

Graphical abstract
Keywords: Immunoliposomes, Transcutaneous immunization, Microneedle, Langerhans cell targeting, Th1-biased immune response
Abstract
Novel Coronavirus is affecting human's life globally and vaccines are one of the most effective ways to combat the epidemic. Transcutaneous immunization based on microneedle (MN) has attracted much attention because of its painlessness, rapidity, high efficiency and good compliance. In this study, CD11c monoclonal antibody-immunoliposomes (OVA@CD11c-ILP) actively targeting to Langerhans cells (LCs) were successfully prepared and were delivered by the microchannels of skin produced by MN to induce an immune response in vivo. OVA@CD11c-ILP could be targeted to LCs by conjugating CD11c monoclonal antibody to the surface of the ILP. OVA@CD11c-ILP promoted the maturation of dendritic cells (DCs) and the uptake and endocytosis of antigen by LCs. Moreover, OVA@CD11c-ILP immunization can significantly inhibit tumor growth and prolong overall survival. Furthermore, a higher antibody’s titer ratio of IgG1/IgG2a indicated that the immune response stimulated by this immunization method was Th1-biased and the liposomes showed Th1-type adjuvant effect. In conclusion, the combination delivery system of immunoliposomes and microneedle can significantly improve the efficiency of antigen presentation and effectively activate cellular immune responses in the body, which is expected to be a promising transdermal immune strategy.
1. Introduction
At the end of 2019, a global pneumonia caused by a novel coronavirus began to sweep the world, led to millions of deaths, and brought huge losses to many countries. On February 10, 2020, the WHO named the disease as COVID-19 and the novel virus as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. Vaccination is one of the most effective ways to contain epidemics and ensure the health of the population [2], [3], [4]. However, the main approach of vaccination is subcutaneous or intramuscular injection, which has some disadvantages [5], [6], such as high injection requirements, easy infection, poor compliance, and limitations of its application in mass vaccination. The workload of vaccination and the probability of side effects, such as infections, is increased. Hence, it has always been the hot issue to search for better compliance, more convenient new vaccine delivery system in pharmaceutics and immunology community.
The skin is the largest immune organ in the human body and is also one of the main routes of stimulating immune response in vivo by external pathogens under natural conditions [7]. Human skin is composed of three layers: the stratum corneum (10–15 μm), the active epidermal layer (50–100 μm) and the dermis. The stratum corneum mainly composed of dense keratinocytes is the foremost obstacle to drug delivery while the epidermis and the dermis contain a large number of antigen-presenting cells (APCs), nerve tissues and vascular tissues [8], [9], [10]. APCs in the skin mainly include dendritic cells (DCs) and immature dendritic cells subtype (Langerhans cells, LCs), and foreign antigens can be effectively uptake, processed and presented by these cells [11]. Furthermore, APCs in the skin effectively trigger the antigen-specific cellular and humoral immune response after moving into draining lymph node. Among APCs, the density of LCs in human skin is about 500–1000 cells/mm2, accounting for 2–3% of epidermal cells, and covering about 25% of the total epidermal area of human skin [12]. Studies have shown that 10 DCs that capture the antigen are sufficient to produce a cytotoxic T lymphocyte response, and 500–1000 DCs can cause a wide range of immune responses [13], [14]. Therefore, the skin is usually considered to be one of the best positions for vaccination [15].
Transcutaneous immunization (TCI) is a new vaccination strategy, which can deliver antigen and adjuvant locally to the skin to decrease the adverse systemic immune response [16]. This method can overcome the drawbacks of injection vaccination, becoming a promising choice [17]. However, the barrier function of the stratum corneum hampers the entry of macromolecular antigens, that is, it is difficult for antigens to pass through the stratum corneum to reach the active epidermal layer [18]. The classic methods to promote the transdermal effect include chemical methods (such as surfactants and liposomes as osmotic enhancers) [19] and physical intervention (such as iontophoresis, electroporation and microneedle) [20]. Microneedle array technology, can overcome the barrier function of the cuticle and promote the effective transdermal delivery of macromolecular antigen, which has aroused widespread research interest [21].
Microneedle (MN) is complicated needle-like structures, machined by micro-electro mechanical systems [22], sized in micrometer and made from silicon [23], polymers or metal [24]. MN become the most efficient transdermal delivery method owing to its accuracy, painless, high efficiency and convenience [25], [26]. MN can be initially utilized in active targeting to active epidermis layer and delivering antigen to active epidermis layer where APCs capture antigen and trigger efficient systemic immune response [27]. Nowadays, many kinds of transdermal immune agents based on microneedle system have entered the stage of clinical trials. For example, two kinds of transdermal immune patches developed by IOMAI Company of the United States, mainly for influenza and traveler diarrhea, had entered phase I and phase II clinical trials respectively [28], [29]. Based on the phase I/II clinical studies, it had been documented that TCI of human adult volunteers with the live attenuated measles vaccine ROUVAX® was safe and poorly reactogenic [30].
Liposomes (LP), sphere-shaped vesicles consisting of one or more phospholipid bilayers, are one of the most successful nano-medical platforms and have exhibited favorable aspects of drug delivery [31]. The success of the liposome-based therapeutics has been attributed to ease of synthesis, biocompatibility and the ability to load both hydrophilic and hydrophobic agents [32], [33]. LP has been widely used as an effective drug carrier for tumor chemotherapy and radiotherapy drugs, gene therapy and tumor imaging diagnosis [34].
Despite numerous researches on the dissolving microneedle and hollow microneedle, applying it with liposomes makes the preparation process complicated. Thus, our study puts the emphasis on elucidating transdermal immunity in mechanism by a clear and direct way, i.e. by the matured technology—Solid microneedle. In this study, a transdermal liposome vaccine delivery system based on microneedle administration to actively target LCs was constructed with ovalbumin (OVA) as the antigen model. The targeting and uptake efficiency of immunoliposomes as well as the immune response transport pathway in vivo was investigated. Transdermal delivery efficiency of macromolecular antigen and its effects on inducing immune response was evaluated. The characteristics of the induced immune response were analysed to look into the mechanism for the enhanced immune response.
