

Abstract
NUT carcinoma (NC) is a rare, aggressive cancer defined by rearrangements of the NUTM1 gene. No routinely effective treatments of NC exist, despite harboring a targetable oncoprotein, most commonly BRD4-NUT. The vast majority of cases are fatal. Poor awareness of the disease is a major obstacle to progress in the treatment of NC. While the incidence likely exceeds that of Ewing sarcoma, and BRD4-NUT heralded the Bromodomain and Extra-Terminal domain (BET) inhibitor class of selective epigenetic modulators, NC is incorrectly perceived as ‘impossibly rare’, and therefore receives comparatively little private or governmental funding or prioritization by pharma. To raise awareness, propagate scientific knowledge, and initiate a consensus on standard and targeted treatment of NC, we held the First International Symposium on NUT Carcinoma on March 3rd, 2021. This virtual event had over eighty attendees from the Americas, Europe, Asia, and Australia. NC patient and family members were represented and shared perspectives. Broadly, the four areas discussed by experts in the field included (1) the biology of NC; (2) standard approaches to the treatment of NC; (3) results of clinical trials using BET inhibitors; and (4) future directions, including novel BET bromodomain inhibitors, combinatorial approaches and immunotherapy. It was concluded that standard chemotherapeutic approaches and first-generation BET bromodomain inhibitors, the latter complicated by a narrow therapeutic window, are only modestly effective in a minority of cases. Nonetheless, emerging second-generation targeted inhibitors, novel rational synergistic combinations and the incorporation of immuno-oncology approaches hold promise to improve the prognosis of this disease.
Keywords: NUT carcinoma, BRD4-NUTM1, BET bromodomain inhibitor, epigenetics, fusion oncogene
Introduction
The First International NUT Carcinoma Symposium was convened with several specific goals. The first goal was to promote advocacy for NUT Carcinoma (NC) by establishing a community of stakeholders invested in the awareness, treatment, and support of NC patients. The NC community is supported through the critical role of the International NUT Carcinoma Registry (www.NMCRegistry.org). Enrollment of patients in this registry allows collection of clinical data and tissue to capture treatments and clinical outcomes, enable full molecular characterization of tumors, and facilitate development of NC models suitable for preclinical and translational work. Advocacy will also require the participation of everyone impacted by or studying in the disease, including patients, family members, basic and translational researchers and clinical providers. The second goal was to establish consensus on standard approaches to treatment of NC, including the use of chemotherapy, radiation therapy, and surgery. The third goal was to promote rapid conduct of clinical trials. The symposium included talks by early drug development leaders on the current status of BET bromodomain inhibition, one of few investigational targeted therapies for NC, as well as additional talks focused on various monotherapies, patient selection, novel chemistry, and innovative combinatorial strategies. The final goal was to establish future directions for NC treatment, with a focus on the incorporation of immuno-oncology approaches.
Landscape and Biology of NUT Carcinoma
Overview.
NUT carcinoma (NC) represents an unmet need, with a list of multiple critical problems including (1) highly aggressive biology, with a median overall survival (OS) of 6.5 months (95%CI=5.8-9.1) (2); (2) prevalence in the adolescent and young adult (AYA) population, although the disease may affect patients of all ages; (3) poor domestic and international awareness of the disease, resulting in substantial underdiagnosis; and (4) absence of effective treatments. The poor awareness of NC is a critical challenge because it has led to poor funding for research and patient care/advocacy; moreover, it is a significant reason for late or misdiagnosis that results in delayed treatment and missed opportunities for curative treatment. The aggressive biology and patient demographics of NC cannot be changed; however, awareness, diagnostic testing, and treatment of NC can be vastly improved. With improved treatment, and development of standards of care for NC, there will be a strong imperative to test for and diagnose NC, which may ultimately translate to improved outcomes. Thus, the Symposium focused primarily on therapeutic approaches for NC, as it is expected that effective treatments will help tackle the other problems of aggressive biology, poor awareness, and under-diagnosis.
NC pathogenesis.
A substantial body of evidence, including the lack of collaborative oncogenic mutations, indicates that NC is driven entirely by NUT-fusion oncoproteins, most commonly BRD4-NUT (2, 3). Other less common drivers of NC include BRD3-NUT (4), NSD3-NUT (5), and ZNF-NUT (6, 7) fusions. BRD4-NUT generally functions to block differentiation and maintain growth of NC cells. It is believed to do so by hijacking the normal functions of native, wild-type BRD4 (8), a transcriptional activator. BRD4 is a member of the BET family of proteins that contain two acetyl-histone-binding bromodomains (BD) and an extra-terminal (ET), protein-interaction domain. All three domains are contained within the canonical BRD4-NUT fusion (9). Through its dual BDs, BRD4 binds to active, acetylated chromatin, and activates transcription through the recruitment of p-TEFb (cyclinT1/CDK9), which phosphorylates RNA polymerase II to ensure transcriptional elongation. (10–12) NUT, unlike the ubiquitously expressed BRD4, is restricted to testes (3) where it functions to promote spermiogenesis (13). It binds to and activates the histone acetyltransferase (HAT) p300, a factor required for enhancer function and the transcription of many genes (14). Thus, the combination of NUT with BRD4 is predicted to super-charge the normal transcriptional activating function of wild-type BRD4 through the recruitment of p300 (8). Indeed, BRD4-NUT, when either endogenously or ectopically expressed, leads to the formation of massive, super-enhancer-like, p300 and acetyl-histone-enriched ‘megadomains’ that are associated with increased transcription of associated genes (15, 16). Across nearly all NCs, megadomain-associated upregulated genes include the well- known proto-oncogenes MYC, and SOX2 (15, 16) (Fig. 1).
Fig. 1.
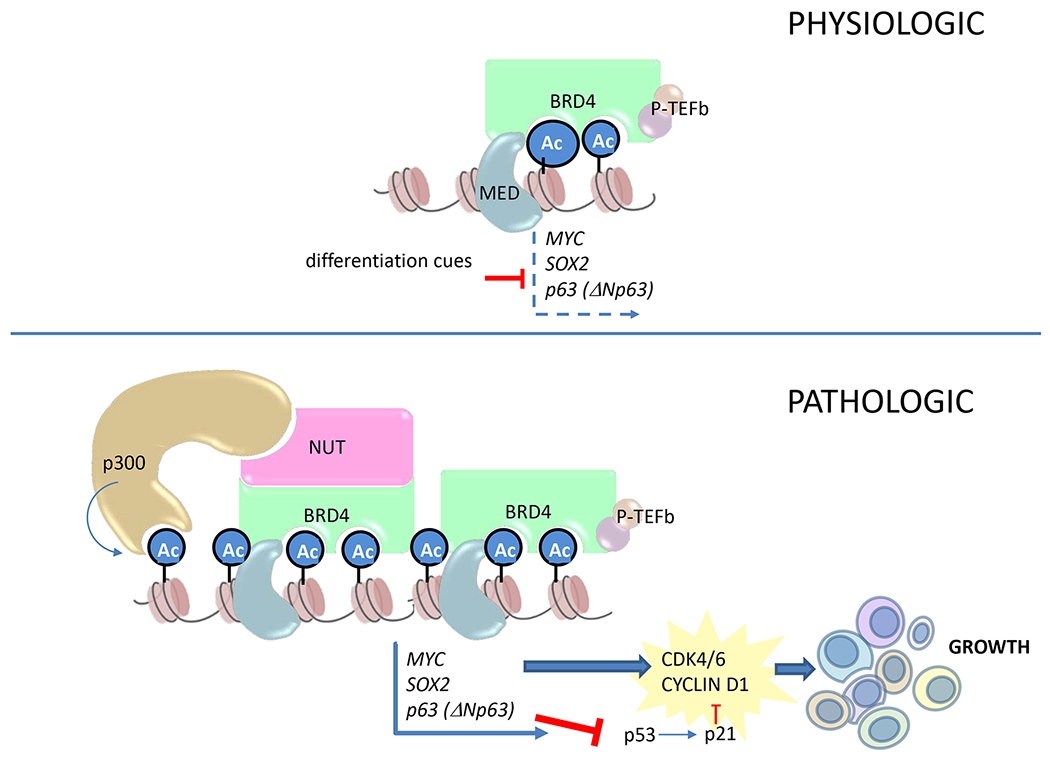
Mechanistic model of how BRD4-NUT drives growth and blocks differentiation in NUT carcinoma. Above, the normal function of BRD4 is to facilitate transcriptional elongation in a manner that can be regulated by changes in cell state, such as pro-differentiative signaling (i.e. TGF-beta signaling). Below, in the pathologic state, when BRD4 is fused to NUT, the recruitment of p300 leads to the formation of hyperacetylated megadomains whose transcriptional activation of oncogenic target genes are resistant to negative regulatory signals due to their large size. Ac, acetyl-lysine; MED, mediator complex. Material from: Eagen K and French CA, Supercharging BRD4 with NUT in carcinoma, Oncogene, 2021, Springer Nature (8).
Paradoxically, histone de-acetylases (HDAC) are required for the maintenance of megadomains. Indeed, HDAC inhibition using small molecule inhibitors such as panobinostat, leads to depletion of BRD4-NUT and acetylated (H3K27Ac) chromatin from megadomains, and increased acetylation in regions that are distant from megadomains (17). It is hypothesized that this occurs through recruitment of BRD4-NUT and p300 to newly acetylated chromatin away from megadomains, causing redistribution of limiting quantities of these and likely other factors, resulting in megadomain collapse.
