

Abstract
Ribonucleotide reductase (RNR) catalyzes the reduction of ribonucleotides to deoxyribonucleotides and is critical for DNA synthesis and repair in all organisms. Its mechanism requires radical transfer along a ~32 Å pathway through a series of proton-coupled electron transfer (PCET) steps. Previous simulations suggested that a glutamate residue (E623) mediates the PCET reaction between two stacked tyrosine residues (Y730 and Y731) through a proton relay mechanism. This work focuses on the adjacent PCET reaction between Y730 and a cysteine residue (C439). Quantum mechanical/molecular mechanical free energy simulations illustrate that when Y730 and Y731 are stacked, E623 stabilizes the radical on C439 through hydrogen bonding with the Y730 hydroxyl group. When Y731 is flipped away from Y730, a water molecule stabilizes the radical on C439 through hydrogen bonding with Y730 and lowers the free energy barrier for radical transfer from Y730 to C439 through electrostatic interactions with the transferring hydrogen but does not directly accept the proton. These simulations indicate that the conformational motions and electrostatic interactions of the tyrosines, cysteine, glutamate, and water strongly impact the thermodynamics and kinetics of these two coupled PCET reactions. Such insights are important for protein engineering efforts aimed at altering radical transfer in RNR.
Graphical Abstract
Introduction
Ribonucleotide reductase (RNR) plays an essential role in DNA synthesis and repair by catalyzing the reduction of ribonucleotides to deoxyribonucleotides1–3 and is a drug target for many cancers.4–5 The active form of class I RNR is a complex composed of two homodimers, denoted α2β2 (Figure 1). A 32 Å pathway of reversible proton-coupled electron transfer (PCET) reactions spanning the α/β subunits generates transient tyrosyl and thiyl radicals that eventually lead to the ribonucleotide reduction reaction.6 On the basis of various experimental studies, the radical transfer pathway has been proposed to involve Y122β ↔ [W48β] ↔ Y356β ↔ Y731α ↔ Y730α ↔ C439α (Figure 1).3, 6 A recently solved cryo-EM structure7 of the α2β2 complex has further elucidated the PCET pathway. Analysis of this structure indicates that α and β are in a pre-turnover state, with an intact PCET pathway, whereas α′ and β′ are in a post-turnover state.
Figure 1.
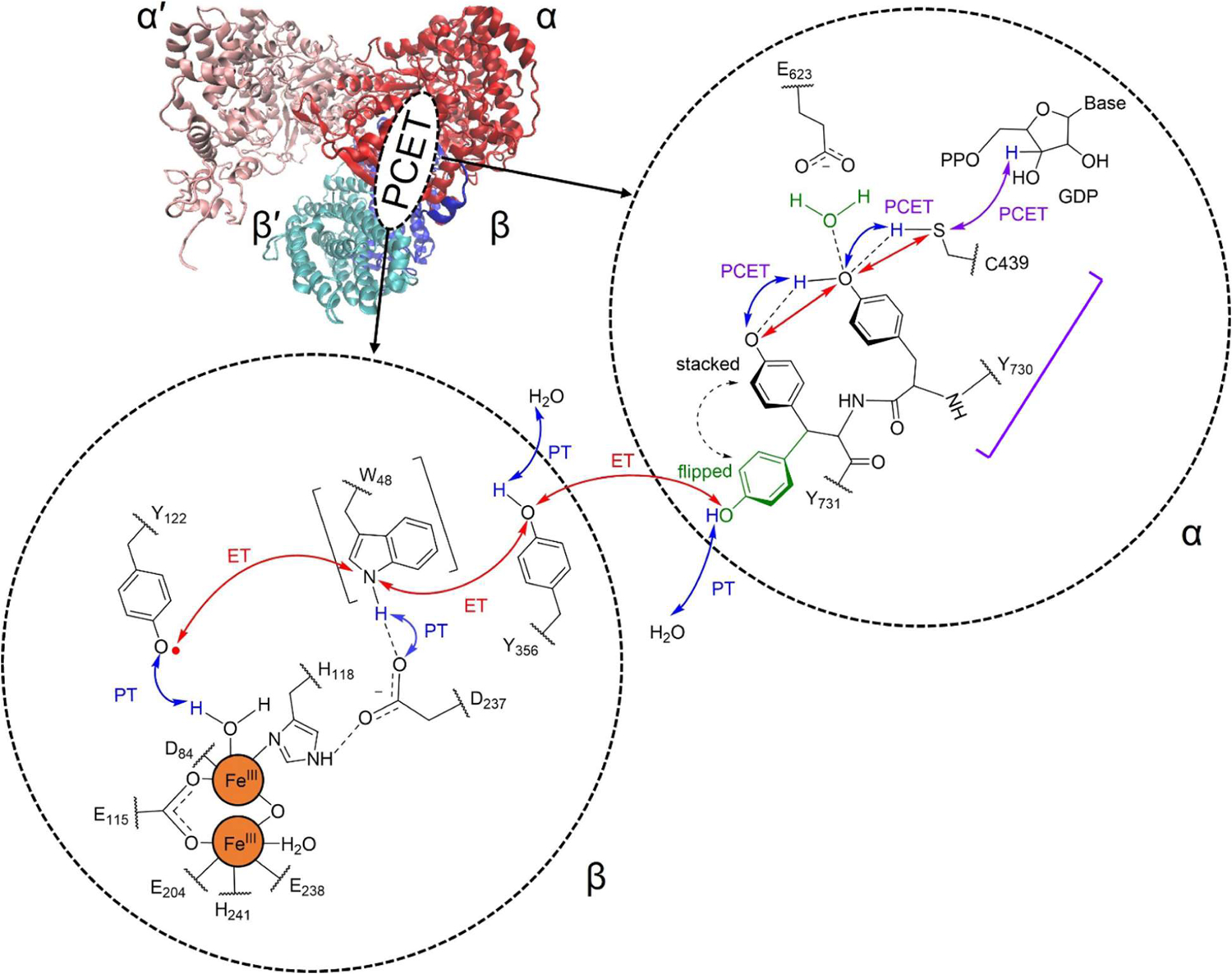
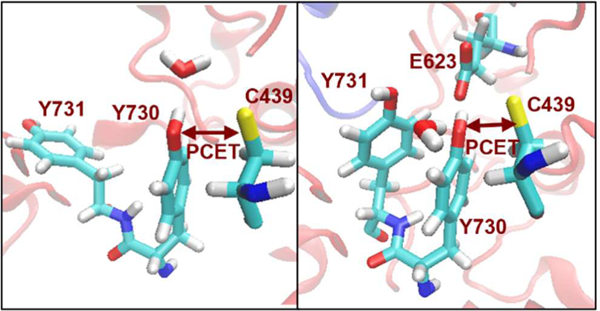
Cryo-EM structure of the α2β2 complex (left) and ordered PCET pathway (right), where ET and PT denote electron transfer and proton transfer, respectively. The participation of W48 is uncertain and therefore is shown in square brackets. Y731 has been shown computationally and experimentally to sample both flipped-out (green) and stacked (black) conformations. The PCET reaction between Y730 and C439 is indicated with a purple square bracket.