2. Materials and methods
2.1. Materials
1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine [methoxy (polyethylene glycol) −2000] (mPEG2000-DSPE) and egg phosphatidylcholine (EPC-3) were purchased from Lipoid company (Germany). 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine [Malermide methoxy (polyethylene glycol)-2000] (Mal-PEG2000-DSPE) was obtained from Creative company (USA). Ovalbumin (OVA), cholesterol (CHO), 2-Iminothiolane hydrochloride (2-IT) and cholera toxin (CT) were purchased from Sigma (USA). Fluorescence-labeled rat anti-mouse monoclonal CD40, CD80, CD86, CD11c antibody and Goat anti-mouse monoclonal IgG-HRP, IgG1-HRP, IgG2a-HRP, IgA-HRP antibody were purchased from eBioscience (USA) and Santa Cruz (USA) respectively. The materials used in cell experiment were obtained from Gibco (USA). Mouse Th1/Th2/Th17 Cytokine Kit was purchased from BD company (USA). EL4 cells and E.G7-OVA cells were kindly gift from the National Key Laboratory of Medical Immunology (Shanghai, People’s Republic of China). Microneedle was donated by Dr. Hua Lin from Nanjing University.
2.2. Preparation and characterization of LC-targeting immunoliposomes
OVA-liposomes were prepared according to previous methods [35]. The preparation procedure of LC-targeting immunoliposomes included two steps. Firstly, the immunoliposomes was prepared by thin-film dispersion method. EPC-3, cholesterol, mPEG2000-DSPE and Mal-PEG2000-DSPE (molar ratio = 2:1:0.008:0.002) were dissolved in anhydrous chloroform. Thereafter, the solvent was evaporated to dryness on a rotary evaporator to obtain a uniform and transparent film. Then 5 mL of the aqueous phase (OVA solution, 0.2 mg/mL) was added to the round bottom flask and mixed with the film to make it fall off to form the liposomes (LP) suspension (Fig. S3a). Next, liposomes were obtained by the film extruder. Subsequently, the liposomes solution was added to the mixed solution of 2-IT and anti-CD11c monoclonal antibody (molar ratio 100:1) and suspended for 1 h (Fig. S1). Then the suspension was purified by the AKTA prime protein purifier and Sepharose CL-4B gel column to remove unencapsulated OVA and un-crosslinked CD11c monoclonal antibody and to obtain the LC-targeting immunoliposomes (OVA@CD11c-ILP) (Fig. S2, S3b). CD19 monoclonal antibody-conjugated immunoliposomes (OVA@CD19-ILP) were also prepared as a control.
Immunoliposomes were observed by laser confocal microscope to prove if the successful preparation of it. OVA@CD11c-ILP solution was smeared on the pre-treated skin of nude mice. After 10 min, the residual solution on the surface was wiped off with PBS. The fluorescence distribution on the skin surface was observed by laser confocal microscope (Leica TCS SP2, Japan). The particle size, PDI and zeta potential of OVA@CD11c-ILP were measured by a dynamic light scattering method and the morphology was observed by transmission electron microscope (TEM) (TecnaiG2 F20 S-Twin; FEI, Hillsboro, USA). The encapsulation efficiency (EE) of immunoliposomes was measured by fluorescence spectrophotometry with FITC-labeled OVA (FITC-OVA) instead of OVA (Fig. S3c) and calculated by the formulae: EE (%) = Amount of OVA in immunoliposomes/Amount of OVA fed initially × 100%.
2.3. Targeting assay in vitro
LCs and DCs were used to evaluate the targeting efficiency of immunoliposomes in vitro. DCs and LCs were extracted respectively from C57BL/6 mice bone marrow and skin by the methods referring to the literature method [36]. DCs or LCs were seeded in a 6-well plate at a density of 1 × 105 cells per well and FITC-OVA@APC-CD11c-ILP and FITC-OVA@APC-CD19-ILP were respectively added to incubate for 30 min. Then DCs or LCs were collected, the fluorescence intensity and the percentage of positive cells were detected by flow cytometry. In addition, CD11c- negative CT-26 cells were incubated with FITC-OVA@APC-CD11c-ILP as the negative control. Correspondingly, immunoliposomes that were treated in the same way for 4 h were observed by the laser confocal microscope.
2.4. DCs uptake assay
DCs derived from mouse bone marrow were selected as the next experimental subject, considering that our immunoliposomes also showed good targeting of DCs and the extraction of LCs derived from skin was difficult. DCs were washed with PBS and resuspended to the density of 1 × 106 cells/mL. FITC-OVA (OVA 2 μg/mL) and FITC-OVA@APC-CD11c-ILP (OVA 2 μg/mL) were added into the above cell suspensions (300 μL) and incubated at 37 °C. The control group was treated with FITC-OVA and incubated at 4 °C. Next, cold PBS was added at 15 min, 30 min, 60 min, 120 min and 180 min respectively to terminate the experiment. After washing twice, fluorescence intensity of cells was detected by flow cytometry (BD FACS Calibur, USA).
2.5. Phenotype and cytokines secretion
DCs were seeded in a 6-well plate at a density of 1 × 106 cells per well. PBS, OVA, OVA@LP, OVA@CD11c-ILP with OVA concentration of 2 μg/mL and LPS (100 ng/mL) were added to the well and incubated for 24 h at 37 °C with 5% CO2. Then, cells and supernatant were collected separately. Cells were washed twice with PBS and resuspended in 100 μL buffers. Cells were labeled with different monoclonal antibodies (CD40, CD80, CD86, Iab) and then washed twice with cold PBS. Expression levels of DCs surface molecules, such as CD40, CD80, CD86 and Iab, were determined by flow cytometry. TNF-α in supernatant was examined by using the BD Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine Kit.
2.6. Skin insertion studies ex vivo
Male BALB/c nude mice were purchased from Shrek Animal Co. Ltd. (Shanghai, China). All animal procedures were approved by the Institutional Animal Care and Use Committee of the Second Military Medical University (SMMU), and the experiments were conformed to the guidelines of the Laboratory Animal Center of SMMU. To evaluate the skin insertion capability of the MN (pyramid, 11 × 11 arrays, 500 μm height, Fig. S6 A&B), pre-treated nude mouse skin was used as an administrative model. The pre-treated nude mouse skin (stratum corneum upward) was placed on the microneedle device (Fig. S6C) and the MN applied on the stratum corneum for 2 min using a force of approximate 10 N (MN can penetrate the stratum corneum and epidermis, but not the dermis). Then the skins were stained with trypan blue or Hematoxylin & eosin and observed by microscope (Optical microscope & Scanning electron microscope).
2.7. Transdermal drug delivery efficiency
The skin of the nude mouse from which subcutaneous tissue and fat were removed was perforated with MN in the same way as in skin insertion studies, and then was fixed between the donor and receptor compartment of the Franz diffusion cell. The receptor compartment was filled with 5 mL PBS buffer (pH 7.4, 32 °C). The donor compartment was added with 0.6 mL of immunoliposomes solution (OVA concentration was 50 μg/mL) and the diffusion area was 0.5 cm2. The transdermal diffusion test was carried out at 32 °C and 300 rpm. When 5 mL of the solution was taken from the receptor compartment at the set time (1, 2, 4, 8, 12 h), the same volume of PBS buffer was supplemented. ELISA kit was used to determine the content of OVA.