It is believed that BRD4-NUT function does not replace, but rather requires that of wild-type BRD4 because there are no documented cases of wild-type BRD4 loss in NC, and protein pull-downs of BRD4-NUT invariably contain high levels of wild-type BRD4 and its interacting proteins, such as p-TEFb (6). These observations have led to a model where BRD4-NUT drives and maintains tumor cell growth by three general mechanisms (8) (Fig. 1). First, it pathologically stabilizes transcription of oncogenic targets, such as MYC and SOX2, through p300-mediated hyperacetylation of associated enhancers (6, 15). Second, it promotes cell cycle progression through MYC-induced activation of cyclins D1 and D2 (18–21). Finally, it inhibits apoptosis through repression of TP53, either by sequestration or upregulation of the dominant negative homologue, δNp63 (22).
This model of BRD4-NUT oncogenic function reveals several mechanistic dependencies, including those on cyclin D1/CDK4/6, histone acetyl-transferase, bromodomain-acetyl-histone binding, and kinase-mediated transcriptional elongation, all of which can be leveraged therapeutically using targeted agents (Fig. 2). Several of these dependencies form the basis of novel approaches to build on BET inhibitor monotherapy.
Fig. 2.
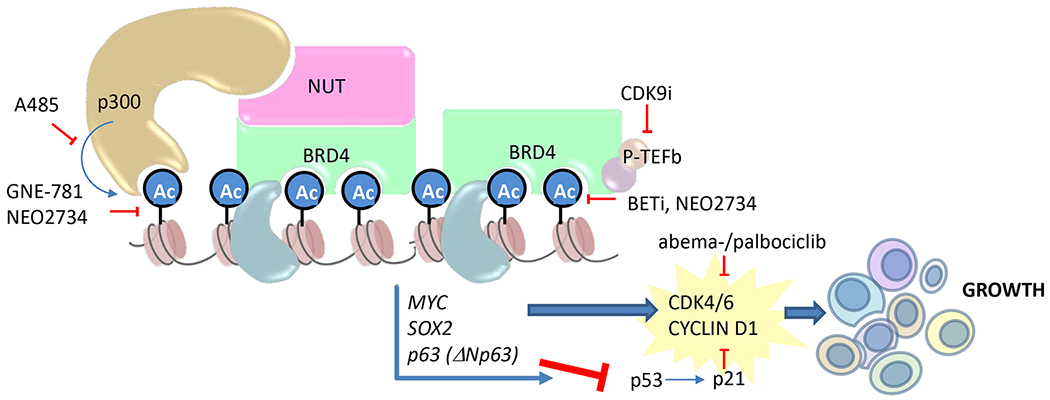
Therapeutic vulnerabilities of NUT carcinoma to small molecule inhibitors. Shown are small molecule inhibitors of p300/CBP histone acetyltransferase (A485), p300/CBP bromodomains (GNE-781), BET bromodomains (BETi), p300/CBP and BET bromodomains (NEO2734), CDK4/6 (abemaciclib and palbociclib), and the CDK9 component of P-TEFb (CDK9i). Material from: Eagen K and French CA, Supercharging BRD4 with NUT in carcinoma, Oncogene, 2021, Springer Nature (8).
Diagnostic considerations.
Despite the widespread under-testing for NUT carcinoma, the diagnosis itself is straightforward and can be made in any pathology laboratory that has the diagnostic anti-NUT rabbit monoclonal antibody, clone C52B1 (Cell Signaling Technologies, Danvers, MA). NUT expression is restricted to testes, and thus expression outside testes can be reliably used to make the diagnosis of NUT carcinoma. Haack et al (23) found that, using a cutoff of ≥ 50%, nuclear NUT immunohistochemical positivity with the C52B1 antibody was 100% specific and 87% sensitive for the diagnosis of NC among a large cohort of primarily carcinomas. While wild-type NUT is expressed in germ cell tumors, its expression using this antibody is only present in ~5% of tumor nuclei. The specificity of NUT immunohistochemistry is so high, that demonstration of NUTM1 rearrangement by molecular testing (fluorescent in situ hybridization (FISH), or next-generation sequencing) is not needed to confirm the diagnosis, though may be useful prognostically by identifying the fusion partner, as discussed below, or to identify very rare non-NC sarcomas with disease-specific NUTM1-fusion partners(24).
Treatment of NC using standard-of-care modalities
Pediatric experience.
NUT carcinoma occurs in patients of all ages, with an age range of 0-81 years (2), yet the median age is 23.6 years and many patients are in their teens at diagnosis. The disease spectrum, in children (i.e., patients ≤ 18 years), including outcomes [OS and progression free survival (PFS)], primary anatomic sites, and NUT translocations, is very similar to that in adults (2). Indeed, while there is a trend for outcomes in children to be slightly better than in adults [HR = 1.5 (95%CI = 1-2.3)], the difference is not significant (p = 0.06) (2).
The use of ifosfamide-based regimens, particularly the Scandinavian Sarcoma Group IX (SSG IX) regimen for Ewing sarcoma, has demonstrated anecdotal successes in pediatric NC, with four long term complete responses (CR) or cures reported (25–27). It is not known which component(s) of this regimen account for activity in pediatric NC.
As an epigenetic disease, NC is an excellent candidate for therapeutic epigenetic modifiers. Indeed, transient responses to the histone deacetylase inhibitor vorinostat have been described (28, 29). However, there has been a long lag time in opening clinical trials to children compared with that for adults. Whereas most adult clinical trials evaluating the use of BET bromodomain inhibitors have been ongoing since 2012, the first phase I clinical trial evaluating BMS-986158 began enrolling children in 2019. The BMS trial has multiple sites open in US and Canada (NCT03936465). On the basis that disease characteristics of NC in children are similar to those observed in adults, particularly with respect to aggressive biology, there was consensus that the age eligibility for novel targeted inhibitor trials enrolling NC patients should be lowered to 12 years.
Thoracic experience.
Among primary lung cancers, NC is rare, with one report citing an incidence of 0.6% of non-glandular lung carcinomas (30). The thorax is the most common site of origin of NCs, comprising ~50% of tumors. In the aforementioned study, survival tree regression identified three statistically distinct risk groups among 124 patients classified by anatomical site and genetics, with the thoracic primary group demonstrating the worst outcomes (n = 67, median OS = 4.4 months, 95% CI = 3.5 to 5.6 months) (2, 31). Diagnostically, NC can be challenging due to its poorly differentiated morphology. Because it is a subtype of squamous cell carcinoma, it is most commonly misdiagnosed as standard poorly differentiated squamous cancer. p63, a squamous lineage marker, is often used to identify NC in the initial diagnostic workup; however, its δNp63 isoform, p40, which is widely considered a superior marker for identification of squamous cancer, is a less sensitive marker than p63 for NC. Therefore, a negative p40 immunohistochemical stain can lead to missing the diagnosis of NC (32). Due to these diagnostic challenges, it is recommended that in non-glandular poorly differentiated carcinomas of the thorax that lack common mutations (i.e. KRAS, EGFR, EML4-ALK, BRAF, etc.), NUT immunohistochemistry be performed, and if positive, that RNA-based next generation sequencing (NGS) be implemented to identify the NUTM1-fusion gene.
Aggressive control of localized disease with surgery and radiation is the most effective treatment of NC (33) and represents the only chance for long-term survival. However, thoracic NC is rarely diagnosed at an early enough stage for local management. Thus, systemic therapy is nearly always required. Cytotoxic chemotherapy regimens that have been used include ifosfamide/etoposide, ifosfamide/etoposide/vorinostat, etoposide/platinum, or platinum/paclitaxel, amongst others. Reports of durable complete responses in pediatric patients using ifosfamide-based regimens(25–27) have led to growing interest in treating thoracic NC patients upfront with this approach, particularly in patients with good performance status.
An emerging concept in NC is the potential incorporation of immune checkpoint inhibitors. Because NC is driven entirely by NUT-fusion oncoproteins, the tumors have only low-to-modest tumor mutation burdens (34, 35), yet a subset of tumors do express PD-L1. Two studies have reported durable disease stability in PD-L1-high NCs treated with nivolumab or pembrolizumab (36, 37).
Despite aggressive conventional approaches to thoracic NC, the consensus is that novel approaches or combinations will be needed to improve currently dismal outcomes.
Head and neck experience.
The head and neck region is the second most common primary site of NCs, comprising 40% of all cases. As with thoracic NC, the most common translocation is BRD4-NUT (~72%), the prognosis is dismal (median OS=9.7 months), and for disseminated cases that cannot be managed with local control using surgery and/or radiation with or without chemotherapy, systemic agents are generally not effective (31, 33). Notably, however, there are four published examples of complete (CR) or partial responses (PR) of localized or disseminated head and neck disease to ifosfamide-based regimens (SSG IX or ifosfamide/etoposide/vorinostat); all patients were < 18 years old (26, 27). For localized disease, upfront complete oncologic surgical resection is associated with significantly improved outcomes, including 2yr PFS (p = 0.01) and OS (p = 0.01), so that this approach is preferred for resectable disease (33).
Based on the above outcome data that underscores the aggressiveness of NC (2, 31, 33), a treatment paradigm for management of head and neck NC was proposed including aggressive upfront surgery and adjuvant chemoradiotherapy (type depends on patient age and performance status) for localized, resectable disease; definitive chemoradiation with or without the incorporation of induction chemotherapy for unresectable localized disease; and systemic chemotherapy with or without palliative radiotherapy for disseminated disease (Fig. 3).