The cryo-EM structure of the active complex has motivated theoretical studies of the PCET pathway in RNR. Our previous molecular dynamics (MD) simulations8 based on this structure illustrated that Y731α, which is located at the interface of the α/β subunits, is conformationally dynamic, consistent with prior spectroscopic studies.9–10 The two dominant conformations correspond to a flipped conformation, in which Y731 is pointing toward the β subunit, and a stacked conformation, in which Y731 is stacked with Y730. Free energy simulations suggested that the stacked and flipped conformations can interconvert at room temperature.8 Moreover, quantum mechanical/molecular mechanical (QM/MM) free energy simulations of the PCET reaction between Y731α and Y730α in the stacked conformation indicated that the nearby E623α mediates this PCET reaction via a proton relay mechanism that involves double proton transfer between the tyrosine residues and E623.11
The goal of the current work is to characterize the adjacent PCET reaction between Y730 and C439. Previous theoretical studies have investigated the role of water in the PCET reaction between Y730 and C439,12–13 as well as between Y731 and Y730.14 ENDOR spectroscopy experiments and accompanying DFT geometry optimizations of active site cluster models indicated that a water molecule is hydrogen bonded to the amino-substituted Y730 tyrosyl radical.12, 15–16 DFT calculations suggested that this water molecule slightly decreases the endothermicity of radical transfer from Y730 to C439 but slightly increases the reaction energy barrier.12 Moreover, DFT calculations on small model systems in the gas phase and QM/MM geometry optimizations in the protein environment using a crystal structure of an isolated NH2Y730-subsituted α subunit17 suggested the possibility of a PCET mechanism involving double proton transfer from cysteine to tyrosine through an intervening water molecule.13 However, these QM/MM calculations were not based on the cryo-EM structure of the active complex and did not include conformational sampling of the protein or entropic contributions. Furthermore, all of these previous DFT studies only considered the stacked conformation of Y731. Nevertheless, these previous studies are valuable in that they predict a water molecule near Y730.
Herein, we investigate the PCET reaction between Y730 and C439 using QM/MM free energy simulations based on the cryo-EM structure of the active complex. These simulations consider the impact of the stacked and flipped Y731 conformations on this PCET reaction. Moreover, we also examine the influence of a nearby water molecule to determine if it mediates this PCET reaction via a proton relay mechanism, analogous to E623 for PCET between Y731 and Y730. Our calculations indicate that this water molecule strongly influences the free energy barrier and reaction free energy for PCET between Y730 and C439 through electrostatic and hydrogen-bonding interactions but does not directly accept the proton.
Methods
We use QM/MM finite temperature string simulations with umbrella sampling18–19 to investigate the PCET reaction between Y730 and C439. This PCET reaction is characterized by the following three reaction coordinates at the proton transfer interface: the distance between the C439 sulfur atom and the transferring hydrogen, the distance between the Y730 oxygen atom and the transferring hydrogen, and the sulfur-oxygen distance (Figure S1). In some cases, a fourth reaction coordinate, defined as the distance between a nearby water oxygen atom and the transferring hydrogen, is also included.
For each system studied, an initial string composed of a series of 20 images was generated to connect approximate reactant and product structures. Each image was equilibrated on the QM/MM potential energy surface by performing 10 ps of MD for the MM region with the QM region frozen followed by at least 100 fs MD for the full system with the reaction coordinates harmonically restrained. After equilibration, the string was evolved using an iterative procedure, where each iteration entailed umbrella sampling for each image with harmonic restraints on the reaction coordinates determined by their average values from the previous iteration. All strings were fully converged according to the specified criteria (Figures S10 and S11). After each string was converged, the minimum free energy path (MFEP) and multidimensional free energy surfaces were computed using all sampling data with the weighted histogram analysis method (WHAM).20–21 Computational details are provided in the SI.
The system preparation and simulation details were adapted from our previous studies of RNR.8, 11 All simulations started with the cryo-EM structure of the α2β2 E.coli RNR complex (PDB ID: 6W4X),7 solvated by explicit solvent molecules. Most of the strings simulated radical transfer between Y730 and C439 in the α subunit, corresponding to the pre-turnover state with the intact PCET pathway, although two strings simulated this reaction in the α′ subunit, corresponding to the post-turnover state for comparison.
For all strings, the QM region included Y731, Y730, and C439. Some of the strings also included a nearby water molecule or nearby residues Q349 or E623. The QM region was described by the ωB97X-D functional22 and the 6–31+G** basis set.23–25 This level of theory has been shown to be consistent with the complete active space self-consistent field with second-order perturbative corrections (CASSCF+NEVPT2) method for calculations on small model systems composed of cysteine and tyrosine (Figure S2). The MM region was described with the AMBER ff14SB force field.26–29 The QM/MM free energy simulations were performed with the AMBER/Q-Chem interface.30–32
As mentioned above, we validated the level of theory for the QM region by benchmarking against ab initio multireference calculations and ensured that all of the strings were fully converged, as supported by plots in the SI. In addition, we propagated two independent strings with flipped Y731 and the same QM region using three and four reaction coordinates, respectively, and qualitatively different initial strings. The agreement between the reaction free energies and free energy barriers for these converged strings illustrates the reproducibility of this approach. Similarly, we simulated analogous strings in the α and α′ subunits and observed consistent reaction free energies and free energy barriers, providing further validation for this approach. Nevertheless, the size of the QM region and the extent of conformational sampling are limited due to the computational expense of these simulations. Moreover, these simulations rely in the initial cryo-EM structure and do not account for relatively slow conformational changes that may be relevant to the radical transfer process.
