-ITD allelic ratio at baseline (62.5%; n = 5 of 8), and had NPM1 comutations (62.5%; n = 5 of 8).
Seven of the 26 patients had stopped gilteritinib therapy within 1 year of treatment initiation, and 19 had continued gilteritinib treatment beyond 1 year of treatment initiation (supplemental Table 3). Reasons for discontinuation of gilteritinib within 1 year were treatment-emergent AEs (n = 4; elevated alanine aminotransferase [ALT]/aspartate aminotransferase [AST] and blood alkaline phosphatase [n = 1]; pneumonia [n = 1], increased blood bilirubin [n = 1], and decreased platelet count [n = 1]), physician decision (n = 1), achievement of CRc after HSCT (n = 1), and progressive disease (n = 1). Six of the 7 patients who stopped gilteritinib within 1 year underwent HSCT, and 4 of these 6 patients resumed gilteritinib post-HSCT. Of the 19 patients who continued gilteritinib treatment beyond 1 year, 12 underwent HSCT, and all 12 resumed gilteritinib therapy post-HSCT. All 19 patients who continued gilteritinib therapy beyond 1 year achieved CR, and 2 of 7 patients who stopped gilteritinib therapy within 1 year achieved CR.
Post-HSCT gilteritinib maintenance therapy
Overall, 40 (62.5%) of the 64 gilteritinib-treated patients who underwent transplantation during the study received post-HSCT maintenance therapy with gilteritinib (median duration of exposure, 9.70 months; IQR, 1.69-28.3 months). Median time to initiation of post-HSCT gilteritinib maintenance therapy from the time of HSCT was 55 days (range, 32-91 days). Seven of the 40 patients who resumed gilteritinib after HSCT had dose decreases due to AEs (thrombocytopenia, pleural effusion, peripheral edema, weight gain, increased blood creatine phosphokinase, pleural thickening, increased ALT, decreased neutrophil count, and hypokalemia). One of these patients had a dose increase from 120-mg to 200-mg gilteritinib prior to HSCT before the dose decrease back to 120 mg immediately after HSCT due to the development of pleural effusion. Among patients in the gilteritinib arm who received post-HSCT gilteritinib maintenance therapy, cumulative 24-month relapse rates were 0% in patients whose response before HSCT was CR (n = 4) or CR/CRh (n = 9) and 18.6% for patients who had a pretransplant response of CRc (n = 20).
Other AML therapies in gilteritinib arm patients who underwent HSCT
Overall, 26 patients in the gilteritinib arm who underwent on-study HSCT received other AML therapies. Of the 64 gilteritinib-treated patients who underwent on-study HSCT, 24 did not resume gilteritinib after HSCT; 15 of these 24 patients received subsequent therapy with other agents, and 9 received HSCT as subsequent therapy. Among the 40 patients who resumed gilteritinib after on-study HSCT, 12 received other AML drug therapies after discontinuing gilteritinib. Of the 15 patients who did not resume gilteritinib after on-study HSCT, 5 received other AML therapy after HSCT. Overall, 80.7% (n = 21 of 26) of transplant patients in the gilteritinib arm relapsed before receiving other AML therapy; the median time from HSCT to initiation of other AML therapy in these patients was 3.5 months.
Other AML therapies (excluding HSCT) administered in transplant patients included high-intensity chemotherapy (46.2%; n = 12 of 26), low-intensity chemotherapy (34.6%; n = 9 of 26), and tyrosine kinase inhibitors (34.6%; n = 9 of 26 [sorafenib, 26.9%, n = 7* of 26; or midostaurin, 11.5%, n = 3* of 26; *includes one patient who received both sorafenib and midostaurin]). For the 24 patients who did not resume gilteritinib after HSCT, other AML drug therapies were administered in 10 patients after a relapse. Five of these patients received subsequent treatment with tyrosine kinase inhibitors: sorafenib alone (n = 1), sorafenib plus chemotherapy (n = 1), midostaurin plus chemotherapy (n = 1), midostaurin plus chemotherapy followed by sorafenib plus chemotherapy (n = 1), and gilteritinib (n = 1).
AEs of interest in patients treated with gilteritinib
The most common AEs of interest were increased levels of ALT and AST (Figure 4; supplemental Figure 3), which were the most frequent AEs of interest associated with gilteritinib during the first and second year of treatment. Most instances of increased ALT and AST levels were grade 1/2 in severity. Compared with the first year of gilteritinib therapy, the second year saw a decline in the incidence of these and other AEs of interest.
Figure 4.

AEs of interest during and after the first year of gilteritinib therapy in patients with R/R FLT3+ AML.