Fig. 3.
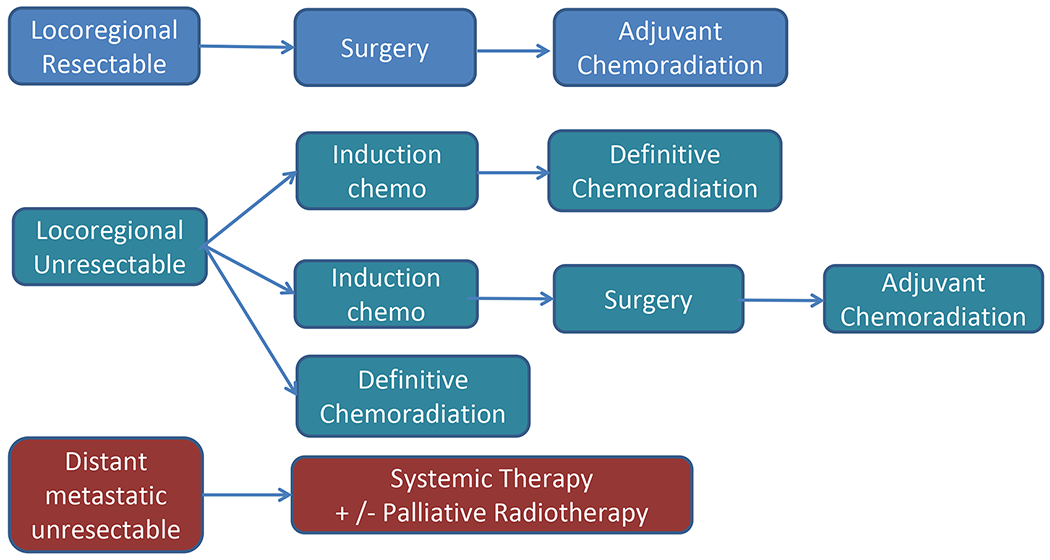
Initial paradigm for treatment of head and neck NUT carcinoma.
Recently it was found that NC can be divided into three statistically significant prognostic risk groups based on the anatomic location and type of NUT fusion, independent of stage. The best prognostic group, and the smallest, includes patients with non-thoracic primaries, primarily head and neck, and non-BRD4-NUT fusions, typically BRD3- and NSD3-NUT (median OS=36.5 months [95%CI=12.5-NR]). The middle group is comprised of patients with non-thoracic, mostly head and neck primaries, and BRD4-NUT fusions (median OS=10 months [95%CI=7-14.6]). The group with the worst prognosis includes patients with thoracic primaries, regardless of the NUT-fusion type (n=67, median OS=4.4 months [95%CI=3.5-5.6]) (2).
Unpublished data from a small cohort of head and neck NC patients, provided by Dr. Glenn Hanna, demonstrated high PD-L1 expression [combined positive score (CPS) > 10] in two of five patients. Based on these findings and those above for thoracic patients, there was a consensus.recommendation to consider the addition of immune checkpoint blockade to the treatment paradigm of non-surgically treated NC patients with PD-L1+ disease.
BET inhibitor monotherapy clinical trial experience
Mechanistic rationale.
BRD4 and BRD4-NUT activate transcription through the recruitment of key factors, including the mediator complex and p-TEFb, to acetylated chromatin in enhancer and promoter regions; the addition of p300 recruitment by NUT is thought to activate transcription above normal levels (8, 15, 16, 38–40). The recruitment of these factors to chromatin is dependent upon the direct binding of the dual bromodomains of BRD4 to acetylated lysine residues on histones (9–11, 41). The bromodomain-acetyl-lysine interaction thus represents a therapeutic vulnerability in cancers that are dependent on BRD4, including NC (Fig. 2). NCs with non-BRD4-NUTM1 fusions, such as those with BRD3-NUTM1, NSD3-NUTM1, or ZNF532-NUTM1, are also BRD4-dependent and sensitive to BET inhibitors because the NUT fusion partners all interact with BRD4 (5, 16, 38, 39). Thus BRD4, BRD3, NSD3, and ZNF532 are all interchangeable as NUT-fusion partners.
In 2010, BET bromodomain inhibitors were first described using NC as the cancer model (42). This class of small molecule inhibitors consist of acetyl-lysine mimetics that competitively inhibit the bromodomain-acetyl-histone interaction, leading to eviction of BET proteins from chromatin. NC, which to date is the only cancer known to be driven by oncogenic BRD4, represents the most robust cancer model in which to assess the anti-oncogenic effects of BET bromodomain inhibition. Indeed, investigators found that treatment of patient-derived NC cells led to rapid dispersion of BRD4-NUT from chromatin, inducing arrested growth and differentiation both in vitro and in vivo (42). The introduction of the first-in-class BET bromodomain inhibitor tool compound, JQ1, in NC led to investigation of other BRD4-dependent cancers and a surge of investigations the normal and pathological biological role of BRD4 in a broad range of normal and pathologic states, primarily cancer (38, 40, 43–58). In the cancer context, upregulation of MYC by BRD4 was found to be targeted by BET bromodomain inhibition, and represents an indirect means by which to target undruggable MYC in MYC-driven cancers (43, 44, 48, 52).
The on-target efficacy in suppressing tumor growth in pre-clinical models of NC, and the demonstration of on-target activity of one clinical BET inhibitor (BETi), birabresib (OTX015, MK-8628, Merck/OncoEthix) (59, 60), administered on expanded access to four NC patients, provided adequate rationale for initial clinical trials (42, 60). To date, several phase 1/2a clinical trials enrolling NC patients for treatment with BET inhibitors have been completed, including those investigating birabresib, molibresib (Glaxo-Smith-Kline, aka iBET-762) (61), RO6870810 (aka TEN-010, Roche, a JQ1 derivative) (62), ODM-207 (Orion Pharma) (63) and BMS-986158 (Bristol-Myers Squibb) (64).
Birabresib.
In the phase 1 study of birabresib, two schedules were explored, including once daily continuous dosing and 7-consecutive-day dosing every 21 days. Forty-six patients with advanced solid malignancies were enrolled (59). The recommended phase 2 dose was 80 mg once daily continuously. Dose-limiting toxicities included thrombocytopenia, hyperbilirubinemia, anorexia and nausea. Of the nine patients with NC, there were 3 partial responses (PR), 3 instances of stable disease (SD) as best response, 2 cases of progressive disease (PD), and one case that was non-evaluable (NE). The duration of response ranged from 1.4 to 8.4 months. Although the translocation partners to NUTM1 were not described, the previous report that demonstrated activity utilizing birabresib on expanded access included four patients with tumors harboring BRD4-NUT fusions (60).
Molibresib.
The molibresib phase 1 enrolled the largest number of NC patients (n = 19) of any trial. In this study, the toxicities were primarily thrombocytopenia (51%), gastrointestinal (diarrhea, emesis, nausea, appetite loss, and dysgeusia, 22%-42%), anemia (22%), and fatigue (20%) (61). Monocyte Chemoattractant Protein-1 (MCP-1) levels in peripheral blood mononuclear cells (PBMC) were found to be depressed in a dose-dependent manner, supporting the idea that MCP-1 can be used as a reliable biomarker of the pharmacodynamic effects of molibresib. While 100mg was considered the maximum tolerated dose, 80 mg was chosen as the recommended phase 2 dose. Results indicated an overall response rate (ORR) of 11% in NC, among which were four PRs (two confirmed), eight patients with SD as best response, two with PD and one with NE disease. While the activity of molibresib in NC patients was overall modest, it was far more so than in patients with non-NC solid tumors (n = 46). Of the four PRs, three patients' tumors harbored BRD3-NUT fusions (61). In the portion of the study evaluating disease-specific expansion cohorts, an additional 12 patients with NC were treated, with three partial responses (one confirmed) and 4 additional patients with stable disease, three of whom had <30% reduction in tumor burden. The NUT fusion proteins associated with these additional cases were not reported.
RO6870810.
Unlike molibresib and birabresib, which are oral agents, RO6870810 is administered subcutaneously, and the final dose chosen in the phase 1 trial was 0.65mg/kg (62). Forty-seven patients were enrolled, eight of whom had NC. In contrast to other BET inhibitors, the most common toxicities were fatigue (42%), decreased appetite (35%) and injection-site erythema (35%). Notably, chronic hepatitis was a toxicity seen in one non-NC patient. CD11b expression in PBMCs was shown to be a reliable biomarker of response to RO6870810. The activity of RO6870810 was similar in NC to other agents in the class: 2 PRs, 4 SD, 1 PD; the ORR therefore 25%, and the median progression-free survival (PFS) was 94 days. Interestingly, both PRs were in patients harboring tumors with non-BRD4-NUT (NSD3-NUT, BRD3-NUT) fusions. The patient with NC harboring NSD3-NUT fusion had thoracic disease and a durable response lasting 797 days. Among the non-NCs, there was one PR in a patient with acinic cell carcinoma of the parotid gland with high MYC expression, supporting the idea that BET bromodomain inhibitors are preferentially active in MYC-driven cancers.
ODM-207.
This phase 1 trial enrolled 35 patients with solid malignancies, four of whom had NC (63). The toxicities closely resembled those with molibresib; however, results indicated very little activity of the compound. Three of the NC patients had PD, and only one had SD.
BMS-986158.