Results and Discussion
We investigated both flipped and stacked Y731 conformations in our simulations of radical transfer from Y730 to C439. Previous spectroscopic experiments9–10, 33 and our recent simulations8 suggest that this interconversion occurs on the nanosecond or faster timescale. Specifically, the free energy barriers for interconversion were estimated to be in the range of ~2 – 5 kcal/mol, and the relative free energies of the flipped and stacked Y731 conformations were estimated to be in the range of 0 – 4 kcal/mol. These experimental and computational results suggest that this conformational transition is faster than radical transfer and that both conformations could be populated at equilibrium at room temperature.8, 10 Thus, although Y731 and Y730 are expected to be stacked during radical transfer from Y731 to Y730, subsequently Y731 could flip out prior to radical transfer from Y730 to C439. For this reason, we considered both the stacked and flipped conformations of Y731.
For flipped Y731, E623 moved ~6–7 Å away from Y730 during equilibration, allowing a water molecule to remain in the region near both Y730 and C439, although it remained closer to Y730 throughout the PCET reaction. Given the potential importance of this water molecule, we included it in the QM region for two of the strings propagated in the flipped conformation but also propagated a third string without including water in the QM region for comparison. The two strings with water in the QM region were propagated with three and four reaction coordinates, respectively. As discussed in the Methods, the three reaction coordinates correspond to the distance between the Y730 oxygen and C439 sulfur as well as the distances between each of these atoms and the transferring hydrogen. The fourth reaction coordinate corresponds to the distance between the water oxygen and the transferring proton.
The fourth reaction coordinate was included to test the hypothesis that water could mediate PCET between Y730 and C439 through a double proton transfer mechanism. In the initial string, we restrained the proton to transfer to water, generating H3O+, as it transfers between C439 and Y730. To stabilize this initial string, each image was equilibrated with the reaction coordinates harmonically restrained according to the procedure described in Methods. During the subsequent iterative procedure, however, the string gradually evolved away from this initial string so that the proton no longer transferred to the water molecule and instead transferred directly from C439 to Y730 (Figure S4). The reaction free energy and free energy barrier associated with the resulting converged string are nearly identical to the free energies obtained with only three reaction coordinates (Table 1). This agreement between two independent strings with different numbers of reaction coordinates and qualitatively different initial strings provides validation for the approach and the results.
Table 1.
Computed Reaction Free Energies (ΔG) and Free Energy Barriers (ΔG‡) for PCET Corresponding to Radical Transfer from Y730 to C439.
The free energy surface and MFEP for the string with flipped Y731 and water in the QM region, generated with four reaction coordinates, is shown in Figure 2A. Analysis of the key distances along the MFEP illustrates that the water molecule moves closer to the proton as it transfers to Y730 and forms a relatively strong hydrogen bond with the hydroxyl group on Y730 in the product (Figure 3A). This water molecule serves as a weak hydrogen bond donor with the radical oxygen on Y730 (~3.2 Å O-O distance, angle of 125°, Figure 4A) for the reactant and serves as a stronger hydrogen bond acceptor with the hydroxyl group on Y730 (~2.8 Å O-O distance, angle of 157°, Figure 4B) for the product. This movement and reorientation of the water molecule also occurs when the water molecule is not in the QM region (Figure S5, but a QM treatment of this water molecule includes the electronic polarization necessary to accurately describe the hydrogen-bonding interactions between the water molecule and Y730. The QM treatment of the water lowers the reaction free energy and free energy barrier by ~12 kcal/mol and 3.5 kcal/mol, respectively (Table 1). Further analysis indicates that this difference in reaction free energy can be attributed in part to the polarization of the QM water by the charges in the MM region and the associated impact on the interaction energies (Table S2). Other factors, such as differences in conformational sampling, may also contribute to this effect. Similar behavior was observed for two strings propagated in the α′ subunit with flipped Y731 (Table 1, Figure S8). For these independent strings in a different subunit, the reaction also became significantly more exoergic when the water was in the QM region, providing validation for this observation.
Figure 2.
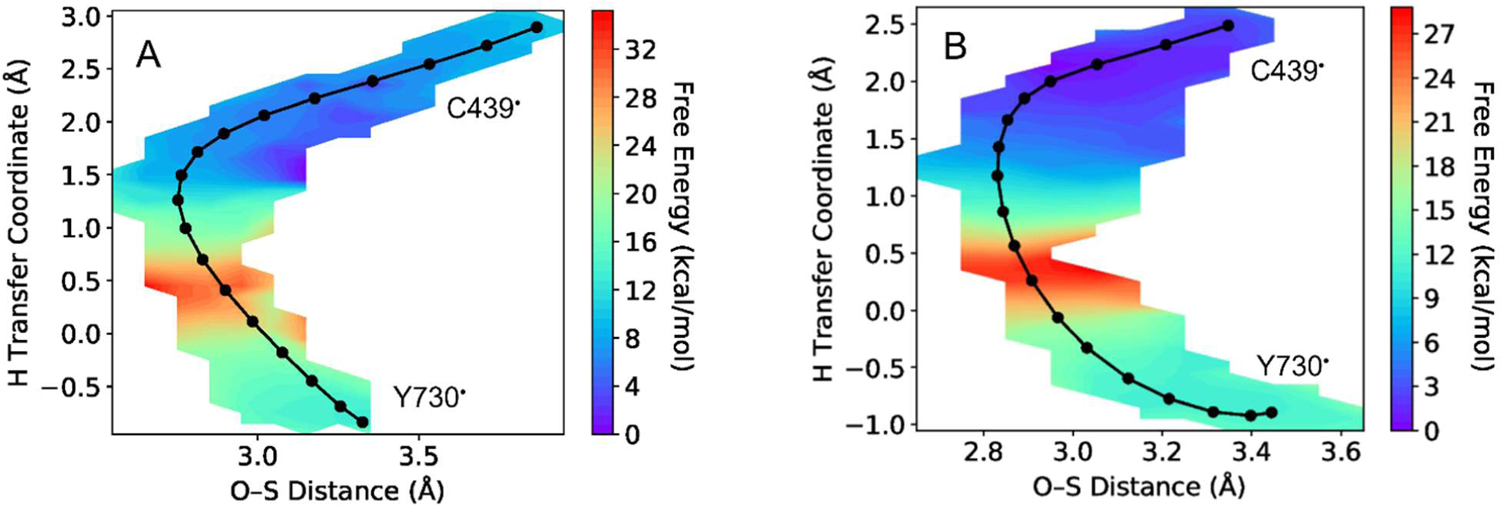
Two-dimensional free energy surfaces for the PCET reaction associated with radical transfer from Y730α to C439α for strings with (A) flipped Y731 and (B) stacked Y731. Both strings have Y731, Y730, C439, Q349, and water in the QM region, as well as E623 for the stacked string, and use four reaction coordinates. The y-axis is the hydrogen transfer coordinate, defined as the difference between the C439 sulfur-hydrogen and Y730 oxygen-hydrogen distances, and the x-axis is the distance between the Y730 oxygen and C439 sulfur. The MFEP is indicated by the black curve. Note that only 18 images rather than 20 images are shown in part A because the first two images were redundant. In part B, the data from the first three iterations of the string was not included to ensure adequate equilibration; this removal changed the reaction free energy and free energy barrier by less than 0.2 kcal/mol.