Fourteen (5.7%) of 246 gilteritinib-treated patients experienced an AE of interest occurring in the first year of gilteritinib therapy that persisted into the second year of treatment. Twelve patients experienced increased ALT/AST levels during both the first and second year of gilteritinib treatment; persistently increased levels of blood creatine phosphokinase (n = 4), blood bilirubin (n = 3), and gamma-glutamyl transferase (n = 2) were also observed. Five patients experienced myalgia and 2 experienced prolonged QT intervals during both years 1 and 2 of gilteritinib therapy.
AEs of interest leading to gilteritinib dose reductions occurred in 5.7% (n = 14 of 246) of patients; dose reductions occurred due to increased ALT level (n = 5), prolonged QT interval (n = 3), increased blood creatine phosphokinase level (n = 2), increased AST level (n = 1), increased blood bilirubin level (n = 1), syncope (n = 1), muscular weakness (n = 1), and cardiac failure (n = 1). Serious AEs of interest occurred in 20.3% (n = 50 of 246) of patients treated with gilteritinib; the most common were increased ALT level (n = 13), increased AST level (n = 10), syncope (n = 7), cardiac arrest (n = 4), cardiac failure (n = 3), myositis (n = 3), pericarditis (n = 3), and pericardial effusion (n = 3). Overall, 5.3% (n = 13 of 246) of patients discontinued gilteritinib because of AEs of interest. Specific AEs of interest leading to treatment discontinuation in >1 patient included increased ALT level (n = 5), increased AST level (n = 4), and cardiac arrest (n = 2). Increased blood bilirubin, increased blood creatine phosphokinase, duodenal perforation, large intestinal perforation, pericardial effusion, and pericarditis each led to discontinuation in individual patients. Overall, 37 gilteritinib-treated patients developed graft-versus-host disease (GVHD); 15 of these patients developed GVHD after resuming gilteritinib therapy. A total of 11 patients developed grade ≥3 GVHD, including 1 fatal case of intestinal acute GVHD.
Differentiation syndrome, PRES, and cutaneous squamous cell carcinoma were reported in 1 patient each in the gilteritinib arm. Differentiation syndrome and PRES occurred during the first year of treatment, and cutaneous squamous cell carcinoma occurred during the second year of treatment. Cardiac AEs of interest that occurred during the first year of treatment with gilteritinib included pericardial effusion (n = 10), pericarditis (n = 5), cardiac arrest (n = 4), cardiac failure (n = 4), congestive cardiac failure (n = 2), ventricular tachycardia (n = 2), and ventricular arrhythmia (n = 1); all 4 cases of cardiac arrest, 2 cases of pericardial effusion, and 1 case of congestive cardiac failure were fatal. All cases of cardiac arrest were unrelated to gilteritinib therapy. During the second year of gilteritinib therapy, cardiac AEs of interest included cardiorespiratory arrest and ventricular tachycardia (both, n = 1) and pericardial effusion (n = 1); none of these AEs was fatal. GI AEs of interest, namely GI perforation, hemorrhage, or obstruction during the first year of gilteritinib therapy, included GI hemorrhage (n = 3), lower GI hemorrhage (n = 2), intestinal perforation (n = 2), and rectal hemorrhage (n = 1); 2 cases of intestinal perforation were fatal. A case of impaired gastric emptying and a case of fatal duodenal perforation occurred during the second year of gilteritinib therapy. AEs of interest related to liver dysfunction during the first year of gilteritinib therapy included hyperbilirubinemia (n = 9; grade 3, n = 4), abnormal liver function (n = 5), and hepatomegaly (n = 2). During the second year of gilteritinib therapy, 2 cases of abnormal liver function (grade 3, n = 1) and 1 case of hepatosplenomegaly were observed.
During the first year of treatment with gilteritinib, 14.2% (n = 35 of 246) of patients experienced AEs of interest leading to dose reduction, with increased ALT (n = 5), neutropenia (n = 4), thrombocytopenia (n = 3), prolonged QT interval (n = 3), decreased neutrophil count (n = 3), and drug eruption (n = 2) being the most common. During the second year of treatment, 8% (n = 4 of 50) of patients experienced AEs of interest leading to dose reduction; these included peripheral edema, increased blood creatine phosphokinase, increased weight, hypokalemia, and pleural effusion (all, n = 1).