While not tested in NC pre-clinically, this agent demonstrated low nM half maximal inhibitory concentration (IC50) in biochemical assays and reduced MYC expression across a wide range of cancer cell types in vitro and in PDX models. The phase 1/2a clinical trial in patients with advanced cancers, including NC, using a five days on, two days off schedule administering a dose of 4.5mg, demonstrated a similar toxicity profile to that of other BET bromodomain inhibitors (64). As with other agents in the class, BMS-986158 induces thrombocytopenia in rats through repression of GATA1(65), a target of BRD2, BRD3, and BRD4 (66). An important outcome of this study was the validation of HEXIM1 and CCR2 levels in PBMCs as biomarkers of the pharmacodynamic effects of BMS-986158, which could extend to other BET bromodomain inhibitors. As with molibresib and RO6870810, the initial report of this agent indicated the most robust clinical benefit in a NC patient with a non-BRD4-NUT (BRD3-NUT) fusion who experienced a durable PFS of 279 days. The overall results of this trial, which is now closed to enrollment of NC patients (n = 7), are forthcoming.
Role of the fusion partner.
Data from the trials of mirabresib, RO6870810 and BMS-986158 raise the possibility that BET inhibitors may be more active in NCs harboring non-BRD4-NUT fusions (Table 1); however, our in vitro data using fluorescent-tagged NUT-fusions suggests activity is not related to differential eviction of various fusion proteins (C.A. French, unpublished). Moreover, differential clinical activity in NCs with non-BRD4-NUT fusions is not supported by birabresib data, where responses have been documented in tumors driven by BRD4-NUT. It is therefore possible that fusion-selective activity may be dependent on the specific compound being investigated, and that not all compounds may effectively dislodge BRD4-NUT from chromatin. Clinical benefit must also be interpreted in light of the site or origin, since tumors originating in the head and neck region overall carry better prognosis.
Table 1:
NC patients with PR or SD as best response on reported bromodomain inhibitor trials.
| Agent | Fusion Partner | Site of origin | Best Overall Response | Duration of Response (mo) | reference |
|---|---|---|---|---|---|
| Birabresib | BRD4-NUT | thoracic | PR | 8 | (60) |
| BRD4-NUT | head & neck | PR | 1 | ||
| BRD4-NUT | thoracic | SD | 1 | ||
| unknown | unknown | PR | 8.4 | (59) | |
| unknown | unknown | PR | 1.4 | ||
| unknown | unknown | PR | 2 | ||
| unknown | unknown | SD | 3 | ||
| unknown | unknown | SD | 1 | ||
| unknown | unknown | SD | unknown | ||
| Molibresib | unknown | thoracic | PR* | 2.7 | (61) |
| BRD3-NUT | thoracic | PR* | 2.3 | ||
| BRD3-NUT | head & neck | PR | 6.5 | ||
| BRD3-NUT | soft tissue | PR | 6.2 | ||
| NSD3-NUT | thoracic | SD | 1.6 | ||
| BRD3-NUT | thoracic | SD | 3.8 | ||
| BRD4-NUT | head & neck | SD | 6.6 | ||
| BRD4-NUT | head & neck | SD | 2.5 | ||
| BRD4-NUT | head & neck | SD | 8.5 | ||
| BRD4-NUT | head & neck | SD | 1.4 | ||
| unknown | unknown | PR x 3** | unknown | (67) | |
| unknown | unknown | SD x 4 | unknown | ||
| RO6870810 | BRD3-NUT | thoracic | PR | 3.8 | (62) |
| NSD3-NUT | thoracic | PR | 26 | ||
| BRD4-NUT | thoracic | SD | 2.9 | ||
| BRD4-NUT | thoracic | SD | 2.1 | ||
| BRD4-NUT | unknown | SD | 1 | ||
| BRD3-NUT | unknown | SD | 2.1 | ||
| BRD4-NUT | unknown | SD | 10.3 | ||
| BMS-986158 | BRD3-NUT | unknown | SD | 9-10*** | (64) |
unconfirmed PR.
Including 2 unconfirmed PRs
Time on treatment
Assessment of tumor response.
In several trial patients with NC, the clinical response to BET inhibitor bromodomain inhibition did not match the metabolic response, as measured by [18F]-fluorodeoxy-glucose (FDG) PET imaging. NC is a MYC-driven cancer, as evidenced by high MYC expression in these tumors, and by the rescue of BRD4-NUT knockdown in part by MYC overexpression in NC cells (41). MYC is widely known to shift tumor metabolism towards glycolysis; thus, it is possible that BET inhibitor treatment can lead to a rapid decrease in glucose uptake that is more reflective of MYC downregulation than true reduction in tumor viability.
Future directions
Novel BET bromodomain inhibitors.
While there are currently no active clinical trials using pan-BET inhibitors, several next generation BET inhibitors are under development that have selective activity for either the BD1 or the BD2 bromodomains.
ABBV-744 and ABBV-075.
ABBV-075 (mivebresib) is a pan-BET bromodomain inhibitor, targeting both bromodomains (BD1 and BD2) of BET proteins. A first-in-human phase 1 trial of mivebresib in non-NC solid malignancies revealed a similar toxicity profile as molibresib; SD was seen in 41% (n=26) of patients with solid tumors (68). Seeking to advance a more tolerable compound, ABBVIE has developed a BD2-selective BETi, ABBV-744, which has 325x higher affinity for BD2 (1.6nM) over BD1 (520 nM) of BRD4. Interestingly, ABBV-744 exhibits much more selectivity based on cancer type than ABBV-075 (69), with selective inhibition of acute myeloid leukemia (AML) and prostate adenocarcinoma cell lines. Consistent with its cell-type selectivity, ABBV-744 has 12.5x less impact on thrombopoiesis (as measured by megakaryocyte colony forming units) than ABBV-075. The toxicity-to-efficacy ratio of ABBV-744 compared with that of ABBV-075 in rats was 20-fold versus 0.5-fold, respectively. These data predict that ABBV-744 will be less toxic at doses that are equally potent. Hematologically, a sharp decline in platelets and reticulocytes occurred in rats treated with ABBV-075, whereas only a slight decline in platelets that subsequently stabilized was observed in animals treated with ABBV-744; additionally, no decline in reticulocytes occurred upon exposure to ABBV-744.
The current clinical plan is to study ABBV-075 in the treatment of myelofibrosis. For ABBV-744, where pre-clinical data indicates an improved therapeutic index, a first-in-human trial investigating its use in AML is ongoing, followed by a study investigating activity in the treatment of myelofibrosis. Preclinical evaluation of BD2-selective BRD4 inhibition with ABBV-774 in NC is ongoing.
BI 894999.
BI 894999 is unique in that it has higher affinity for BD1 (5nM) over BD2 (41nM). It has shown activity in liquid and solid cancers in vitro and has demonstrated low nM IC50 (1-2nM) in four NC models (three BRD4-NUT and one BRD3-NUT) in vitro. Investigators at Boehringer Ingelheim (BI) have used immunofluorescence (IF) visualization of BRD3/4-NUT to score eviction of BRD-NUT from chromatin in NC cell lines. This is seen as dispersion of BRD-NUT from nuclear foci to a fine granular, even distribution within the nucleus. BRD-NUT foci observed by fluorescence microscopy represent megadomains. Using this assay, BI 894999 evicts BRD4-NUT from chromatin at 30 nM, compared with 1µM for molibresib.
For human dosing of BI 894999, a schedule of 1 week on, 1week off, for a 28-day cycle, was established as the optimal schedule based on pharmacokinetic exposures (PK). Increased levels of HEXIM1, a repressor of p-TEFb (11, 70, 71), was determined to be a robust biomarker of response to BI 894999.
Additional Targeted Approaches and BET Inhibitor Combinations
p300 BDi + BETi.
The single bromodomain of p300/CBP mediates acetylated protein-p300 interactions, including those with histones and non-histone proteins (72–74). The French lab demonstrated that combining GNE-781 (75), a selective inhibitor of the p300/CBP bromodomain, and the BET bromodomain inhibitor OTX015 synergistically inhibited NC growth (76). Treatment of several NC cell lines with the novel dual p300/CBP and BET bromodomain inhibitor, NEO2734 (76), potently inhibited growth and induced differentiation of NC cells in vitro (76). Transcriptomic analyses indicated that combined inhibition of the p300/CBP bromodomain with BET bromodomain inhibition potentiates the effects of BET bromodomain inhibition alone (76), rather than inducing a distinct set of transcriptional changes. In two of three disseminated NC xenograft models, clinically relevant and well-tolerated doses of NEO2734 provided greater growth inhibition, tumor regression, and a significant survival benefit compared with a lead clinical BET inhibitor or with etoposide and cisplatin chemotherapy (76). The working model is that p300/CBP bromodomain inhibition enhances the effects of BET bromodomain inhibition by disrupting BRD4-NUT protein complexes, and by reducing p300 and BRD4-NUT chromatin association, as well as H3K27Ac levels (a p300/CBP-specific mark) levels. Ongoing work is planned to continue to dissect the mechanism of synergism.
Similar experiments have been conducted by Boehringer Ingelheim investigators to determine whether the selective p300/CBD bromodomain inhibitors CCS1477 (77) or GNE-781 (75) are synergistic with BI 894999. In vitro synergism by Bliss scoring was observed across a wide dose range in Ty-82, a NC cell line harboring a BRD4-NUT fusion. In contrast to previous reports(78), the combination of a p300/CBP histone acetyltransferase inhibitor, A485, with BI 894999, was not synergistic, suggesting that the bromodomain of p300/CBD, as opposed to its catalytic activity, may play a more pivotal role in the BRD4-NUT oncogenic mechanism. In xenograft studies utilizing NC cell lines Ty-82 (BRD4-NUT) and 10326 (BRD3-NUT), the combination of BI 894999 and CCS1477 demonstrated synergistic growth inhibition with regression, which was not achieved with either compound alone.
