

Abstract
Allogeneic hematopoietic stem cell transplantation (aHSCT) is complicated by Graft Versus Host Disease (GVHD) which causes immune dysfunction and further delays immune reconstitution via effects on primary and secondary lymphoid organs. Treatments to prevent GVHD and improve immune recovery following aHSCT are needed. Post-transplant cyclophosphamide (PTCy) is a well-established and clinically widely used method for GVHD prophylaxis after HLA-matched as well as haploidentical aHSCT, and also a promising strategy in the setting of mismatched unrelated donor aHSCT. Recently, regulatory T cells (Tregs), a critical subset for immune homeostasis and tolerance induction, are being evaluated for use as GVHD prophylaxis in experimental models and clinical trials. Natural killer (NK) cells are one of the first lymphoid populations to reconstitute following aHSCT and are important mediators of protective immunity against pathogens and also critical for limiting post-transplant relapse of hematologic cancers. Several reports have noted that a delay in NK cell recovery may occur following allogeneic experimental mouse HSCT as well as after clinical aHSCT. Here we examined how two treatment strategies, PTCy and donor expanded Tregs (TrED), in an experimental MHC-matched allogeneic HSCT affected NK recovery. Experiments showed that both strategies improved NK cell numbers, with PTCy slightly better vs TrED, early after aHSCT (1 month) compared to untreated aHSCT recipients. Importantly, NK cell interferon gamma (IFNγ) production and cytotoxic function, reflected by CD107 expression as well as in vivo killing of NK-sensitive tumor cells, were improved using either PTCy or TrED vs. control aHSCT recipients. In conclusion, both prophylactic treatments were found to be beneficial for NK recovery and NK cell function following MHC-matched minor antigen mismatched experimental HSCT. Improved NK recovery could help provide early immunity towards tumor and pathogens in these transplant recipients.
Keywords: aHSCT, GVHD, PTCy, Treg, TL1A, IL-2, NK cells
Introduction
In addition to providing immunity against infections and anti-tumor activity dependent on the specific cancer, NK cells eliminate various populations of host cells which diminish HVG, promote engraftment as well as reduce GVHD1–6. Allogeneic HSCT (aHSCT) requires GVHD prophylaxis. Following aHSCT, NK cells are one of the earliest lymphoid populations to recover quantitatively, which make them critical early mediators of post-aHSCT outcomes, including opportunistic infection7–9. Within the first month following aHSCT, NK cells reach levels approximating normal in the blood of aHSCT patients, but it takes several months to acquire phenotypic and functional characteristics found in healthy donors10, 11. Moreover, the incidence of acute GVHD was reported to be associated with a delayed expansion of the NK compartment, therefore, treatments which promote NK cell recovery could be useful to improve these transplants8.
The use of post-transplant cyclophosphamide (PTCy) was initially reported to diminish HVG responses and promote engraftment in mouse studies12. Subsequent work by Luznik and colleagues demonstrated that administration of cyclophosphamide on post-aHSCT days 3 and 4 was an effective GVHD prophylactic treatment for both MHC-matched and MHC-mismatched aHSCT; this approach is now widely utilized in many centers for transplants involving a variety of donor and recipient genetic disparities13–17. Moreover, PTCy is becoming a viable option in patients as a sole GVHD prophylactic treatment after ablative conditioning18–23.
CD4+Foxp3+ Treg cells are a non-redundant regulatory population critical for immunologic homeostasis and self-tolerance24. The suppressive capacity of these cells has made them an attractive option for regulation of allogeneic transplants including HSCT where they are highly effective at ameliorating GVHD25–28. Our laboratory has reported in vivo manipulation of Tregs through stimulation of the TNFRSF25 receptor alone and together with low dose IL-229. Recently we analyzed the use of both PTCy and Tregs in murine aHSCT models and reported both diminished GVHD and prolonged survival30. Interestingly, although both treatments ameliorated GVHD, we found that the use of expanded donor Tregs facilitated earlier thymopoetic recovery compared to PTCy therapy30.
Russo et al., evaluated NK cell reconstitution following PTCy administration in patients receiving unmanipulated haploidentical grafts (containing high numbers of mature NK cells)31. They observed a robust proliferation of donor derived NK cells immediately after aHSCT. Infusion of PTCy led to a marked reduction in NK cells with subsequent recovery to approximately pre-HSCT numbers by day 30. Recently, Rambaldi et al., showed significant delays in NK cell recovery in the first 3 months after haplo-HCT with PTCy compared with matched related/unrelated HCT with conventional GVHD prophylaxis32. Importantly, McCurdy et al. reported that early NK cell recovery is the main determinant for overall survival after haploidentical and HLA-matched BMT using PTCy as GVHD prophylaxis33.
Based on the above studies and our own findings involving thymus-derived immune reconstitution, we were interested in assessing NK recovery in murine aHSCT to evaluate how these two GVHD prophylaxis strategies may affect reconstitution within the NK compartment. Notably, both strategies were found to improve reconstitution of NK cell numbers, with PTCy tending to facilitate greater recovery early post-transplant (within 4 weeks), when compared to untreated recipients; these augmented numbers of NK cells were functional, reflected by in vivo killing of NK-sensitive tumors.
Materials and Methods
Mice.
C3H.SW (Stock: 000438), C57BL/6J (B6, Stock: 000664), BALB/c (Stock: 000651) mice and B6-CD45.1 breeders (Stock: 002014) were purchased from The Jackson Laboratory. All mice were maintained in specific pathogen-free housing at the University of Miami and given autoclaved food and water ad libitum. Mice were used at 12–16 weeks of age. All animal use procedures were approved by the University of Miami institutional animal care and use committee.