Figure 3.
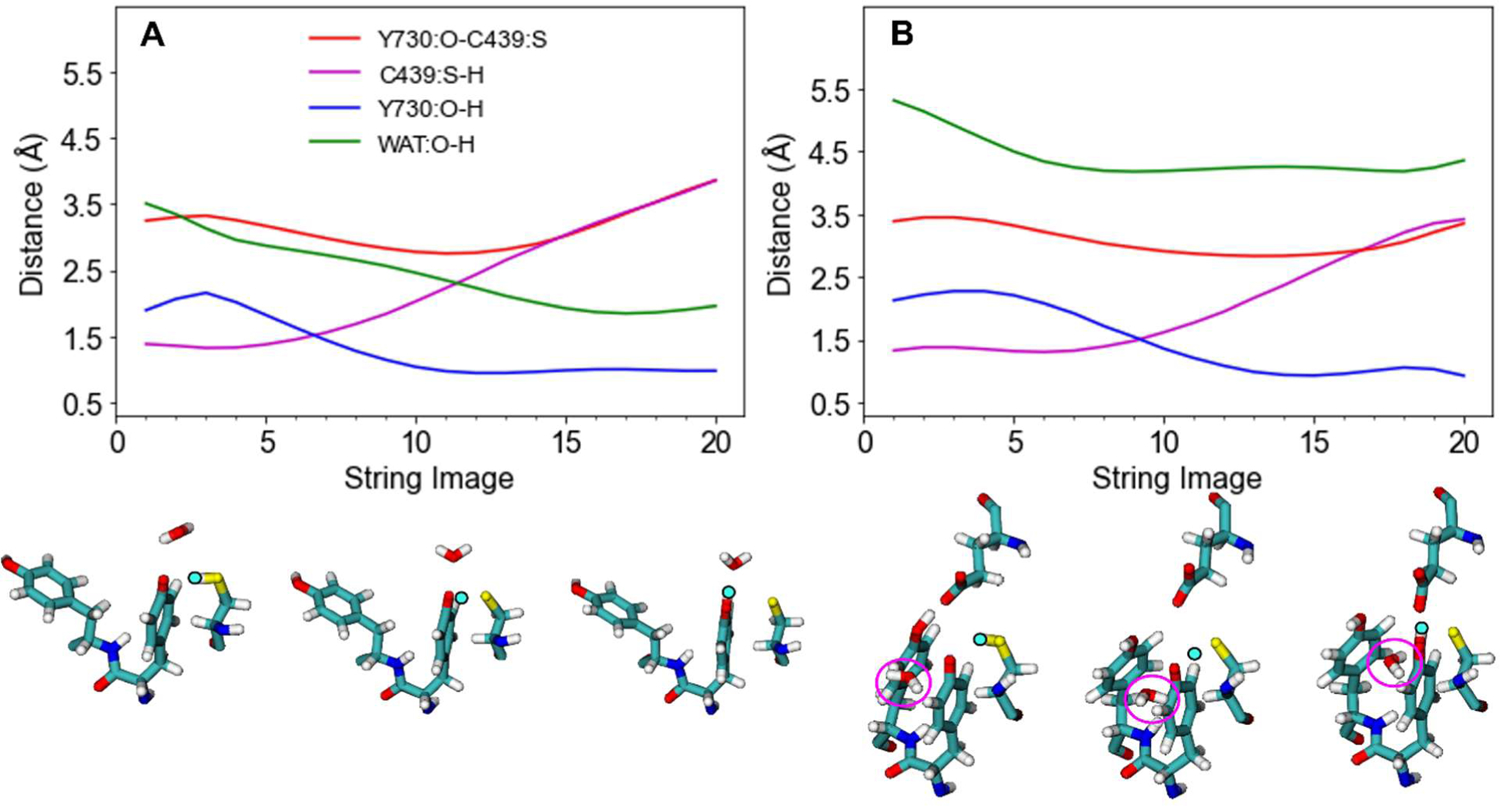
Average distances between the Y730 oxygen and C439 sulfur, as well as between the transferring hydrogen and the C439 sulfur, Y730 oxygen, and water oxygen. These distances were obtained from the final iteration of the converged string with (A) flipped Y731 or (B) stacked Y731. Both strings have Y731, Y730, C439, Q349, and water in the QM region, as well as E623 for the stacked string, and use four reaction coordinates. Configurations corresponding to the reactant, top of the barrier, and product with the transferring hydrogen shown in bright cyan and water circled in magenta for part (B) are also shown.
Figure 4.