2.8. Promotion of transdermal penetration
High-efficient promotion of transdermal penetration was crucial factors to determine the application of MN. The skin of nude mice treated with MN was incubated with immunoliposomes solution for 4 and 12 h respectively. The skin was homogenized and a mixed solution (saline: isopropanol = 9:1) was used to ultrasonically extract OVA for 1 h. Then the extract was centrifuged and the OVA in the supernatant was determined by the ELISA kit.
2.9. Distribution of antigen in the skin
To further verify the promoting effect of MN treatment on the transdermal delivery of macromolecular antigens, confocal microscopy was used to observe the distribution of immunoliposomes in the skin. Immunoliposomes (FITC-OVA@APC-CD11c-ILP) were prepared by FITC-OVA (FITC-labeled OVA) and APC-CD11c (APC-labeled CD11c). The FITC-OVA@APC-CD11c-ILP solution was evenly coated on the skin of nude mice that had processed through the MNs treatment, and further incubated at 32 °C for 4 h. Subsequently, the remaining solution of immunoliposomes on the skin surface was removed. The nude mouse skin was placed on a glass slide and immediately observed under a laser confocal microscope. Different depths of fluorescence in the skin were scanned with excitation wavelengths of 488 nm (FITC) and 630 nm (APC). Skin with no MNs treatment was used as control.
2.10. Transfer pathway in vivo
To study the immune transport pathway of immunoliposomes in vivo, C57BL/6 mice (male, 6–8 weeks old, 18–20 g) were used as animal models. Mice skin on the left lower abdomen was shaved and exposed for 24 h Then mice were anesthetized and the MN were applied to the exposed skin area of mice. FITC-OVA@APC-CD11c-ILP solution (100 μL) was evenly smeared on the exposed skin area and the mice were kept in an upward posture for 30 min. Mice were sacrificed after the MNs transdermal administration for 24 h. The spleen and the draining lymph nodes near the immune site were removed to make frozen sections and were observed under laser confocal microscope.
2.11. Animal immunization
C57BL/6 mice were purchased from Shrek Animal Co. Ltd. (Shanghai, China). All animal procedures were approved by the Institutional Animal Care and Use Committee of the Second Military Medical University (SMMU), and the experiments were conformed to the guidelines of the Laboratory Animal Center of SMMU. C57BL/6 mice (male, 6–8 weeks old, 18–20 g, n = 6) were inoculated subcutaneously with 25 μg of OVA e.c. with MNs, OVA@CD11c-ILP e.c. (5 μg of OVA), OVA@LP (5 μg of OVA) with MNs, OVA@CD11c-ILP (5 μg of OVA) with MNs, and OVA@CD11c-ILP (5 μg of OVA) with MNs plus 25 μg CT, or PBS as a negative control and 25 μg of OVA s.c. as a positive control. It was important to note that CT was added in a single control group at similar doses to establish a positive control for the adjuvanted vaccines. Before immunization, mice were exposed about 4 cm2 area of skin on the left lower abdominal area shaved and were kept for 24 h. Subsequently, the MNs were applied to the abdomen of anesthetized mice and removed after 2 min. After the primary immunization (incubating for 30 min per mouse), mice were boosted intracutaneously with the same dose of the corresponding immunogens at week 1, 2, 3, respectively (Fig. S7). Samples of blood were taken from the eyes of mice on the 7, 14, 28, and 35 days after immunization to detect the specific antibody titer.
2.12. Anti-tumor effects in vivo after immunization
Immune studies in vivo were performed in C57BL/6 mice that had been inoculated with T lymphoma cells (E.G7-OVA). Seven days after the last immunization, the xenograft model was established by subcutaneously injecting a E.G7-OVA cell suspension (1 × 106 cells) into the left lower abdominal area of mice (transdermal administration area). Tumor size and survival of mice were monitored and recorded every two days.
2.13. ELISs
To investigate the proliferation of OVA-specific T cells in mice after transdermal immunization with MN, splenocytes from each group of mice were obtained by aseptic operation 10 days after the last immunization and labeled with CFSE. CFSE labeled splenocytes suspension was spread on 96 well plate (5 × 105 cells/well). E.G7-OVA cells inactivated by Co60 irradiation (15,000 rads) were co-cultured with splenocytes in the ratio of 1:5. RPMI 1640 medium was used as negative control. Each well was supplemented with 200 μL RPMI 1640 culture medium containing 10% FBS. After 3 days of incubation, the cells were collected, and the splenocytes proliferation was evaluated by CFSE dilution using flow cytometry.
To further verify the type of immune response, the level of cytokine in the supernatant of splenocytes culture was measured. OVA (10 μg/mL) was added into splenocyte suspension (5 × 105 cells/mL) and incubated for 24 h. The contents of IL-2 and IFN-γ in culture supernatant were measured by the BD Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine Kit.
CFSE and PI double staining methods were used in cytotoxic T lymphocyte (CTL) killing test. Mice splenocytes suspension (2 × 107 cells/mL) was added with 4 × 106 cells/mL inactivated E.G7-OVA cells, and then 20 U/mL mouse IL-2 was added to the culture for 7 days. E.G7-OVA cells were isolated and removed by density gradient centrifugation of lymphocyte separation solution and the obtained splenocytes were used as CTL to test the killing ability to E.G7-OVA cells. CFSE-labeled E.G7-OVA cells and EL4 cells were used as target cells and added into 96 well plates (1 × 104 cells/well), respectively. Next, effector cells (the stimulated mice splenocytes) were added and cultured overnight at three different target ratios (50:1, 25:1, and 12.5:1). The cells were collected and labeled with PI. The proportion of PI positive cells in CFSE stained cells was detected by flow cytometry and the specific CTL killing effect of target cells was observed.
2.14. Statistical analysis
Statistical analysis of data was performed using software SPSS 21.0. Student's test (α = 0.05) was performed when comparing two different conditions, and ANOVA was used when comparing multiple groups. Data were presented as mean ± standard deviation (SD) and P < 0.05 was considered to indicate a statistically significant difference.