CDK9 inhibition.
In a high throughput kinase screen, CDK9 was identified as a synthetic lethal target in NC cell lines (79). Knockdown of CDK9 or CCNT1, its binding partner in the p-TEFb complex, or treatment with the tool CDK9 inhibitor, LDC67, inhibited growth and induced apoptosis of NC cells, but not of the non-NC lung adenocarcinoma cell line, A549. These effects correlated with dephosphorylation of RNA polymerase II (RNA PolII), the substrate of CDK9. Moreover, treatment with LDC67 led to decreased PolII occupancy at multiple gene bodies, including MYC, and inhibited transcriptional elongation. Given the essential role of CDK9 in cellular homeostasis, and the adverse effects resulting from systemic CDK9 inhibition, CDK9 remains a difficult drug target (80). The targeting of MYC upregulation in NC by LDC67 suggests that this cancer may be responsive to small molecule inhibitors of CDK9, either alone or in combination with other agents. Notably, combined BET bromodomain and CDK9 inhibition has been shown to be synergistic in a variety of tumor models (58, 81–84).
CDK4/6i + BETi.
A rescue screen was performed testing the ability of a panel of guide RNAs (gRNA) targeting 500 tumor suppressor targets, and the overexpression of 400 oncogenic genes (open reading frames, ORF), to confer growth advantage of a BRD4-NUT cell line, 10-15 (41), in the presence of the BETi, JQ1 (42, 85). The hits that conferred rescue fell into several categories, including the following: MYC pathway, the receptor tyrosine kinase (RTK) signaling, cell cycle (cyclin D1/RB), TGF-beta signaling, Protein kinase A (PKA) signaling, and KLF4. Because it is a targetable resistance mechanism, follow-on studies focused on inhibiting CDK4/6, the kinases which, in complex with cyclin D1, inactivate Rb through phosphorylation. It was found that palbociclib, a licensed CDK4/6 inhibitor (Pfizer), synergistically inhibited NC cell growth when combined with JQ1, in vitro. In mouse xenografts using the 10-15 model, only the combination of JQ1 and palbociclib, and not either agent alone, resulted in tumor regression. Moreover, the combination, administered for twenty-one days, resulted in a significant survival benefit compared with the monotherapies. Pharmacodynamic (PD) analyses found that cycle arrest, as measured by Ki-67, E2F1, and cyclin A, occurred only in the combination group, and was the case for necroptosis, detected by Westing blotting for phosphorylated RIP1. Unfortunately, it was also observed that when combination treatment stopped, there was rapid tumor re-growth, so that all mice were dead by day 35. This finding indicated that the combination of CDK4/6i and BETi leads to reversible NC cell quiescence, and that necroptosis may occur in only a subset of treated cells. Overall, however, the findings identify a potent and feasible agent that can be combined with BETi compounds in the treatment of NC. An NCI-CTEP-sponsored clinical trial is planned combining the BET bromodomain inhibitor ZEN3694 and the CDK4/6 inhibitor abemaciclib (CTEP 10509).
DNA-damaging agents.
Studies investigating non-NC cancer models indicate that BET bromodomain inhibition sensitizes cells to DNA damage or inhibitors of DNA repair through a variety of mechanisms. For example, these agents may compromise homologous recombination (HR) repair by reducing transcription of BRCA1, RAD51 or CtIP, the latter critical for DNA double-strand break end resection, sensitizing HR-proficient breast and ovarian cancer models to PARP inhibition and reversing PARP inhibitor resistance (86, 87). In ovarian cancer models it has also been shown that BET bromodomain inhibition suppresses TOPBP1 and WEE1 expression (88). TOPBP1 is required for HR, so that there is sensitization to PARP inhibition along with defective G2/M checkpoint control that promotes mitotic catastrophe (88). These mechanistic considerations have prompted a study investigating the combination of the BET bromodomain inhibitor ZEN3694 with talazoparib in triple-negative breast cancer (NCT03901469).
In addition, BRD4 has been implicated in non-homologous end joining-mediated repair of double-strand DNA breaks in prostate cancer, so that BET bromodomain inhibition sensitizes cells to ionizing radiation (89). Similarly, treatment with BET bromodomain inhibition resulted in a prolonged delay in DNA repair in response to irradiation in non-small cell lung cancer models, associated with upregulation of p21Waf1/Cip1 (90). Effects on both HR and NHEJ likely account for preclinical synergism of BET bromodomain inhibition with DNA damaging chemotherapy such as cisplatin or etoposide, and provide the rationale for the upcoming NCI-CTEP-sponsored trial of ZEN3694 combined with cisplatin/etoposide in NC patients (CTEP 10507).
In addition to these mechanistic considerations, two studies have recently demonstrated that BRD4 inhibition can directly induce DNA damage in cancer cells (91, 92). BRD4 activates effective transcriptional elongation via its interaction with pTEFb/CDK9; inhibition of BRD4 results in pausing of RNA Pol II, leading to an accumulation of RNA-DNA hybrids (R-loops) at sites of BRD4 occupancy, causing transcription-replication conflicts (i.e., collision events) and DNA damage. Reduced expression of TOPBP1 also prevents activation of the ATR-CHK1 pathway despite the consequent replication stress, leading to apoptotic cell death during S phase and mitotic catastrophe. Taken together, these findings further support combinations of BET bromodomain inhibitors with replication stress-inducing DNA damaging agents.
Novel immuno-oncology approaches to consider in NUT carcinoma.
Leveraging tumor immunogenicity and the immune micro-environment.
Cancer cells of types that harbor high tumor mutational burden (TMB) compared with normal somatic cells present mutated peptides that act as neoantigens to stimulate the immune system, specifically CD4+ and CD8+ T-cells. Dr. Catherine Wu and colleagues have taken advantage of this property and vaccinated patients with high-risk melanoma after surgical resection with neoantigens known to be expressed in those tumors. Patients had durable immunological responses to the personal neoantigen vaccinations with lengthy disease-free intervals. Those who progressed had developed resistance to immune infiltration through expression of PD-L1; these patients had complete responses when anti-PD1 antibodies (nivolumab or pembrolizumab) were administered (93). This study demonstrated the feasibility of this approach and provided rationale for the use of neoantigen vaccines in conjunction with immune checkpoint blockade. NUT carcinoma may harbor antigenic tumor-specific neoantigens created by NUT-fusion proteins. This possibility could be investigated by characterizing the immunopeptidome of NC cells to identify MHC-associated peptides. However, it will also be important to fully characterize the immune microenvironment and immune evasion mechanisms of NC prior to consideration of vaccines for this cancer.
Synthetic lethal and resistance interactions with BET bromodomain inhibitors with a focus on the immune response.
Unbiased approaches to explore determinants of sensitivity and resistance to BET bromodomain inhibitors have been utilized by the laboratory of Dr. Kornelia Polyak, using models of ER-, PR-, Her2/neu-negative (triple-negative) breast cancer (TNBC). Unlike NC, TNBC is not driven by recurrent oncogene mutations, and has a heterogenous epigenomic, transcriptomic, genomic, proteomic, and immune landscape (94). The immune response is important in TNBC outcomes. For example, the presence of tumor infiltrating lymphocytes (TILs), or immunologically “hot” tumors, predict response to treatment. As with many other cancers, PD-L1 can be expressed in TNBC as an immune evasion mechanism.
Investigations have shown that BRD4 downregulation can slow the growth of TNBC. (95) The Polyak lab performed a CRISPR screen to identify genes whose knock-out is synthetically lethal when combined with BETi (96). Interestingly, the top hits overlap with those identified in the Liao screen in NC (85) (CDK4, MYC, CCND1), and proteins known to interact with BRD4-NUT in NC (16) (BRD2, EP300, and Mediator proteins). One of the top resistance hits, RB1, was also identified in the Liao screen, whereas others, CDKN1A (97), SPOP (98), ARID1A (99), BRD7 (96, 100), and proteasomal subunits (101), have been described in other cancers. Validating some of the CRISPR screen hits, a small molecule screen identified palbociclib (CDK4/6) and paclitaxel as top synergy hits in TNBC in vitro and in vivo (96).
The above findings led the Polyak lab to test the in vivo efficacy of combining the synergistic duo of BETi and paclitaxel, with anti-PD1, considering that combined taxane and immune checkpoint blockade is now a mainstay of PD-L1+ metastatic TNBC. This triplet combination significantly reduced tumor volume and metastases, and increased survival of mice-bearing TNBC (Aleckovic and Polyak, unpublished). Interestingly, the triplet therapy induced apoptosis of tumor cells while also increasing infiltration of B and T cells, and MHCII-high macrophages. Based on these promising results, a clinical trial investigating the safety and efficacy of the BETi compound, ZEN-3694, the PD-1 antibody, pembrolizumab and paclitaxel is being initiated in the treatment of TNBC. These findings and the planned clinical translation may ultimately have relevance in NC, where the combination of a taxane with BETi may synergistically inhibit NC cell proliferation while also engaging the immune response.
Consensus statements for diagnosis and standard treatment modalities.
NUT immunohistochemistry is adequate for diagnosis and should be considered in any patient with a poorly differentiated carcinoma.