Cell lines.
RMA and RMA-S cell lines were kindly provided by Dr. Eli Gilboa (University of Miami, Miami FL)34,35. Tumor cells were maintained in IMDM media supplemented with 10% FCS plus antibiotics (PS) and used for in vivo cytotoxicity assays.
Bone marrow transplantation.
For MHC-matched (C3H.SW to B6) and syngeneic (B6 to B6) transplants, B6-WT mice received 10.5 Gy total body irradiation (TBI) (Cs137 source) on day 0. Three hours later, irradiated mice were injected iv with 7×106 TCD-BM and pooled splenocytes plus lymph node cells containing 2×106 CD8+ T cells from C3H.SW mice (MHC-matched) or 10×106 non-TCD BM plus 5×106 splenocytes from B6 mice (syngeneic). T cell depletion was performed using HO134 hybridoma supernatant (αThy1.2) and rabbit complement (Cedarlane Labs, Burlington, NC). Mice were monitored 3x per week for weight loss and clinical score as previously described29.
Cyclophosphamide treatment.
Cyclophosphamide (50 mg/kg) was administered ip on days +3 and +4 following aHSCT14, 15.
Treg expansion protocol.
TL1A-Ig was generated as described previously36. TL1A-Ig (50 μg) was administered ip on post-aHSCT days 1–4 and free human IL-2 (10,000 U) was given ip on days 4–629. Mice were sacrificed on day 7 and Treg expansion confirmed by Foxp3 staining (flow cytometry). Spleen and LN cells were collected from donors and used in HSCTs.
Flow cytometry.,
Peripheral blood, lymphoid tissue and BM were collected from transplant recipients at indicated time points post-HSCT. Lymphoid organs and BM were prepared into single-cell suspensions. Peripheral blood was collected in heparinized tubes. Peripheral blood mononuclear cells (PBMC) were isolated by standard Ficoll density gradient centrifugation. Next, 106 cells were pre-blocked with anti-mouse CD16/CD32 and stained with different antibody combinations. The following commercial mAbs from BD Biosciences, Biolegend or eBioscience/ThermoFisher to the indicated molecules were used for flow cytometry: CD4, CD8, CD3, CD19, NK1.1 (B6), Nkp46, CD122, CD27, CD11b, CD107, Ki67, Foxp3, IFNγ. For intracellular cytokine staining, splenic single cell suspensions were stimulated in complete media containing 10% FBS, CD107, PMA (50 ng/mL), ionomycin (500 ng/mL) and brefeldin A for 5h. After surface staining, cells were fixed using Cytofix/Cytoperm and Perm/Wash Buffers (BD Biosciences) according to the manufacturer’s instructions. Permeabilized samples were then stained for 1h for cytokine detection. Intracellular staining for Foxp3 and Ki67 was performed using the Foxp3 Transcription Factor Staining Set from eBioscience/ThermoFisher Scientific according to the manufacturer’s instructions. Flow cytometry analyses was performed on a BD LSRII Flow Cytometer. The data was analyzed using FlowJo version 10 software (TreeStar).
In vivo cytotoxicity assay for NK cells.
One-month post-aHSCT mice were inoculated iv with 10×106 RMA plus 10×106 RMA-S (ratio 1:1) cells labeled with low (0.4 μM) and high (4 μM) CellTrace Violet (CTV) (Invitrogen/Thermo Fisher Scientific), respectively. 5h later the mice were sacrificed, and cytotoxicity was assessed in spleen and blood by gating on the CTV positive cells. Cytotoxicity (% specific killing) was calculated using the following formula: [1-(RMA-Ssample/RMAcontrol)/(RMA-Scontrol/RMAsample)]×10037. As controls, CTV labeled cells cultured in vitro for the same amount of time were used.
Statistical analysis.
Numbers of animals per group and statistical tests utilized are described in the figure legends. All figure panels include data sets obtained from individual animals. Graphing and statistical analysis were done using GraphPad Prism 9 (La Jolla, CA). Statistical differences between two experimental groups were determined using two-tailed unpaired t tests. For experiments comparing more than two groups, data were analyzed using a one- (bar graphs) or two-way (kinetic graphs) ANOVA with Bonferroni correction for multiple comparisons. For survival analyses, a Log-rank (Mantel-Cox) test was performed. Significance indicated by * p < 0.05, ** p < 0.01, *** p < 0.001, ns=non-significant. Data shown are means ± SD.
Results and Discussion
To begin to assess the NK compartment in recipients following aHSCT, we utilized an MHC-matched (H2kb), non-MHC-mismatched donor – recipient strain combination (C3H.SW→B6) that results in mild GVHD and no lethality (Fig S1A, B). Nonetheless, we observed a clear deficit in the NK compartment early post-aHSCT as defined by both the percentage and numbers of NK cells (defined by an NKp46+CD122+CD3−CD19− phenotype) in the spleens of B6 recipients compared to syngeneic (B6→B6) transplant recipients (Fig 1A, B). This defect was observed 2–4 weeks post-aHSCT (Fig 1A, B).
Fig. 1.: NK cell recovery in recipients treated with expanded donor Tregs or PTCy is improved following aHSCT.