3. Results and discussion
3.1. Preparation and characterization of ILP
In this study, the thin-film dispersion and extrusion method was utilized to prepare OVA-loaded CD11c monoclonal antibody immunoliposomes (OVA@CD11c-ILP) [37]. LCs-targeting immunoliposomes were constructed by covalently binding the CD11c monoclonal antibody to the surface of the liposomes. As shown in Fig. S4, the red fluorescence of APC (representing ILP coated with APC-labeled anti-CD11c monoclonal antibody; Fig. S4b) and the green fluorescence of FITC (representing FITC-labeled OVA encapsulated in ILP; Fig. S4c) on the skin surface were completely overlapped (Fig. S4d), which suggested that immunoliposomes effectively encapsulating OVA antigen and were successfully connected with CD11c monoclonal antibody. The binding rate of CD11c monoclonal antibody and entrapment efficiency (EE) of immunoliposomes were 77.6% ± 3.2% and 52.67% ± 1.41%, respectively. As shown in Fig. S5, the microscopic morphology of OVA@CD11c-ILP was presented as favorable spherical-like nanoparticles with a narrow size distribution (PDI = 0.108 ± 0.014). Moreover, a smaller hydrodynamic diameter of OVA@CD11c-ILP (170.2 ± 1.3 nm) and a larger Zeta potential (from −29.5 ± 0.6 mV) hinted a stable and condensed structure of OVA@CD11c-ILP.
3.2. Targeting to and uptake of OVA@CD11c-ILP by LCs or DCs in vitro
According to the results of flow cytometry (Fig. 2 A and B), the binding rate of OVA@CD11c-ILP to CD11c-positive DCs was 86.65% ± 1.78%, while that of OVA@CD19-ILP was only 2.54% ± 0.97%. Meanwhile, the binding rate of OVA@CD11c-ILP to CD11c-positive LCs was 95.74% ± 1.33%. Furthermore, the binding rate of OVA@CD11c-ILP to CD11c-negative CT-26 cells was about 4.04% ± 0.92%. These results suggested that the immunoliposomes modified with CD11c monoclonal antibody showed good specific targeting to both LCs and DCs.
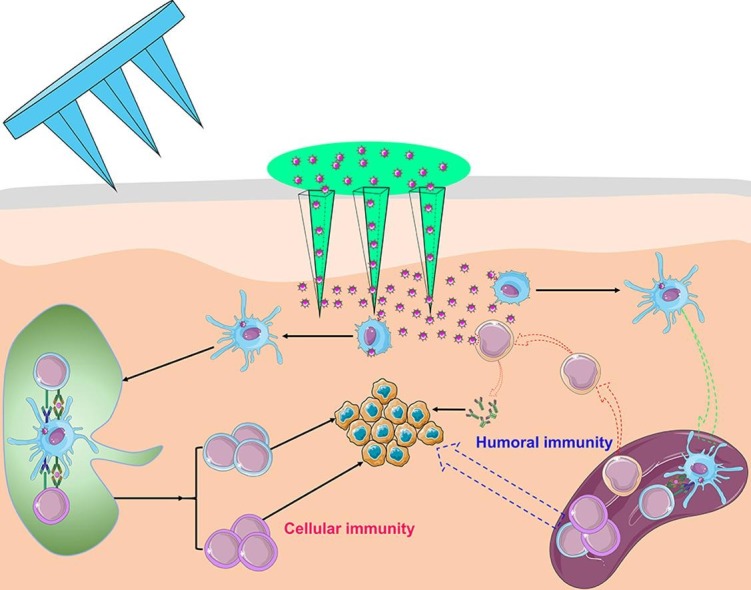
Fig. 1.

Schematic illustration of LC-targeting immunoliposomes application process and its mechanism.
Fig. 2.

Targeting assay in vitro. FACS assays (A): DCs being labeled with CD11c (a), DCs co-incubated with OVA@CD19-ILP (b), CT-26 cells co-incubated with OVA@CD11c-ILP (c), DCs co-incubated with OVA@CD11c-ILP (d), LCs co-incubated with CD11c (e) or OVA@CD11c-ILP (f). Targeting efficiency of OVA@CD11c-ILP in vitro (B). Targeting to LCs or DCs was evaluated by laser confocal microscopy (C): LCs incubated with OVA@CD11c-ILP (a); DCs incubated with OVA@CD11c-ILP (b); DCs incubated with OVA@CD19-ILP (c); CT-26 cells incubated with OVA@CD11c-ILP (d). The data were presented as mean ± sd (n = 3), *P<0.05, **P<0.01, ***P<0.001.
As shown in Fig. 2C, after co-incubation of OVA@CD11c-ILP with LCs or DCs, the red fluorescence was observed obviously on the surface of LCs or DCs while the green fluorescence was mainly distributed in the cytoplasm (Fig. 2C, a or b), indicating that the antigen was effectively delivered into LCs or DCs. As the control group, no corresponding fluorescence was detected on the surface of DCs in the OVA@CD19-ILP co-incubation group (Fig. 2C, c). And then there was no red fluorescence on the surface of CD11c- negative CT-26 cells incubated with OVA@CD11c-ILP (Fig. 2C, d). The results of laser confocal microscopy confirmed that the binding of CD11c immunoliposomes with LCs or DCs was induced by surface modified CD11c monoclonal antibody, which further proved that the OVA@CD11c-ILP could be specifically targeted to LCs or DCs in vitro.
DCs derived from mouse bone marrow were selected as the next experimental subject, considering that our immunoliposomes also showed good targeting of DCs. DCs can uptake external antigens and initiate antigen-specific immune responses. Thus, the ability of antigen uptake is critical to the immune function of DCs. Fig. 3 A showed that, as compared with free OVA, the fluorescence intensity of FITC-labeled OVA antigen was greatly improved by immunoliposomes, indicating that immunoliposomes can effectively promote the uptake of the antigen by DCs. In Fig. 3B, the fluorescence intensity of DCs uptaking immunoliposomes loaded with antigen gradually decreased with time, which was significantly different from the kinetic curve of OVA uptake. These results suggested that the specific binding of CD11c on DCs surface to CD11c monoclonal antibody on immunoliposomes surface may be the primary reason for DCs to uptake the antigen, which had reached the maximum binding within 15 min. Meanwhile, the encapsulation of antigens in immunoliposomes can significantly increase the uptake efficiency of antigens in a short time, which means that this delivery strategies can increase the antigen presentation efficiency of antigen-presenting cells to a certain extent.
Fig. 3.

Uptake of OVA@CD11c-ILP by DCs in vitro. Kinetics of OVA uptake (A): OVA treated DCs at 4 °C (a); OVA treated DCs at 37 °C (b); OVA@CD11c-ILP treated DCs at 37 °C (c). Uptake kinetic curve of OVA@CD11c-ILP or OVA (B). The data were presented as mean ± sd (n = 3).