Given the frequency with which NC involves the thorax, next-generation sequencing (NGS) should be performed on all thoracic tumors with methodology that can detect NUTM1 rearrangement.
All patients should be encouraged to enroll in the NUT Carcinoma Registry (www.NMCRegistry.org).
For patients with localized disease, aggressive surgical and/or radiation therapy approaches are appropriate.
An ifosfamide-based regimen is suitable for upfront chemotherapy in the advanced or recurrent setting, but only in patients with good performance status.
Multiple individual factors may influence initial choice of chemotherapy, so that platinum- and taxane-based regimens may also be appropriate.
It is reasonable to consider addition of immune checkpoint blockade to chemotherapy, particularly in disease with documented high tumor mutation burden or with PD-L1-positivity.
The status quo is largely unsatisfactory and better drugs are needed to effectively treat NC.
Consensus statements on BET inhibitor monotherapy in NC.
BET inhibitor monotherapy has produced modest benefit in a minority of patients.
In trials of molibresib, RO6870810 and BMS-986158 where translocation data are available, most NC patients who have experienced PRs have had tumors harboring non-BRD4-NUT translocations. This observation requires further investigation to determine whether BET bromodomain inhibitor activity is biochemically related to the NUT-fusion.
Responses to birabresib have been reported in patients with NC harboring BRD4-NUT fusions, suggesting compound-specific differences in the inhibition of various NUT-fusion proteins.
Prolonged clinical benefit may also be related to less aggressive disease, such as that arising in the head and neck region, as has previously been described (2) and must be considered when clinical trial results are interpreted.
FDG-PET scanning may not be a reliable measure of tumor response to BET inhibitor-based treatment.
Consensus statements on novel approaches for NC treatment.
Eligibility in trials of novel agents or novel combinations for NC patients should be lowered to 12 years.
Combined chemotherapy and BET bromodomain inhibitor clinical trials should have high priority based on the known effects of bromodomain inhibition on the expression of DNA repair proteins and proteins involved in the DNA damage response. CTEP 10507 is an NCI-CTEP-sponsored study that will soon launch combining etoposide and cisplatin with the BET bromodomain inhibitor ZEN3694.
Preclinical data support the evaluation of CDK9 inhibition in NC, with several selective drugs currently in early phase clinical trials (KB-0742, NCT04718675; TP1287, NCT03604783(1))
Additional combinatorial approaches supported by preclinical data include combined BET bromodomain and CDK4/6 inhibition, to be investigated in an NCI-CTEP-sponsored study of ZEN3694 and abemaciclib (CTEP 10509), as well as combined BET bromodomain inhibition and p300 inhibition, to be addressed in a planned Phase 1 study of NEO2734.
The immune microenvironment of NC should be comprehensively characterized to describe the immune populations that are present and to understand whether they result in an immunosuppressive phenotype. An immunocompetent mouse model of the disease may help model appropriate targeted and immunological combinatorial approaches, including those involving immune checkpoint blockade.
Investigation of whether NUT fusion proteins result in immunogenic neoantigens against which personalized vaccines can be developed to prevent recurrence after local therapy is another area of research deserving prioritization.
Translational relevance.
The over-riding purpose of this first multi-national meeting of experts was to bring together diverse scientific and clinical perspectives to lay the groundwork towards improving conventional and investigational treatment of NUT carcinoma. As an epigenetic disease driven by the only known oncogene encoding BRD4, much is to be gained for NUT carcinoma and more common cancers driven by the BRD4-p300/CBP axis through rationale targeting of this mechanism. The presentations included herein summarize scientific, clinical, and pre-clinical progress towards this goal with a robust discussion of novel strategies using state-of-the-art selective small molecule inhibitors, alone and in combination. An important outcome of this conference was the establishment of consensus statements on diagnosis and standard treatment modalities, BET inhibitor monotherapy, targeted approaches and BET inhibitor combinations, and novel approaches for NUT carcinoma treatment.
Patient and Family Perspective.
Physician scientist attendees were grateful for the shared perspective of NC survivors and family members of patients stricken with NC, including David Hale and Catherine Crews, who were previously successfully treated for NC, and Colleen McGrath Richards, the wife and caregiver of her husband, Ryan, who died from NC shortly after their daughter was born. Ms. Crews maintains a NUT Carcinoma Support Page (https://www.facebook.com/nutmidlinecarcinoma/) that has served as a critical resource for patients and families dealing with this rare, alienating disease. There are over 200 supporting members of this highly international Facebook page. Unfortunately, most NC members have died; however, there are approximately thirty live members currently, who describe the page as a “life-line” because it addresses the critical need for connection and the ability to fight isolation, a feeling uniquely experienced by NC patients and their families because of the rarity of the disease. A growing number of people associated with the NC Facebook page plan to form a coalition to increase the awareness of NC; this could occur through the formation of foundations, but also through industry collaboration (102).
Acknowledgments
We thank all of the patients and families who attended and contributed to the Symposium, as well as the additional 61 clinicians and scientists who also participated. We are grateful for the assistance of Erin Boudreau and Devan A. Boucher of the Dana-Farber Cancer Institute, who administratively organized the Symposium. We thank the McDevitt Strong Foundation, Friends of Jay Dion Memorial Classic, and the Ryan Richards Foundation for their support that made this meeting possible. We are grateful to the National Institutes of Health for their support of the International NUT Midline Carcinoma Registry (grant no. CA124633 to C.A. French).
Authors’ disclosures of potential conflicts of interest.
K. Bennett is an employee of and owns stock in AbbVie.
N. G. Chau has received institutional research funding from BMS, GSK, Merck, and Roche, and reports consultancy for Bayer, BMS, Eisai, Lilly, Merck, and Roche.
M. Cheng is a paid consultant/advisory board member for AstraZeneca, Mirati, Inivata, and Boehringer Ingelheim, and has received honoraria from The Lynx Group (supported by Bristol-Myers Squibb), PCME (supported by Lilly and Merck), WebMD (supported by AstraZeneca), and funding for travel from Daiichi Sankyo, AstraZeneca, Genzyme. He reports research funding from Palleon Pharmaceuticals.
S. Coker is an employee of and owns stock in Bristol Myers Squibb (BMS).
S. DuBois has received funding for travel from Loxo Oncology, Roche, and Salarius, and reports previous funded consultancy for Loxo Oncology. He has received compensation for advisory boards for Amgen and Bayer and has received study drug from BMS.
C. A. French reports grants and personal fees from Boehringer Ingelheim and Glaxo-Smith-Kline outside the submitted work, and research funding from Epigenetix.
C. L. Hann reports consultant/advisory fees from Amgen, AstraZeneca, AbbVie, BMS, Janssen and Genentech. She has received research funding (to insutition) from AbbVie, AstraZeneca, BMS and Genentech.
G. J. Hanna reports research funding from ACCRF, Actuate Therapeutics, ASCO Conquer Cancer Foundation, Bicara, Bristol-Myers Squibb, Elevar, Exicure, Gateway for Cancer Research, Genentech, GlaxoSmithKline, Kartos Therapeutics, Kite Pharma, KSQ Therapeutics, Kura Oncology, NantKwest/Altor Bioscience, Regeneron, Repertoire, Sanofi Genzyme, and Secura Bio, V Foundation. He has served as paid consultant or advisory board member of Bicara, Bio-Rad, Boxer Capital, Bristol-Myers Squibb, Exicure, General Catalyst, Kura Oncology, Maverick Therapeutics, Merck, Naveris, Prelude, Rain Therapeutics, Regeneron, Remix, Sanofi Genzyme, and SIRPant.
U.M. Lauer has served on the advisory boards of Amgen, Boehringer Ingelheim, IPSEN Pharma, Novartis and Themis Biosciences and has received drugs from Boehringer Ingelheim.
S. Liao reports no relevant disclosures.
Christophe Massard reports consultant/advisory fees from Amgen, Astellas, Astra Zeneca, Bayer, BeiGene, BMS, Celgene, Debiopharm, Genentech, Ipsen, Janssen, Lilly, MedImmune, MSD, Novartis, Pfizer, Roche, Sanofi, Orion. He is principal/sub-Investigator of clinical trials for Abbvie, Aduro, Agios, Amgen, Argen-x, Astex, AstraZeneca, Aveo pharmaceuticals, Bayer, Beigene, Blueprint, BMS, Boeringer Ingelheim, Celgene, Chugai, Clovis, Daiichi Sankyo, Debiopharm, Eisai, Eos, Exelixis, Forma, Gamamabs, Genentech, Gortec, GSK, H3 biomedecine, Incyte, Innate Pharma, Janssen, Kura Oncology, Kyowa, Lilly, Loxo, Lysarc, Lytix Biopharma, Medimmune, Menarini, Merus, MSD, Nanobiotix, Nektar Therapeutics, Novartis, Octimet, Oncoethix, Oncopeptides AB, Orion, Pfizer, Pharmamar, Pierre Fabre, Roche, Sanofi, Servier, Sierra Oncology, Taiho, Takeda, Tesaro, Xencor.