Discussion
This follow-up of the ADMIRAL trial confirms the survival benefit associated with gilteritinib previously reported at the time of primary analysis and shows that ∼11% of patients in the gilteritinib arm survived for at least 2 years in an ongoing remission. Most frequently, these patients were aged <65 years and underwent HSCT after initial gilteritinib therapy that led to CRc, followed by post-HSCT gilteritinib maintenance therapy. However, a smaller number who discontinued gilteritinib also remained in remission at 2 years. Estimated OS rates at 1, 2, and 3 years were higher in the gilteritinib arm than in the SC arm, although the small number of patients alive at 2 or 3 years limits validity of cross-arm comparisons. Although the primary analysis of the ADMIRAL trial reported superior OS with gilteritinib, relatively few patients had long-term follow-up, and the long-term survival trajectories for the gilteritinib and SC arms were likely to overlap with time. With a median follow-up of >3 years, our analysis suggests that gilteritinib may be associated with superior survival compared with SC for years after randomization. This finding provides the first evidence that an FLT3 inhibitor administered in the salvage setting can afford an ongoing and absolute survival benefit over SC in patients with R/R FLT3+ AML.
We previously reported that survival in ADMIRAL was superior with gilteritinib compared with SC, even after censoring for transplantation. With a longer follow-up, most (∼62%) gilteritinib arm patients remaining in initial remission after at least 2 years from randomization underwent HSCT while in CRc followed by gilteritinib maintenance. A detailed analysis of the role of HSCT, pretransplant response, and posttransplant maintenance with gilteritinib as a predictor of survival is beyond the scope of the current article. The small sample size and lack of randomization preclude any definitive statements regarding the utility of posttransplant gilteritinib or comparisons with no posttransplant maintenance therapy.
Administration of FLT3-targeted therapy in the posttransplant AML setting has been evaluated in studies of quizartinib10,11 or sorafenib.6,7,12,13 A consistent finding among these trials is prolonged protection from relapse and improvement in progression/disease-free survival or OS among patients who received these agents in the posttransplant setting.6,7,10-12 However, most patients in trials of posttransplant sorafenib were in first CR6,7,12 and therefore had a relatively lower relapse risk compared with the R/R AML population. Thus, caution is warranted when extrapolating findings from trials of posttransplant sorafenib therapy after first CR to those from studies of posttransplant FLT3 inhibitor therapy in R/R AML. In the current study, 2-year cumulative relapse rates were low in patients achieving CR before allogeneic HSCT. The benefit of post-HSCT gilteritinib maintenance in first remission is currently being assessed in an ongoing prospective randomized trial (#NCT02997202). However, for the aforementioned reasons, the risk–benefit ratio of gilteritinib maintenance therapy could be different in the context of first remission vs remission in the R/R AML setting, and the optimal duration of gilteritinib maintenance therapy may differ in these 2 scenarios.
The most common AEs of interest associated with gilteritinib were increased liver enzyme (ALT or AST) levels. Only one patient experienced differentiation syndrome, PRES, or secondary cutaneous malignancy. Because symptoms of differentiation syndrome can be nonspecific and might have been captured by other AEs (eg, fever, pulmonary infiltrate or pneumonia, peripheral edema, pleural effusion, and pericardial effusion), close monitoring for differentiation syndrome is warranted during gilteritinib therapy, including considerations for corticosteroid use. No new significant safety signals related to gilteritinib therapy emerged during the second year of treatment; however, a small risk remains for late-occurring cardiac events, liver dysfunction, and GI events. Compared with the first year of gilteritinib therapy, the overall incidence of AEs declined during the second year of treatment. Although many long-term gilteritinib-treated patients also received HSCT, an interaction between hepatic or GI AEs and HSCT with or without subsequent GVHD was not evaluated. AEs of interest occurred more frequently with gilteritinib compared with SC, but this does not necessarily suggest that gilteritinib was more toxic than SC, as the short duration of SC (1-2 cycles) confounds direct comparison during follow-up. Prior analysis from ADMIRAL showed that, when corrected for therapy duration, gilteritinib was associated with fewer toxicities overall and less frequent severe and treatment-related AEs than SC.1
The strength of any post hoc analysis is limited and may be subject to reporting bias. Analysis of survival across the treatment arms at defined time points was neither prespecified nor was the study powered to address this question. Other limitations of this analysis include the small number of patients evaluated for pretransplant and posttransplant outcomes and the lack of a second randomization and/or placebo group to quantify the benefits of posttransplant maintenance for either arm. There also could be imbalances with respect to transplant-preparative regimens, donors, and other factors that contribute to posttransplant survival, for which we did not control. Because adjustments for multiple comparisons for subgroup analyses were not made, the results herein should not be inferred as definitive treatment effects. A major reason for missing data are that the vast majority of patients in the SC arm discontinued within the first 2 treatment cycles, resulting in an insufficient number of bone marrow samples for assessment of remission status beyond cycle 2. As such, follow-up data related to the incidence of relapse, posttransplant outcomes, and maintenance therapy were not available for a considerable number of SC-treated patients. Finally, FLT3 mutation clearance before or after HSCT was not systematically evaluated, which, based on data from earlier studies of gilteritinib,14,15 likely had an impact on response and survival profiles.