3.3. Maturation of DCs induced by immunoliposomes
Dendritic cells exist in two basic functional states: immature DCs and mature DCs. The mature DCs is often used functionally to designate immunogenic DCs, expressing high levels of MHC-II, adhesion and co-stimulatory molecules [38]. As shown in Fig. 4 A, as compared to OVA-pulsed DCs, OVA@LP (OVA encapsulated in liposomes) and OVA@CD11c-ILP could more effectively stimulate the expression of Iab, CD40, CD80, CD86 on DCs surface. Maturation of DCs phenotype was beneficial to DC-mediated immune response.
Fig. 4.

Maturation of DCs by immunoliposomes in vitro. FACS assays of maturation-related factors on the surface of DCs (A). TNF-α production of DCs stimulated by different additives (B). The data were presented as mean ± sd (n = 3), *P<0.05, **P<0.01, ***P<0.001.
TNF-α is a major immunomodulatory and proinflammatory factor that can promote the migration of DCs from the skin to the draining lymph node and that can stimulate the proliferation and maturation of T cells, which are one of the characteristics of functional maturation of DCs [39]. As shown in Fig. 4B, OVA and OVA@LP could not stimulate DCs to produce TNF-α, whereas OVA@CD11c-ILP effectively stimulated DCs to produce TNF-α. Therefore, immunoliposomes may be able to effectively promote the functional maturation of DCs.
3.4. Skin insertion studies ex vivo
It is essential for MN to penetrate the stratum corneum to enable drug delivery into the skin. To evaluate the insertion capability of the MN in this work, 500 μm height and 11 × 11 MN arrays were used for skin insertion tests (Fig. S6A, B). The results of trypan blue and H&E staining showed that the MNs penetrated the stratum corneum (Fig. 5 A) and created microchannels with a diameter of approximately 50 μm and a depth of approximately 100 μm in the skin (Fig. 5B). The results were further confirmed by scanning electron microscope (SEM) (Fig. 5C, D), indicating that the MN can penetrate the stratum corneum to reach the dermis, which was beneficial for the immunoliposomes to pass through the stratum corneum to reach the dermis via the microchannels.
Fig. 5.

Microscope imaging of skin examined by trypan blue staining (A), H&E staining (B) or SEM imaging (C, D) after MN administration. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. Transdermal drug delivery ex vivo
In order to investigate the transdermal permeation by MN for immunoliposomes, OVA levels were measured by ELISA. The time-dependent curve of OVA permeation in the receptor solution of the MN treatment group was shown in Fig. 6 A, demonstrating that the skin permeation of OVA encapsulated by immunoliposomes was greatly increased after MN administration. As shown in Fig. 6B, as compared with the no MN treatment groups, the OVA retention in the skin after MN administration was significantly increased (P<0.01) and the content of OVA in the skin at 12 h was significantly higher than that at 4 h (P < 0.05). The results suggested that MN treatment can effectively increase the amount of skin permeation and retention of macromolecular antigens encapsulated by immunoliposomes, and both of them gradually increase with time, indicating that MN treatment was better in promoting percutaneous permeation.
Fig. 6.

Time-dependent penetration curve of OVA treated with MN (A). Amounts of OVA retention in skin were measured by ELISA (B). The data were presented as mean ± sd (n = 3), *P<0.05, **P<0.01, ***P<0.001.
As shown in Fig. 7 , immunoliposomes could penetrate into the skin through the microchannels produced by MN. The skin was composed of stratum corneum, epidermis and dermis. There are a large number of immune cells (such as LCs and DCs) distributed in the epidermis and dermis [7]. The thickness of the stratum corneum and epidermis is approximately 10–15 µm and 100 µm respectively, while a large amounts of fluorescence of the monoclonal antibody (red fluorescence) on the surface of the immunoliposomes and OVA (green fluorescence) encapsulated by immunoliposomes could be observed at 130 µm. Next, fluorescence could be observed even at a depth of about 200 μm. Therefore, the antigen-loaded immunoliposomes can be effectively delivered into the skin epidermis and upper dermis through MN, which further proved that the delivery of immunoliposomes via MN can achieve localized delivery of the epidermis and dermis.
Fig. 7.

Transdermal drug delivery by MN. Laser confocal microscopic images (×10 objective) of nude mouse skin at different depth. The yellow circles indicated for microchannels produced by MN. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. Transfer pathway of antigen-loaded immunoliposomes in vivo
24 h after percutaneous administration of immunoliposomes plus microneedle system, the frozen sections of draining lymph nodes (LN) and spleen were observed by laser confocal microscopy (Fig. 8 ). Obviously, there were plenty of red fluorescence (CD11c-ILP) and green fluorescence (OVA) distribution both in LN and spleen, indicating that LCs-targeting immunoliposomes may effectively transport the immunoliposomes and antigen to secondary immune organs, which may serve as an important experimental basis for immunoliposomes to stimulate an effective immune response.
Fig. 8.

Laser confocal images of nodes sections in draining lymph and spleen.
3.7. Anti-tumor effects in vivo after immunization of immunoliposomes
The antitumor efficacy of OVA@CD11-ILP and other counterparts was evaluated in E.G7-OVA tumor bearing mouse model. Immune strategies included epicutaneous immunization (e.c.) and immunization (s.c.) respectively. As shown in Fig. 9 A, the tumor volumes were periodically monitored. On the eighth day after inoculation, tumor nodules were observed in the mice treated with PBS, OVA e.c. + MN, OVA@CD11c-ILP e.c., OVA@LP e.c. + MN. While only part of the mouse developed tumor after 12 days in groups of OVA@CD11c-ILP e.c. + MN and OVA s.c.. No tumor was found in the mice immunized with OVA@CD11c-ILP e.c. + MN + CT (cholera toxin) until 14 days later. When compared with the negative control group, OVA@CD11c-ILP e.c., OVA e.c. + MN and OVA@LP e.c. + MN, the tumor of the OVA@CD11c-ILP e.c. + MN group appeared later and grew significantly slower. Notably, one of the mice in OVA@CD11c-ILP e.c. + MN group did not grow tumors by the end of the 60-day experiment. It was speculated that this may be mainly due to the difference in the strength of the immune response triggered by the OVA@CD11c-ILP e.c. + MN immunization in different individuals of the mouse. The tumor growth trend of OVA s.c. group and OVA@CD11c-ILP e.c. + MN + CT group was similar to that of OVA@ CD11c-ILP e.c. + MN group, which suggested that OVA@CD11c-ILP delivered by MNs and subcutaneous injection of OVA can exert the same immune effect (P > 0.05).