S. A. Piha-Paul receives clinical trial research support/grant funding through the institution from the following sources: AbbVie, Inc.; ABM Therapeutics, Inc.; Acepodia, Inc; Alkermes; Aminex Therapeutics; Amphivena Therapeutics, Inc.; BioMarin Pharmaceutical, Inc; Boehringer Ingelheim; Bristol Myers Squib; Cerulean Pharma, Inc.; Chugai Pharmaceutical Co., Ltd; Curis, Inc.; Cyclacel Pharmaceuticals; Daiichi Sankyo; Eli Lilly; ENB Therapeutics; Five Prime Therapeutics; F-Star Beta Limited; F-Star Therapeutics; Gene Quantum; Genmab A/S; GlaxoSmithKline; Helix BioPharma Corp.; HiberCell, Inc.; Immunomedics, Inc.; Incyte Corp.; Jacobio Pharmaceuticals Co., Ltd.; Lytix Biopharma AS; Medimmune, LLC.; Medivation, Inc.; Merck Sharp and Dohme Corp.; Novartis Pharmaceuticals; Pieris Pharmaceuticals, Inc.; Pfizer; Principia Biopharma, Inc.; Puma Biotechnology, Inc.; Purinomia Biotech, Inc. Rapt Therapeutics, Inc.; Seattle Genetics; Silverback Therapeutics; Synlogic Therapeutics; Taiho Oncology; Tesaro, Inc.; TransThera Bio; NCI/NIH; P30CA016672 – Core Grant (CCSG Shared Resources). She has also worked as a consultant for CRC Oncology.
K. Polyak reports having served on the scientific advisory boards of Vividion Therapeutics, Acrivon Therapeutics, and Scorpion Therapeutics, and has a sponsored research agreement with Novartis. She is a paid consultant for TwoXAR, has received honoraria from AstraZeneca, and has equity in Scorpion Therapeutics.
Ravi Salgia reports being on speakers’ bureaus for Astra-Zeneca and Merck.
G.I. Shapiro has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin, Concarlo Holdings, Syros, Zentalis, CytomX Therapeutics, Blueprint Medicines, Kymera Therapeutics, Janssen and Xinthera. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell.
M. L. Sos is a co-founder and shareholder of PearlRiver Bio (now a Centessa Pharmaceuticals company), and received research funding from PearlRiver Bio (now a Centessa Pharmaceuticals company).
A. Stathis reports institutional research funding from, MEI Pharma, Merck, Bayer, Roche, Novartis, Pfizer, ADC Therapeutics, and Eli Lilly, and consulting fees from Bayer, Eli Lilly, Roche, and Novartis.
U. Tontsch-Grunt is an employee of Boehringer Ingelheim RCV GmbH & Co KG.
M. Trucco reports no relevant disclosures.
C.J. Wu is an equity holder of BioNTech and receives research funding from Pharmacyclics.
References
- 1.Beesley AH, Stirnweiss A, Ferrari E, Endersby R, Howlett M, Failes TW, et al. Comparative drug screening in NUT midline carcinoma. Br J Cancer. 2014;110(5):1189–98. Epub 2014/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chau NG, Ma C, Danga K, Al-Sayegh H, Nardi V, Barrette R, et al. An anatomical site and genetic-based prognostic model for patients with nuclear protein in testis (NUT) midline carcinoma: Analysis of 124 patients. JNCI Cancer Spectr. 2020;4(2):pkz094. Epub 2020/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003;63(2):304–7. [PubMed] [Google Scholar]
- 4.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, et al. BRD-NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27(15):2237–42. Epub 2007/10/16. [DOI] [PubMed] [Google Scholar]
- 5.French CA, Rahman S, Walsh EM, Kuhnle S, Grayson AR, Lemieux ME, et al. NSD3-NUT fusion oncoprotein in nut midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014;4(8):928–41. Epub 2014/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alekseyenko AA WE, Zee BM, Pakozdi T, Hsi P, Lemieux ME, Dal Cin P, Ince TA, Kharchenko PV, Kuroda MI, French CA. Ectopic protein interactions within BRD4 chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017;114(21):E4184–E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiota H, Elya JE, Alekseyenko A, Chou PM, Gorman SA, Barbash O, et al. ‘Z4’ complex member fusions in NUT carcinoma: implications for a novel oncogenic mechanism. Mol Cancer Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eagen KP, French CA. Supercharging BRD4 with NUT in carcinoma. Oncogene. 2021. Epub 2021/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein BRD4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A. 2003;100(15):8758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein BRD4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–34. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein BRD4. Mol Cell. 2005;19(4):535–45. [DOI] [PubMed] [Google Scholar]
- 12.Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol. 2014;21(12):1047–57. Epub 2014/11/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiota H, Barral S, Buchou T, Tan M, Coute Y, Charbonnier G, et al. NUT directs p300-dependent, genome-wide H4 hyperacetylation in male germ cells. Cell Rep. 2018;24(13):3477–87 e6. [DOI] [PubMed] [Google Scholar]
- 14.Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J, et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNA Pol II recruitment, and pause release. Mol Cell. 2021. Epub 2021/03/26. [DOI] [PubMed] [Google Scholar]
- 15.Alekseyenko AA, Walsh EM, Wang X, Grayson AR, Hsi PT, Kharchenko PV, et al. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015;29(14):1507–23. Epub 2015/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alekseyenko AA, Walsh EM, Zee BM, Pakozdi T, Hsi P, Lemieux ME, et al. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017;114(21):E4184–E92. Epub 2017/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiota H AA, Wang ZA, Filic I, Knox TM, Luong NM, Huang Y, Scott DA, Jones KL, Gokhale PC, Lemieux ME, Cole PA, Kuroda MI, French CA. Chemical screen identifies diverse and novel HDAC inhibitors as repressors of NUT function: implications for NUT carcinoma pathogenesis and treatment. Mol Cancer Res. 2021. Epub Jul 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, et al. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Mycdependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001;15(16):2042–7. Epub 2001/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18(19):5321–33. Epub 1999/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ. Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene. 1994;9(12):3635–45. Epub 1994/12/01. [PubMed] [Google Scholar]
- 21.Mateyak MK, Obaya AJ, Sedivy JM. C-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999;19(7):4672–83. Epub 1999/06/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2(3):305–16. Epub 1998/10/17. [DOI] [PubMed] [Google Scholar]
- 23.Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB, et al. Diagnosis of nut midline carcinoma using a NUT-specific monoclonal antibody. Am J Surg Pathol. 2009;33(7):984–91. Epub 2009/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickson BC, Sung YS, Rosenblum MK, Reuter VE, Harb M, Wunder JS, et al. NUTM1 gene fusions characterize a subset of undifferentiated soft tissue and visceral tumors. Am J Surg Pathol. 2018;42(5):636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mertens F, Wiebe T, Adlercreutz C, Mandahl N, French CA. Successful treatment of a child with t(15;19)-positive tumor. Pediatr Blood Cancer. 2007;49(7):1015–7. Epub 2006/01/26. [DOI] [PubMed] [Google Scholar]
- 26.Leeman R, Pinkney K, Bradley JA, Ruiz R, DuBois SG, French C, et al. NUT carcinoma without upfront surgical resection: A case report. J Pediatr Hematol Oncol. 2020. Epub 2020/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storck S, Kennedy AL, Marcus KJ, Teot L, Vaughn J, Gnekow AK, et al. Pediatric NUT-midline carcinoma: Therapeutic success employing a sarcoma based multimodal approach. Pediatr Hematol Oncol. 2017:1–7. Epub 2017/10/19. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz BE, Hofer MD, Lemieux ME, Bauer DE, Cameron MJ, West NH, et al. Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer Res. 2011;71(7):2686–96. Epub 2011/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maher OM, Christensen AM, Yedururi S, Bell D, Tarek N. Histone deacetylase inhibitor for NUT midline carcinoma. Pediatr Blood Cancer. 2015;62(4):715–7. [DOI] [PubMed] [Google Scholar]
- 30.Sholl LM, Nishino M, Pokharel S, Mino-Kenudson M, French CA, Jänne PA, et al. Primary pulmonary NUT midline carcinoma: Clinical, radiographic, and pathologic characterizations. J Thorac Oncol. 2015;10(6):951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauer DE, Mitchell CM, Strait KM, Lathan CS, Stelow EB, Luer SC, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18(20):5773–9. Epub 2012/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuda K, Kashima J, Yatabe Y. The isoform matters in NUT carcinoma: A diagnostic pitfall of p40 immunohistochemistry. J Thorac Oncol. 2020;15(10):e176–e8. Epub 2020/09/29. [DOI] [PubMed] [Google Scholar]
- 33.Chau NG, Hurwitz S, Mitchell CM, Aserlind A, Grunfeld N, Kaplan L, et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer. 2016;122(23):3632–40. Epub 2016/08/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JK, Louzada S, An Y, Kim SY, Kim S, Youk J, et al. Complex chromosomal rearrangements by single catastrophic pathogenesis in NUT midline carcinoma. Ann Oncol. 2017;28(4):890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stirnweiss A, Oommen J, Kotecha RS, Kees UR, Beesley AH. Molecular-genetic profiling and high-throughput in vitro drug screening in NUT midline carcinoma-an aggressive and fatal disease. Oncotarget. 2017;8(68):112313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie XH, Wang LQ, Qin YY, Lin XQ, Xie ZH, Liu M, et al. Clinical features, treatment, and survival outcome of primary pulmonary NUT midline carcinoma. Orphanet J Rare Dis. 2020;15(1):183. Epub 2020/07/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis A, Mahar A, Wong K, Barnet M, Kao S. Prolonged disease control on nivolumab for primary pulmonary NUT carcinoma. Clin Lung Cancer. 2020. Epub 2020/12/23. [DOI] [PubMed] [Google Scholar]