In conclusion, patients with R/R FLT3+ AML in the ADMIRAL trial continue to benefit from long-term therapy with gilteritinib years after randomization, and an ongoing survival benefit may be afforded by gilteritinib therapy. Both continued gilteritinib treatment and post-HSCT maintenance therapy with gilteritinib seemed to sustain remission achieved with this drug or with HSCT. The safety profile of gilteritinib seems stable at 2 years, with no new clinically significant safety signals.
Supplementary Material
The online version of this article contains a data supplement.
{kind=link}
Acknowledgments
Medical writing/editorial support was provided by Kalpana Vijayan, Cheryl Casterline, and Elizabeth Hermans, of Peloton Advantage, LLC, an OPEN Health company, and funded by the study sponsor.
This study was funded by Astellas Pharma, Inc.
Footnotes
Researchers may request access to anonymized participant-level data, trial-level data, and protocols from Astellas-sponsored clinical trials at www.clinicalstudydatarequest.com. The Astellas criteria on data sharing may be accessed at https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.E.P., N. Hasabou, and M.J.L. contributed to the study design; and all authors contributed to the acquisition, analysis, and interpretation of study data; critically reviewed the manuscript; and provided final approval.
Conflict-of-interest disclosure: A.E.P. reports grants, personal fees, and nonfinancial support from Astellas, during the conduct of the study; grants, personal fees, and nonfinancial support from Daiichi Sankyo; grants and personal fees from AbbVie and Actinium Pharmaceuticals; personal fees from Agios, Loxo, LLS/Beat AML, and Forma; nonfinancial support from Arog; personal fees and nonfinancial support from New Link Genetics, Novartis, Takeda, and Jazz; and grants from Bayer and Biomed Valley Discoveries, outside the submitted work. R.A.L. reports consulting from Amgen, Novartis, Celgene, ARIAD, CVS Caremark, Astellas Pharma, DAVA Pharmaceuticals, and Epizyme; patents from UpToDate, Inc.; and research funding from Daiichi Sankyo, Celgene, Astellas Pharma, Novartis, Rafael Pharmaceuticals, Cellectis, and Forty Seven. N.A.P. reports consulting and advisory fees from Alexion, Pfizer, Celgene, Agios Pharmaceuticals, Blueprint Medicines, Incyte, Novartis, Bristol Myers Squibb, CTI BioPharma, PharmaEssentia, Constellation Pharmaceuticals, Cogent Biosciences, and AbbVie; and research funding granted to his institution from Pfizer, Celgene, CTI BioPharma, Boehringer Ingelheim, Astellas Pharma, Daiichi Sankyo, Sunesis Pharmaceuticals, Jazz Pharmaceuticals, Astex Pharmaceuticals, Genentech, AI Therapeutics, Samus Therapeutics, Arog Therapeutics, and Kartos Therapeutics. S.S. reports consulting or advisory fees from AbbVie, Astellas Pharma, Jazz Pharmaceuticals, Kite, a Gilead company, Novartis, and Pfizer; and research funding at an institutional level from AbbVie, Astellas Pharma, Inc., Celator/Jazz, Celgene, Daiichi Sankyo, Karyopharm Therapeutics, Menarini, Novartis, and Sunesis Pharmaceuticals. E.S.W. reports consulting fees from MacroGenics, PTC Therapeutics, Genentech, AbbVie, Jazz Pharmaceuticals, and BMS; and speakers bureau from Pfizer and Stemline. G.J.S. reports stock holdings for BMS, Pfizer, and J&J; research funding from Agios, Amgen, Incyte, Novartis, Celgene, AbbVie, Actinium, Ariad, Celator, Constellation, Cyclacel, Daiichi Sankyo, Deciphera, DeltaFly, BMS, Forma, Fujifilm, Gamida, Genentech-Roche, Geron, Jazz Pharmaceuticals, Karyopharm, Kite pharma, Mateon, Onconova, Pfizer, Regimmune, Samus, Sangamo, Tolero, and Trovagene; research grants from Astellas Pharma, Agios, Amgen, Incyte, Novartis, Celgene, AbbVie, Actinium, Ariad, Celator, Constellation, Cyclacel, Daiichi Sankyo, Deciphera, DeltaFly, BMS, Forma, Fujifilm, Gamida, Genentech-Roche, Geron, Jazz Pharmaceuticals, Karyopharm, Kite Pharma, Mateon, Onconova, Pfizer, Regimmune, Samus, Sangamo, Tolero, and Trovagene; speakers bureau from Incyte, Celgene, Sanofi, Gilead, and Stemline; and consultancy from Novartis, AstraZeneca, Amgen, Ono