Fig. 9.

Tumor size (A) and survival curves (B) of immunized C57BL/6 mice. The data were presented as mean ± sd (n = 6), *P<0.05, **P<0.01, ***P<0.001.
As shown in Fig. 9B, all mice in groups of PBS, OVA e.c. + MN, OVA@CD11c-ILP e.c., OVA@LP e.c. + MN were died between day 22 and day 40. However, 50% of mice in the immunoliposomes combined with microneedle transdermal immunization group still survived on day 33, indicating that this method of immunization can prolong the survival time of tumor-bearing mice.
3.8. Induction of immune response to OVA
The specific humoral immune response of mice in each group was studied. The titers of specific anti-OVA IgG, IgG1, IgG2a and IgA in serum were determined by double-antibody sandwich ELISA on the 7, 14, 28, 35 days of immunization. No antibody was detected on the seventh day and the results of serum detection on the 14th, 28th and 35th days were recorded. As shown in Fig. 10 , the production of OVA-specific antibody in mice after the fourth immunization showed that the mice in OVA@CD11c-ILP e.c. + MN group produced higher titer of various types of antibodies than those of negative control group, OVA@CD11c-ILP e.c group, OVA@LP e.c. + MN group and OVA e.c. + MN group (P < 0.05). The antibody titer of the positive control group (OVA s.c.) and OVA@CD11c-ILP e.c. + MN + CT group was higher (P < 0.01). We speculated that it may be due to the fact that subcutaneous injection could guarantee less antigen loss, and our vaccine combination administration method may lose some antigen. Moreover, in OVA@CD11c-ILP e.c. + MN + CT group, it may be that CT exerts an adjuvant effect to enhance the immune response effect, which just made up for this loss of antigen. These results indicated that the immunoliposomes combined with microneedle could effectively stimulate antigen-specific humoral immune response in mice and cholera toxin (CT) as an immune adjuvant can effectively improve the immune response of the body.
Fig. 10.

OVA-specific serum IgG (A), IgG1 (B), IgG2a (C), and IgA (D) antibodies titers were analyzed by ELISA. The data were presented as mean ± sd (n = 6), *P<0.05, **P<0.01, ***P<0.001.
Th1 type cellular immune response plays a more important role in the anti-tumor and anti-viral immune response. More significantly, a higher IgG1/IgG2a antibody titer ratio appeared in the OVA@CD11c-ILP e.c + MN group of mice (Fig. 11 A), which indicated the immune response triggered in this way was Th1-polarized that was more conducive to cellular immunity. Correspondingly, although anti-tumor effect of it was not obvious, compared with the group of OVA e.c. + MN, OVA@LP e.c. + MN group had a higher titer of IgG1/IgG2a, which indicated that liposomes might have Th1 type adjuvant effect.
Fig. 11.

Polarization of the IgG subclass response is illustrated as ratio of IgG2a over IgG1 titers at day 35 (A). OVA-specific proliferation of splenocytes from transcutaneous immunized mice (B). OVA-specific IFN-γ (C) or IL-2 (D) secretion of splenocytes from immunized mice. The data were presented as mean ± sd (n = 6), *P<0.05, **P<0.01, ***P<0.001.
To further verify the Th1-type immune response, the proliferation of OVA antigen-specific T cells was detected by flow cytometry after the spleen cells of immunized mice were stimulated with inactivated E.G7-OVA cells (Fig. 11B). The results showed that the specific proliferation of T cells was stronger in the OVA@CD11c-ILP e.c. + MN group than in other control groups, such as PBS, OVA e.c. + MN, OVA@CD11c-ILP e.c., OVA@LP (P<0.05). Moreover, the specific proliferation of T cells in OVA@CD11c-ILP e.c. + MN group was similar to that in OVA s.c. group (P > 0.05). These data preliminarily suggested that this immunization method could stimulate antigen-specific Th1-type cellular immune response.
Flow cytometry was used to detect the production of IFN-γ and IL-2 in the supernatant of splenocytes from immunized mice. It was shown that the levels of IFN-γ in the supernatant of splenocytes of OVA@CD11c-ILP e.c. + MN group were significantly higher than that of other groups except the OVA@CD11c-ILP e.c. + MN + CT group (P < 0.01) (Fig. 11C). IFN-γ is an important Th1-type cytokine and a sign that T cells were fully activated [40], which further indicates that immunoliposomes percutaneous immunization can effectively stimulate cellular immune response. Th1 cytokine IL-2 can induce activation and proliferation of T cells in vivo [41]. Furthermore, more IL-2 was produced in OVA@CD11c-ILP e.c. + MN + CT group and OVA s.c. group, while the other groups did not produce IL-2 significantly (Fig. 11D). The possible reason was that the specific immune response induced by immunoliposomes was faster, while the specific immune response induced by subcutaneous injection and percutaneous immunity combined with CT was relatively slow and long-term, which was also consistent with the results of tumor bearing experiment.
The essence of immunotherapy is to induce immune response in patients, and the key to immune response was to activate cytotoxic T lymphocyte (CTL) and maximize the function of CTL. The CTL killing experiment was carried out by isolating spleen cells from immunized mice to testify the efficiency of immunotherapy against tumor. It showed that the effector cells from the OVA@CD11c-ILP e.c. + MN group showed significant killing activity against E.G7-OVA cells, while the effector cells in PBS group, OVA e.c. + MN, OVA@CD11c-ILP e.c., and OVA@LP e.c. + MN group had no obvious specific killing effect (Fig. 12 A). EL4 cells did not express OVA antigen, so the effector cells of all experimental groups could not kill EL4 cells, indicating that the killing activity of effector cells was OVA antigen specific (Fig. 12B). These results indicated that OVA-specific CTL could be induced in mice through the percutaneous immunization of immunoliposomes mediated by microneedle, which effectively improved the proliferation and killing ability of CTL cells, and thus contributed to the greatly improved anti-tumor efficiency of immunotherapy.
Fig. 12.

OVA-specific CTL activity after transcutaneous immunization with immunoliposomes and MN was evaluated by using E.G7-OVA cells (A) or EL4 cells (B) as targets. The data were presented as means ± sd (n = 6), *P<0.05, **P<0.01, ***P<0.001.