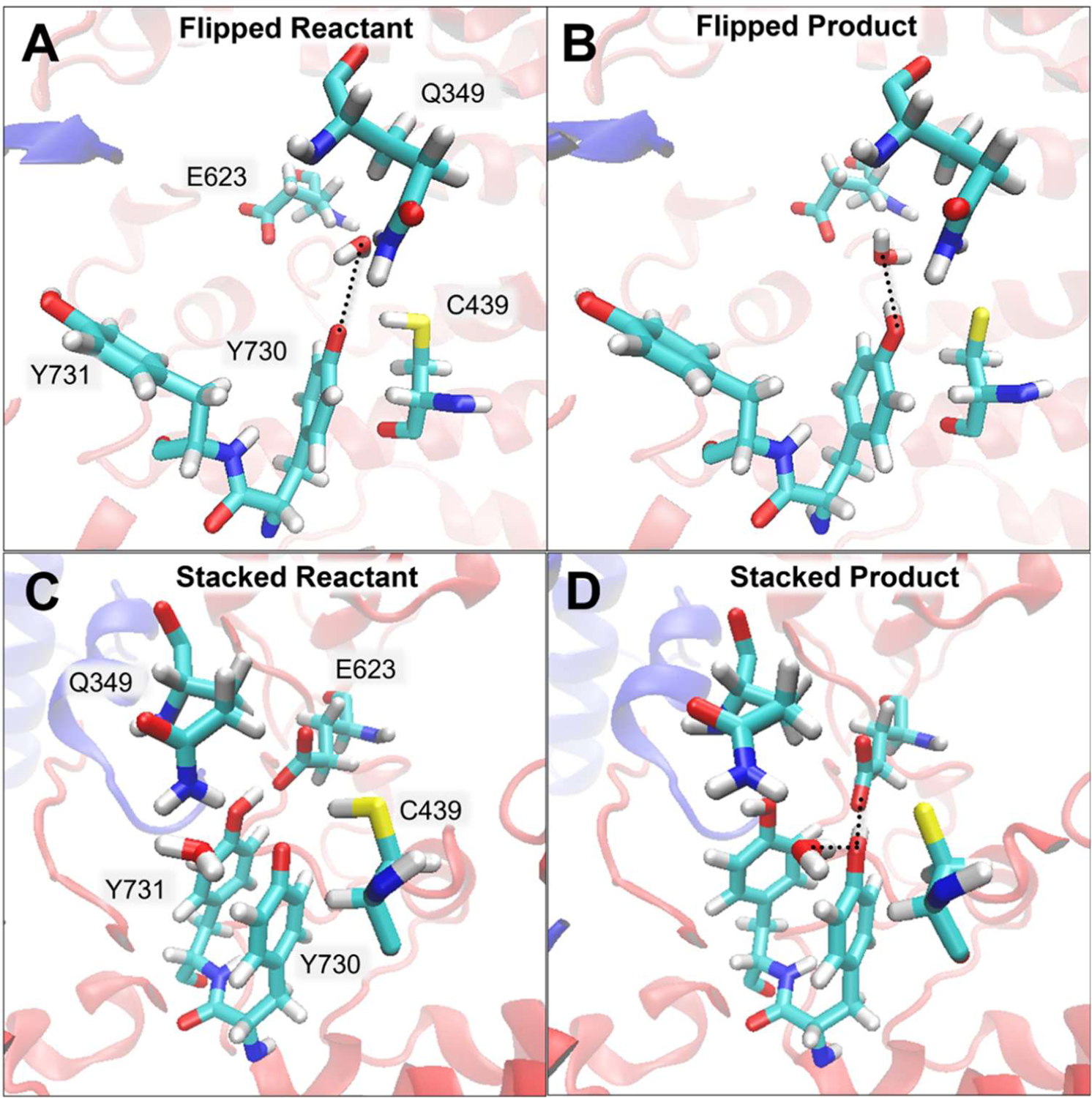
Reactants (A and C) and products (B and D) of configurations with flipped Y731 (A and B) and stacked Y731 (C and D) for radical transfer from Y730 to C439 in the α subunit. These configurations were obtained from the converged strings with Y731, Y730, C439, Q349, and water in the QM region, as well as E623 for the stacked string, using four reaction coordinates. Analyses of the hydrogen bonds, labeled with dashed black lines, are provided in Table S3.
When Y731 is stacked with Y730, the region around Y730 is more crowded and E623 is close to both tyrosine residues. In this case, water is prevented from interacting strongly with Y730 (Table S3). Given this structural complexity, we explored three different types of strings in the stacked conformation: (1) with water interacting on the E623 side of Y730, (2) without water interacting directly with Y730, and (3) with water interacting on the Q349 side of Y730. Introducing water on the E623 side of Y730, where it was located for the strings with the flipped Y731 conformation (Figure 4A), disrupts the stacking interaction and causes E623 and Q349 to move apart. We propagated such a string with only Q349 added to the QM region and obtained similar thermodynamics as obtained for flipped Y731 when only Q349 is added to the QM region (Table 1, Figure S3A and S6A,B).
The string for the stacked Y731 conformation in the absence of nearby water was propagated with both Q349 and E623 in the QM region. In this case, the radical transfer from Y730 to C439 becomes exoergic by 5 kcal/mol (Table 1, Figure S3B and S6C,D). This effect is qualitatively similar to the effect of treating the water molecule quantum mechanically for flipped Y731. In this case, E623 forms a strong hydrogen bond with the hydroxyl group of Y730 (~2.8 Å O-O distance, angle of 169°) to stabilize the product. The free energy barrier is not significantly influenced when E623 is treated quantum mechanically for stacked Y731, however, in contrast to the lowering of this free energy barrier when the water is treated quantum mechanically for flipped Y731. This difference arises because water is able to shift its position and stabilize the configurations at the top of the barrier through electrostatic interactions with the transferring proton (middle configuration in lower part of Figure 3A), whereas E623 remains too far away to enable this stabilization (Table S3).
The string for the stacked Y731 conformation with water introduced on the Q349 side was propagated with Q349, E623, and the water molecule in the QM region (Figure 4C,D). In the initial string, water was restrained to remain close to Y730 (~3.0 Å O-O distance), followed by equilibration of all images with this restraint using the procedure described in Methods. As the string evolved, this water moved away from the Y730 radical in the reactant (Figure 4C). In the product (Figure 4D), this water serves as a weak hydrogen bond donor with Y730 (~3.3 Å O-O distance, angle of 109°), while E623 serves as a strong hydrogen bond acceptor with Y730 (~2.6 Å O-O distance, angle of 135°). These hydrogen-bonding interactions significantly stabilize the product (Table S3). Although the water does not interact strongly with the transferring proton (Figure 3B), the free energy barrier is lower for this string, most likely due to enhanced hydrogen-bonding interactions among the water, Q349, D334, and Y413 (Figure S7). The converged string has a similar free energy barrier (14.8 kcal/mol) and reaction free energy (−9.4 kcal/mol) as the strings with flipped Y731α and a QM water (Table 1).