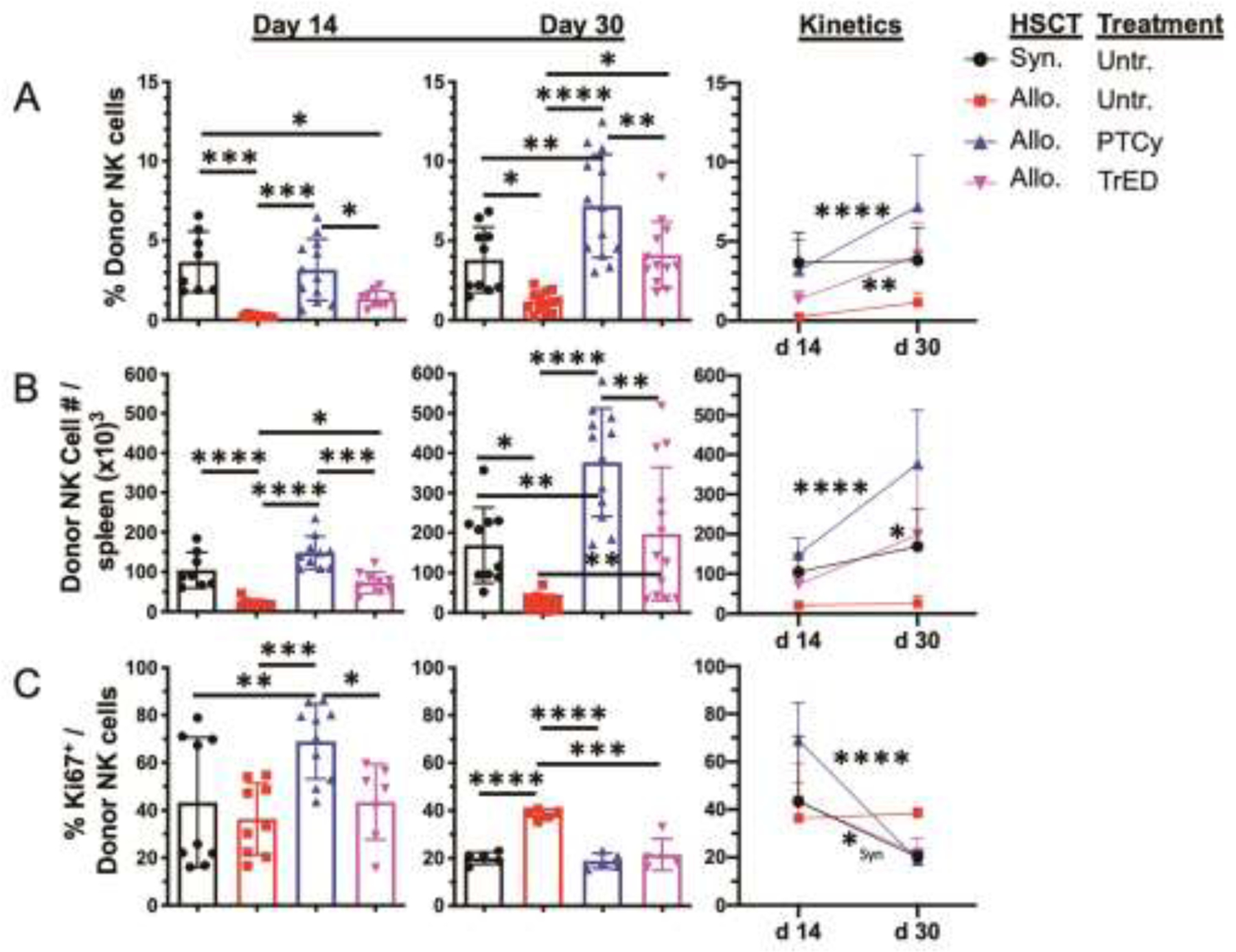
A HSCT utilizing a C3H.SW → B6 donor/recipient mouse model involving a minor MHC mismatch was performed on day 0. Lethally irradiated (10.5 Gy on day 0) B6 mice received 7×106 TCD C3H.SW BM cells and spleen+LN cells from expanded (TL1A-Ig/IL-2; TrED group) or untreated C3H.SW (GVHD and PTCy group) donor mice adjusted to contain 2×106 CD8 T cells. Cyclophosphamide was given on day 3 and 4 post-HSCT at 50 mg/kg ip. For the syngeneic HSCT 10×106 non-TCD BM cells plus 5×106 spleen cells from B6 mice were transplanted. (A) Percent (%) donor derived NK cells (CD3−CD19−NKp46+CD122+) on day 14 and 30 post HSCT in spleens are depicted. (B) Donor derived NK cell number (#) per spleen is shown on day 14 and 30 post HSCT. (C) Proliferation of donor NK cells (% Ki67) in spleen on day 14 and 30. n = 5–12 per group per time point. Data in column graphs are expressed as means ± SD and were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Data in kinetics graphs were analyzed by two-way ANOVA with Bonferroni correction for multiple comparisons. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. Data are pooled from 3 independent experiments, respectively.
Next, to examine the effect of GVHD prophylaxis on the NK compartment, groups of animals were transplanted with either donor cells from Treg expanded mice (TrED) or cells from unmanipulated mice that received cyclophosphamide on days 3 and 4 post-transplant (PTCy) at 50mg/kg/d (Fig 1). Notably, both frequency and numbers of total NK cells were elevated in recipients administered either PTCy or receiving TrED cells (Fig 1A,B).