Pharma, and Kaiser Permanente. G.M. reports grant funding and consultancy fees from Amgen, Ariad, Incyte, Pfizer, Roche, Celgene, Janssen, AbbVie, and Novartis. J.S. reports personal fees from Astellas, Jazz Pharmaceuticals, AbbVie, Daiichi Sankyo, and Pfizer; and grants and personal fees from Novartis, outside the submitted work. P.M. reports research support from Pfizer, AbbVie, and Daiichi Sankyo; consultancy from Celgene, Pfizer, and AbbVie; and speakers bureau from Astellas, Novartis, and Janssen. C.R. reports research grants from AbbVie, Amgen, Novartis, Celgene, Jazz Pharmaceuticals, Agios, Chugai, MaatPharma, Astellas, Roche, and Daiichi Sankyo; and an advisory role for AbbVie, Sunesis, Janssen, Jazz Pharmaceuticals, Novartis, Celgene, Otsuka, Astellas, Daiichi Sankyo, MacroGenics, Roche, and Pfizer. S.-S.Y. reports consulting from Janssen, Takeda, Amgen, and Celgene/Jazz; honoraria from Novartis; and research funding from Kyowa Kirin, Roche/Genentech, and Yuhan. S.C. reports subsidy from Astellas, Ono Pharmaceutical Co., Kyowa-Kirin, Takeda, Sanofi, Chugai Pharmaceutical Co., and BMS. H.-J.K. reports honoraria from AbbVie; honoraria and consultancy from Amgen, AML Hub, Astellas, Celgene, Daiichi Sankyo, Novartis, SL VaxiGen, Yuhan, Janssen; and grants from BL&H, outside the submitted work. Q.L., N. Hasabou, and R.T. are employees of Astellas. M.J.L. reports grants and personal fees from Astellas and Fujifilm; and personal fees from Daiichi Sankyo, Amgen, and Menarini. The remaining authors declare no competing financial interests.
*Correspondence: Alexander E. Perl, 12-154 South Tower, Perelman Center for Advanced Medicine, Division of Hematology/Oncology, Perelman School of Medicine at the University of Pennsylvania, 3400 Civic Center Blvd, Philadelphia, PA 19104; e-mail: alexander.perl@pennmedicine.upenn.edu.
REFERENCES
- 1.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728-1740. [DOI] [PubMed] [Google Scholar]
- 2.Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984-997. [DOI] [PubMed] [Google Scholar]
- 3.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. [DOI] [PubMed] [Google Scholar]
- 4.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38(26):2993-3002. [DOI] [PubMed] [Google Scholar]
- 7.Xuan L, Wang Y, Huang F, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020;21(9):1201-1212. [DOI] [PubMed] [Google Scholar]
- 8.Cheson BD, Bennett JM, Kopecky KJ, et al. ; International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia . Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia [published correction appears in J Clin Oncol. 2004;22(3):576]. J Clin Oncol. 2003;21(24):4642-4649. [DOI] [PubMed] [Google Scholar]
- 9.Murphy KM, Levis M, Hafez MJ, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5(2):96-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganguly S, Cortes JE, Krämer A, et al. Clinical outcomes in patients with FLT3-ITD-mutated relapsed/refractory acute myelogenous leukemia undergoing hematopoietic stem cell transplantation after quizartinib or salvage chemotherapy in the QuANTUM-R trial. Transplant Cell Ther. 2021;27(2):153-162. [DOI] [PubMed] [Google Scholar]
- 11.Sandmaier BM, Khaled S, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93(2): 222-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunner AM, Li S, Fathi AT, et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol. 2016;175(3):496-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tschan-Plessl A, Halter JP, Heim D, Medinger M, Passweg JR, Gerull S. Synergistic effect of sorafenib and cGvHD in patients with high-risk FLT3-ITD+AML allows long-term disease control after allogeneic transplantation. Ann Hematol. 2015;94(11):1899-1905. [DOI] [PubMed] [Google Scholar]
- 14.Altman JK, Perl AE, Hill JE, Rosales M, Bahceci E, Levis MJ. The impact of FLT3 mutation clearance and treatment response after gilteritinib therapy on overall survival in patients with FLT3 mutation-positive relapsed/refractory acute myeloid leukemia. Cancer Med. 2021;10(3):797-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.