In all of these strings, E623 was deprotonated on the basis of its pKa value of ~4.2,34 although the protein environment is expected to modulate this value to some extent. Our previous simulations of PCET between Y731 and Y730 indicated that E623 mediates proton transfer by a proton relay mechanism in which it accepts and then donates the proton.11 In the present simulations of PCET between Y730 and C439, however, E623 is not close enough to the region between Y730 and C439 to mediate proton transfer. Even when E623 was included in the QM region for the strings with stacked Y731, E623 never accepted the proton during the PCET reaction between Y730 and C439. In contrast, the deprotonated E623 always accepted the proton in our previous simulations of the PCET reaction between Y731 and Y730.11 On the other hand, in the present simulations of radical transfer from Y730 to C439 with stacked Y731, E623 remained close enough to Y730 to hydrogen bond to Y730 when the radical was on C439. These simulations investigated the two PCET steps independently, but we cannot exclude a concerted Y731 to Y730 to C439 radical transfer mechanism, which could be studied in the future.
Analysis of the spin densities of both the flipped and stacked Y731 conformations indicates that the electron and proton move concertedly during radical transfer from Y730 to C439. Figure 5 illustrates that the radical is localized on Y730 when the proton is bonded to C439 in the reactant and is localized on C439 when the proton is bonded to Y730 in the product. For configurations near the top of the barrier along the MFEP, the radical is delocalized over both Y730 and C439, and the proton is approximately at the midpoint between the Y730 oxygen and the C439 sulfur.
Figure 5.
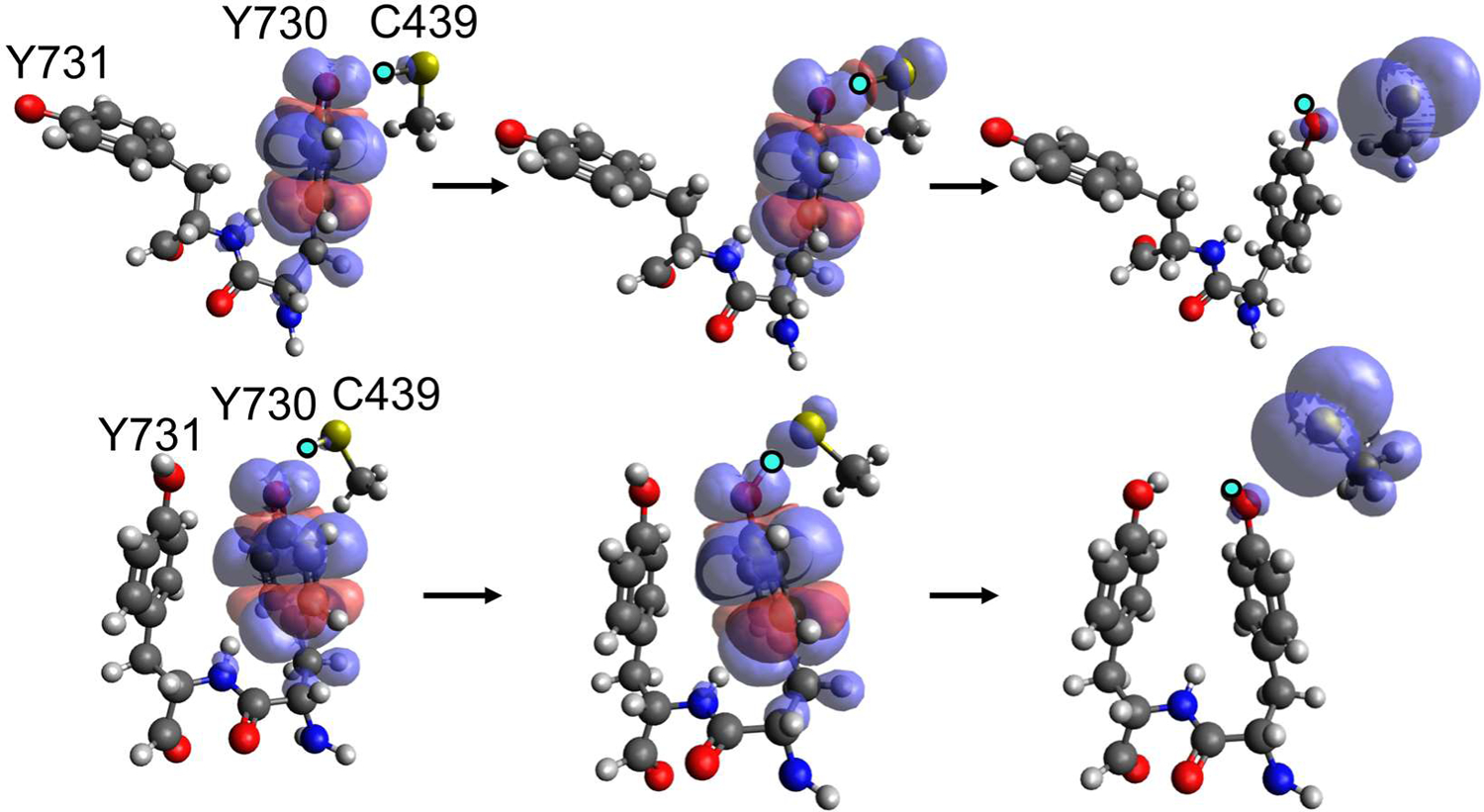
Spin densities computed for representative configurations along the MFEP from the converged strings for flipped (top) and stacked (bottom) Y731. The configurations correspond approximately to the reactant (left), top of the barrier (middle), and product (right). The spin densities were calculated with the complete active space self-consistent field (CASSCF) method in the gas phase using an (11e, 10o) active space and the 6–31+G** basis set. The transferring proton is shown in bright cyan. The analogous figure computed with DFT/ωB97X-D is given in Figure S9.