Although both treatment strategies augmented the NK compartment, PTCy treatment yielded improved NK recovery as reflected by significantly increased frequencies and total numbers (Fig 1A, B). The percentages and numbers of NK cells also increased over time post-aHSCT in the PTCy and TrED transplanted recipients compared to untreated aHSCT recipients or syngeneic HSCT recipients (Fig 1A, B – right panels). Since one potential explanation for why PTCy-treated recipients had higher NK cell levels would be higher proliferation rates, we assessed NK proliferation by examining Ki67 staining at both time-points. First, Ki67 expression in NK cells was higher in both PTCy and TrED mice at Day 14 compared with their levels at Day 28, consistent with the increased numbers of NK cells in both groups of recipients. Second, the higher levels of Ki67 expression were present after two weeks in NK cells of PTCy treated vs TrED recipients (Fig 1C–left panels). This latter observation is consistent with the findings of greater percentage and numbers of NK cells in the former groups of recipients. Investigation of the marrow compartment revealed a similar pattern of percentage and numbers of donor NK cells two weeks post-aHSCT in the groups examined (Fig S1C).
One prior clinical study noted that PTCy treatment of recipients of haploidentical and HLA-matched transplants caused an early (~1 month) delay in NK recovery in the former group32. In the experimental studies here using MHC-matched recipients, PTCy treatment (and as noted above, use of TrED donors) increased recovery of the NK compartment compared to untreated aHSCT recipients. Interestingly, the findings here using the MHC-matched experimental aHSCT regarding NK cell proliferation post-transplant identified elevated proliferation within several weeks following PTCy treatment as reported following haplo-matched clinical transplants31.
To more granularly assess the NK compartment, established subsets of NK cells were evaluated as defined by phenotypic markers identifying Immature (CD27+CD11b−), M1 (CD27+CD11b+) and M2 (CD27−CD11b−) populations38 (Fig 2). Two weeks post-HSCT, only syngeneic transplant recipients had greater levels and numbers of M2 versus immature NK cells in contrast to all aHSCT recipients whose compartments consisted of primarily immature NK cells (Fig 2A, B). Analysis of NK subsets indicated that untreated aHSCT recipients had lower numbers of all NK subsets compared to syngeneic recipients as well as PTCy and TrED treated groups (Fig 2B). M2 NK cells, the most mature and cytolytic subset, are important in reducing GVHD39. Notably, the M2 NK frequency and numbers increased between 2 and 4 weeks post-HSCT in PTCy and TrED treated allogeneic recipients as well as syngeneic transplanted animals in contrast to untreated aHSCT recipients. (Fig 2C). Examination of the immature and M1 subset numbers indicated a trend toward higher levels in the treated groups compared to untreated aHSCT recipients although differences did not reach statistical significance (Fig S1D). One implication of these observations was that the functional capabilities of the total NK compartment in these groups likely differ due to the overall numbers and perhaps function of individual NK cells.