The observations that the immunoliposomes targeting to LCs stimulated cellular immunity of mice in vivo indicated that, after MN transcutaneous immunization, the immunoliposomes antigen delivery system greatly activated the T cell with antigenic specificity, induced Th1 factors to generate CTL with OVA antigenic specificity, which led to enhanced cellular immune response. Correspondently, although cholera toxin virus as transdermal adjuvant or injected subcutaneously can trigger Th1 type response, its mechanism may be different from transcutaneous immunization of LCs-targeting immunoliposomes alone.
4. Conclusion
Immunoliposomes transdermal vaccine delivery system based on microneedle to actively target LCs had been successfully constructed. Immunoliposomes can actively target LCs and promote the uptake and presentation of antigens by LCs. Transdermal penetration and skin retention of immunoliposomes and its encapsulated macromolecular antigens can be greatly increased by microneedle. The main mechanism was that the microchannels were created by MN on the skin (the first step delivery), then immunoliposomes reach the epidermis and dermis through microchannels, and immunoliposomes that had been taken up by LCs reached the draining lymph node and spleen (the secondary step delivery), thereby triggering effective cellular and humoral immunity. Further, tumor growth of immunoliposomes-treated mice was slow and the survival time was significantly prolonged. It was more significant to find that the immune response stimulated by immunoliposomes combined with microneedle was Th1-biased and the liposomes might have Th1 type adjuvant effect. This study provides a new idea for the transdermal delivery of vaccines and is expected to be a promising transdermal immune delivery system.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by grants from Ministry of Science and Technology Project (Grant No. 20SWAQK24) and Shanghai Sailing Program (Grant No. 21YF1457700).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejpb.2022.06.004.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Kim E., Erdos G., Huang S., Kenniston T.W., Balmert S.C., Carey C.D., Raj V.S., Epperly M.W., Klimstra W.B., Haagmans B.L., Korkmaz E., Falo L.D., Gambotto A. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine. 2020;55:102743. doi: 10.1016/j.ebiom.2020.102743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.di Pietrantonj C., Rivetti A., Marchione P., Debalini M.G., Demicheli V. Vaccines for measles, mumps, rubella, and varicella in children. Cochrane Database System. Rev. 2020;2020 doi: 10.1002/14651858.CD004407.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maruggi G., Zhang C., Li J., Ulmer J.B., Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019;27(4):757–772. doi: 10.1016/j.ymthe.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piot P., Larson H.J., O’Brien K.L., N’kengasong J., Ng E., Sow S., Kampmann B. Immunization: vital progress, unfinished agenda. Nature. 2019;575(7781):119–129. doi: 10.1038/s41586-019-1656-7. [DOI] [PubMed] [Google Scholar]
- 5.Hervé C., Laupèze B., del Giudice G., Didierlaurent A.M., da Silva F.T. The how’s and what’s of vaccine reactogenicity. npj Vaccines. 2019;4 doi: 10.1038/s41541-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Y., Lei K., Tang L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers E.S., Vukmanovic‐Stejic M. Skin barrier immunity and ageing. Immunology. 2020;160(2):116–125. doi: 10.1111/imm.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwiecien K., Zegar A., Jung J., Brzoza P., Kwitniewski M., Godlewska U., Grygier B., Kwiecinska P., Morytko A., Cichy J. Architecture of antimicrobial skin defense. Cytokine Growth Factor Rev. 2019;49:70–84. doi: 10.1016/j.cytogfr.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Kashem S.W., Haniffa M., Kaplan D.H. Antigen-presenting cells in the skin. Annu. Rev. Immunol. 2017;35(1):469–499. doi: 10.1146/annurev-immunol-051116-052215. [DOI] [PubMed] [Google Scholar]
- 10.Rippa A.L., Kalabusheva E.P., Vorotelyak E.A. Regeneration of Dermis: Scarring and Cells Involved. Cells. 2019;8(6):607. doi: 10.3390/cells8060607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balan S., Saxena M., Bhardwaj N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019 doi: 10.1016/bs.ircmb.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Pogorzelska-Dyrbuś J., Szepietowski J.C. Density of Langerhans Cells in Nonmelanoma Skin Cancers: A Systematic Review. Mediators Inflamm. 2020;2020:1–10. doi: 10.1155/2020/8745863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bal S.M., Ding Z., van Riet E., Jiskoot W., Bouwstra J.A. Advances in transcutaneous vaccine delivery: Do all ways lead to Rome? J. Control. Release. 2010;148(3):266–282. doi: 10.1016/j.jconrel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 14.Simerska P., Moyle P., Olive C., Toth I. Oral Vaccine Delivery – New Strategies and Technologies. Curr. Drug Deliv. 2009;6 doi: 10.2174/156720109789000537. [DOI] [PubMed] [Google Scholar]
- 15.Wculek S.K., Cueto F.J., Mujal A.M., Melero I., Krummel M.F., Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 16.Pielenhofer J., Sohl J., Windbergs M., Langguth P., Radsak M.P. Current Progress in Particle-Based Systems for Transdermal Vaccine Delivery. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaurav M., Minz S., Sahu K., Kumar M., Madan J., Pandey R.S. Nanoparticulate mediated transcutaneous immunization: Myth or reality. Nanomed. Nanotechnol. Biol. Med. 2016;12(4):1063–1081. doi: 10.1016/j.nano.2015.12.372. [DOI] [PubMed] [Google Scholar]
- 18.van Smeden J., Bouwstra J.A. Stratum Corneum Lipids: Their Role for the Skin Barrier Function in Healthy Subjects and Atopic Dermatitis Patients. Curr. Probl. Dermatol. (Switzerland). 2016;49 doi: 10.1159/000441540. [DOI] [PubMed] [Google Scholar]