Previous studies combining ENDOR spectroscopy with DFT calculations suggested that water could hydrogen bond to the 3-aminotyrosine radical.12, 16 According to these studies, water hydrogen bonds to the 3-aminotyrosine radical from the Q349 side, which is opposite the amino group.16 In our simulations, water hydrogen bonds to the Y730 radical in the flipped Y731 conformation on the E623 side and does not hydrogen bond to the Y730 radical in the stacked Y731 conformation. These differences between our simulations and the previous studies could arise from several factors. The main factor is that the aminotyrosine used in both the experiments and DFT calculations of the previous studies12, 16 could have a different hydrogen-bonding environment than the canonical tyrosine used in our simulations. Moreover, the DFT calculations used to interpret the ENDOR data were based on either a structure of RNR with the aminotyrosine substitution12 or an isolated amino tyrosyl,16 whereas our simulations are based on the cryo-EM structure7 without this substitution. In a separate combined ENDOR and DFT study,15 water was found to hydrogen bond to Y730 when the radical is on Y731. This experimental finding is consistent with our simulations that show Y730 hydrogen bonding to water when the radical is on C439 for both the stacked and flipped conformations.
Previous experimental and theoretical studies also examined other properties relevant to radical transfer from Y730 to C439. Spectroscopic experiments with selenocysteine substituted at position 439 in RNR did not directly observe the selenocysteine radical or the formation of deoxynucleotide product.35 However, the observation of thiyl radicals is challenging due to high reactivity and broadening of the EPR signal.36 In an earlier study, electrochemical experiments on glutathione, which contains a cysteine residue,37 were interpreted to suggest that radical transfer from tyrosine to cysteine is approximately isoergic or slightly endoergic, but such model systems may not be representative of the RNR environment. Previous DFT calculations found that this radical transfer is slightly endoergic (~3 kcal/mol),12 but these calculations used small cluster models based on an earlier crystal structure38 without the intact PCET pathway and did not include entropic effects.
The QM/MM free energy simulations presented herein are based on the α/β subunits of the cryo-EM structure with a fully intact PCET pathway considered to be in the pre-turnover state.7 This structure is expected to favor forward radical transfer, and conformational changes could occur during radical transfer that are not accessible on the simulation timescales. For these reasons, the finding that radical transfer from Y730 to C439 is exoergic may only be valid in this region of conformational space. On the other hand, the qualitative conclusions, such as the role of water, are expected to be more broadly applicable.
Conclusion
We performed QM/MM free energy simulations of the radical transfer reaction from Y730 to C439. Our results show that the hydrogen-bonding interaction between a water molecule and Y730 plays an important role in lowering the free energy barrier and enhancing the exoergicity. The nearby E623 residue plays a similar but less pronounced role in stabilizing the product through hydrogen-bonding interactions with Y730 when Y731 is stacked with Y730. Understanding each PCET step along the pathway is critical for eventual simulation of the entire radical transfer process in this biochemically important enzyme. Furthermore, this study suggests that mutation of E623 will not only directly impact radical transfer between Y731 and Y730, but also may indirectly impact radical transfer between Y730 and C439. Such fundamental insights are relevant to drug design efforts.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant Number R35 GM139449. This work utilized the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562).39 This work used resources on Expanse at the San Diego Supercomputer Center through allocation TG-MCB120097. C.R.R was supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE1752134 and in part by the National Institutes of Health (5T32GM06754 3–12). We thank Alexander Soudackov for technical assistance and helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website. Simulation details, computational benchmarking data, additional string analysis (PDF)
REFERENCES
- (1).Stubbe J; van Der Donk WA, Protein Radicals in Enzyme Catalysis. Chem. Rev 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (2).Stubbe J; Nocera DG; Yee CS; Chang MCY, Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev 2003, 103, 2167–2202. [DOI] [PubMed] [Google Scholar]
- (3).Minnihan EC; Nocera DG; Stubbe J, Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Aye Y; Li M; Long M; Weiss R, Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [DOI] [PubMed] [Google Scholar]
- (5).Misko TA; Liu YT; Harris ME; Oleinick NL; Pink J; Lee HY; Dealwis CG, Structure-guided design of anti-cancer ribonucleotide reductase inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry 2019, 34, 438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Greene BL; Kang G; Cui C; Bennati M; Nocera DG; Drennan CL; Stubbe J, Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem 2020, 89, 45–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kang G; Taguchi AT; Stubbe J; Drennan CL, Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Reinhardt CR; Li P; Kang G; Stubbe J; Drennan CL; Hammes-Schiffer S, Conformational Motions and Water Networks at the α/β Interface in E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 2020, 142, 13768–13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kasanmascheff M; Lee W; Nick TU; Stubbe J; Bennati M, Radical transfer in E coli ribonucleotide reductase: a NH2Y731/R411A-α mutant unmasks a new conformation of the pathway residue 731. Chem. Sci 2016, 7, 2170–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Greene BL; Taguchi AT; Stubbe J; Nocera DG, Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc 2017, 139, 16657–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Reinhardt CR; Sayfutyarova ER; Zhong J; S., H.-S., Glutamate Mediates Proton-Coupled Electron Transfer Between Tyrosines 730 and 731 in Escherichia coli Ribonucleotide Reductase. J. Am. Chem. Soc 2021, 143, 6054–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Argirević T; Riplinger C; Stubbe J; Neese F; Bennati M, ENDOR Spectroscopy and DFT Calculations: Evidence for the Hydrogen-Bond Network Within α2 in the PCET of E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 2012, 134, 17661–17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen X; Ma G; Sun W; Dai H; Xiao D; Zhang Y; Qin X; Liu Y; Bu Y, Water Promoting Electron Hole Transport between Tyrosine and Cysteine in Proteins via a Special Mechanism: Double Proton Coupled Electron Transfer. J. Am. Chem. Soc 2014, 136, 4515–4524. [DOI] [PubMed] [Google Scholar]
- (14).Kaila VRI; Hummer G, Energetics of Direct and Water-Mediated Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2011, 133, 19040–19043. [DOI] [PubMed] [Google Scholar]
- (15).Nick TU; Lee W; Koßmann S; Neese F; Stubbe J; Bennati M, Hydrogen Bond Network between Amino Acid Radical Intermediates on the Proton-Coupled Electron Transfer Pathway of E. coli α2 Ribonucleotide Reductase. J. Am. Chem. Soc 2015, 137, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hecker F; S. J. M., Detection of Water Molecules on the Radical Transfer Pathway of Ribonucleotide Reductase by 17O Electron–Nuclear Double Resonance Spectroscopy. J. Am. Chem. Soc 2021, 143, 7237–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Minnihan EC; Seyedsayamdost MR; Uhlin U; Stubbe J, Kinetics of Radical Intermediate Formation and Deoxynucleotide Production in 3-Aminotyrosine-Substituted Escherichia coli Ribonucleotide Reductases. J. Am. Chem. Soc 2011, 133, 9430–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rosta E; Nowotny M; Yang W; Hummer G, Catalytic mechanism of RNA backbone cleavage by ribonuclease H from quantum mechanics/molecular mechanics simulations. J. Am. Chem. Soc 2011, 133, 8934–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ganguly A; Thaplyal P; Rosta E; Bevilacqua PC; Hammes-Schiffer S, Quantum mechanical/molecular mechanical free energy simulations of the self-cleavage reaction in the hepatitis delta virus ribozyme. J. Am. Chem. Soc 2014, 136, 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Souaille M; Roux B. t., Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun 2001, 135, 40–57. [Google Scholar]
- (21).Grossfield A. WHAM: the weighted histogram analysis method., WHAM version 2.0.9.