Fig. 2.: Donor NK cell subset recovery.
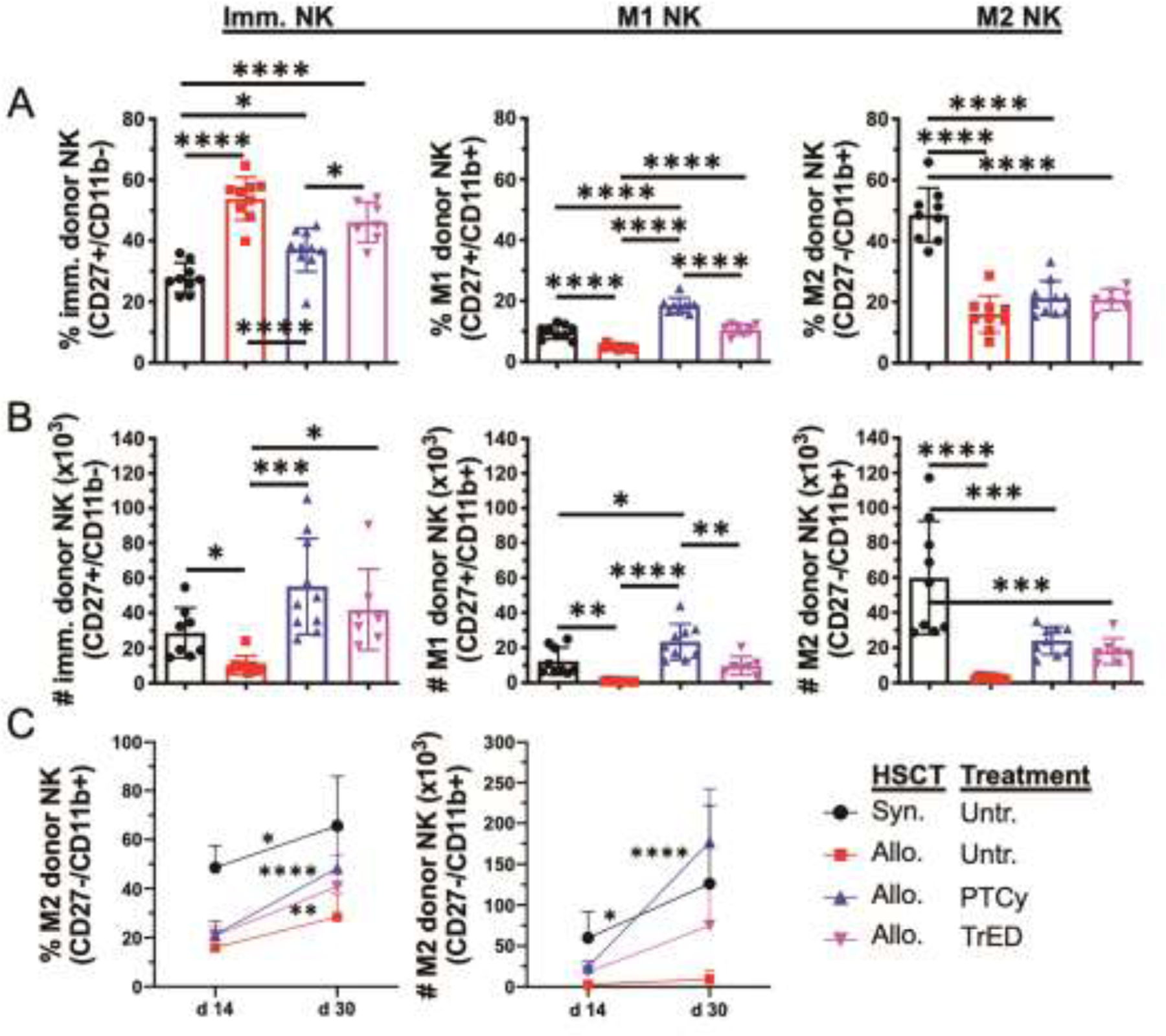
A HSCT was performed as in Fig 1. Donor NK cell subsets (immature [imm.] NK cells = CD27+/CD11b−; M1 NK cells = CD27+/CD11b+; M2 NK cells = CD27−/CD11b+) were examined in recipient spleens 14 days post HSCT (C3H.SW → B6). (A) Percent (%) and (B) Numbers (#) per spleen are shown (C) Kinetics of the M2 subset over time (% and #). Data are pooled from 3 independent experiments with n = 7–13 per group per time point. Data in column graphs are expressed as means ± SD and were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Data in kinetics graphs were analyzed by two-way ANOVA with Bonferroni correction for multiple comparisons. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
To address this hypothesis, we assessed the function of NK cells present in untreated and treated groups of recipient mice post-HSCT by examining interferon gamma (IFNγ) production and expression of the degranulation marker CD10740. Following in vitro stimulation with PMA + ionomycin, the frequency of splenic IFNγ-producing NK cells was significantly greater in allogeneic vs syngeneic HSCT recipients two and four weeks post-HSCT (Fig 3A). Since immature NK cells dominated the early reconstituting compartment (Fig 2B), their IFNγ production was assessed, and this same pattern in the allogeneic and syngeneic groups was detected (data not shown). The frequency of IFNγ+ NK cells increased over the first month post-transplant; therefore, NK cells from all groups of transplanted mice were capable of producing this effector cytokine (Fig 3A). However, at day 30 we did not detect functional cytokine (IFNγ) or cytotoxic impairment (see below) in PTCy or TrED treated MHC-matched recipients following HSCT compared to untreated HSCT recipients. Within this context, Rambaldi et. al. reported immature NK cells from PTCy-treated MHC-mismatched and MHC-matched recipients were functionally impaired (based on IFNγ, TNFα production) two months following clinical aHSCT, although these responses were compared to healthy donor NK cells32. It should be noted that another study reported that reconstituting NK cells - after in vitro stimulation - were capable of cytokine production similar to healthy controls beginning at one-month post-HSCT (HLA-matched reduced intensity conditioning)41.
Fig. 3.: Functional assessment of NK cells in vitro and in vivo following minor mismatch aHSCT.
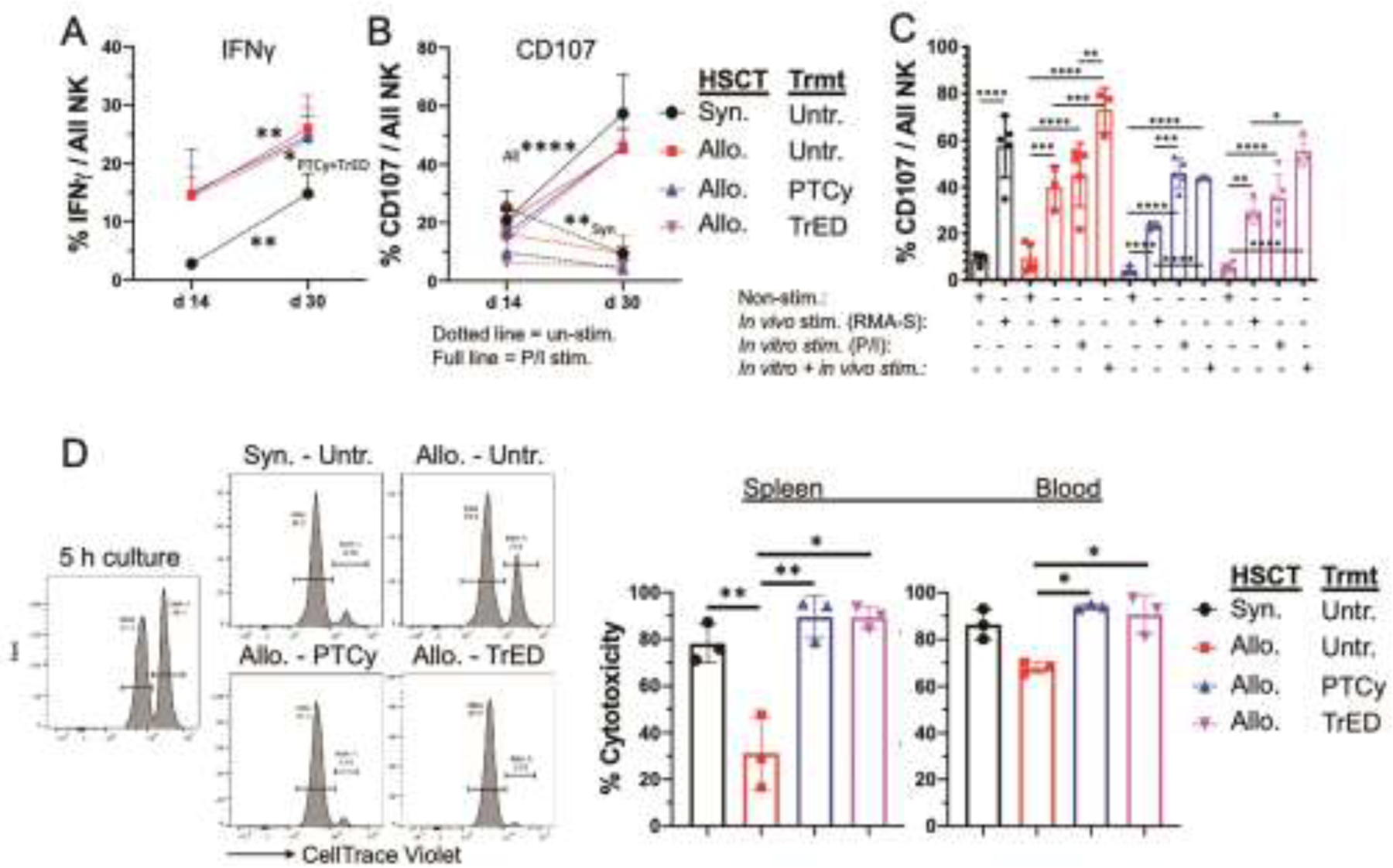