- 19.Yang L., Wu L., Wu D., Shi D., Wang T., Zhu X. Mechanism of transdermal permeation promotion of lipophilic drugs by ethosomes. Int. J. Nanomed. 2017;12 doi: 10.2147/IJN.S134708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang G., Zhang Y., Gu Z. Punching and electroporation for enhanced transdermal drug delivery. Theranostics. 2018;8(13):3688–3690. doi: 10.7150/thno.27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuan-Mahmood T.-M., McCrudden M.T.C., Torrisi B.M., McAlister E., Garland M.J., Singh T.R.R., Donnelly R.F. Microneedles for intradermal and transdermal drug delivery. Eur. J. Pharm. Sci. 2013;50(5):623–637. doi: 10.1016/j.ejps.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García J., Ríos I., Fonthal Rico F. Design and analyses of a transdermal drug delivery device (TD3) Sensors (Switzerland). 2019;19(23):5090. doi: 10.3390/s19235090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffin P., Elliott S., Krauer K., Davies C., Rachel Skinner S., Anderson C.D., Forster A. Safety, acceptability and tolerability of uncoated and excipient-coated high density silicon micro-projection array patches in human subjects. Vaccine. 2017;35(48):6676–6684. doi: 10.1016/j.vaccine.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 24.Sabri A.H., Kim Y., Marlow M., Scurr D.J., Segal J., Banga A.K., Kagan L., Lee J.B. Intradermal and transdermal drug delivery using microneedles – Fabrication, performance evaluation and application to lymphatic delivery. Adv. Drug Deliv. Rev. 2020;153:195–215. doi: 10.1016/j.addr.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Rabiei M., Kashanian S., Samavati S.S., Jamasb S., McInnes S.J.P. Nanomaterial and advanced technologies in transdermal drug delivery. J. Drug Target. 2020;28(4):356–367. doi: 10.1080/1061186X.2019.1693579. [DOI] [PubMed] [Google Scholar]
- 26.Hao Y., Li W., Zhou XingLi, Yang F., Qian ZhiYong. Microneedles-based transdermal drug delivery systems: A review. J. Biomed. Nanotechnol. 2017;13(12):1581–1597. doi: 10.1166/jbn.2017.2474. [DOI] [PubMed] [Google Scholar]
- 27.Zaric M., Ibarzo Yus B., Kalcheva P.P., Klavinskis L.S. Microneedle-mediated delivery of viral vectored vaccines. Expert Opin. Drug Del. 2017;14(10):1177–1187. doi: 10.1080/17425247.2017.1230096. [DOI] [PubMed] [Google Scholar]
- 28.Combadière B., Vogt A., Mahé B., Costagliola D., Hadam S., Bonduelle O., Sterry W., Staszewski S., Schaefer H., van der Werf S., Katlama C., Autran B., Blume-Peytavi U., Kallas E.G. Preferential amplification of CD8 effector-T cells after transcutaneous application of an inactivated influenza vaccine: A randomized phase I trial. PLoS ONE. 2010;5(5):e10818. doi: 10.1371/journal.pone.0010818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frech S.A., DuPont H.L., Bourgeois A.L., McKenzie R., Belkind-Gerson J., Figueroa J.F., Okhuysen P.C., Guerrero N.H., Martinez-Sandoval F.G., Meléndez-Romero J.HM., Jiang Z.-D., Asturias E.J., Halpern J., Torres O.R., Hoffman A.S., Villar C.P., Kassem R.N., Flyer D.C., Andersen B.H., Kazempour K., Breisch S.A., Glenn G.M. Use of a patch containing heat-labile toxin from Escherichia coli against travellers’ diarrhoea: a phase II, randomised, double-blind, placebo-controlled field trial. The Lancet. 2008;371(9629):2019–2025. doi: 10.1016/S0140-6736(08)60839-9. [DOI] [PubMed] [Google Scholar]
- 30.Etchart N., Hennino A., Friede M., Dahel K., Dupouy M., Goujonhenry C., Nicolas J., Kaiserlian D. Safety and efficacy of transcutaneous vaccination using a patch with the live-attenuated measles vaccine in humans. Vaccine. 2007;25(39-40):6891–6899. doi: 10.1016/j.vaccine.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Akbarzadeh A., Rezaei-Sadabady R., Davaran S., Joo S.W., Zarghami N., Hanifehpour Y., Samiei M., Kouhi M., Nejati-Koshki K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013;8 doi: 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee W., Im H.-J. Theranostics Based on Liposome: Looking Back and Forward. Nucl. Med. Mol. Imag. 2019;53(4):242–246. doi: 10.1007/s13139-019-00603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M., Du C., Guo N.a., Teng Y., Meng X., Sun H., Li S., Yu P., Galons H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019;164:640–653. doi: 10.1016/j.ejmech.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Sabnani M.K., Rajan R., Rowland B., Mavinkurve V., Wood L.M., Gabizon A.A., La-Beck N.M. Liposome promotion of tumor growth is associated with angiogenesis and inhibition of antitumor immune responses. Nanomed. Nanotechnol. Biol. Med. 2015;11(2):259–262. doi: 10.1016/j.nano.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Chen M.-C., Lai K.-Y., Ling M.-H., Lin C.-W. Enhancing immunogenicity of antigens through sustained intradermal delivery using chitosan microneedles with a patch-dissolvable design. Acta Biomater. 2018;65:66–75. doi: 10.1016/j.actbio.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Ortner U., Inaba K., Koch F., Heine M., Miwa M., Schuler G., Romani N. An improved isolation method for murine migratory cutaneous dendritic cells. J. Immunol. Methods. 1996;193(1):71–79. doi: 10.1016/0022-1759(96)00058-0. [DOI] [PubMed] [Google Scholar]
- 37.Li N., Peng L.H., Chen X., Nakagawa S., Gao J.Q. Effective transcutaneous immunization by antigen-loaded flexible liposome in vivo. Int. J. Nanomed. 2011;6 doi: 10.2147/IJN.S26152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reis e Sousa C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006;6(6):476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 39.Akdis M., Aab A., Altunbulakli C., Azkur K., Costa R.A., Crameri R., Duan S.u., Eiwegger T., Eljaszewicz A., Ferstl R., Frei R., Garbani M., Globinska A., Hess L., Huitema C., Kubo T., Komlosi Z., Konieczna P., Kovacs N., Kucuksezer U.C., Meyer N., Morita H., Olzhausen J., O'Mahony L., Pezer M., Prati M., Rebane A., Rhyner C., Rinaldi A., Sokolowska M., Stanic B., Sugita K., Treis A., van de Veen W., Wanke K., Wawrzyniak M., Wawrzyniak P., Wirz O.F., Zakzuk J.S., Akdis C.A. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016;138(4):984–1010. doi: 10.1016/j.jaci.2016.06.033. [DOI] [PubMed] [Google Scholar]
- 40.Li C., Zhu D., Zhao Y., Guo Q., Sun W., Li L., Gao D., Zhao P. Dendritic Cells Therapy with Cytokine-Induced Killer Cells and Activated Cytotoxic T Cells Attenuated Th2 Bias Immune Response. Immunol. Invest. 2020;49(5):522–534. doi: 10.1080/08820139.2019.1696360. [DOI] [PubMed] [Google Scholar]
- 41.Safar H.A., Mustafa A.S., Amoudy H.A., El-Hashim A., Rawkins A. The effect of adjuvants and delivery systems on Th1, Th2, Th17 and Treg cytokine responses in mice immunized with Mycobacterium tuberculosis-specific proteins. PLoS ONE. 2020;15(2):e0228381. doi: 10.1371/journal.pone.0228381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.