- (22).Mardirossian N; Head-Gordon M, omegaB97X-V: a 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys 2014, 16, 9904–24. [DOI] [PubMed] [Google Scholar]
- (23).Hehre WJ; Ditchfield R; Pople JA, Self-consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- (24).Hariharan PC; Pople JA, The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- (25).Clark T; Chandrasekhar J; Spitznagel GW; Schleyer PVR, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3–21+G basis set for first-row elements, Li–F. J. Comput. Chem 1983, 4, 294–301. [Google Scholar]
- (26).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA, A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (27).Cheatham TE; Cieplak P; Kollman PA, A Modified Version of the Cornell et al. Force Field with Improved Sugar Pucker Phases and Helical Repeat. J. Biomol. Struct. Dyn 1999, 16, 845–862. [DOI] [PubMed] [Google Scholar]
- (28).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C, Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gotz AW; Clark MA; Walker RC, An extensible interface for QM/MM molecular dynamics simulations with AMBER. J. Comput. Chem 2014, 35, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Shao Y; Gan Z; Epifanovsky E; Gilbert AT; Wormit M; Kussmann J; Lange AW; Behn A; Deng J; Feng X; Ghosh D; Goldey MB; Horn PR; Jacobson L; Kaliman I; Khaliullin RZ; Kus T; Landau A; Liu J; Proynov EI; Rhee YM; Richard RM; Rohrdanz MA; Steele RP; Sundstrom E; Woodcock HL; Zimmerman PM; Zuev D; Albrecht BJ; Alguire EC; Austin B; Beran GJ; Bernard YA; Berquist EJ; Brandhorst K; Bravaya KB; Brown ST; Casanova D; Chang C; Chen Y; Chien S; Closser KD; Crittenden DL; Diedenhofen M; Distasio RA; Do H; Dutoi AD; Edgar RG; Fatehi S; Fusti-Molnar L; Ghysels A; Golubeva-Zadorozhnaya A; Gomes J; Hanson-Heine MW; Harbach PH; Hauser AW; Hohenstein EG; Holden ZC; Jagau T; Ji H; Kaduk BJ; Khistyaev K; Kim J; Kim J; King RA; Klunzinger PE; Kosenkov D; Kowalczyk T; Krauter CM; Lao KU; Laurent AD; Lawler KV; Levchenko SV; Lin CY; Liu F; Livshits E; Lochan RC; Luenser A; Manohar P; Manzer SF; Mao S; Mardirossian N; Marenich AV; Maurer SA; Mayhall NJ; Neuscamman E; Oana CM; Olivares-Amaya R; O’Neill DP; Parkhill JA; Perrine TM; Peverati R; Prociuk A; Rehn DR; Rosta E; Russ NJ; Sharada SM; Sharma S; Small DW; Sodt AJ; Stein T; Stück D; Su Y; Thom AJ; Tsuchimochi T; Vanovschi V; Vogt L; Vydrov OA; Wang T; Watson M; Wenzel J; White AF; Williams CF; Yang J; Yeganeh S; Yost S; You Z; Zhang IY; Zhang X; Zhao Y; Brooks BR; Chan GK; Chipman DM; Cramer CJ; Goddard WA; Gordon MS; Hehre WJ; Klamt A; Schaefer HF; Schmidt MW; Sherrill CD; Truhlar DG; Warshel A; Xu X; Aspuru-Guzik A; Baer R; Bell AT; Besley NA; Chai J; Dreuw A; Dunietz BD; Furlani TR; Gwaltney SR; Hsu C; Jung Y; Kong J; Lambrecht DS; Liang W; Ochsenfeld C; Rassolov VA; Slipchenko LV; Subotnik JE; Van Voorhis T; Herbert JM; Krylov AI; Gill PM; Head‐Gordon M, Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Molecular Physics 2015, 113, 184–215. [Google Scholar]
- (33).Ravichandran K; Minnihan EC; Lin Q; Yokoyama K; Taguchi AT; Shao J; Nocera DG; Stubbe J, Glutamate 350 Plays an Essential Role in Conformational Gating of Long-Range Radical Transport in Escherichia coli Class Ia Ribonucleotide Reductase. Biochemistry 2017, 56, 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lide DR, CRC Handbook of Chemistry and Physics. Internet Version 2005 ed; CRC Press: Boca Raton, FL, 2005. [Google Scholar]
- (35).Greene BL; Stubbe J; Nocera DG, Selenocysteine Substitution in a Class I Ribonucleotide Reductase. Biochemistry 2019, 58, 5074–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lassmann G; Kolberg M; Bleifuss G; Gräslund A; Sjöberg B-M; Lubitz W, Protein thiyl radicals in disordered systems: A comparative EPR study at low temperature. Phys. Chem. Chem. Phys 2003, 5, 2442–2453. [Google Scholar]
- (37).Madej E; Wardman P, The Oxidizing Power of the Glutathione Thiyl Radical as Measured by its Electrode Potential at Physiological pH. Arch. Biochem. Biophys 2007, 462, 94–102. [DOI] [PubMed] [Google Scholar]
- (38).Eriksson M; Uhlin U; Ramaswamy S; Ekberg M; Regnström K; Sjöberg B-M; Eklund H, Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure 1997, 5, 1077. [DOI] [PubMed] [Google Scholar]
- (39).Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD, XSEDE: accelerating scientific discovery. Comput. Sci. Eng 2014, 16, 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.