The (C3H.SW → B6 experimental transplant model was performed as in Fig 1. (A) Kinetics of IFNγ – producing NK cells on day 14 and day 30 is shown after 5 h of in vitro stimulation with PMA (50 ng/ml) and Ionomycin (500 ng/ml) in the presence of 1 μl/ml BD GolgiStop. n = 5 per group. (B,C) Expression of the degranulation marker CD107 after in vitro and in vivo stimulation. (B) CD107 expression on unstimulated (dotted line) and in vitro stimulated NK cells (full line; 5 h, PMA+Ionomycin stimulation as in A) on day 14 and 30. n = 4–5 per group. (C) CD107 expression on NK cells on day 30 after in vivo (1×106 iv injected RMA-S cell for 18 h), in vitro (5 h PMA + Ionomycin as in A) or double stimulation (in vivo + in vitro). n = 3–5 per group. Data are pooled from 2 independent experiments (A-C). (D) In vivo cytotoxicity assay. On day 30 post HSCT, 10×106 NK resistant RMA and 10×106 NK sensitive RMA-S cells, respectively, were labeled with low (0.4 uM) and high (4 uM) amounts of CellTrace Violet (CTV) and injected iv into recipients. 5 h later, the killing of the NK sensitive RMA-S cells was quantified in blood and spleen. Representative histograms and graphs are shown from one of 2 experiments. n = 3 per group. Data in kinetics graphs are expressed as means ± SD and were analyzed by two-way ANOVA with Bonferroni correction for multiple comparisons. Data in column graphs are expressed as means ± SD and were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Next, CD107 expression was examined in unstimulated and PMA + ionomycin stimulated NK cells. While unstimulated cells expressed low levels which decreased further between 2 weeks and one-month post-HSCT, the CD107 levels in stimulated cells significantly increased in NK cells from all groups of transplanted mice (Fig 3B). To evaluate CD107 expression after in vivo stimulation, we used NK-sensitive RMA-S cells. We posit the upregulation of CD107 on NK cells examined resulted from RMA-S cell presence in all transplanted groups (Fig 3C). These observations support the notion there is no endogenous defect in cytotoxic function in NK cells from all groups of transplanted mice one-month post-HSCT.
To directly assess overall NK function in each group of transplanted mice, in vivo cytotoxicity was examined (Fig 3D). Therefore, CellTrace Violet labeled NK-sensitive (RMA-S; MHC I deficient) and NK-resistant (RMA) cells were adoptively transferred at a ratio 1:1 into recipients and after 5h the relative levels of RMA-S/RMA were evaluated to determine the level of killing. These studies demonstrated that the cytotoxic capacity of the NK compartments (in spleen and peripheral blood) in both PTCy and TrED treated recipients were greater versus that in untreated transplant recipients (Fig 3D). These findings correlated with the numbers of NK cells detected in the three groups of recipient animals, i.e. untreated animals had the lowest numbers of total NK cells and both PTCy and TrED treated mice contained significantly higher numbers of all splenic NK cell subset compartments (Fig 1B, 2B). The reconstitution of the peripheral NK compartment in an independent experimental MHC-matched aHSCT was previously reported to be delayed during the first month post-transplant in untreated animals as observed in the present studies42. Regarding NK cytotoxic function in those untreated recipients, cytotoxicity was examined 2 weeks post-HSCT. Results in the MHC-matched model examined here clearly indicated that PTCy and TrED treatment improved cytotoxic NK function by one-month post-aHSCT.