- 38.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–33. Epub 2011/10/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The BRD4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011;31(13):2641–52. Epub 2011/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. Epub 2013/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grayson AR, Walsh EM, Cameron MJ, Godec J, Ashworth T, Ambrose JM, et al. MYC, a downstream target of BRD4-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene. 2014;33(13):1736–42. Epub 2013/04/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. Epub 2010/09/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–74. Epub 2011/09/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150(4):673–84. Epub 2012/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109(47):19408–13. Epub 2012/11/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B-cell lymphoma. Cancer Cell. 2013;24(6):777–90. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, et al. Bet inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8(8):e72967. Epub 2013/09/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and bet inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190(7):3670–8. Epub 2013/02/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing BRD4 inhibition of TAT-transactivation. Nucleic Acids Res. 2013;41(1):277–87. Epub 2012/10/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19(22):6183–92. Epub 2013/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4(12):2419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510(7504):278–82. Epub 2014/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–71. Epub 2015/09/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2015. Epub 2015/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinemann A, Cullinane C, De Paoli-Iseppi R, Wilmott JS, Gunatilake D, Madore J, et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget. 2015;6(25):21507–21. Epub 2015/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker EK, Taylor S, Gupte A, Sharp PP, Walia M, Walsh NC, et al. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci Rep. 2015;5:10120. Epub 2015/05/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol. 2018:JCO2018782292. [DOI] [PubMed] [Google Scholar]
- 60.Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord JP, de La Motte Rouge T, et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX-015/MK-8628. Cancer Discov. 2016;6(5):492–500. Epub 2016/03/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectrum. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shapiro GI, LoRusso P, Dowlati A, K TD, Jacobson CA, Vaishampayan U, et al. A phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with nut carcinoma, other solid tumours, or diffuse large b-cell lymphoma. Br J Cancer. 2020. Epub 2020/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ameratunga M, Brana I, Bono P, Postel-Vinay S, Plummer R, Aspegren J, et al. First-in-human phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br J Cancer. 2020;123(12):1730–6. Epub 2020/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hilton J, Cristea MC, Voskoboynik M, Postel-Vinay S, Edenfield W, Gavai A, et al. Initial results from a phase I/IIa trial evaluating BMS-986158, an inhibitor of the bromodomain and extra-terminal (BET) proteins, in patients (pts) with advanced cancer. Annals of Oncology.29 Suppl 8(VIII134). [Google Scholar]
- 65.Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266–70. Epub 2000/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stonestrom AJ, Hsu SC, Jahn KS, Huang P, Keller CA, Giardine BM, et al. Functions of BET proteins in erythroid gene expression. Blood. 2015;125(18):2825–34. Epub 2015/02/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cousin S BJ, Braña Garcia I, de Bono JS, Le Tourneau C, Moreno V, Trigo J, Hann CL, Azad AA, Im SA, Cassier PA, French CA, Italiano A, Keedy VL, Plummer R, Sablin MP, Hemming ML, Ferron-Brady G, Wyce A, Khaled A, Datta A, Foley SW, McCabe MT, Wu Y, Horner T, Kremer BE, Dhar A, O’Dwyer P, Shapiro G, Piha-Paul S. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of NUT carcinoma and other cancers: Results of a phase I/II open-label, dose escalation study. International Journal of Cancer. 2022. Mar 15;150(6):993–1006. doi: 10.1002/ijc.33861. [DOI] [PubMed] [Google Scholar]
- 68.Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, et al. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin Cancer Res. 2019;25(21):6309–19. Epub 2019/08/20. [DOI] [PubMed] [Google Scholar]
- 69.Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578(7794):306–10. Epub 2020/01/24. [DOI] [PubMed] [Google Scholar]
- 70.Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem. 2012;287(2):1090–9. Epub 2011/11/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin X, Huang X, Uziel T, Hessler P, Albert DH, Roberts-Rapp LA, et al. HEXIM1 as a robust pharmacodynamic marker for monitoring target engagement of BET family bromodomain inhibitors in tumors and surrogate tissues. Mol Cancer Ther. 2016. [DOI] [PubMed] [Google Scholar]
- 72.Zucconi BE, Makofske JL, Meyers DJ, Hwang Y, Wu M, Kuroda MI, et al. Combination targeting of the bromodomain and acetyltransferase active site of p300/CBP. Biochemistry. 2019;58(16):2133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim JH, Cho EJ, Kim ST, Youn HD. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol. 2005;12(5):423–8. [DOI] [PubMed] [Google Scholar]
- 74.Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. 2008;283(45):30725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L, et al. GNE-781, a highly advanced potent and selective bromodomain inhibitor of cyclic adenosine monophosphate response element binding protein, binding protein (CBP). J Med Chem. 2017;60(22):9162–83. [DOI] [PubMed] [Google Scholar]
- 76.Morrison-Smith CD, Knox TM, Filic I, Soroko KM, Eschle BK, Wilkens MK, et al. Combined targeting of the BRD4-NUT-p300 axis in nut midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol Cancer Ther. 2020;19(7):1406–14. Epub 2020/05/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021;11(5):1118–37. Epub 2021/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Zegar T, Lucas A, Morrison-Smith C, Knox T, French CA, et al. Therapeutic targeting of p300/CBP hat domain for the treatment of NUT midline carcinoma. Oncogene. 2020;39(24):4770–9. Epub 2020/05/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bragelmann J, Dammert MA, Dietlein F, Heuckmann JM, Choidas A, Bohm S, et al. Systematic kinase inhibitor profiling identifies CDK9 as a synthetic lethal target in NUT midline carcinoma. Cell Rep. 2017;20(12):2833–45. Epub 2017/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang L, Ruiz P, Ito T, Sellers WR. Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell. 2021;39(4):466–79. Epub 2021/01/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Richter GHS, Hensel T, Schmidt O, Saratov V, von Heyking K, Becker-Dettling F, et al. Combined inhibition of epigenetic readers and transcription initiation targets the EWS-ETS transcriptional program in ewing sarcoma. Cancers (Basel). 2020;12(2). Epub 2020/02/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerlach D, Tontsch-Grunt U, Baum A, Popow J, Scharn D, Hofmann MH, et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene. 2018;37(20):2687–701. Epub 2018/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moreno N, Holsten T, Mertins J, Zhogbi A, Johann P, Kool M, et al. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget. 2017;8(49):84986–95. Epub 2017/11/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife. 2015;4:e06535. Epub 2015/06/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liao S, Maertens O, Cichowski K, Elledge SJ. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev. 2018;32(17–18):1188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med. 2017;9(400). Epub 2017/07/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401–16 e8. Epub 2018/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21(12):3398–405. Epub 2017/12/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X, Baek G, Ramanand SG, Sharp A, Gao Y, Yuan W, et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep. 2018;22(3):796–808. Epub 2018/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang J, Wang Y, Mei H, Yin Z, Geng Y, Zhang T, et al. The bet bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett. 2017;391:141–51. Epub 2017/02/02. [DOI] [PubMed] [Google Scholar]
- 91.He DD, Shang XY, Wang N, Wang GX, He KY, Wang L, et al. BRD4 inhibition induces synthetic lethality in ARID2-deficient hepatocellular carcinoma by increasing DNA damage. Oncogene. 2022. Epub 2022/01/13. [DOI] [PubMed] [Google Scholar]
- 92.Lam FC, Kong YW, Huang Q, Vu Han TL, Maffa AD, Kasper EM, et al. BRD4 prevents the accumulation of r-loops and protects against transcription-replication collision events and DNA damage. Nat Commun. 2020;11(1):4083. Epub 2020/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. Epub 2017/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019;9(2):176–98. Epub 2019/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–7. Epub 2016/01/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shu S, Wu HJ, Ge JY, Zeid R, Harris IS, Jovanovic B, et al. Synthetic lethal and resistance interactions with BET bromodomain inhibitors in triple-negative breast cancer. Mol Cell. 2020;78(6):1096–113 e8. Epub 2020/05/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shi C, Yang EJ, Liu Y, Mou PK, Ren G, Shim JS. Bromodomain and extra-terminal motif (bet) inhibition is synthetic lethal with loss of SMAD4 in colorectal cancer cells via restoring the loss of MYC repression. Oncogene. 2021;40(5):937–50. Epub 2020/12/10. [DOI] [PubMed] [Google Scholar]
- 98.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23(9):1063–71. Epub 2017/08/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berns K, Caumanns JJ, Hijmans EM, Gennissen AMC, Severson TM, Evers B, et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene. 2018;37(33):4611–25. Epub 2018/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clegg MA, Bamborough P, Chung CW, Craggs PD, Gordon L, Grandi P, et al. Application of atypical acetyl-lysine methyl mimetics in the development of selective inhibitors of the bromodomain-containing protein 7 (BRD7)/bromodomain-containing protein 9 (BRD9) bromodomains. J Med Chem. 2020;63(11):5816–40. Epub 2020/05/16. [DOI] [PubMed] [Google Scholar]
- 101.Chen J, Nelson C, Wong M, Tee AE, Liu PY, La T, et al. Targeted therapy of TERT-rearranged neuroblastoma with BET bromodomain inhibitor and proteasome inhibitor combination therapy. Clin Cancer Res. 2021;27(5):1438–51. Epub 2020/12/15. [DOI] [PubMed] [Google Scholar]
- 102.Pompilus F, Ciesluk A, Marquis P, Griebsch I, Voorhaar M. Understanding the patient experience in nut carcinoma: Qualitative interviews with patients and caregivers to develop a conceptual framework. Oncol Ther. 2021. Epub 2021/08/07. [DOI] [PMC free article] [PubMed] [Google Scholar]