In addition to GVHD prophylaxis treatment, the presence of virus and tumor post-HSCT has been reported to impact the NK compartment. For example, studies have been reported examining the effect of CMV reactivation which accelerated the emergence of mature NK cells following HLA-haploidentical TCD HSCT43. Tumor presence in un-transplanted patients has also been reported to affect NK cells. A relationship of NK cells to myeloid malignancies (CML, AML, MDS) was detected, observing an inverse correlation of NK levels and tumor burden and some studies reported a selective loss of immature NK cells(reviewed in 44). Additionally, transcriptomic analysis recently indicated NK function was affected by AML tumor presence45. While the studies here did not examine NK reconstitution in the presence of tumor, further investigation will be required to begin dissecting the individual contributions of tumor presence plus treatment for GVHD prophylaxis.
In summary, the NK cell compartment in the experimental model studied here, as in the clinical setting, recovers robustly following PTCy treatment31, 32. Notably, our studies suggest that the total numbers and functional capability of NK cells post-aHSCT were improved compared with untreated HSCT recipients using either PTCy or TrED as GVHD prophylaxis, thereby strengthening anti-pathogen and tumor immune function. These observations further suggest that since mouse NK and Treg cells can promote donor hematopoietic engraftment, these treatments likely facilitate beneficial transplant outcomes mediated by these critical cellular compartments3, 25, 46, 47.
Supplementary Material
Highlights.
PTCy and Treg treatment for GVHD prophylaxis improves NK reconstitution post-MHC matched aHSCT
Effector function of the NK compartment is augmented after PTCy or Treg treatment
Acknowledgements
We are grateful to Dr. Eli Gilboa for providing the RMA and RMA-S cell lines and Despina Kolonias for assistance with these studies. We also thank the staff at the Division of Veterinary Resources at the University of Miami Miller School of Medicine for their outstanding animal maintenance and care of the mice used in these studies as well as the Sylvester Comprehensive Cancer Center Flow Cytometry Core for technical support.
Funding.
This work was supported by funds from the NIH RO1EY024484-06, R01EY030283-01, R41AI149916-01 (RBL); Kalish Family Foundation and the Applebaum Foundation (KVK); and the Sylvester Comprehensive Cancer Center (RBL, KVK). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure Statement
RBL is a compensated consultant/advisory board member for and equity holder in Heat Biologics. KVK is an ad hoc consultant for Janssen, Novartis, Genentech/Roche, Kite, Takeda, Iovance, United Healthcare, Avacta Therapeutics and Kiadis. None are directly relevant to the efforts of this study. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability:
Data reported in the manuscript will be shared under the terms of a Data Use Agreement and may only be used for approved proposals. Requests may be made to: dwolf@med.miami.edu.
References
- 1.Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115:4293–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asai O, Longo DL, Tian ZG, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest. 1998;101:1835–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy WJ, Bennett M, Kumar V, Longo DL. Donor-type activated natural killer cells promote marrow engraftment and B cell development during allogeneic bone marrow transplantation. J Immunol. 1992;148:2953–2960. [PubMed] [Google Scholar]
- 4.Hu B, He Y, Wu Y, et al. Activated allogeneic NK cells as suppressors of alloreactive responses. Biol Blood Marrow Transplant. 2010;16:772–781. [DOI] [PubMed] [Google Scholar]
- 5.Barao I, Hanash AM, Hallett W, et al. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:5460–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonetta F, Alvarez M, Negrin RS. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front Immunol. 2017;8:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogonek J, Kralj Juric M, Ghimire S, et al. Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol. 2016;7:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ullrich E, Salzmann-Manrique E, Bakhtiar S, et al. Relation between Acute GVHD and NK Cell Subset Reconstitution Following Allogeneic Stem Cell Transplantation. Front Immunol. 2016;7:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Witte MA, Sarhan D, Davis Z, et al. Early Reconstitution of NK and gammadelta T Cells and Its Implication for the Design of Post-Transplant Immunotherapy. Biol Blood Marrow Transplant. 2018;24:1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ullah MA, Hill GR, Tey SK. Functional Reconstitution of Natural Killer Cells in Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol. 2016;7:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komanduri KV, St John LS, de Lima M, et al. Delayed immune reconstitution after cord blood transplantation is characterized by impaired thymopoiesis and late memory T-cell skewing. Blood. 2007;110:4543–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luznik L, Engstrom LW, Iannone R, Fuchs EJ. Posttransplantation cyclophosphamide facilitates engraftment of major histocompatibility complex-identical allogeneic marrow in mice conditioned with low-dose total body irradiation. Biol Blood Marrow Transplant. 2002;8:131–138. [DOI] [PubMed] [Google Scholar]
- 13.Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luznik L, Bolanos-Meade J, Zahurak M, et al. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood. 2010;115:3224–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross D, Jones M, Komanduri K, Levy RB. Antigen and lymphopenia-driven donor T cells are differentially diminished by post-transplantation administration of cyclophosphamide after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2013;19:1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganguly S, Ross DB, Panoskaltsis-Mortari A, et al. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood. 2014;124:2131–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw BE, Jimenez-Jimenez AM, Burns LJ, et al. National Marrow Donor Program-Sponsored Multicenter, Phase II Trial of HLA-Mismatched Unrelated Donor Bone Marrow Transplantation Using Post-Transplant Cyclophosphamide. J Clin Oncol. 2021;39:1971–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanakry JA, Kasamon YL, Bolanos-Meade J, et al. Absence of post-transplantation lymphoproliferative disorder after allogeneic blood or marrow transplantation using post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant. 2013;19:1514–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanakry CG, O’Donnell PV, Furlong T, et al. Multi-institutional study of post-transplantation cyclophosphamide as single-agent graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation using myeloablative busulfan and fludarabine conditioning. J Clin Oncol. 2014;32:3497–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanakry CG, Tsai HL, Bolanos-Meade J, et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-matched BMT for AML, ALL, and MDS. Blood. 2014;124:3817–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacoby E, Chen A, Loeb DM, et al. Single-Agent Post-Transplantation Cyclophosphamide as Graf-versus-Host Disease Prophylaxis after Human Leukocyte Antigen-Matched Related Bone Marrow Transplantation for Pediatric and Young Adult Patients with Hematologic Malignancies. Biol Blood Marrow Transplant. 2016;22:112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.George B, Pn N, Devasia AJ, et al. Post-Transplant Cyclophosphamide as Sole Graft-versus-Host Disease Prophylaxis Is Feasible in Patients Undergoing Peripheral Blood Stem Cell Transplantation for Severe Aplastic Anemia Using Matched Sibling Donors. Biol Blood Marrow Transplant. 2018;24:494–500. [DOI] [PubMed] [Google Scholar]
- 23.Williams L, Cirrone F, Cole K, Abdul-Hay M, Luznik L, Al-Homsi AS. Post-transplantation Cyclophosphamide: From HLA-Haploidentical to Matched-Related and Matched-Unrelated Donor Blood and Marrow Transplantation. Front Immunol. 2020;11:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. [DOI] [PubMed] [Google Scholar]
- 25.Hanash AM, Levy RB. Donor CD4+CD25+ T cells promote engraftment and tolerance following MHC-mismatched hematopoietic cell transplantation. Blood. 2005;105:1828–1836. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hefazi M, Bolivar-Wagers S, Blazar BR. Regulatory T Cell Therapy of Graft-versus-Host Disease: Advances and Challenges. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldmann H Regulatory T cells and transplantation tolerance: Emerging from the darkness? Eur J Immunol. 2021;51:1580–1591. [DOI] [PubMed] [Google Scholar]
- 29.Wolf D, Barreras H, Bader CS, et al. Marked in Vivo Donor Regulatory T Cell Expansion via Interleukin-2 and TL1A-Ig Stimulation Ameliorates Graft-versus-Host Disease but Preserves Graft-versus-Leukemia in Recipients after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2017;23:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf D, Bader CS, Barreras H, et al. Superior immune reconstitution using Treg-expanded donor cells versus PTCy treatment in preclinical HSCT models. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russo A, Oliveira G, Berglund S, et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: dynamics and clinical implications. Blood. 2018;131:247–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rambaldi B, Kim HT, Reynolds C, et al. Impaired T- and NK-cell reconstitution after haploidentical HCT with posttransplant cyclophosphamide. Blood Adv. 2021;5:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCurdy SR, Radojcic V, Tsai HL, et al. Signatures of GVHD and relapse after posttransplant cyclophosphamide revealed by immune profiling and machine learning. Blood. 2022;139:608–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ljunggren HG, Ohlen C, Hoglund P, Franksson L, Karre K. The RMA-S lymphoma mutant; consequences of a peptide loading defect on immunological recognition and graft rejection. Int J Cancer Suppl. 1991;6:38–44. [DOI] [PubMed] [Google Scholar]
- 35.Nair SK, Snyder D, Gilboa E. Cells treated with TAP-2 antisense oligonucleotides are potent antigen-presenting cells in vitro and in vivo. J Immunol. 1996;156:1772–1780. [PubMed] [Google Scholar]
- 36.Khan SQ, Tsai MS, Schreiber TH, Wolf D, Deyev VV, Podack ER. Cloning, expression, and functional characterization of TL1A-Ig. J Immunol. 2013;190:1540–1550. [DOI] [PubMed] [Google Scholar]
- 37.Quah BJ, Wijesundara DK, Ranasinghe C, Parish CR. Fluorescent target array T helper assay: a multiplex flow cytometry assay to measure antigen-specific CD4+ T cell-mediated B cell help in vivo. J Immunol Methods. 2013;387:181–190. [DOI] [PubMed] [Google Scholar]
- 38.Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol. 2018;9:1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meinhardt K, Kroeger I, Bauer R, et al. Identification and characterization of the specific murine NK cell subset supporting graft-versus-leukemia- and reducing graft-versus-host-effects. Oncoimmunology. 2015;4:e981483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. [DOI] [PubMed] [Google Scholar]
- 41.Pical-Izard C, Crocchiolo R, Granjeaud S, et al. Reconstitution of natural killer cells in HLA-matched HSCT after reduced-intensity conditioning: impact on clinical outcome. Biol Blood Marrow Transplant. 2015;21:429–439. [DOI] [PubMed] [Google Scholar]
- 42.Bunting MD, Varelias A, Souza-Fonseca-Guimaraes F, et al. GVHD prevents NK-cell-dependent leukemia and virus-specific innate immunity. Blood. 2017;129:630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muccio L, Bertaina A, Falco M, et al. Analysis of memory-like natural killer cells in human cytomegalovirus-infected children undergoing alphabeta+T and B cell-depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica. 2016;101:371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlsten M, Jaras M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front Immunol. 2019;10:2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crinier A, Dumas PY, Escaliere B, et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell Mol Immunol. 2021;18:1290–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. [DOI] [PubMed] [Google Scholar]
- 47.Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on engraftment, graft-versus-tumor, and graft-versus-host responses. Biol Blood Marrow Transplant. 2009;15:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in the manuscript will be shared under the terms of a Data Use Agreement and may only be used for approved proposals. Requests may be made to: dwolf@med.miami.edu.